
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton exchange membranes with perfluorobenzenesulfonic acid groups for vanadium redox flow battery applications
Xiaoting Xuea,
Peiru Lvb,
Jingshuai Yang *ab and
Patric Jannasch*a
*ab and
Patric Jannasch*a
aDepartment of Chemistry, Lund University, P.O. Box 124, SE-22100 Lund, Sweden. E-mail: patric.jannasch@chem.lu.se
bDepartment of Chemistry, College of Sciences, Northeastern University, Shenyang, 110819, China. E-mail: yjs@mail.neu.edu.cn
First published on 4th February 2026
Abstract
Proton exchange membranes (PEMs) are critical to the performance of vanadium redox flow batteries (VRFBs). Still, conventional perfluorosulfonic acid membranes such as Nafion® suffer from poor ion selectivity and high cost. In this study, we introduce PEMs prepared from four types of poly(arylene perfluorobenzenesulfonic acid)s, synthesized via polyhydroxyalkylations of biphenyl (BP) or p-terphenyl (TP) with pentafluorobenzaldehyde (BA) or perfluoroacetophenone (AP), named by their monomer contents (e.g., sBPBA). The combination of rigid and ether-free polymer backbones and densely distributed highly acidic sulfonic acid groups led to high proton conductivity and improved ion selectivity. In addition, the –CF3 substitution in the PEMs derived from perfluoroacetophenone likely increased the free volume and enhanced chemical stability. These membranes displayed a reduced area resistance and markedly lower vanadium ion permeability compared with Nafion®115. In VRFB single-cell tests, the membranes sBPBA, sTPBA, and sBPAP consistently delivered higher voltage and energy efficiencies than Nafion®115 across 40–160 mA cm−2, with the former PEM achieving the highest energy efficiency at all current densities. Long-term cycling demonstrated outstanding stability for sBPAP (∼99.5% CE, ∼82% EE over 450 cycles), moderate stability for sTPAP (∼98% CE over 250 cycles), and rapid performance degradation for sBPBA and sTPBA, prepared from pentafluorobenzaldehyde. This study demonstrates that CF3-containing poly(arylene perfluorophenyl) PEMs, with only a small fraction of the perfluoroalkyl (PFAS) content of Nafion®, are promising candidates for high-efficiency long-term VRFB operation. It also provides a clear molecular design framework for developing advanced membrane materials.
1. Introduction
Escalating challenges concerning fossil fuel depletion and environmental issues drive considerable worldwide efforts to develop and invest in renewable energy sources such as solar, wind, and hydroelectric power, which are pivotal for building a sustainable future.1–3 However, the inherent intermittency of renewable energy creates significant challenges in delivering a consistent power supply. This discrepancy between energy generation and consumption underscores the urgent need for efficient energy storage systems to reliably integrate renewable sources into modern power grids.4,5 Among various energy storage technologies, vanadium redox flow batteries (VRFBs) have attracted considerable attention due to their scalability, long cycle life, and rapid response, making them one of the most promising candidates for grid-scale applications.6–8In VRFBs, the membrane serves as a critical component that separates the two half-cells, facilitates fast proton transport between the electrodes, and mitigates vanadium ion crossover.9,10 Proton exchange membranes (PEMs) are typically functionalized with sulfonic acid groups to promote electrolyte uptake and ionic conduction while maintaining electrochemical separation.11 Perfluorosulfonic acid PEMs such as Nafion® are most widely employed due to their accessibility, high proton conductivity, mechanical robustness, and excellent chemical stability under the strongly acidic and oxidative conditions characteristic of VRFB operation.12,13 However, the widespread use of Nafion® is limited by a high cost and relatively poor selectivity toward vanadium ions.14,15 Excessive vanadium crossover through Nafion® membranes leads to electrolyte imbalance, reduced coulombic efficiency, and capacity fading, ultimately compromising cycling stability and limiting the long-term viability of VRFB systems.16
As shown in Scheme 1, hydrocarbon-based PEMs, such as sulfonated poly(aryl ether sulfone)s (Scheme 1a) and sulfonated polyimides, have attracted considerable attention as cost-effective and environmentally benign alternatives to perfluorinated membranes. These materials typically exhibit lower vanadium ion crossover compared to Nafion®, owing to their denser structures and tailored hydrophilic/hydrophobic phase separation.6,17 However, their oxidative stability remains a concern, especially under exposure to VO2+ species.18 Aromatic ether bonds have been identified as primary sites of oxidative degradation, where chain scission is triggered by electrophilic oxidants or reactive oxygen species, with the hydrophilic phase domain enhancing the accessibility of these species.19–21 This leads to the formation of structural defects followed by a subsequent decline in mechanical integrity and proton conductivity. In addition to sulfonated poly(aryl ether sulfone)s and sulfonated polyimides, polybenzimidazole (PBI) membranes (Scheme 1b) have emerged as another class of promising hydrocarbon-based PEMs due to their exceptional thermal and chemical stability. PBIs are electrically insulating in their pristine form,6 However, upon exposure to acidic aqueous electrolytes, they are protonated to form benzimidazolium cations along the PBI chain, and the membrane consequently absorbs the electrolyte solution.22,23 Sulfuric acid-doped PBI membranes thus possess positively charged, narrow hydrophilic domains (0.5–2 nm), which effectively suppress vanadium ion crossover through a size-exclusion mechanism.24,25 Despite these advantages, the absence of intrinsic ion-exchange groups in PBI limits the overall proton conductivity.26 To address this issue, sulfonic acid functionalities have recently been introduced into the PBI backbone. Sulfonated PBI membranes (Scheme 1c) have been found to achieve conductivity enhancements of up to an order of magnitude relative to non-sulfonated variants, without compromising selectivity or stability in VRFB operation.24
 | ||
| Scheme 1 Selection of previously reported sulfonated polymers. Key: (a) sulfonated poly(aryl ether sulfone); (b) polybenzimidazole; (c) sulfonated polybenzimidazole; (d–f) sulfonated polyphenylene; (g and h) sulfonated poly(arylene-alkane)s, and (i) synthetic pathway to poly(arylene perfluorophenylsulfonic acid)s reported in the present work via polyhydroxyalkylation, and subsequent thiolation and oxidation. | ||
Aromatic all-carbon-backbone polymer membranes have attracted growing interest as advanced PEM candidates.27 For example, Cornelius and co-workers first reported on polyphenylene-based PEMs,28 followed by structural refinements from the Holdcroft29 and Miyatake30 groups, who synthesized sulfonated variants via polymerization of sulfonated aromatic monomers. Despite promising properties, these polymers are synthetically challenging due to their poor solubility in polar solvents and the difficulty of achieving high molecular weights using conventional transition metal-catalysed polycondensation reactions (Scheme 1d–f). To overcome these limitations, our group and others have developed polyphenylene-based PEMs via a superacid-catalyzed Friedel–Crafts polycondensation reaction.31–34 These membranes, classified as poly(arylene-alkane)s, incorporate both phenyl–phenyl and phenyl-alkylene linkages. The inclusion of flexible alkylene units addresses the mechanical brittleness often associated with rigid all-aromatic polymers, without sacrificing oxidative or thermal stability. Bae and colleagues27 have further advanced this class of materials by synthesizing sulfonated poly(arylene-alkane)s via polyhydroxyalkylation. In their method, sulfonic acid groups were introduced in a one-pot SN2 reaction by converting bromoalkyl side chains to sulfonates, followed by oxidation. While this approach is both scalable and synthetically efficient, the resulting alkyl sulfonic acid groups possess relatively low acidity, limiting their proton conductivity (Scheme 1h). Nevertheless, all-carbon-backbone aromatic polymer membranes, devoid of ether linkages, may offer superior oxidative durability in the harsh electrochemical environments of redox flow batteries. As an example, we recently synthesized sulfonated poly(isatin-p-terphenyl) membranes (Scheme 1g) by introducing sulfonated side chains in a graft reaction with 1,3-propane sultone. These membranes exhibited a favorable balance of mechanical robustness, oxidative stability, and low vanadium permeability.32 However, as with other alkylsulfonated systems, the moderate acidity of the sulfoalkyl groups limited the proton conductivity.
We have previously developed sulfonated poly(arylene alkane) PEMs (PTPF-Phenyl-SA) via polyhydroxyalkylation of commercially available p-terphenyl and perfluoroacetophenone, followed by post-polymerization sulfonation through a thiolation–oxidation sequence that efficiently and selectively replaces the para-fluorine atoms of the pentafluorophenyl rings with sulfonic acid groups.33,34 Notably, the resulting membrane demonstrated proton conductivity and VRFB performance comparable to, or even surpassing, that of Nafion®, suggesting that the electron-withdrawing perfluorophenyl substituents enhance sulfonic acid dissociation and proton transport.31 In the present study, shown in Scheme 1i, we have extended this work by also synthesizing and studying a series of sulfonated poly(arylene perfluorophenyl)s via polyhydroxyalkylation of pentafluorobenzaldehyde and perfluoroacetophenone, with biphenyl and p-terphenyl, respectively, to systematically investigate the influence of polymer backbone structure on the performance and stability of PEMs for VRFBs. The four membranes were subsequently evaluated with regard to their electrochemical performance and long-term cycling stability in VRFBs. Finally, the structure–property–performance relationships were established, and highly efficient PEMs functionalized with perfluorobenzenesulfonic acid groups were developed for VRFBs.
2. Experimental section
2.1 Materials
Biphenyl (BP, 99%) was obtained from Acros Organics. p-Terphenyl (TP, 98%), pentafluorobenzaldehyde (PFBA, 98%), sodium hydrosulfide (NaSH, ≥90%), sodium chloride (NaCl), sodium hydroxide (NaOH) and N,N-dimethylacetamide (DMAc) were purchased from Sigma-Aldrich. Trifluoromethanesulfonic acid (TFSA, ≥99%) was supplied by Tokyo Chemical Industry, and perfluoroacetophenone (PFAP, 98%) was received from Apollo Scientific. Hydrogen peroxide (H2O2, 30%), sulfuric acid (H2SO4, 98%), hydrochloric acid (HCl, 37%), and glacial acetic acid (≥99.7%) were purchased from VWR. N-Methyl-2-pyrrolidone (NMP), dichloromethane (DCM, anhydrous), and methanol (analytical grade) were obtained from Sigma-Aldrich or VWR and used without further purification.2.2 Synthesis of sulfonated poly(arylene perfluorophenyl)s and membrane preparation
Four polymers, namely poly(p-terphenyl perfluoroacetophenone) (TPAP), poly(biphenyl perfluoroacetophenone) (BPAP), poly(p-terphenyl perfluorobenzaldehyde) (TPBA), and poly(biphenyl perfluorobenzaldehyde) (BPBA), were synthesized via a modified previously published method.35–37 The synthesis of BPBA is described below. In a 50 mL round-bottom flask equipped with a mechanical stirrer, BP (0.97 g, 6.28 mmol) was dissolved in DCM (30 mL) under stirring at room temperature. PFBA (1.74 g, 8.85 mmol) was then added, and the reaction mixture was cooled to 0 °C in an ice bath. TFSA (4.00 mL, 45.31 mmol) was added dropwise over several minutes under continuous stirring. The polymerization was maintained at 0 °C until a pronounced increase in viscosity was observed. At this point, the reaction mixture was slowly added to methanol under vigorous stirring to precipitate the polymer. The resulting white fibrous solid was collected by filtration, washed thoroughly with methanol, and dried under vacuum at room temperature for 24 h.Substitution of the para-fluorine atom in the pentafluorophenyl rings with sulfonic acid groups was carried out following our previously reported procedure.34 The polymer (1.00 g) was first dissolved in DMAc (30 mL), and then NaSH (0.75 g) was added. The reaction mixture was stirred at room temperature for 24 h before precipitating the polymer in 1 M aqueous HCl under vigorous stirring. The resulting yellow solid was collected by filtration, washed thoroughly with 1 M aqueous HCl, followed by deionized water, and finally dried under vacuum. The thiolated polymer was then suspended in glacial acetic acid (40 mL), and H2O2 (28 mL) and H2SO4 (4 mL) were added. The mixture was stirred at 50 °C for 24 h to complete the oxidation of the thiol groups to sulfonic acid. The crude product was purified by dialysis (MWCO = 3500 Da), first against deionized water for 24 h, then against 0.01 M NaCl solution for 24 h, and finally against deionized water for another 24 h. The dialyzed solution was concentrated by rotary evaporation, and the final product was dried in a vacuum oven at room temperature. The resulting sulfonated poly(arylene perfluorophenyl) polymers were named sTPBA, sBPBA, sTPAP, and sBPAP, respectively.
NMP solutions of the sulfonated polymers (5 wt%) were prepared and magnetically stirred until clear and homogeneous. The resulting solutions were filtered through 0.5 µm PTFE syringe filters into Petri dishes, before casting membranes by solvent evaporation at 80 °C for at least 2 days in a ventilated oven. The resulting transparent membranes were carefully peeled off and washed thoroughly with deionized water to remove any residual solvent. The membranes were subsequently kept in refluxing 1 M aqueous H2SO4 for 2 h, followed by boiling in deionized water for 2 h to remove remaining free acid.
2.3 Polymer characterization
Nuclear magnetic resonance (NMR) spectra were recorded using a Bruker DRX-400 spectrometer, and Fourier-transform infrared (FTIR) spectra were obtained using a Thermo Fisher Scientific FT-IR instrument (Thermo Fisher Scientific Inc., Waltham, USA). All FTIR spectra were collected at room temperature in the wavenumber range 4000–550 cm−1, using 16 scans at a resolution of 4 cm−1.The molecular weights of the precursor polymers were determined using a Malvern OMNISEC system equipped with a refractive index detector, a TGuard Org Guard column (10 × 4.6 mm) as the guard column, and two T6000M general mixed organic columns (300 × 8.0 mm) as the analytical columns. Tetrahydrofuran (THF) was used as eluent at 35 °C at a flow rate of 1 mL min−1. Calibration was performed using narrow dispersity polystyrene standards.
Thermal decomposition of the polymer samples was investigated by thermogravimetric analysis (TGA) using a TA Instruments Q500 analyzer in the range 50–600 °C at a heating rate of 10 °C min−1 under nitrogen atmosphere. The glass transition temperature (Tg) of the precursor polymers was measured by differential scanning calorimetry (DSC) using a TA Instruments Q2000 in the temperature range of 50 to 350 °C.
2.4 Membrane characterization
Atomic force microscopy (AFM) images were obtained by a Bruker Icon Atomic Force Microscope using AC240TS-R3 tips. The morphology and phase separation of dry PEMs in the proton form were analyzed by small-angle X-ray scattering (SAXS) using a SAXLAB ApS (JJ-Xray, Denmark).The ion exchange capacity (IEC) was determined by acid–base titration. Before weighing, the membranes (in the proton form) were dried under vacuum at 50 °C for 2 days. Then, the membranes were kept in a 1 M NaCl solution at 60 °C for ion exchange. After two days, the solutions were titrated with ∼0.01 M NaOH solutions (with precisely known concentrations) using phenolphthalein as an indicator. The IEC value was then calculated as:
 | (1) |
The in-plane proton conductivity of the membranes was recorded by electrochemical impedance spectroscopy (EIS) under fully immersed conditions at 20 °C using a sealed two-probe cell. Measurements were performed in the frequency range 101 to 107 Hz at a 50 mV amplitude, using a Novocontrol BDC40 high-resolution dielectric analyzer (version 1.01S) equipped with a Novocool cryostat.
The acid uptake, which reflects the total mass increase of the PEM due to the absorption of both H2SO4 and water, is a key parameter for evaluating physicochemical performance. The dimensions (L0, W0, D0) and mass (m0) of the dried membranes were first measured. At room temperature, membranes were soaked in a 3 M aqueous H2SO4 solution. After 48 h, the membranes were taken out, wiped with filter paper, and weighed again. The final dimensions (L1, W1, D1) and weight (m1) were recorded. Acid uptake (AU%) and area and volume swelling (AS% and VS%) were calculated as:
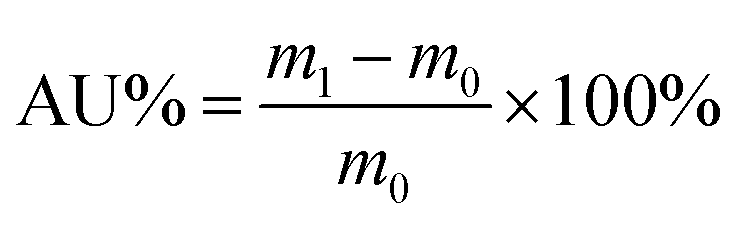 | (2) |
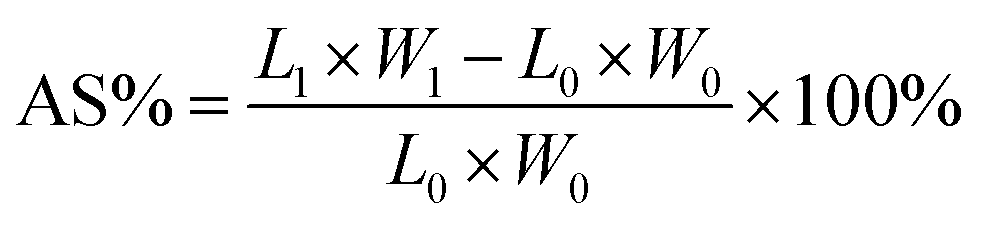 | (3) |
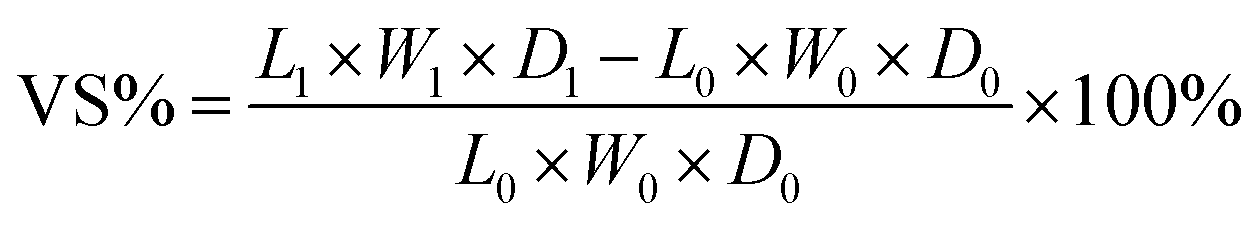 | (4) |
The area resistance (AR) was determined by EIS measurement using an electrochemical workstation (VersaSTAT 3, Princeton) spanning frequencies from 101 to 108 Hz. An H-type electrolytic cell filled with 3 M aqueous H2SO4 solution was used. AR was calculated according to eqn (5), where R0 is the resistance of sulfuric acid solution alone, and R1 is the resistance of the cell with the membrane:
| AR = (R1−R0) × A | (5) |
The vanadyl ion (VO2+) permeability was evaluated using a diffusion cell with an effective area of 1.69 cm2. Both chambers contained 80 mL of 3 M aqueous H2SO4, with equal concentrations (1.5 M) of VOSO4 and MgSO4 to balance the osmotic pressure. Magnetic stirring was applied to reduce concentration polarization. The concentration of VO2+ in the MgSO4 compartment was periodically measured using a PerkinElmer Lambda 750 s UV-visible spectrometer. The permeability (P) was calculated as:
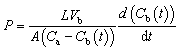 | (6) |
The ion selectivity of each membrane was calculated based on both AR and P. The ion selectivity was calculated as:
 | (7) |
The chemical stability was determined according to a previously reported method.38 The membrane was immersed in a 1.5 M VO2+/3 M H2SO4 aqueous solution for around 12 h. Next, the membrane was removed and rinsed with deionized water, followed by drying and re-weighing. For long-term testing, the procedure was repeated periodically, and weight retention was used to quantify the oxidative degradation.
The mechanical properties of the polymers were evaluated by measuring the stress–strain properties using a TA Instruments Q800 dynamic mechanical analyzer (DMA). Rectangular samples (approx. 0.4 cm × 2 cm, 40–60 µm in thickness) were cut out and mounted between the two clamps before applying a 0.01 N preload force. The measurements were then conducted with a ramping force of 0.3 N min−1 at 31 °C in a controlled force mode. Membranes were measured in the hydrated state by removing samples immersed in water before immediate mounting and measurement.
2.5 VRFB single battery performance and durability tests
VRFB evaluations were conducted following protocols described in our previous reports.11,39 Graphite felt electrodes with an effective area of 9 cm2 were used. Charge–discharge cycling was performed, employing a Neware CT-4008 battery testing setup (5 V/12 A) at current densities between 40 and 160 mA cm−2. For long-term durability evaluation, the cell was operated at 100 mA cm−2. To mitigate electrode corrosion and minimize hydrogen evolution, the voltage of the single VRFB cell was limited to a range of 1.00 to 1.65 V throughout the test. Coulombic efficiency (CE), voltage efficiency (VE), and energy efficiency (EE) were calculated according to eqn (7)–(9), respectively:
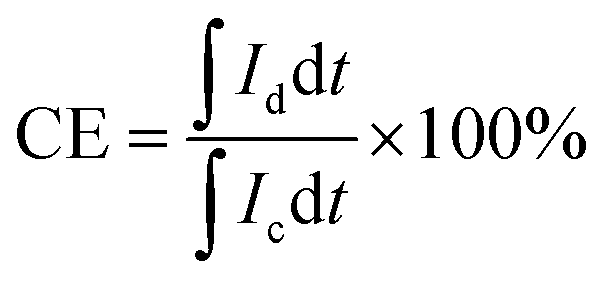 | (8) |
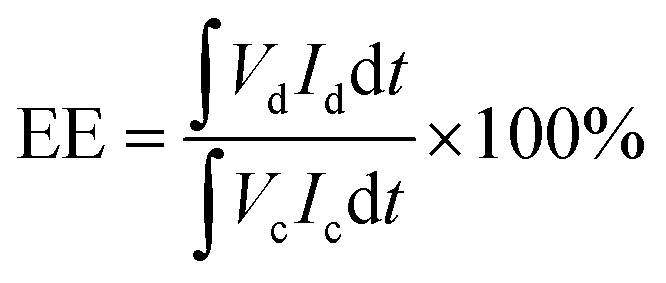 | (9) |
 | (10) |
3. Result and discussion
3.1 Polymer synthesis and membrane fabrication
The chemical structures of the polymers before and after the thiolation–oxidation reaction were confirmed by 1H and 19F NMR spectroscopy. As shown in Fig. 1a, the precursor polymers TPBA, BPBA, TPAP, and BPAP exhibited characteristic 1H NMR signals (recorded in CDCl3) between 7.0 and 8.0 ppm, corresponding to the aromatic protons on the benzene rings. Additionally, distinct signals at 6.00 and 5.97 ppm in the spectra of TPBA and BPBA were assigned to the methine protons on the tertiary carbon atoms along the backbone. As expected, the polarity of the polymers changed after sulfonation, and they became insoluble in chloroform. Therefore, the sulfonated sTPBA, sBPBA, sTPAP, and sBPAP polymers were dissolved in deuterated DMSO-d6 before characterization by 1H NMR spectroscopy. As shown in Fig. 1b, all the characteristic 1H signals were retained but moved to slightly higher chemical shifts. For example, the signals belonging to the methine linkage in sTPBA and sBPBA were observed at 6.08 and 6.04 ppm, respectively. 19F NMR spectra were also recorded to confirm the complete sulfonation of the TPBA and BPBA polymers. As shown in Fig. S1a, the precursor polymers displayed a sharp resonance near −150 ppm, which was assigned to the para-fluorine atoms on the pentafluorophenyl rings. Fig. S1b–d shows that this peak completely disappeared after the sulfonation, indicating complete substitution of the para-fluorine atoms by sulfonic acid groups. Moreover, the electron-withdrawing nature of the sulfonic acid groups led to deshielding of the fluorine atoms in the aromatic region, resulting in downfield shifts of the corresponding signals. Further confirmation of sulfonic acid grafting was obtained by FTIR spectroscopy. As shown in Fig. S2, the FTIR spectra of sBPAP displayed characteristic absorption bands at 1032 and 1065 cm−1, which were attributed to the symmetric and asymmetric stretching vibrations of the sulfonic acid (–SO3H) groups, respectively. These bands were absent from the spectrum of the unmodified BPAP. Consequently, both NMR and IR data confirm the successful synthesis of the four precursor polymers and their sulfonated derivatives. | ||
| Fig. 1 1H NMR spectra of the precursor (a) and sulfonated (b) polymers recorded using CDCl3 and DMSO-d6 solutions, respectively. | ||
SEC analysis showed that the four precursor polymers TPBA, BPBA, TPAP, and BPAP had number-average molecular weights (Mn) of 39, 86, 46, and 51 kg mol−1, respectively (Table S1). The stress–strain properties of dry precursor polymers were measured in controlled force mode with a ramping force of 0.3 N min−1 at 31 °C (Fig. S3). The precursor polymers exhibited tensile strengths ranging from 32 to 60 MPa, with strains of break between 18 and 60%. These results indicated that all the polymers exhibited sufficiently high molecular weights to fabricate membranes with adequate mechanical robustness. A clear relationship between the mechanical properties and the molecular weight was observed. BPBA had the highest molecular weight (Mn = 86 kg mol−1) and showed the greatest tensile strength and elongation, whereas the other three polymers, with rather similar molecular weights, displayed comparable mechanical properties. The sulfonated polymers were not soluble in THF. However, the chain length of the four polymers can be expected to remain the same after sulfonation, because it involves substitution and oxidation reactions that are unlikely to cause chain scission or crosslinking of the present polymer.
Since the sulfonated sTPBA, sBPBA, sTPAP, and sBPAP samples exhibited excellent solubility in common polar aprotic solvents such as dimethylformamide (DMF), dimethyl sulfoxide (DMSO), and NMP, flexible and transparent PEMs were readily obtained by casting from NMP solutions. As shown in Fig. 2, all the PEMs (sTPBA, sBPBA, sTPAP, and sBPAP) had a uniform transparent appearance with a light-yellow coloration.
 | ||
| Fig. 2 Photographic images of various pristine(down) and sulfonated membranes (up). | ||
3.2 Physicochemical characterization
The morphology of the PEMs in the dry state was investigated by SAXS. All sulfonated membranes exhibited ionomer peaks, indicating the presence of ionic aggregation and microphase separation (Fig. 3a). sBPBA and sBPAP showed scattering features centred near ∼0.31 Å−1 (d ≈ 20.27 Å), whereas sTPBA and sTPAP displayed peaks at ∼0.28 Å−1(d ≈ 22.24 Å). Hence, sBPBA and sBPAP exhibited a lower d-spacing than sTPBA and sTPAP, most probably due to the shorter length of the biphenyl unit, which provided a higher IEC and a shorter inter-acid distance along the backbone compared to the p-terphenyl-containing polymers.40,41 Notably, the ionomer peaks of sTPBA and sBPBA were more pronounced and sharp, possibly because the absence of the bulky –CF3 substituents gave a more flexible polymer backbone. This leads to stronger and more regular ionic aggregation (ionic clustering). In contrast, the chain stiffness introduced by –CF3 groups broadened the distribution of the ionic domain sizes, resulting in weaker, less defined, SAXS features. To complement the SAXS results, AFM was employed to probe the surface morphologies of dry membranes in the proton form. As shown in Fig. 3b, the AFM phase images revealed an efficient phase separation and well-defined phase domains for all samples, with bright and dark regions corresponding to hydrophobic and hydrophilic phases, respectively. Combined, the SAXS and AFM results consistently demonstrated the formation of distinctly phase-separated nanoscale structures, while also highlighting the influence of the backbone architecture on the phase structure. | ||
| Fig. 3 (a) SAXS profiles of dry sTPBA, sBPBA, sTPAP, and sBPAP PEMs in the H+, (b) AFM images, (c) TGA curves in N2 at a heating rate of 10 °C min−1, (d) mass retention in 1.5 M VO2+ and 3 M H2SO4 solution at room temperature of sTPBA, sBPBA, sTPAP, and sBPAP PEMs. | ||
The mechanical properties of the PEMs are important for the assembly and application of electrochemical devices. As shown in Fig. S4a, sBPAP membrane could undergo multiple folding and twisting cycles in the hydrated state. Similarly, the sTPAP membrane demonstrates excellent structural integrity under various deformations (Fig. S4b), including longitudinal, transverse, diagonal, and radial folding, as well as smooth out-of-plane bending and partial self-overlap. Notably, no cracking or permanent crease formation was observed in either sample. These observations demonstrate the mechanical robustness of sulfonated poly(arylene perfluorophenyl) membranes during handling and assembly. To further characterize their mechanical properties, we measured the stress–strain response of sBPAP and sTPAP under a ramp load of 0.3 N min−1 at 31 °C (Fig. S4c). The 0.5% secant modulus values were determined to be 260 and 453 MPa for sTPAP and sBPAP, respectively. These results confirmed that the membranes possessed sufficient mechanical strength and flexibility for practical use in electrochemical devices. A systematic investigation of long-term durability and vanadium permeability evolution under repeated mechanical deformation will be pursued in future work.
The TGA traces of all the PEMs are shown in Fig. 3c. For comparison, the TGA curves of the corresponding precursor polymers (TPBA, BPBA, TPAP, and BPAP) are shown in Fig. S5a. All precursor polymers possessed excellent thermal stability, with the onset of decomposition occurring at approximately Td,onset = 430 °C for TPAP and BPAP, and at around 500 °C for TPBA and BPBA. As expected, the PEMs had a lower thermal stability than the precursor polymers, owing to the presence of the thermally sensitive sulfonic acid groups. As shown in Fig. 3c, all PEMs in the proton form displayed a characteristic two-step decomposition process. The initial weight loss, starting just above 200 °C, corresponded to the degradation and loss of the sulfonic acid groups. The second, more significant, mass loss arose from the decomposition of the polymer backbones. For sTPAP and sBPAP, containing –CF3 groups, the degradation of the backbone began just above 400 °C, while for sTPBA and sBPBA, with methine protons, the decomposition occurred at temperatures exceeding 500 °C. Thus, sTPAP and sBPAP showed a degradation step between 400 and 450 °C, attributed to the cleavage of the –CF3 groups, followed by decomposition of the aromatic backbone above 500 °C. These results collectively highlight the influence of both the sulfonation and the backbone composition on the thermal stability of the poly(arylene perfluorophenylsulfonic acid) PEMs. Although VRFBs operate near room temperature, TGA characterization remains highly important for polymer electrolyte membranes. Differences in thermal stability clearly reflect variations in polymer backbone structures and indirectly confirm the successful synthesis of the different polymers. For example, polymers containing terphenyl units generally exhibit higher thermal stability than those incorporating biphenyl units. Moreover, TGA data provides a useful indication of intrinsic chemical stability. Higher decomposition temperatures generally reflect stronger and more oxidation-resistant backbones, which may correlate with slower degradation in the acidic and oxidizing vanadium electrolyte. DSC analysis revealed glass transitions at Tg = 298 and 307 °C for BPBA and BPAP, respectively (Fig. S5b). No glass transition was detected for TPBA and TPAP up to their respective thermal decomposition temperatures, confirming the exceptionally rigid backbone of this polymer based on p-terphenyl and pentafluorobenzaldehyde. As expected, no glass transition was detected for any of the poly(arylene perfluorophenylsulfonic acid) PEMs up to the decomposition temperature.
The chemical stability of polymer membranes indicates their capacity to resist the harsh chemical environments encountered in VRFBs.42 Polymers degrade due to attack by highly oxidative VO2+ ions in the electrolyte leads to poor stability of the membranes. In the present study, the PEMs were immersed in a 1.5 M VO2+ and 3.0 M H2SO4 aqueous solution at room temperature to assess the chemical stability of the membranes. Fig. 3d shows the mass retention (%) of the sTPBA, sBPBA, sTPAP, and sBPAP membranes during the chemical stability measurement, and also includes photographs of the membranes before and after the 80 h oxidative testing period. The small initial weight loss observed for all membranes within the first 12 h is attributed to an early-stage conditioning process in the strongly acidic and oxidative VO2+/H2SO4 environment, which may involve the release of trace extractable species as well as minor swelling and deswelling effects during the initial immersion and drying cycle.43,44 As seen after the 80 h period, sTPAP retained 93% of its initial mass and was the only membrane to maintain its initial shape without fragmentation. The sBPAP membrane was fragmented and retained 88% of the initial mass. In contrast, both sTPBA and sBPBA membranes disintegrated into small pieces and only retained 49% and 48%, respectively, of their initial mass. Fig. S6 shows 1H NMR spectra of all membranes after immersion in an aqueous 1.5 M VO2+/3 M H2SO4 solution for 80 h. It was clearly observed that the main chains of sBPBA and sTPBA underwent significant oxidative degradation, with new peaks appearing in the aromatic region. In contrast, the sBPAP and sTPAP membranes showed no substantial changes in the aromatic region. The reason was likely that the methine (>CH–) linkages in the backbone of these membranes are prone to oxidation, leading to backbone cleavage. In contrast, the sTPAP and sBPAP membranes contained no methine linkages and instead contained –CF3 groups and thus exhibited a markedly higher stability under the strongly acidic and highly oxidative conditions than sTPBA and sBPBA. The –CF3 groups may induce enhanced oxidative resistance by their electron-withdrawing character, thus stabilizing the membranes against radical attack.45,46
3.3 Acid uptake, area resistance, and vanadium ion permeability
The in-plane proton conductivity of sTPBA, sBPBA, sTPAP, sBPAP, and Nafion® 115 immersed in deionized water was measured by EIS at 20 °C. As shown in Fig. 4a, the sBPBA membrane exhibited a much higher proton conductivity (152 mS cm−1) than the sBPAP membrane (97 mS cm−1), most likely due to the higher IEC of the sBPBA membrane (2.54 vs. 2.15 mequiv. g−1). The same trend was also shown by the terphenyl-based sTPBA and sTPAP membranes. It should be noted that the high mass density of fluorinated moieties makes gravimetric comparisons conservative. A volumetric IEC analysis, though limited here by the difficulties of thin membrane density measurement, would likely reveal a closer parity in the true fixed charge density required for percolation in these distinct polymer architectures.47–50 However, the presence of aqueous sulfuric acid as the electrolyte solution in VRFBs implies that membrane performance depends little on the intrinsic conductivity in water, but much more on the amount of electrolyte solution absorbed. The sulfuric acid solution absorbed by the membrane contributes greatly to the ion transport and thus plays a significant role in the overall electrochemical performance of VRFBs. | ||
| Fig. 4 (a) Proton conductivity in water and the IEC values of the PEMs, (b) acid uptake and swelling, (c) AR and vanadium ion permeability, (d) ion selectivity of the PEMs, and (e) scheme of the proposed mechanism of proton transfer and vanadium ion suppression of the poly(arylene perfluorophenylsulfonic acid) membranes. | ||
Fig. 4b shows the AU, AS, and VS of the PEMs after immersion in 3 M aqueous H2SO4 solution at room temperature. Generally, both the AS and VS of the membranes increased with the acid uptake. For example, the sBPAP membrane had the highest AU value (60%), correspondingly exhibiting the highest VS (38%). When replacing the biphenyl units with terphenyl ones, the sTPAP membrane displayed a lower AU (44%), mainly due to the lower IEC and lower hydrophilicity of the sTPAP PEM. When the perfluoroacetophenone monomer was replaced by pentafluorobenzaldehyde in the polymerizations, the AU values of the corresponding sBPBA and sTPBA PEMs decreased to 38 and 13%, respectively. These results indicated that the –CF3 groups present in the backbones of sBPAP and sTPAP increased the membrane's AU, possibly because the bulky and hydrophobic –CF3 substituents perturb chain packing and create additional free volume around the sulfonic acid groups.51,52 Higher AU could result in a lower AR, which significantly impacts both voltage and energy efficiencies of VRFBs. Fig. 4c shows the AR and vanadium ion permeability of the present membranes with Nafion®115 included as a reference. The latter was characterized under identical experimental conditions as used for sTPAP, sBPAP, sTPBA, and sBPBA. All PEMs, except Nafion®115, had comparable thicknesses of approximately 80 µm to ensure a fair comparison. Compared to Nafion®115, which showed an AR of 0.60 Ω cm2, all the poly(arylene perfluorophenylsulfonic acid) membranes displayed lower ARs, reflecting a more efficient ion conduction. Among them, sBPAP exhibited the lowest AR at 0.18 Ω cm2, consistent with its high acid uptake, indicating excellent ion transport properties. The other three membranes had AR values close to 0.44 Ω cm2.
The low AR of sBPAP may come at the cost of increased vanadium ion permeability. As previously reported, high vanadium ion permeability can result in severe self-discharge and a decline in CE, which significantly limits the application in VRFBs.53,54 As shown in Fig. 4c, the sBPAP membrane exhibited a higher vanadium ion permeability value (8.48 × 10−7 cm2 min−1) than the sTPBA membrane (4.16 × 10−8 cm2 min−1), which also demonstrated the lowest permeability. The vanadium ion permeability values of sTPBA and sBPBA were lower than those of the sBPAP membrane, indicating a higher vanadium ion blocking capacity. Nevertheless, the sBPAP membrane still displayed approximately one order of magnitude lower vanadium ion permeability than the Nafion®115 membrane (6.94 × 10−6 cm2 min−1). The excellent vanadium ion-blocking performance of poly(arylene perfluorophenylsulfonic acid) membranes most probably arose from their rigid backbones, dense chain packing, and finely phase-separated morphologies, which combined restrict the diffusion pathways of vanadium ions.
Ion selectivity is a key parameter governing the performance of VRFBs, as it quantifies the ability of the membrane to facilitate proton transport while suppressing vanadium ion crossover. To comprehensively evaluate membrane performance, the ion selectivity of each membrane was calculated based on both AR and vanadium ion permeability. As depicted in Fig. 4d, the sTPBA membrane demonstrated the highest ion selectivity, reaching up to 4.53 × 105 S min cm−3, because of its relatively low AR and permeability of vanadium ions. The sTPAP (8.07 × 104 S min cm−3), sBPAP (4.72 × 104 S min cm−3), and sBPBA (2.03 × 105 S min cm−3) membranes all exhibited significantly higher ion selectivity than Nafion®115 (0.31 × 103 S min cm−3). The outstanding ion selectivity indicated that protons migrated faster than vanadium ions within the poly(arylene perfluorophenylsulfonic acid) membranes, as seen in Fig. 4e.
3.4 VRFB and cycling performance
Fig. 5a shows the charge–discharge curves at 100 mA cm−2 of VRFB cells assembled with the poly(arylene perfluorophenylsulfonic acid) membranes and Nafion®115. Membranes with higher AR generally have higher charging voltages and lower discharging voltages, resulting in shorter charging times. It can be observed from Fig. 5a that all poly(arylene perfluorophenylsulfonic acid) membranes, except the sTPAP membrane, had lower charging voltages and higher discharging voltages compared to Nafion®115. Fig. 5b–d present the CE, VE, and EE of the same set of membranes, including Nafion®115, measured at current densities ranging from 40 to 160 mA cm−2. The VE of the poly(arylene perfluorophenylsulfonic acid) membranes and Nafion®115 increased with decreasing current density, due to more pronounced ohmic polarization and overpotential at higher current densities.55,56 Notably, the VE values of all poly(arylene perfluorophenylsulfonic acid) membranes, except for sTPAP, consistently exceed those of Nafion®115 across the entire current density range. This was clearly due to their lower AR values. As seen in Fig. 4c, sTPAP exhibited a lower AR than Nafion®115 in the ex situ measurements. This inconsistency suggested that AR values obtained from ex situ methods may not fully represent the effective resistance encountered during cell operation. Hence, in situ measurements directly in the flow cell environment should provide a more comprehensive evaluation, as the data account for the combined contributions of the membrane, electrode interfaces, and interfacial contact resistances.31,57,58 | ||
| Fig. 5 VRFB performance of cells assembled with the different PEMs: (a) charge–discharge curves at 100 mA cm−2, (b , c and d) VE, CE, and EE values, respectively, measured at 40−160 mA cm−2. Nafion®115 was evaluated under the same conditions used for the other membranes. | ||
Fig. 5c shows the CE results of VRFB cells assembled with the present PEMs measured at different current densities. As the current density increased, the CE values of poly(arylene perfluorophenylsulfonic acid) membranes and Nafion®115 slightly improved. This was because the charge–discharge time shortened with increasing current density, which reduced the number of vanadium ions, that crossed over during one cycle.26 Furthermore, the CE values of the sBPAP and sTPBA membranes were higher than for Nafion®115, which was attributed to the higher vanadium ion resistance of the former membranes. At high current density (80−160 mA cm−2), the CE values of sBPBA were comparable to those of Nafion®115, and decreased significantly at 60 and 40 mA cm−2. The CE values of the sBPBA membrane gradually decreased with increasing battery operation time, especially under low current densities (40−60 mA cm−2). This decline was likely due to high vanadium ion permeability or poor chemical stability. Moreover, sBPBA showed a relatively low ex situ permeability (Fig. 4c), but the lowest CE. This discrepancy likely arose under cell operating conditions, where its high intrinsic proton conductivity and increased electro-osmotic water drag facilitated the transport of vanadium species through hydrated ionic pathways.59 Alternatively, its limited chemical stability led to partial backbone degradation or domain swelling, which may create additional routes for crossover.
The EE of a VRFB is a combination of CE and VE and is jointly determined by these values. As is shown in Fig. 5d, the EE of the VRFB cells containing sBPBA, sTPBA, and sBPAP was higher than that of Nafion®115 at 40–160 mA cm−2. The cell based on the sBPBA membrane achieved the highest EEs at all current densities, reflecting its high ion selectivity. Correspondingly, sBPBA displayed the lowest charging voltages, the highest discharging voltages, and the highest VE values from both the charge–discharge curves (Fig. 5a) and the VE values measured over 40–160 mA cm−2 (Fig. 5b). This indicated that sBPBA had the lowest AR, consistent with its higher IEC and comparatively large electrolyte uptake (appox. 40%), which together facilitate efficient ion transport. Consequently, the superior short-term performance of sBPBA relative to sBPAP can be attributed to its high intrinsic proton conductivity and reduced voltage losses during operation. The sTPAP cell displayed lower EEs than the Nafion®115 cell at 100–160 mA cm−2, but higher EEs at 40−80 mA cm−2, obviously because of the higher AR and lower vanadium ion permeability compared to Nafion®115.
In addition to characterizing the poly(arylene perfluorophenylsulfonic acid) membranes at varying current densities, we also evaluated the long-term VRFB cycling stability of these PEMs and Nafion®115 without balancing the electrolyte during the VRFB operation. Fig. 6 shows the cycling performance and the discharge capacity retention rate during long-term VRFB cycling. It should be noted that, although the sBPBA-based cell displayed the highest EE, the efficiencies of the sBPBA and sTPBA membranes decreased significantly within the first 50 cycles of battery operation, mainly due to poor chemical stability. In contrast, the VRFBs assembled with the sTPAP and sBPAP membranes demonstrated superior cycling stability compared with sTPBA and sBPBA, which indicated that the presence of the –CF3 group in the polymer backbones of the former PEMs (or absence of the methine hydrogens) was critical to ensure chemical stability and high VRFB cycling performance. The CE of the cell equipped with Nafion®115 was around 98%, while the EE decreased from 80 to 71% after 250 cycles, indicating degradation in performance over time, likely due to increased vanadium ion crossover. The sTPAP membrane maintained a CE of ∼98%, while EE decreased slightly from 77 to 75% over 250 cycles, still matching Nafion®115 in long-term stability. Finally, the sBPAP membrane exhibited outstanding electrochemical stability, maintaining a CE of ∼99.5% and a stable EE of ∼82% over 450 cycles. This high performance was attributed to the membrane's high ion selectivity and robust polymer architecture that minimized vanadium ion crossover. Consequently, due to its outstanding VRFB performance and cycling stability, the sBPAP membrane can be considered a promising candidate component for VRFBs and other electrochemical energy devices. A concise summary of structural features, physicochemical properties, and VRFB performance for all membranes is provided in Table S2.
 | ||
| Fig. 6 Long-term stability of VRFB cells assembled with the different PEMs: (a) cycling performance at 100 mA cm−2 and (b). Discharge capacity retention. Nafion®115 was evaluated under the same conditions used for the other membranes. | ||
The poly(arylene perfluorophenylsulfonic acid) PEMs incorporate different fluorinated structural motifs. However, the fluorine content is only a fraction of that in Nafion®. Among the four polymers studied, only sBPAP and sTPAP contain –CF3 substituents, formally designated as perfluoroalkyl moieties (PFAS) under current regulatory definitions (EU REACH proposal, 2023; OECD, 2021).60,61 As discussed above, the presence of these groups significantly improves the stability of the PEMs. In contrast, the fluorophenyl groups are not considered as PFAS. These groups impart a strong electron-withdrawing effect to enhance acid strength, increasing desulfonation resistance, and promoting the formation of well-organized ionic clusters within the membrane matrix. sBPAP contains seven fluorine atoms per repeat unit, of which only the three in the –CF3 group meet the structural definition of PFAS. Hence, the PFAS-related [F]/[C] ratio is 0.15, compared with approximately 2.0 for Nafion®. Consequently, sBPAP has a PFAS content corresponding to ∼1/13 of that in Nafion®. Accordingly, sBPBA and sTPBA can be regarded as PFAS-free PEMs. In summary, while the present poly(arylene perfluorophenylsulfonic acid) membranes contain limited and strategically positioned fluorinated groups, their environmental and regulatory impact is expected to be far less significant than that of fully perfluorosyulfonic acid PEMs such as Nafion®.
4. Conclusions
Poly(arylene perfluorophenylsulfonic acid) PEMs, prepared by superacid-mediated polyhydroxyalkylations and a thiolation-oxidation process, exhibited enhanced proton conductivity and ion selectivity compared with Nafion®115. Using pentafluorobenzaldehyde and perfluoroacetophenone in the polyhydroxyalkylation produced PEMs with methine (Ph3C–H) and Ph3C–CF3 linkages in the backbone, respectively. The latter PEMs had sulfonic acid groups with increased acidity and a higher chemical stability, but allowed higher V(IV) permeability than the analogues with methine groups. In VRFB evaluations, the sBPBA, sTPBA, and sBPAP PEMs consistently delivered higher voltages and energy efficiencies than Nafion®115 across 40–160 mA cm−2, driven by reduced area resistance and improved ion selectivity. Among them, sBPBA achieved the highest energy efficiencies at all current densities, while sBPAP and sTPBA also surpassed Nafion®115 in coulombic efficiency. However, both sBPBA and sTPBA exhibited poor chemical stability, most likely due to the poor oxidative stability of the methine linkages, and the efficiencies declined markedly within the first 50 cycles. In contrast, sTPAP maintained a CE of ∼98% and an EE decline from 77 to only 75% over 250 cycles, comparable to Nafion®115 in long-term stability. Notably, the sBPAP PEM demonstrated exceptional electrochemical stability, maintaining a CE of ∼99.5% and an EE of ∼82% over 450 cycles. These results highlight that –CF3-containing polymers enable stable cycling and high efficiency in VRFBs. These findings demonstrate that PEMs based on ether-free poly(arylene perfluorophenylsulfonic acid)s containing Ph3C–CF3 linkages (devoid of methine linkages) enable stable, high-efficiency VRFB cycling, and provide a clear structure–performance framework for next-generation membrane design. The sBPBA and sTPBA membranes delivered very good performance in the first 30 cycles. If it is possible to overcome the stability issues, these PEMs could be especially attractive for VRFB applications, since they contain less fluorine and can be synthesized from cheaper monomers. While sBPBA and sTPBA exhibit excellent short-term performance, their limited chemical stability remains a challenge. Addressing this limitation could make these low-fluorine and cost-effective PEMs particularly promising for practical VRFB applications.Author contributions
Xiaoting Xue: conceptualization, methodology, investigation, validation, writing – original draft. Peiru Lv: investigation, validation, data curation. Jingshuai Yang: conceptualization, methodology; validation, writing – review & editing; supervision, project administration. Patric Jannasch: writing – review & editing; supervision; project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): membrane photographs, NMR and FTIR spectra, stress–strain data, and TGA traces. See DOI: https://doi.org/10.1039/d5ta08577b.Acknowledgements
We thank the Swedish Energy Agency (grants 50519-1, 45057-1, 37806-3, and 45515-3) for financial support, and Si Chen for help with the AFM analysis.References
- M. Shin, H. Lim, K. Hyun and Y. Kwon, Appl. Energy, 2025, 381, 125225 Search PubMed.
- Q. Yang, H. Lin, X. Niu, Y. Xu, Z. Wu, L. Tan, D. Li, J. Xi and L. Liu, Small, 2025, 21, 2505051 Search PubMed.
- Y. Huang, J. Xu, X. Huang, X. He, J. Cao, C. Jia and M. Ding, ACS Sustain. Chem. Eng., 2024, 12, 12837–12844 CrossRef CAS.
- D. Bordignon, N. Zatta and A. Trovò, J. Power Sources, 2025, 653, 237605 Search PubMed.
- A. Castillo and D. F. Gayme, Energy Convers. Manag., 2014, 87, 885–894 Search PubMed.
- M. M. Ikhsan, S. Abbas, X. H. Do, H. Y. Ha, K. Azizi and D. Henkensmeier, Adv. Energy Mater., 2025, 15, 2400139 Search PubMed.
- X. Li, L. Yu, L. Liu and J. Xi, J. Energy Storage, 2024, 86, 111251 Search PubMed.
- L. Yang, Z. Fan, F. Cui, T. Wu, T. Fang, Y. Guo, L. Tian, B. Pang, G. He and X. Wu, Electrochim. Acta, 2025, 526, 146190 CrossRef CAS.
- A. Wang, C. Breakwell, F. Foglia, R. Tan, L. Lovell, X. Wei, T. Wong, N. Meng, H. Li, A. Seel, M. Sarter, K. Smith, A. Alvarez-Fernandez, M. Furedi, S. Guldin, M. M. Britton, N. B. McKeown, K. E. Jelfs and Q. Song, Nature, 2024, 635, 353–358 CrossRef CAS PubMed.
- Y. Zhao, L. Liu, X. Qiu and J. Xi, Electrochim. Acta, 2019, 303, 21–31 Search PubMed.
- M. Zhang, P. Lv, L. Wang, J. Wang and J. Yang, ACS Appl. Polym. Mater., 2025, 7, 3164–3173 Search PubMed.
- B. Jiang, L. Wu, L. Yu, X. Qiu and J. Xi, J. Membr. Sci., 2016, 510, 18–26 Search PubMed.
- S. Liu, L. Wang, B. Zhang, B. Liu, J. Wang and Y. Song, J. Mater. Chem. A, 2015, 3, 2072–2081 Search PubMed.
- B. Jiang, L. Yu, L. Wu, D. Mu, L. Liu, J. Xi and X. Qiu, ACS Appl. Mater. Interfaces, 2016, 8, 12228–12238 Search PubMed.
- Z. Zhao, Q. Dai, S. Huang, W. Lu, Y. Chen, J. Zheng, S. Zhang, S. Li and X. Li, Chin. Chem. Lett., 2024, 35, 109231 CrossRef CAS.
- X. Lou, B. Lu, M. He, Y. Yu, X. Zhu, F. Peng, C. Qin, M. Ding and C. Jia, J. Membr. Sci., 2022, 643, 120015 Search PubMed.
- Y. Xia, H. Yu, C. Yuan and Y. Wang, Chem. Eng. J., 2021, 425, 131448 CrossRef CAS.
- X. Michel Myures, S. Suresh and G. Arthanareeswaran, J. Power Sources, 2024, 591, 233818 Search PubMed.
- J. Li, X. Yuan, S. Liu, Z. He, Z. Zhou and A. Li, ACS Appl. Mater. Interfaces, 2017, 9, 32643–32651 Search PubMed.
- J. F. M. Sousa, J. Pina, C. Gomes, L. D. Dias, M. M. Pereira, D. Murtinho, P. Dias, J. Azevedo, A. Mendes, J. S. Seixas de Melo, A. A. C. C. Pais, M. Pineiro and A. J. M. Valente, Mater. Today Commun., 2021, 29, 102781 CrossRef CAS.
- S. K. Hanson, R. T. Baker, J. C. Gordon, B. L. Scott and D. L. Thorn, Inorg. Chem., 2010, 49, 5611–5618 CrossRef CAS PubMed.
- X. L. Zhou, T. S. Zhao, L. An, L. Wei and C. Zhang, Electrochim. Acta, 2015, 153, 492–498 CrossRef CAS.
- B. G. Thiam and S. Vaudreuil, J. Electrochem. Soc., 2021, 168, 070553 CrossRef.
- T. T. Bui, M. Shin, S. Abbas, M. M. Ikhsan, X. H. Do, A. Dayan, M. R. Almind, S. Park, D. Aili, J. Hjelm, J. Hwang, H. Y. Ha, K. Azizi, Y. Kwon and D. Henkensmeier, Adv. Energy Mater., 2025, 15, 2401375 Search PubMed.
- R. Ye, D. Henkensmeier, S. J. Yoon, Z. Huang, D. K. Kim, Z. Chang, S. Kim and R. Chen, J. Electrochem. Energy Convers. Storage, 2018, 15, 010801 Search PubMed.
- X. Ren, L. Zhao, X. Che, Y. Cai, Y. Li, H. Li, H. Chen, H. He, J. Liu and J. Yang, J. Power Sources, 2020, 457, 228037 CrossRef CAS.
- M. K. Pagels, S. Adhikari, R. C. Walgama, A. Singh, J. Han, D. Shin and C. Bae, ACS Macro Lett., 2020, 9, 1489–1493 CrossRef CAS PubMed.
- C. H. Fujimoto, M. A. Hickner, C. J. Cornelius and D. A. Loy, Macromolecules, 2005, 38, 5010–5016 CrossRef CAS.
- T. J. G. Skalski, B. Britton, T. J. Peckham and S. Holdcroft, J. Am. Chem. Soc., 2015, 137, 12223–12226 CrossRef CAS PubMed.
- J. Miyake, R. Taki, T. Mochizuki, R. Shimizu, R. Akiyama, M. Uchida and K. Miyatake, Sci. Adv., 2017, 3, eaao0476 Search PubMed.
- A. Khataee, H. Nederstedt, P. Jannasch and R. W. Lindström, J. Membr. Sci., 2023, 671, 121390 Search PubMed.
- J. Yang, P. Lv, W. Tang and Q. Wang, ACS Appl. Polym. Mater., 2024, 6, 10727–10737 Search PubMed.
- H. Gong, H. Nederstedt, S.-Y. Choi and P. Jannasch, Solid State Ionics, 2025, 423, 116837 Search PubMed.
- N. R. Kang, T. H. Pham and P. Jannasch, ACS Macro Lett., 2019, 8, 1247–1251 Search PubMed.
- X. Hu, Y. Ao, Y. Gao, B. Liu and C. Zhao, J. Membr. Sci., 2023, 687, 122102 CrossRef CAS.
- T. Wei, Y. Zhao, Z. Ren, Y. Han, H. Zhang and Z. Shao, Sustain, 2024, 3, 100021 Search PubMed.
- L. I. Olvera, M. T. Guzmán-Gutiérrez, M. G. Zolotukhin, S. Fomine, J. Cárdenas, F. A. Ruiz-Trevino, D. Villers, T. A. Ezquerra and E. Prokhorov, Macromolecules, 2013, 46, 7245–7256 Search PubMed.
- Z. Dong, M. Di, L. Hu, L. Gao, X. Yan, X. Ruan, X. Wu and G. He, J. Membr. Sci., 2020, 608, 118179 Search PubMed.
- N. Shi, G. Wang, Q. Wang, L. Wang, Q. Li and J. Yang, Chem. Eng. J., 2024, 489, 151121 Search PubMed.
- M. Zharnikov, A. Küller, A. Shaporenko, E. Schmidt and W. Eck, Langmuir, 2003, 19, 4682–4687 Search PubMed.
- X. Lu, C. He, P. Liu and A. C. Griffin, J. Polym. Sci. Part Polym. Chem., 2005, 43, 3394–3402 Search PubMed.
- D. Chen and M. A. Hickner, Phys. Chem. Chem. Phys., 2013, 15, 11299–11305 RSC.
- D. Lu, L. Wen, F. Nie and L. Xue, RSC Adv., 2016, 6, 6029–6037 RSC.
- Z. Peng, Z. Ren, S. Chen, Y. Zhao, P. Jannasch and J. Yang, Int. J. Hydrogen Energy, 2025, 188, 151884 CrossRef CAS.
- J. Jin, C. M. Topping, S. Chen, J. Ballato, S. H. Foulger and D. W. Smith Jr., J. Polym. Sci. Part Polym. Chem., 2004, 42, 5292–5300 Search PubMed.
- L. Chu and F.-L. Qing, Acc. Chem. Res., 2014, 47, 1513–1522 CrossRef CAS PubMed.
- Y. S. Kim and B. S. Pivovar, Annu. Rev. Chem. Biomol. Eng., 2010, 1, 123–148 Search PubMed.
- X. Wu, X. Wang, G. He and J. Benziger, J. Polym. Sci. Part B Polym. Phys., 2011, 49, 1437–1445 CrossRef CAS.
- J. Rozière and D. J. Jones, Annu. Rev. Mater. Res., 2003, 33, 503–555 Search PubMed.
- Y. S. Kim, B. Einsla, M. Sankir, W. Harrison and B. S. Pivovar, Polymer, 2006, 47, 4026–4035 Search PubMed.
- Y. Dai, M. D. Guiver, G. P. Robertson and Y. S. Kang, Macromolecules, 2005, 38, 9670–9678 Search PubMed.
- T. Corrado and R. Guo, Mol. Syst. Des. Eng., 2020, 5, 22–48 RSC.
- J. Ye, L. Xia, H. Li, F. P. G. de Arquer and H. Wang, Adv. Mater., 2024, 36, 2402090 Search PubMed.
- T. Ban, M. Guo, Y. Wang, J. Ma, X. Wang, Z. Wang and X. Zhu, J. Mater. Chem. A, 2023, 11, 24013–24025 RSC.
- Q. Wang, Z. Zhang, P. Lv, Z. Peng and J. Yang, Chem. Eng. J., 2025, 505, 58922 Search PubMed.
- Y. Zhang, H. Liu, M. Liu, X. Ma, D. Liang, P. Qian and J. Yan, Int. J. Hydrog. Energy, 2024, 53, 229–238 Search PubMed.
- J. Shi, H. Zhang, H. Yu, Y. Xu, S. Xu, L. Sheng, X. Feng and X. Wang, eTransportation, 2024, 20, 100321 Search PubMed.
- Y. Yang, Q. Wang, S. Xiong and Z. Song, Inorg. Chem. Front., 2024, 11, 4049–4079 Search PubMed.
- K. Oh, M. Moazzam, G. Gwak and H. Ju, Electrochim. Acta, 2019, 297, 101–111 Search PubMed.
- Reconciling terminology of the universe of per- and polyfluoroalkyl substances, https://www.oecd.org/en/publications/reconciling-terminology-of-the-universe-of-per-and-polyfluoroalkyl-substances_e458e796-en.html, (accessed October 17, 2025).
- Per- and polyfluoroalkyl substances (PFAS) - ECHA, https://echa.europa.eu/hot-topics/perfluoroalkyl-chemicals-pfas, (accessed October 17, 2025).
| This journal is © The Royal Society of Chemistry 2026 |