
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Poly-cyclodextrin modified graphene oxide for PFAS removal from drinking water
Andrea
Trifoglio†
 *ae,
Angela
Pintus†
af,
Sara
Khaliha
a,
Francesco
Blancato
a,
Marco
Agnes
a,
Lucrezia
Aversa
b,
Roberto
Verucchi
b,
Tainah Dorina
Marforio
eg,
Nina
Burduja
cd,
Giuseppe
Nocito
c,
Antonino
Mazzaglia
c,
Matteo
Calvaresi
eg and
Manuela
Melucci
*a
*ae,
Angela
Pintus†
af,
Sara
Khaliha
a,
Francesco
Blancato
a,
Marco
Agnes
a,
Lucrezia
Aversa
b,
Roberto
Verucchi
b,
Tainah Dorina
Marforio
eg,
Nina
Burduja
cd,
Giuseppe
Nocito
c,
Antonino
Mazzaglia
c,
Matteo
Calvaresi
eg and
Manuela
Melucci
*a
aInstitute for Organic Synthesis and Photoreactivity (ISOF-CNR), Bologna, Italy. E-mail: manuela.melucci@cnr.it
bInstitute of Materials for electronics and Magnetism (IMEM-CNR), c/o Fondazione Bruno Kessler, Trento, Italy
cInstitute for Nanostructured Materials (ISMN-CNR), Messina, Italy
dDepartment of Chemical, Biological, Pharmaceutical and Environmental Sciences (ChiBioFarAm), University of Messina, Messina, Italy
eDepartment of Chemistry ‘Giacomo Ciamician’, University of Bologna, Italy
fDipartimento di Physics, University of Modena e Reggio Emilia, Modena, Italy
gIRCCS Azienda Ospedaliero – Universitaria di Bologna, Preclinical & Translational Research in Oncology Lab (PRO), Bologna, Italy
First published on 21st November 2025
Abstract
Per- and polyfluoroalkyl substances (PFASs) are persistent organic pollutants of growing concern due to their widespread occurrence in drinking water and resistance to conventional remediation technologies. Granular activated carbon (GAC), the current benchmark adsorbent, exhibits limited efficiency toward short- and medium-chain PFASs and slow adsorption kinetics. Here, we report the synthesis of a graphene oxide (GO) material covalently grafted with an amino-functionalized β-cyclodextrin polymer crosslinked with epichlorohydrin (GO-Poly-βCD). The hybrid material was characterized and evaluated for the adsorption of a mixture of PFASs with varying chain lengths (CF)3–9 and functional groups under environmentally relevant conditions. GO-Poly-βCD outperformed pristine GO, showing enhanced affinity for medium-chain PFAS and removal efficiencies up to 97% for PFHpA (CF)6 and PFHxS (CF)6 after only 15 min of contact. At neutral pH, PFPeA (CF)4 removal reached 44%, compared to no removal by GO, while under acidic conditions its removal increased to 80%. Notably, PFBA (CF)3, which was not removed at neutral pH, exhibited 31% removal under acidic conditions. Molecular dynamics simulations revealed a cooperative adsorption mechanism in which PFAS molecules are stabilized through pH-responsive conformational rearrangements that strengthen interactions between the hybrid material and the contaminants, primarily via van der Waals and hydrophobic forces. Compared to GAC, GO-Poly-βCD achieved a higher overall PFAS uptake (4.0 µg g−1vs. 1.3 µg g−1 for GAC) within the same contact time, demonstrating its potential for rapid and efficient PFAS removal in real drinking water treatment applications.
Introduction
Per- and poly-fluoro alkyl substances (PFASs) have attracted global concern due to their widespread use in everyday products, their environmental and biological persistence and widely reported effects on human health and ecosystems.1–3 PFASs are a class of synthetic organo-fluorine compounds characterized by the presence of fully or partially fluorinated alkyl-chains with a carboxylate or sulphonate ending group.4 PFASs are widely used as additives in food packaging, coatings, non-stick cookware, waterproof clothing and firefighting foams.5 The disposal of everyday products inevitably results in the release of PFASs into various environmental compartments and, ultimately, into drinking water sources through multiple pathways.1 The C–F chemical bond being very stable, their persistency in the environment is exceptional making PFASs the so-called “forever chemicals”.6 The Environmental Working Group reported that in 2018 over 16 million people in the United States were exposed to PFAS contaminated drinking water with concentrations exceeding 10 ng L−1.3 The removal of PFASs from drinking water remains one of the key challenges in water purification. Among the treatment strategies currently implemented in municipal drinking water plants, adsorption is the most effective approach. In particular, granular activated carbon (GAC) represents the most widely employed adsorbent for PFAS mitigation.7–9 GAC is a non-specific adsorbent that exhibits moderate affinity toward long-chain PFASs but limited effectiveness for their short- and medium-chain counterparts.10,11 Moreover, its adsorption process is characterized by slow, diffusion-limited kinetics, which represents a significant drawback under real plant flow conditions.12,13 New technologies for GAC replacement or integration are currently a matter of great interest. Among emerging materials, graphene oxide (GO) nanosheets have attracted considerable attention as promising adsorbents for the removal of emerging contaminants from drinking water, offering new opportunities for advanced treatment technologies.10,14 The most relevant features of GO are its large specific surface area and the abundance of tunable oxygen-containing functional groups on the nanosheet surface, which enable versatile covalent functionalization.15GO nanosheets have previously been explored for PFAS removal using a two-step approach involving batch adsorption followed by tandem microfiltration.10 Furthermore, some of us demonstrated that incorporating GO into polysulfone hollow fiber membranes (now commercially available as Graphisulfone®16) provides a new integrated filter model combining adsorption and ultrafiltration mechanisms and allowing the removal of long-chain PFASs from drinking water.13 As a step forward, it has been demonstrated that the covalent functionalization of GO nanosheets enables the tuning of adsorption selectivity and significantly enhances the removal of contaminants that exhibit poor affinity for unmodified GO, including organic emerging pollutants with performances overcoming those achieved by GAC.10,17,18 For instance, functionalization of GO with N,N-dimethylethylenediamine (GO-DMEN),19 provided nanosheets with high affinity for medium- and long-chain PFASs. Studies based on theoretical modelling demonstrated that adsorption depends on multiple interactions including hydrophobic, electrostatic and van der Waals forces.13 In particular, the interaction between non-functionalized GO and PFASs is hindered by electrostatic repulsion between the negatively charged nanosheets and the negatively charged PFAS molecules.16 Hence, PFASs with longer hydrophobic chains are more efficiently adsorbed, whereas shorter, more hydrophilic PFASs are not removed.
Cyclodextrins are cyclic oligosaccharides made up of glucose subunits linked by α-1,4-glycosidic bonds. They have a toroidal structure with a hydrophobic inner cavity and a hydrophilic outer surface.20 Cyclodextrins,21,22 in particular β-cyclodextrins23,24 (i.e., consisting of seven glucose subunits), can form inclusion complexes with PFASs through non-covalent forces such as van der Waals interactions and hydrophobic effects. Some of us recently demonstrated that covalent modification of GO with an amino-derived βCD and a C6 aliphatic linker to GO nanosheets enables the efficient removal of perfluorobutanoic acid (PFBA, (CF)3)25 thanks to the formation of a sandwich-like complex, where PFBA is stably confined between the GO nanosheet and the βCD cavity. Interestingly, βCD can serve as a monomer for the synthesis of βCD-based polymers through various types of cross-linkers.26 PFAS adsorption has been demonstrated for βCD polymers containing fluorinated aromatic cross-linkers,27,28 which raise environmental concerns similar to those of the targeted PFASs.29 Moreover, the partial water solubility of poly-cyclodextrins makes their direct application as sorbents under real conditions unrealistic.30 To overcome this limitation, we propose here a novel polymeric material insoluble in water and based on a non-fluorinated chain (Poly-βCD from now on) and its use for PFAS removal from drinking water (Fig. 1). GO-Poly-βCD adsorption mechanism was investigated by combining adsorption experiments under different pH conditions with molecular dynamics (MD) simulations.
 | ||
| Fig. 1 Chemical structure of GO-Poly-βCD and Poly-βCD. | ||
Experimental section
Materials
GO was purchased from Layer One (Norway) and used without further purification. Before using, GO was sonicated in ultrapure water to exfoliate the bulk material into a monolayer (>99%) with a lateral size of few micrometers. GAC was purchased from CABOT Norit Spa (Ravenna, Italy, Norit GAC 830 AF, MB index min 240 mg g−1, BET surface area >1000 m2 g−1) and used without further purification. PFAS standards were purchased from Agilent Technologies (Santa Clara, CA, US). The experiments on PFASs were carried out by using polypropylene vials. Poly-βCD was kindly provided by Cyclolab (Budapest).Synthesis of poly-β-cyclodextrin (Poly-βCD)
Poly-βCD was provided as the chlorohydrated salt of amino β-cyclodextrin polymer crosslinked with epichlorohydrin (average MW = 25 kDa) and with 70% of β-cyclodextrin content. The salt was treated with NaHCO3 and dialysed for 72 hours in ultrapure water then lyophilized and used in the next synthetic step.Synthesis and purification of GO-Poly-βCD
The synthesis of GO-Poly-βCD was carried out via an epoxide ring-opening reaction.25Briefly, 100 mg of GO was dispersed in 30 mL of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:EtOH solution by sonication for 2 hours to obtain a stable aqueous suspension. Separately, 200 mg of Poly-βCD-NH2 was dissolved in 100 mL of distilled water and added to the GO suspension (GO:Poly-βCD mass ratio 1:2, total volume 130 mL). The resulting mixture was refluxed at 80 °C for 12 hours. Upon completion of the reaction, the crude product was purified by centrifugation (9000 rpm, 15 minutes per cycle, eight cycles), and the final material was obtained by lyophilization.
:1 H2O:EtOH solution by sonication for 2 hours to obtain a stable aqueous suspension. Separately, 200 mg of Poly-βCD-NH2 was dissolved in 100 mL of distilled water and added to the GO suspension (GO:Poly-βCD mass ratio 1:2, total volume 130 mL). The resulting mixture was refluxed at 80 °C for 12 hours. Upon completion of the reaction, the crude product was purified by centrifugation (9000 rpm, 15 minutes per cycle, eight cycles), and the final material was obtained by lyophilization.
Synthesis and purification of GO-Control
The synthesis of GO-Control was carried out under the same conditions as GO-Poly-βCD, but in the absence of Poly-βCD-NH2. Briefly, 100 mg of GO was dispersed in 130 mL of a 1:1 H2O:EtOH solution by sonication for 2 h to obtain a stable suspension. The mixture was then refluxed at 80 °C for 12 h. After completion of the reaction, the crude product was purified by centrifugation (9000 rpm, 15 min per cycle, eight cycles) and lyophilized.
Characterization
ATR-FTIR spectra were recorded with an Agilent Cary 630 FTIR spectrophotometer, and the spectra are expressed by wavenumber (cm−1).TGA was conducted with a PerkinElmer Thermogravimetric Analyzer TGA 4000 by PerkinElmer, in an air atmosphere, from 30 °C to 800 °C, with a scanning temperature of 10 °C min−1.
Elemental analysis was performed on powder materials by using an Elementar Unicube Elemental analyser, method GRAPHITE.
XPS was performed using the Mg-Ka emission at 1253.6 eV of a non-monochromatized X-ray photon source. Photoelectrons were analysed using a VSW HA100 electron energy analyser, resulting in a total energy resolution of 0.86 eV. Core level BEs were referred to the Au 4f7/2 core level signal (at 84.0 eV), obtained from a sputtered gold surface. The photoemission line shapes of selected core levels were analyzed using Voight line shape deconvolution, after background subtraction of a Shirley function. The typical precision for energy peak position is ±0.05 eV, while the uncertainty for FWHM is <±5% and for area evaluation it is ±5%. GO, GO-Control, and GO-Poly-βCD samples were mounted directly on the sample holder, while the Poly-βCD-NH2 powder was supported on carbon tape.
Dynamic light scattering (DLS)
The mean hydrodynamic diameter (DH), width of distribution (polydispersity index, PDI) and ζ-potential were measured with a Malvern Zetasizer Nano ZS (Malvern Instruments, Malvern, UK), equipped with a He–Ne laser (λ = 633 nm, 4 mW). Measurements were performed using the non-invasive backscattering technique (NIBS) with an angle of 173° to the incident beam. For freshly prepared dispersions, 45 acquisitions at a temperature of 25 ± 1 °C for each measurement were collected. Deconvolution of the measured correlation curve (distribution of relaxation times and the cumulant analysis) into a dimensional average intensity distribution was achieved using the Laplace inversion algorithm.Colloidal stability
The colloidal stability of GO-Poly-βCD was evaluated with an adapted turbidimetric method for a 250 µg mL−1 dispersion in aqueous 1 mM NaCl. The sample was placed in a 1 cm quartz cuvette inside a Jasco V-770 (Jasco Co., Tokyo, Japan) Peltier temperature-controlled UV/Vis-NIR scanning spectrophotometer immediately after preparation. Experiments were performed by recording the extinction values at 790 nm every 5 min in a time interval from 0 to 600 min at 25 °C, respectively. Results were plotted against time and fitted using the OriginPro 2021 software with the equation | (1) |
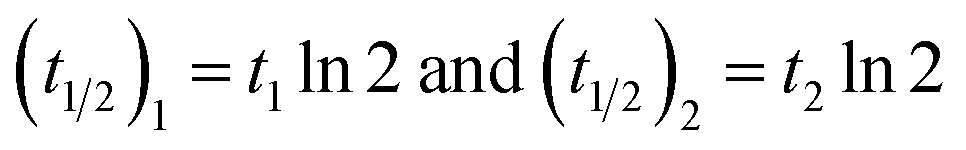 | (2) |
Adsorption experiments
25 mg of powdered materials (GO or GO-Poly-βCD) was sonicated in 25 mL of tap water for 2 hours. A mixture containing ten PFASs (125 µL, 100 µg L−1) was then added to the suspensions to achieve a final concentration of 0.5 µg L−1 each in a total volume of 25 mL. Similarly, 25 mg of GAC was directly added to 25 mL of tap water, followed by the addition of 125 µL of the ten-PFAS mixture (100 µg L−1), resulting in the same final PFAS concentration (0.5 µg L−1). All samples were subjected to gentle agitation for 15 minutes, then centrifuged (10.000 rpm, 10 minutes) and analyzed by UPLC-MS/MS.PFAS quantification
Samples containing PFASs were analyzed by UPLC-MS/MS (ACQUITY UPLC H-Class PLUS coupled to a XEVO TQS Micro mass detector, Waters). A 1 mL aliquot of each sample was used for automated injection. Chromatographic separation was performed on a Waters Acquity UPLC CSH Phenyl–Hexyl column (1.7 µm, 2.1 × 100 mm) with a Waters Isolator Column (2.1 × 50 mm). The column temperature was maintained at 34 °C, the flow rate was 0.3 mL min−1, and the injection volume was 40 µL. Total run times were 8 min for PFBA and 21 min for the mixture of eight PFASs. The mobile phase consisted of a biphasic gradient: 2 mM NH4OAc in ultrapure water:methanol (95:5) as phase A, and 2 mM NH4OAc in methanol as phase B (for further details, see SI Section 4).
Computational modelling
All MD simulations were carried out using the Amber22 software suite. The PFAS molecules were parameterized using the generalized AMBER force field (GAFF), and atomic partial charges were derived using the restrained electrostatic potential (RESP) fitting procedure after geometry optimization at the HF/6-31G* level of theory. The GO-Poly-βCD model-system was generated by covalently grafting a representative Poly-βCD onto a GO surface, modelled according to experimental XPS data. Different protonation states of the GO-Poly-βCD system were used to mimic pH 4, 7, and 10, where surface groups were protonated or deprotonated accordingly. Complexes of GO-Poly-βCD with PFASs were solvated in an explicit TIP3P water box, ensuring a 20 Å buffer from the solute to the box edge. The systems were neutralized by adding appropriate counterions. Energy minimization was performed in two steps: first with restraints on solute atoms to relax the solvent environment, and second without restraints to minimize the entire system. Subsequently, each system was equilibrated under NPT conditions at 298 K for 10 ns using periodic boundary conditions and an Andersen thermostat. Production MD simulations were then run for 100 ns under constant temperature and pressure conditions. To evaluate binding affinities, Molecular Mechanics-Generalized Born Surface Area (MM-GBSA) calculations were performed and the total binding free energy (ΔGbinding) was decomposed into van der Waals (VDW), electrostatic (EEl), non-polar solvation (ESURF), and entropic (TΔS) contributions. Entropic contributions were estimated via normal mode analysis. The cavities reported in this study were identified using ChimeraX software (version 1.10.1) with the Cavity tool.Results and discussion
Synthesis and characterization
The synthesis of GO-Poly-βCD was carried out via the epoxide ring-opening reaction of exfoliated GO nanosheets,25 using Poly-βCD-NH2 obtained by crosslinking of mono-6-azido β-CD in the presence of epichlorohydrin and subsequent reduction of azido to amino groups31 (Scheme 1). Under the reaction conditions (80 °C, 16 h), GO nanosheets are partially reduced, thus to clarify the role of Poly-βCD, a control material (GO-Control) was prepared by treating GO under the same conditions as those used for GO-Poly-βCD synthesis, but in the absence of Poly-βCD. The resulting materials were purified from unreacted materials through repeated washing and centrifugation steps with water until the supernatant reached neutral pH. The lyophilized products were then characterized and used for the targeted adsorption experiments. The surface chemical properties of pristine GO, GO-Control, GO-Poly-βCD, and Poly-βCD-NH2 were investigated by X-ray photoelectron spectroscopy (XPS). Long-range spectra (Fig. S1) revealed the presence of carbon, oxygen, and nitrogen in almost all samples; therefore, the corresponding core levels (C 1s, N 1s, and O 1s) were analyzed in detail. Fig. 2a and b reports the C 1s and N 1s core levels of all samples while O 1s core level is shown in Fig. S2. GO and GO-Control surfaces were found to be almost identical, exhibiting the typical C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C sp2 component (285.0 eV), ascribed to the aromatic graphene framework, together with C–O, CO, and COOR features (287.0–287.1 eV, 288.4–288.7 eV, and 289.3 eV, respectively). The N 1s intensity was very low (<1% atomic concentration) and was detected only in GO-Control. Its deconvolution yielded a C–NH–C signal (graphitic nitrogen) at 400.1 eV, as previously reported,32 and two additional components at 398.5 eV and ∼402.0 eV, which can be assigned to pyridinic nitrogen32 and to amino groups,25 respectively, although the latter attribution is not straightforward since different N-based functionalities contribute in this range. Poly-βCD-NH2 displayed a completely different C 1s line shape, dominated by C–O bonds at 286.4 eV, and a single N 1s peak corresponding to primary amines at 399.8 eV. Upon functionalization of GO, a complete change in the C 1s line shape was observed, along with an increase in the intensity of high-binding-energy N 1s components. The relative contribution of graphitic and amino-related components in N 1s increased from 43.1% in pristine GO to 74.8% in GO-Poly-βCD. These changes are compatible with the covalent functionalization of GO with poly-βCD.19,25 Thermogravimetric analysis (TGA) supported the successful covalent bonding of GO with Poly-βCD. The TGA profile of GO-Poly-βCD (Fig. 2c) exhibited a distinct inflection point at 333 °C, clearly visible as a peak in the derivative thermogravimetric (DTG) curve. This thermal event was absent in pristine GO and closely matched the degradation onset of pure Poly-βCD (Fig. 2e), which showed a similar DTG peak at 342 °C. The slight shift in decomposition temperature suggested covalent interaction between GO and Poly-βCD, which altered the thermal stability of the polymer moieties. Additionally, the increased weight loss observed in the 200–400 °C range for GO-Poly-βCD compared to pristine GO (Fig. 2d) supported the presence of organic functional groups from the grafted polymer. Elemental Analysis (EA) was used to estimate the bulk composition of the synthesised materials and to evaluate the Poly-βCD loading (Table S1). GO-Poly-βCD exhibits a higher nitrogen content compared to pristine GO (0.3% vs. 0.08%), ascribed to the presence of amine functionalities in the glucose units. Under the reaction conditions (16 hours at 80 °C), the GO nanosheets tend to undergo partial reduction15 (GO-Control), leading to a decreased oxygen content (24% vs. 30.8%) and, consequently, an increased C/O ratio (1.4 vs. 1.3). By monitoring the increase in nitrogen content, it was possible to estimate the Poly-βCD loading. Starting from a βCD/epichlorohydrin ratio of 7:3 in the starting material, the molecular formula of the Poly-βCD repetitive unit was determined to be C67H129NO42. Given a total of 239 atoms per repetitive unit, the theoretical nitrogen content was calculated as 0.42%, which is in excellent agreement with the experimentally determined value (Table S1). The Poly-βCD loading in GO-Poly-βCD was found to be 55% as it can be estimated using eqn (3), where nN is the number of nitrogen atoms involved (one in this case), nTotal is the total number of atoms per polymer unit (239) and the % refers to that of atomic nitrogen in GO-Poly-βCD and GO-Control (see SI Section 1 for more details):
C sp2 component (285.0 eV), ascribed to the aromatic graphene framework, together with C–O, CO, and COOR features (287.0–287.1 eV, 288.4–288.7 eV, and 289.3 eV, respectively). The N 1s intensity was very low (<1% atomic concentration) and was detected only in GO-Control. Its deconvolution yielded a C–NH–C signal (graphitic nitrogen) at 400.1 eV, as previously reported,32 and two additional components at 398.5 eV and ∼402.0 eV, which can be assigned to pyridinic nitrogen32 and to amino groups,25 respectively, although the latter attribution is not straightforward since different N-based functionalities contribute in this range. Poly-βCD-NH2 displayed a completely different C 1s line shape, dominated by C–O bonds at 286.4 eV, and a single N 1s peak corresponding to primary amines at 399.8 eV. Upon functionalization of GO, a complete change in the C 1s line shape was observed, along with an increase in the intensity of high-binding-energy N 1s components. The relative contribution of graphitic and amino-related components in N 1s increased from 43.1% in pristine GO to 74.8% in GO-Poly-βCD. These changes are compatible with the covalent functionalization of GO with poly-βCD.19,25 Thermogravimetric analysis (TGA) supported the successful covalent bonding of GO with Poly-βCD. The TGA profile of GO-Poly-βCD (Fig. 2c) exhibited a distinct inflection point at 333 °C, clearly visible as a peak in the derivative thermogravimetric (DTG) curve. This thermal event was absent in pristine GO and closely matched the degradation onset of pure Poly-βCD (Fig. 2e), which showed a similar DTG peak at 342 °C. The slight shift in decomposition temperature suggested covalent interaction between GO and Poly-βCD, which altered the thermal stability of the polymer moieties. Additionally, the increased weight loss observed in the 200–400 °C range for GO-Poly-βCD compared to pristine GO (Fig. 2d) supported the presence of organic functional groups from the grafted polymer. Elemental Analysis (EA) was used to estimate the bulk composition of the synthesised materials and to evaluate the Poly-βCD loading (Table S1). GO-Poly-βCD exhibits a higher nitrogen content compared to pristine GO (0.3% vs. 0.08%), ascribed to the presence of amine functionalities in the glucose units. Under the reaction conditions (16 hours at 80 °C), the GO nanosheets tend to undergo partial reduction15 (GO-Control), leading to a decreased oxygen content (24% vs. 30.8%) and, consequently, an increased C/O ratio (1.4 vs. 1.3). By monitoring the increase in nitrogen content, it was possible to estimate the Poly-βCD loading. Starting from a βCD/epichlorohydrin ratio of 7:3 in the starting material, the molecular formula of the Poly-βCD repetitive unit was determined to be C67H129NO42. Given a total of 239 atoms per repetitive unit, the theoretical nitrogen content was calculated as 0.42%, which is in excellent agreement with the experimentally determined value (Table S1). The Poly-βCD loading in GO-Poly-βCD was found to be 55% as it can be estimated using eqn (3), where nN is the number of nitrogen atoms involved (one in this case), nTotal is the total number of atoms per polymer unit (239) and the % refers to that of atomic nitrogen in GO-Poly-βCD and GO-Control (see SI Section 1 for more details):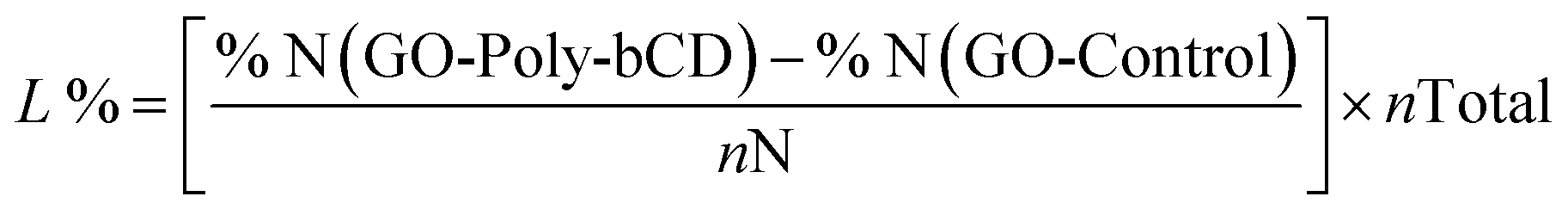 | (3) |
 | ||
| Scheme 1 Synthetic route to GO-Poly-βCD. | ||
 | ||
| Fig. 2 XPS core level spectra of (a) C 1s and (b) N 1s of GO, GO Control, GO-Poly-βCD and Poly-βCD-NH2 (for reference) and TGA profiles of (c) GO-Poly-βCD, (d) pristine GO and (e) Poly-βCD. | ||
Fig. 3 shows the attenuated total reflection Fourier-transform infrared spectroscopy (ATR-FTIR) spectra of GO-Poly-βCD in comparison with pristine GO and Poly-βCD. All GO-based samples exhibited a broad band in the 3700–3000 cm−1 region, associated with O–H and N–H stretching vibrations (in the case of GO-Poly-βCD), and the C–H stretching bands of Poly-βCD between 2920 and 2800 cm−1 (in the case of Poly-βCD and GO-Poly-βCD) confirming the presence of the polymer. In the 1800–1500 cm−1 region, GO-Poly-βCD displayed the characteristic carbonyl stretching bands of GO at 1710 and 1610 cm−1, indicative of CO vibrations in carboxylic groups. The band of Poly-βCD at 1636 cm−1 was no longer visible after functionalization, consistent with the chemical modification of the amine groups. Overall, the spectral features confirm the coexistence of both GO and Poly-βCD in the hybrid material.
 | ||
| Fig. 3 ATR-FTIR spectra of GO (black), Poly-βCD (red) and GO-Poly-βCD (blue). | ||
The colloidal stability of GO-Poly-βCD was assessed by turbidimetry and Dynamic Light Scattering (DLS) (Fig. S4, S5 and Table S4). Turbidimetric analysis of 250 µg mL−1 dispersions revealed a two-phase exponential decay with half-lives of ∼35 and 883 min, indicating polydispersity with at least two particle populations. DLS at 62.5 µg mL−1 confirmed a predominant fraction with hydrodynamic diameter, DH ≈ 100 nm alongside larger aggregates (∼570 nm), with ζ-potential ∼−28 mV ensuring stability. pH-dependent studies showed enhanced stability under alkaline conditions, while at pH 2–3 protonation of GO surface groups led to aggregation and increased DH, evidencing partial destabilization.
PFAS adsorption
GO-Poly-βCD nanosheets were tested as sorbents under batch conditions for a mixture of ten PFAS molecules (Fig. 4a), varying in chain length ((CF)3–9) and end-group substitution (sulfonate and carboxylate). | ||
| Fig. 4 (a) Molecular structure of the PFAS molecules, with different chain lengths ((CF)3–9) and end-group substitution (sulfonate and carboxylate) studied in this work. (b) Removal of PFAS mixture at pH 7 (orange) from GO-Poly-βCD compared to GO-Control (blue) and unmodified GO (grey). (c) Experiments at pH 4 (yellow), pH 7 (orange) and pH 10 (green). | ||
The concentration of the PFAS mixture was selected to reflect environmentally relevant levels commonly found in contaminated water sources (0.1–3 µg L−1) and the pH of the tested solution was that of tap water (pH 7). To simulate real-field applications, the adsorption contact time was set to 15 minutes, in line with the typical contact time between water and GAC in municipal water treatment plants (approximately 10–20 minutes). The removal of each contaminant was estimated by UPLC-MS-MS analysis (details in the SI, Section 4). Fig. 4b compares the PFAS removal efficiency of pristine GO, GO-Poly-βCD, and GO-Control after 15 minutes of contact. It can be seen that GO-Poly-βCD outperforms pristine GO for short- and medium-chain PFASs, achieving up to 97% removal for long-chain PFASs with (CF)6–9 length. Medium-chain PFASs ((CF)4–6) are also effectively removed by GO-Poly-βCD, with removal efficiencies exceeding 80%. On the other hand, PFPeA ((CF)4) was adsorbed by GO-Poly-βCD with removal up to 44%, compared to 0% and 11% observed for GO and GO-Control respectively, while PFBA ((CF)3), one of the most persistent PFASs, was not removed at all. Pristine GO showed negligible adsorption for short- and medium-chain PFASs ((CF)3–7), with increasing removal observed for long-chain PFASs, ranging from 14% to 76%. GO-Control, which exhibits a slightly higher degree of reduction than pristine GO (C/O = 1.4 vs. 1.3), showed modest removal of medium-chain PFASs ((CF)4–7) and increasing removal for long-chain PFASs ((CF)8–9), reaching up to 59% for PFDA. Overall, GO-Poly-βCD demonstrates consistently higher adsorption selectivity toward medium-chain PFASs compared to both unmodified GO and GO-Control. This is clearly evidenced by the adsorption of (CF)4–6 chains, which are removed by GO-Poly-βCD with efficiencies ranging from 44% to 97%, whereas GO-Control shows only limited removal, with a maximum of 20% for PFHpA ((CF)6), and pristine GO exhibits negligible removal across this range. Poly-βCD polymers carry positive charges due to multiple protonated amine moieties, which interact with the negatively charged, deprotonated carboxylate groups of GO. As a result, pH variations can strongly influence the adsorption behavior of the GO-Poly-βCD system. We therefore compared the removal performances at pH 4, 7, and 10 (Fig. 4c). Adsorption at pH 4 is consistently higher than at pH 7, with the most pronounced improvements observed for short- and medium-chain PFASs. At pH 4, PFBS, PFHxA, and PFHpA were removed with exceptional efficiencies of 95%, 94%, and 97%, respectively. PFPeA, which exhibited 44% removal at pH 7, showed an increased removal of 80% at pH 4.
Notably, PFBA, which was not removed at neutral pH, was adsorbed at acidic pH with a removal efficiency of 31%. Under basic conditions, the system exhibited substantially reduced adsorption: short- and medium-chain PFASs ((CF)3–6) were removed with negligible to poor efficiency, while long-chain PFASs ((CF)7–9) showed only moderate adsorption, with a maximum removal of 58% for PFHxS, significantly lower than the near-quantitative removal observed under neutral and acidic conditions.
Mechanistic insights of adsorption
To elucidate the binding mechanism responsible for the enhanced removal of PFASs by GO-Poly-βCD at pH 7, molecular dynamics (MD) simulations were performed using perfluorohexanoic acid (PFHxA) as a representative case study. To investigate the specific role of Poly-βCD moieties grafted onto the GO surface, the interactions of PFHxA with both GO and GO-Poly-βCD were analyzed. Five distinct adsorption sites for PFHxA were identified in the GO-Poly-βCD system (Fig. 5), designed to capture a range of interaction environments: adsorption on GO (Case 1), interaction with Poly-βCD (Case 2), encapsulation within the cyclodextrin cavity (Case 3), and sandwich interaction with both GO and Poly-βCD (Case 4). Five independent MD simulations, each 100 ns in length, were carried out from these starting geometries to dynamically sample the interactions between PFHxA and the nanosorbent. The binding free energy between PFHxA and GO-Poly-βCD was calculated in each case using the MM-GBSA approach (Table 1). PFHxA exhibited weak binding to GO (ΔGbinding = −1.9 kcal mol−1 for pristine GO, −2.8 kcal mol−1 for GO-Poly-βCD, Case 1). Similarly, interactions between PFHxA and Poly-βCD alone were very weak (Case 2, –1.8 kcal mol−1). | ||
| Fig. 5 Schematic representation of the interactions between PFHxA and GO-Poly-βCD, highlighting the different adsorption sites. | ||
| PFHxA@ | VDW | E El | E SURF | ΔHbinding | ΔS | ΔGbinding |
|---|---|---|---|---|---|---|
| GO | −16.0 | 4.8 | −1.2 | −12.4 | −10.5 | −1.9 |
| Case 1 | −18.7 | 4.2 | −1.4 | −15.9 | −13.1 | −2.8 |
| Case 2 | −15.2 | 4.4 | −2.7 | −13.4 | −11.6 | −1.8 |
| Case 3 | −29.7 | 6.0 | −4.3 | −28.0 | −15.6 | −12.4 |
| Case 4 | −35.2 | 8.0 | −5.1 | −32.3 | −16.7 | −15.6 |
Encapsulation of PFHxA within the βCD cavity of the Poly-βCD polymer (Case 3) significantly enhanced binding (ΔGbinding = −12.4 kcal mol−1). Notably, the strongest interaction was observed in Case 4, where PFHxA is sandwiched between Poly-βCD and GO, interacting with both components of the nanosorbent, yielding a ΔGbinding of −15.6 kcal mol−1. This value demonstrates that Poly-βCD is more effective than monomeric βCD derivatives (PFBA:βCD = −12.3 kcal mol−1).25 The high affinity arises from a favorable combination of interaction forces, primarily driven by van der Waals interactions, which contribute −35.2 kcal mol−1. These strong dispersion forces result from the close contact between PFHxA and both the GO surface and the overlying Poly-βCD. The electrostatic contribution is slightly unfavorable (Eel = 8.0 kcal mol−1), likely due to repulsion between the negatively charged head group of PFHxA and the GO surface, as well as desolvation of the PFAS head group.
In contrast, non-polar solvation contributions were favorable (ESURF = −5.1 kcal mol−1), reflecting burial of the PFAS aliphatic chain within the cleft formed by Poly-βCD and the GO sheet. The entropy term (ΔS = −16.7 kcal mol−1) represents a binding penalty due to the conformational restriction of PFHxA upon adsorption. These results indicate that stable adsorption and retention of PFASs require the combined action of both Poly-βCD and GO moieties, highlighting the necessity of both components for effective pollutant capture and supporting a cooperative “sandwich-like” mechanism as the most favorable binding mode. The binding energies of all investigated PFASs on GO-Poly-βCD were also calculated, reproducing the experimentally observed trends (Fig. S3).
To assess the influence of pH on PFAS adsorption, MD simulations were performed for a model system (PFHxA adsorption on GO-Poly-βCD) at pH 4, 7, and 10, and the corresponding binding free energies were calculated (Table 2). A net decrease in binding affinity was observed with increasing pH: ΔGbinding = −19.9, −15.6, and −10.6 kcal mol−1 at pH 4, 7, and 10, respectively, consistent with the experimental removal trend (Fig. 4c). Electrostatic repulsion (Eel) between PFHxA and GO-Poly-βCD increases as pH rises, since PFHxA remains deprotonated across the studied range (pKa = −0.16). The improved adsorption under acidic conditions is primarily attributed to stronger van der Waals interactions, which are unexpectedly influenced by the pH. Structural analysis revealed a pH-dependent conformational change of the nanosystem, affecting the accessibility and stability of PFAS adsorption sites (Fig. 6). At pH 4, Poly-βCD remains closely associated with GO through hydrogen bonding, forming tight binding clefts (365.04 Å2) for PFHxA.
| PFHxA@ GO-Poly-βCD | VDW | E El | E SURF | ΔHbinding | ΔS | ΔGbinding |
|---|---|---|---|---|---|---|
| pH 4 | −37.8 | 7.8 | −5.1 | −35.2 | −15.3 | −19.9 |
| pH 7 | −35.2 | 7.9 | −5.1 | −32.3 | −16.7 | −15.6 |
| pH 10 | −31.2 | 10.6 | −3.2 | −23.7 | −13.1 | −10.6 |
 | ||
| Fig. 6 Sketch of PFHxA@GO-Poly-βCD and relative cleft formation and calculated area at (a) pH 4, (b) pH 7 and (c) pH 10. | ||
These clefts become less well-defined at pH 7 (443.94 Å2), while at pH 10, Poly-βCD partially detaches from the GO surface, disrupting the interaction network and dismantling the clefts responsible for PFHxA adsorption. Consequently, under basic conditions, PFHxA remains largely exposed on the GO surface and is only partially covered by the polymer, breaking the sandwich-like adsorption site, reducing the synergistic binding effect, and resulting in overall weaker interactions. This indicates that the adsorption of PFHxA—and PFASs in general—on GO-Poly-βCD is primarily hydrophobically driven. This behavior is also reflected in the non-polar solvation term (ESURF), which becomes less stabilizing at higher pH (ESURF = −3.2 kcal mol−1 at pH 10 vs. −5.1 kcal mol−1 at pH 4). As with van der Waals interactions, ESURF depends directly on the contact area between the adsorption site and the PFAS, i.e., their shape complementarity.
Although it was initially hypothesized that pH would primarily modulate electrostatic interactions between PFAS and the GO-Poly-βCD surface—particularly involving the protonated amines of Poly-βCD—our findings indicate that the critical factor is the pH-dependent conformational rearrangement of the hybrid material. At lower pH, stronger interactions between GO and Poly-βCD generate well-defined clefts that facilitate efficient guest inclusion and encapsulation of PFAS.
To further confirm the pivotal role of Poly-βCD in modulating the adsorption behavior of the hybrid material under different pH conditions, the removal performance of pristine GO was also evaluated across pH 4, 7, and 10 (Fig. S6). Short- and medium-chain PFASs (CF3–6) exhibited negligible removal at all tested pH values. A modest improvement was observed at pH 4, with removal increasing to 16% for PFHpA and 29% for PFHxS. This can be attributed to the partial neutralization of the negative surface charge of GO under acidic conditions, slightly reducing electrostatic repulsion with the anionic PFASs. For long-chain PFASs (CF7–9), removal efficiency increased more substantially under acidic conditions, reaching up to 91% in the case of PFDA. At neutral and basic pH, only minor improvements were observed for PFNA, while PFOS and PFDA showed higher removal efficiencies, though they never exceeded 80%. The gradual increase in adsorption with chain length under all pH conditions reflects the stronger hydrophobic interactions of longer-chain PFASs. Overall, the negligible adsorption of pristine GO under most conditions highlights the essential role of Poly-βCD in stabilizing PFASs and corroborates the experimental and computational findings described above.
PFAS adsorption of GO-Poly-βCD vs. GAC
To evaluate the potential of GO-Poly-βCD for drinking water treatment, its adsorption performance was compared with that of GAC, representing the current industrial sorbent benchmark widely employed in treatment plants for tertiary and quaternary water treatment as well as in point-of-use purification systems.The comparison was performed in tap water at neutral pH, with contact time of 15 minutes, conditions that closely reflect real-world applications. Fig. 7 shows the removal efficiencies of GO-Poly-βCD and GAC, expressed as micrograms of PFAS removed per gram of sorbent (µg g−1). The data clearly indicate that GO-Poly-βCD outperforms GAC in PFAS adsorption. While GAC achieves a maximum removal of 0.2 µg g−1 for most of the selected PFASs, GO-Poly-βCD reaches up to 0.5 µg g−1 within the same contact time. For PFPeA ((CF)4), GAC removes 0.1 µg g−1, slightly lower than the 0.2 µg g−1 achieved by GO-Poly-βCD. The shortest-chain PFAS, PFBA, is only minimally adsorbed by GAC (0.07 µg g−1) and remains undetectable for GO-Poly-βCD. The advantage of GO-Poly-βCD becomes even more pronounced when considering total PFAS removal, which reaches 4 µg g−1 compared to 1.3 µg g−1 for GAC. In addition to higher removal efficiencies, GO-Poly-βCD exhibits rapid adsorption kinetics, achieving significant PFAS uptake within just 15 min. This feature is particularly relevant for practical drinking water purification, including both household point-of-use filters and industrial treatment systems operating at short contact times.
 | ||
| Fig. 7 PFAS removal performances of GO-Poly-βCD and GAC. | ||
Conclusions
In conclusion, we report the synthesis of a novel GO material covalently grafted with poly-β-cyclodextrin crosslinked with epichlorohydrin. The resulting hybrid is insoluble in water, unlike previously reported β-cyclodextrin-based polymers that often exhibit partial solubility, making it more water-processable and suitable for practical applications in aqueous environments. GO-Poly-βCD material adsorption performances were evaluated on a mixture of PFASs with varying chain lengths ((CF)3–9) and acidic and sulfonic functional groups. Batch adsorption experiments demonstrated that GO-Poly-βCD outperforms pristine GO in the removal of medium-chain PFASs, with remarkable efficiencies of 97% for PFHxA and PFHpA ((CF)6–7) and 80% for PFPeA ((CF)4), whereas pristine GO and GO-Control exhibited negligible adsorption in this range. Compared to GAC, GO-Poly-βCD achieved higher overall PFAS uptake within 15 minutes of contact, highlighting its superior performance under field-relevant conditions. Molecular dynamics simulations revealed that PFAS adsorption is governed by a synergistic mechanism, in which contaminants are stabilized through a pH-responsive conformational arrangement of GO grafted with poly-βCD. Overall, these findings provide new insights into the design of hybrid nanomaterials for PFAS remediation and underscore the potential of poly-β-cyclodextrin-based GO materials as efficient and sustainable sorbents for drinking water treatment.Author contributions
Andrea Trifoglio: methodology, investigation, reviewing editing, writing original draft. Angela Pintus: methodology, investigation. Sara Khaliha: methodology, investigation. Marco Agnes: methodology. Lucrezia Aversa: methodology, investigation. Roberto Verucchi: methodology, investigation. Nina Burduja: investigation. Giuseppe Nocito: methodology, investigation and review original draft. Antonino Mazzaglia: methodology and review original draft. Tainah Dorina Marforio: investigation, methodology, formal analysis. Matteo Calvaresi: methodology, investigation. Manuela Melucci: coordination, conceptualization, validation, writing original draft.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): GO-Poly-βCD loading calculation, X-ray photoelectron spectroscopy (XPS), binding affinities, UPLC-MS/MS details, colloidal stability studies, and PFAS adsorption from GO at different pH values. See DOI: https://doi.org/10.1039/d5ta08410e.Acknowledgements
MM and MA thank Dr I. Manet and Dr A. Kovtun for their scientific support and Cyclolab for kindly providing Poly-βCD-NH2. The authors gratefully acknowledge the support of this work by the projects Life-Remembrance, ENV/IT/001001 Life Resource and Environment LIFE20 ‘Give plastic wastes from the production of hollow-fiber membranes a second life’, project PNRR MUR project ECS_00000033_ECOSISTER-ATOS and project Capitale Naturale e Risorse per il futuro dell’Italia (FOE 2020, DTA.AD005.314). M.A. thanks European Union's Horizon Europe Research and Innovation Programme under the MSCA grant agreement Bicyclos No. 101130235. Views and opinions expressed are however those of the author(s) only and do not necessarily reflect those of the European Union. TDM was supported by Fondazione Umberto Veronesi.Notes and references
- Nat. Water, 2023, 1, 993 Search PubMed.
- O. S. Arvaniti and A. S. Stasinakis, Sci. Total Environ., 2015, 524–525, 81–92 CrossRef CAS.
- M. Sun, E. Arevalo, M. Strynar, A. Lindstrom, M. Richardson, B. Kearns, A. Pickett, C. Smith and D. R. U. Knappe, Environ. Sci. Technol. Lett., 2016, 3, 415–419 CrossRef CAS.
- S. Garg, J. Wang, P. Kumar, V. Mishra, H. Arafat, R. S. Sharma and L. F. Dumée, J. Environ. Chem. Eng., 2021, 9, 105784 CrossRef CAS.
- J. Gardiner, Aust. J. Chem., 2015, 68, 13–22 CrossRef CAS.
- S. Dolui, D. Kumar, S. Banerjee and B. Ameduri, Acc. Mater. Res., 2021, 2, 242–251 Search PubMed.
- Z. Du, S. Deng, Y. Bei, Q. Huang, B. Wang, J. Huang and G. Yu, J. Hazard. Mater., 2014, 274, 443–454 CrossRef CAS.
- T. H. Boyer, Y. Fang, A. Ellis, R. Dietz, Y. J. Choi, C. E. Schaefer, C. P. Higgins and T. J. Strathmann, Water Res., 2021, 200, 117244 CrossRef CAS.
- P. McCleaf, S. Englund, A. Östlund, K. Lindegren, K. Wiberg and L. Ahrens, Water Res., 2017, 120, 77–87 CrossRef CAS PubMed.
- S. Khaliha, A. Bianchi, A. Kovtun, F. Tunioli, A. Boschi, M. Zambianchi, D. Paci, L. Bocchi, S. Valsecchi, S. Polesello, A. Liscio, M. Bergamini, M. Brunetti, M. Luisa Navacchia, V. Palermo and M. Melucci, Sep. Purif. Technol., 2022, 300, 121826 CrossRef CAS.
- E. Gagliano, M. Sgroi, P. P. Falciglia, F. G. A. Vagliasindi and P. Roccaro, Water Res., 2020, 171, 115381 CrossRef CAS PubMed.
- A. M. Kennedy, A. M. Reinert, D. R. U. Knappe, I. Ferrer and R. S. Summers, Water Res., 2015, 68, 238–248 CrossRef CAS PubMed.
- S. Khaliha, F. Tunioli, L. Foti, A. Bianchi, A. Kovtun, T. D. Marforio, M. Zambianchi, C. Bettini, E. Briñas, E. Vázquez, L. Bocchi, V. Palermo, M. Calvaresi, M. L. Navacchia and M. Melucci, Environ. Sci.:Water Res. Technol., 2024, 10, 1097–1107 RSC.
- S. Khaliha, T. D. Marforio, A. Kovtun, S. Mantovani, A. Bianchi, M. Luisa Navacchia, M. Zambianchi, L. Bocchi, N. Boulanger, A. Iakunkov, M. Calvaresi, A. V. Talyzin, V. Palermo and M. Melucci, FlatChem, 2021, 29, 100283 CrossRef CAS.
- S. Guo, S. Garaj, A. Bianco and C. Ménard-Moyon, Nat. Rev. Phys., 2022, 4, 247–262 Search PubMed.
- M. Melucci, L. Bocchi, M. Zambianchi and V. Palermo, Nat. Water, 2025, 3, 369–371 CrossRef CAS.
- S. Mantovani, S. Khaliha, L. Favaretto, C. Bettini, A. Bianchi, A. Kovtun, M. Zambianchi, M. Gazzano, B. Casentini, V. Palermo and M. Melucci, Chem. Commun., 2021, 57, 3765–3768 Search PubMed.
- S. Mantovani, S. Khaliha, T. D. Marforio, A. Kovtun, L. Favaretto, F. Tunioli, A. Bianchi, G. Petrone, A. Liscio, V. Palermo, M. Calvaresi, M. L. Navacchia and M. Melucci, Chem. Commun., 2022, 58, 9766–9769 RSC.
- A. Trifoglio, S. Mantovani, S. Khaliha, A. Kovtun, T. D. Marforio, M. Calvaresi and M. Melucci, Nanoscale, 2025, 17, 12124–12133 RSC.
- G. Crini, Chem. Rev., 2014, 114, 10940–10975 CrossRef CAS PubMed.
- A. H. Karoyo, A. S. Borisov, L. D. Wilson and P. Hazendonk, J. Phys. Chem. B., 2011, 115, 9511–9527 CrossRef CAS PubMed.
- P. Lo Nostro, I. Santoni, M. Bonini and P. Baglioni, Langmuir, 2003, 19, 2313–2317 CrossRef CAS.
- H. Tatsuno and S. Ando, J. Phys. Chem. B., 2006, 110, 25751–25760 CrossRef CAS PubMed.
- A. H. Karoyo, P. S. Sidhu, L. D. Wilson, P. Hazendonk and A. Borisov, J. Phys. Chem. C., 2015, 119, 22225–22243 Search PubMed.
- F. Tunioli, T. D. Marforio, L. Favaretto, S. Mantovani, A. Pintus, A. Bianchi, A. Kovtun, M. Agnes, V. Palermo, M. Calvaresi, M. L. Navacchia and M. Melucci, Chem.–Eur. J., 2023, 29, e202301854 Search PubMed.
- Y. Miyah, N. El Messaoudi, M. Benjelloun, J. Georgin, D. S. P. Franco, M. El-habacha, O. A. Ali and Y. Acikbas, Carbohydr. Polym., 2025, 350, 122981 CrossRef CAS.
- L. Xiao, Y. Ling, A. Alsbaiee, C. Li, D. E. Helbling and W. R. Dichtel, J. Am. Chem. Soc., 2017, 139, 7689–7692 CrossRef CAS PubMed.
- A. Alsbaiee, B. J. Smith, L. Xiao, Y. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS.
- J.-L. Sun, H. Zeng and H.-G. Ni, Chemosphere, 2013, 90, 1751–1759 CrossRef CAS.
- C. Belenguer-Sapiña, E. Pellicer-Castell, A. R. Mauri-Aucejo, E. F. Simó-Alfonso and P. Amorós, Nanomaterials, 2021, 11, 1 Search PubMed.
- A.-M. Hada, M. Potara, S. Astilean, A. Cordaro, G. Neri, M. Malanga, A. Nostro, A. Mazzaglia, A. Scala and A. Piperno, Carbohydr. Polym., 2022, 293, 119736 CrossRef CAS PubMed.
- X. Deng, A. Verdaguer, T. Herranz, C. Weis, H. Bluhm and M. Salmeron, Langmuir, 2008, 24, 9474–9478 Search PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |