
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegulating Ni oxidation states through ruthenium incorporation in Ni based catalysts
Laura Mallón†
a,
Laurent Peres†b,
Nicolas Rivasc,
Alba Garzón Manjón‡
c,
Cristina Scheuc,
Marcos Gil-Sepulcread,
Olaf Rüdiger d,
Serena DeBeerd,
Nuria Romerob,
Jérôme Esvane,
Jordi García-Antóna,
Luis Rodríguez-Santiagof,
Xavier Solans-Monfortf,
Roger Bofilla,
Karine Philippot*b,
Laia Francàs*a and
Xavier Sala*a
d,
Serena DeBeerd,
Nuria Romerob,
Jérôme Esvane,
Jordi García-Antóna,
Luis Rodríguez-Santiagof,
Xavier Solans-Monfortf,
Roger Bofilla,
Karine Philippot*b,
Laia Francàs*a and
Xavier Sala*a
aDepartament de Química, Unitat de Química Inorgànica, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain. E-mail: laia.francas@uab.cat; xavier.sala@uab.cat
bCNRS, LCC (Laboratoire de Chimie de Coordination), UPR8241, University of Toulouse, UPS, INPT, Toulouse, Cedex 4 F-31077, France. E-mail: karine.philippot@lcc-toulouse.fr
cMax Planck Institute for Sustainable Materials GmbH, Max-Planck-Str. 1, 40237 Düsseldorf, Germany
dMax Planck Institute for Chemical Energy Conversion, Stiftstrasse 34-36, D-45470 Mülheim an der Ruhr, Germany
eInstitut Carnot – Centre Inter-universitaire de Recherche et d’Ingénierie des Matériaux, INP-ENSIACET, CNRS, Université de Toulouse, 118, route de Narbonne, 31062 Toulouse, France
fDepartament de Química, Universitat Autònoma de Barcelona, Cerdanyola del Vallès, 08193 Barcelona, Spain
First published on 28th January 2026
Abstract
NiFe-based materials are state-of-the-art electrocatalysts for water oxidation at alkaline pH. Several strategies to improve their activity have been reported, amongst which Ru-incorporation has appeared as a suitable approach. In this work, three Ni based nanomaterials have been prepared through organometallic synthesis and surface-decorated with small (sub-nanometric) Ru clusters (Ru(L)@Ni-NW) or large (ca. 8 nm) Ru nanoparticles (Ru(H)@Ni-NW and Ru(HH)@Ni-NW). As model systems, Ru(L)@Ni-NW and Ru(H)@Ni-NW have been thoroughly characterized by a complementary set of advanced techniques, including atom probe tomography, X-ray absorption spectroscopy, X-ray photoelectron spectroscopy and high-angle annular dark-field scanning transmission electron microscopy. Our study reveals that Ru nanoparticles remain unstable under electrocatalytic oxygen evolution reaction (OER) conditions, leaching from the Ni based NW surface. In contrast, sub-nanometric Ru clusters remain stable on the Ni based NWs and modify the Ni oxidation states at the surface sites, outperforming the counterparts that contain no Ru or Ru nanoparticles. The spectroelectrochemical and DFT modelling results suggest the interaction between the Ru sub-nanometric clusters and the Ni sites as the origin of the stabilization of Ni at higher oxidation states, boosting the OER efficiency under both Fe-containing (unpurified electrolyte) and Fe-free (purified electrolyte) conditions.
1. Introduction
Harnessing and storing solar energy in chemical bonds is a powerful alternative to current non-renewable and pollutant energy sources.1 Amongst all the different possible strategies to achieve this objective, using green electricity to split water to generate H2 (as a clean fuel) and O2 is an attractive approach.2,3 However, the extraction of four electrons and four protons from two water molecules to form an oxygen–oxygen double bond is a thermodynamically uphill and kinetically slow reaction, requiring the use of efficient catalysts.4,5 Thus, from the anode perspective, the fine tuning of water oxidation catalysts to enhance their performance towards oxygen evolution is the cornerstone in the field. Fe-containing nickel-based materials are state-of-the-art electrocatalysts when performing water oxidation (OER) under basic conditions (pH 14). Over ten years, several studies demonstrated that the exceptional activity of Ni-based catalysts originates from the spontaneous incorporation of trace amounts of Fe from the electrolyte.6–11 Since then, different studies have been devoted to understand the role of the incorporated Fe in the catalysis, increasing the conductivity,6 acting as active species7,8 or synergistically working with the Ni centers to evolve O2.9–11 However, this issue is still a subject of debate in the literature.12–17 These studies also revealed that (1) there was a strong dependence of catalytic activity on the degree of Fe incorporation, with ca. 25% representing the optimal fraction;7 (2) the regulation of the Ni(III/II) redox potential was clearly observed, with Ni(II) being more stable after Fe incorporation.6,18,19Among the different strategies that have been used to further increase the activity of Ni-based catalysts,20 the incorporation of metal elements has been shown to be a promising route.21–24 Several studies confirmed the improvement of the electrocatalytic water oxidation activity of Ni-based nanomaterials by Ru incorporation via both atomically distributed Ru (single atoms)21,22,25 or Ru/RuO2 nanoparticles (NPs) (Table S1).23,26 The high OER activity reported when incorporating RuO2 NPs has been widely attributed to synergistic effects between Ru and Ni. It has been reported that at a RuO2/NiO(OH) interface,26 dissociation of both H2O and OH− (which is not optimal in NiO(OH) or in RuO2 separately) can be coupled and optimized to produce the −OOH intermediate, which can be further deprotonated to release O2. This has been attributed to the ability of NiO(OH) to favour the dissociation of water molecules27 and that of RuO2 to dissociate OH− moieties.28,29 It has also been reported that Ru can aid the electron transfer process between the water substrate and the intermediate species, facilitating O–O bond formation at basic pH.23 On the other hand, when dispersed single atoms are incorporated into Ni-based catalysts, a change in the electronic structure of the resulting catalysts is detected.21,22,25 In all these studies, DFT calculations reveal a change in the Ni d band position, thus provoking the optimization of the adsorption energy of intermediate species. In addition, a recent report has related the incorporation of Ru single atoms into Ni-based catalysts with the stabilization of Ni high oxidation states and dynamic surface reconstruction.25 All these studies demonstrate that the effect of incorporated Ru depends on its size and morphology, with Ru ranging from single atoms to nanoparticles, influencing Ni-based catalysts in different ways, from directly promoting O–O bond formation to modulating oxidation states and the electronic structure. However, despite the number of reports devoted to the effect of Ru incorporation on the OER performance of Ni-based electrocatalysts, no study has yet compared the effect of Ru species of different sizes and morphologies (i.e., clusters and NPs) on a single Ni-based substrate. Different Ru morphologies, from single atoms to clusters or NPs, can interact with the Ni host in fundamentally distinct ways, potentially altering the electronic structure, oxidation states and the adsorption energies of reaction intermediates, or determining the stability of the hybrid materials under electrocatalytic turnover. The thorough structural and electronic characterization of the interface of different morphologies of incorporated Ru species within a given Ni-based material is thus of particular interest to unravel the structure–function relationships that govern OER efficiency and enable the rational design of improved OER electrocatalysts.
In this work, we report the synthesis of three Ru-incorporating Ni-based nanomaterials that consist of Ni-based nanoworms (NWs) post-functionalized with three different amounts of Ru (0.4 wt%, 3.2 wt% and 6.4 wt%), which have been prepared through the organometallic synthetic method,30 allowing a fine control of the nanostructure (Ni NWs, Ru NPs or subnanometric Ru clusters) and surface composition. The nanomaterials containing 0.4 and 3.2 wt% Ru have been fully characterized by a complementary set of advanced microscopy and spectroscopic techniques, revealing that Ru is present as small (sub-nanometric) clusters finely distributed throughout all the Ni based surface in the low Ru containing material, while ca. 8 nm NPs are present in the high Ru containing one. The 6.4 wt% Ru containing nanomaterial was not fully characterized due to the similar Ru NP structure formed in the 3.2 wt% system. When the two selected nanomaterials are tested for the electrochemical OER under alkaline conditions, the low Ru incorporating Ni-based electrocatalyst outperforms both the non-incorporating and the high Ru-incorporating counterparts. The reasons behind this superior performance are discussed in view of the structural and electronic features of the synthesized nanomaterials. Our results highlight the importance of tuning the size and nature of the incorporated Ru to modulate the interaction with the Ni catalyst, adjusting the influence of the incorporated metal on the Ni electronic structure and enhancing the resulting OER performance.
2. Experimental section
2.1. Reagents
All procedures concerning the synthesis and preparation of samples were carried out using standard Schlenk tubes, Fisher-Porter glassware and vacuum line techniques or with the use of a glove-box (MBraun) under an argon atmosphere. Tetrahydrofuran (THF) was obtained from Carlo Erba, purified using purification MBraun SPS-800 equipment and degassed with three freeze–pump–thaw cycles before use. Absolute anhydrous ethanol (Carlo Erba, ACS reagent) was dried over a molecular sieve and degassed by Ar bubbling before use. Bis(1,5-cyclooctadiene)nickel(0) [Ni(COD)2] (>98%, Strem Chemicals) and [Ru(Me-allyl)2(COD)] (Sigma-Aldrich) were stored under argon inside a glovebox (MBraun). Deionized water was obtained from Millipore (MilliQ, 18.2 MΩ cm−1; Millipore, Bedford, MA). Ar and H2 were purchased from Air Liquide (Alphagaz).2.2. Synthetic procedures
2.3. Characterization techniques
Transmission electron microscopy (TEM) and high resolution transmission electron microscopy (HRTEM) analyses were performed at the “Centre de microcaractérisation Raimond Castaing, CNRS UAR 3623, Toulouse” by using a JEOL JEM 1400 operating at 120 kV with a point resolution of 2.0 Å. High resolution analyses were conducted using a JEOL JEM 2100F equipped with a Field Emission Gun (FEG) operating at 200 kV with a point resolution of 2.3 Å and a JEOL JEM-ARM200F Cold FEG (cold field emission gun) operating at 200 kV with a point resolution of 1.9 Å and coupled to an EDX spectrometer and an electron energy loss spectrometer (EELS).![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 117 and 8333 eV, respectively. A step size of 0.2 eV was used in the XANES and EXAFS regions (1 s integration time). Final spectra were processed and normalized using the Athena program, included in the DEMETER package.33
117 and 8333 eV, respectively. A step size of 0.2 eV was used in the XANES and EXAFS regions (1 s integration time). Final spectra were processed and normalized using the Athena program, included in the DEMETER package.332.4. Computational details
For each model, different types of OH groups of the Ni(OH)2 surface were identified as a function of the distance from and position of the Ru clusters. For these different H atoms of Ni(OH)2, the feasibility of their oxidation was evaluated through a proton coupled electron transfer (PCET) process described as
| *OH → *O + H+ + 1e− | (1) |
The ΔG0 values for the PCET step described in eqn (1) were computed by using the computational standard hydrogen electrode as defined by Rossmeisl, Nørskov and co-workers, which allows replacing a proton and an electron with half a hydrogen molecule at U = 0 V vs. SHE.48 Then, under standard conditions, the free energy of the reaction (eqn (1)) can be calculated as the free energy of
| *OH → *O + ½H2 | (2) |
For this reaction, ΔG0 can be approximated as ΔG at U = 0, pH = 0, P = 1 bar and T = 298.15 K. Thus, ΔG0 = ΔE + ΔZPE – TΔS, where ΔE is calculated using DFT and vZPE and ΔS were calculated using the DFT computed vibrational frequencies. Entropy contributions for H2 were obtained from the tabulated values. At a pH different from 0 the free energy of the H+ ions can be corrected by considering the concentration dependence: ΔGpH (pH) = −kT ln[H+]. Thus, the free energy of the reaction is calculated as
| ΔG0(pH) = ΔG0(pH = 0) + ΔGpH(pH) | (3) |
3. Results and discussion
3.1. Synthesis of the Ru-incorporating Ni-based nanomaterials
The Ru-incorporating Ni-based nanomaterials were synthesized by an organometallic approach30 following a two-step procedure (Scheme 1). First, nickel nanoworms (Ni-NWs) were produced by hydrogenation of the [Ni(COD)2] complex (COD = 1,5-cyclooctadiene), in ethanol/THF (10% v/v), under 3 bar of H2 at 70 °C, as previously reported.31 These Ni-NWs are made of agglomerated Ni NPs, which provide a porous structure to the Ni-based nanomaterial.31 Second, Ru incorporation was performed on top of the pre-synthesized Ni-NWs by reduction of the [Ru(Me-allyl)2(COD)] (Me-allyl = 2-methylallyl) complex as a Ru source. In this second step, the pre-synthesized Ni-NWs were first mixed with a THF solution of the Ru precursor for 3 days at room temperature under vigorous stirring to ensure the impregnation of the precursor all over the Ni-NW nanomaterials. Then, the Ru-impregnated Ni-NW nanomaterials were exposed to 3 bar of H2 at 50 °C to induce the reduction of the Ru precursor (Fig. 1). For comparative purposes, the Ru impregnation was performed at three different Ru loadings (1, 5 and 10 wt%, theoretical values), yielding three Ru-incorporating nanomaterials, the one corresponding to low (L) Ru content, Ru(L)@Ni-NW, the one corresponding to the high (H) Ru content (5%), Ru(H)@Ni-NW, and the one corresponding to very high (HH) Ru content (10%), Ru(HH)@Ni-NW, respectively. All nanomaterials were allowed to passivate under slow atmospheric air diffusion inside a closed vial for 3 weeks before running the electrocatalytic tests. This passivation step ensured homogeneous and comparable catalysts by minimizing variations in air exposure that could modify the active surface and compromise reproducibility in OER measurements. The latter is a key parameter when studying catalytic processes.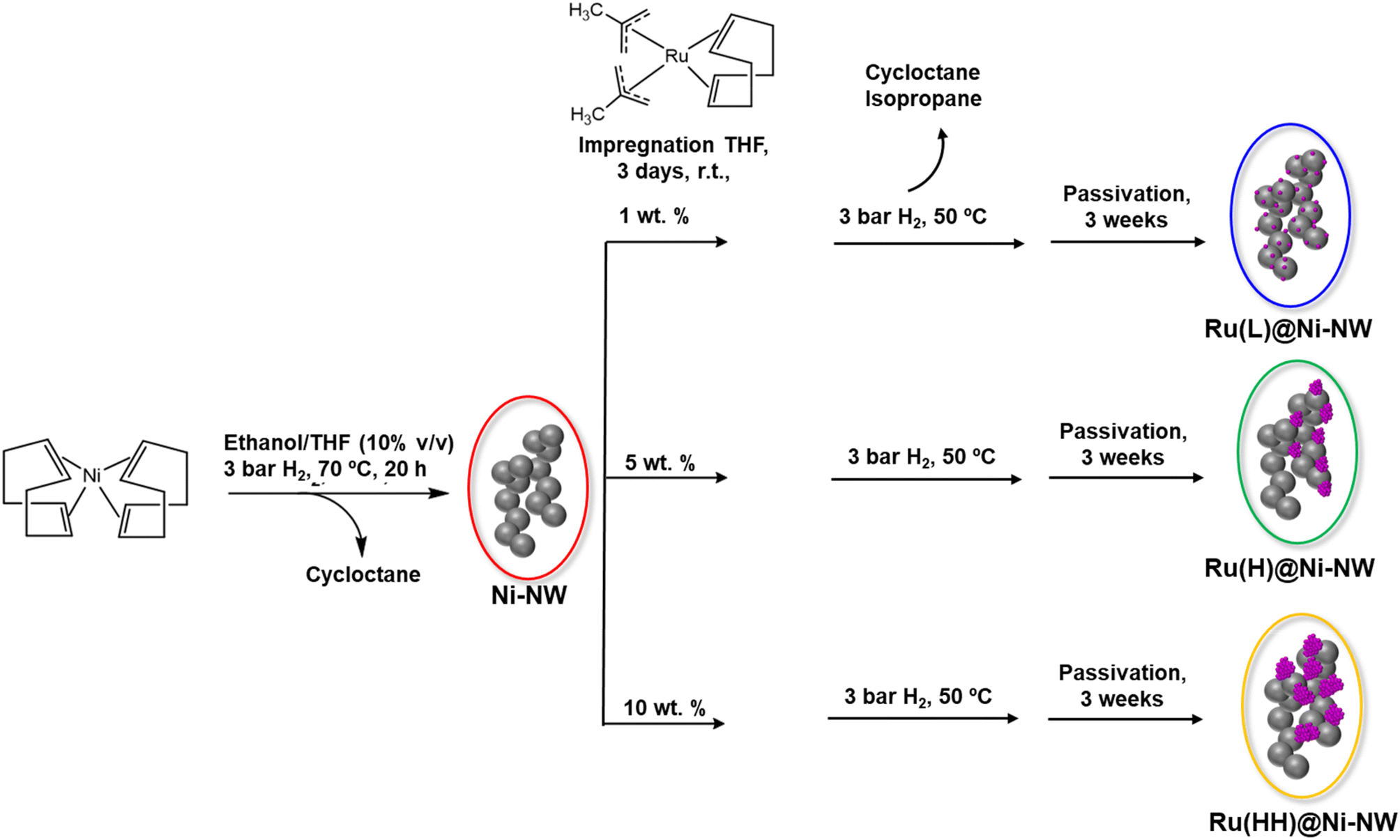 | ||
| Scheme 1 Organometallic synthesis of pristine Ni-NWs and three Ru@Ni-NW nanomaterials. | ||
 | ||
| Fig. 1 From top to bottom: Ni-NW characterization by TEM (a) and SEM (b); Ru(L)@Ni-NW characterization by TEM (c), SEM (d) and EDX analysis (e); Ru(H)@Ni-NW characterization by TEM (f), SEM (g) and EDX mapping of Ni Kα1 (red) and Ru Lα1 (green) (h); Ru(HH)@Ni-NW characterization by TEM (i), SEM (j) and EDX mapping of Ni Kα1 (red) and Ru Lα1 (green) (k). The arrows in (f) and (i) show the presence of small Ru NPs. | ||
3.2. Structural characterization of Ni-NWs, Ru(L)@Ni-NW and Ru(H)@Ni-NW
State-of-the-art techniques such as transmission electron microscopy (TEM), scanning electron microscopy (SEM), high-angle annular dark-field scanning transmission electron microscopy coupled with energy dispersive X-ray spectroscopy (HAADF-STEM- EDX), powder X-ray diffraction (PXRD), inductively coupled plasma optical emission spectroscopy (ICP-OES), X-ray photoelectron spectroscopy (XPS), atomic probe tomography (APT) and X-ray absorption spectroscopy (XAS) were used to structurally and chemically characterize the Ni-NW, Ru(L)@Ni-NW and Ru(H)@Ni-NW nanomaterials. For Ru(HH)@Ni-NW only TEM and SEM characterization was performed due to the encountered similarity to Ru(H)@Ni-NW.The HAADF-STEM analysis of Ni-NWs, Ru(L)@Ni-NW and Ru(H)@Ni-NW (Fig. S2) confirmed the TEM results. The quantity of the Ru precursor mixed with the Ni-NWs appeared to have a direct impact on the structural characteristics and arrangement of the Ru atoms within the Ni-NWs. Thus, while the HAADF-STEM analysis of the Ru-incorporating Ni-NW nanomaterial prepared with the low Ru concentration, Ru(L)@Ni-NW, did not show the presence of Ru NPs, that of the nanomaterial prepared with high Ru incorporation, Ru(H)@Ni-NW, clearly showed the presence of Ru NPs (Fig. S2 and 1f).
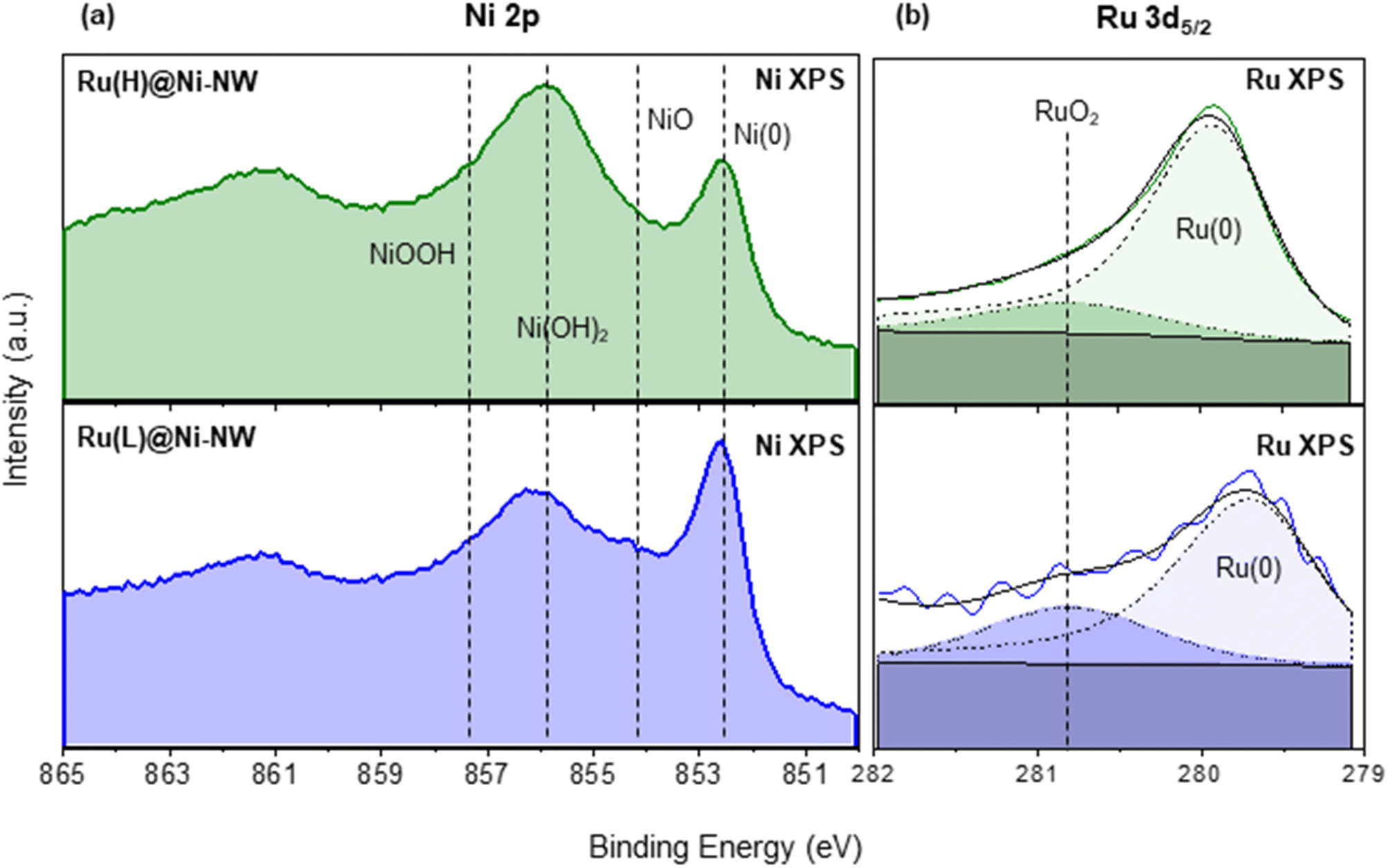 | ||
| Fig. 2 Ni 2p3/2 XPS spectra of Ru(L)@Ni-NW (blue) and Ru(H)@Ni-NW (green) (a). Ru 3d5/2 XPS spectra of Ru(L)@Ni-NW (blue) and Ru(H)@Ni-NW (green) (b). Metallic-Ru component (Ru 3d5/2 279.7–280.3 eV, dashed black), RuO2-component (Ru 3d5/2 280.8 eV, dotted black), and envelope (bold black). | ||
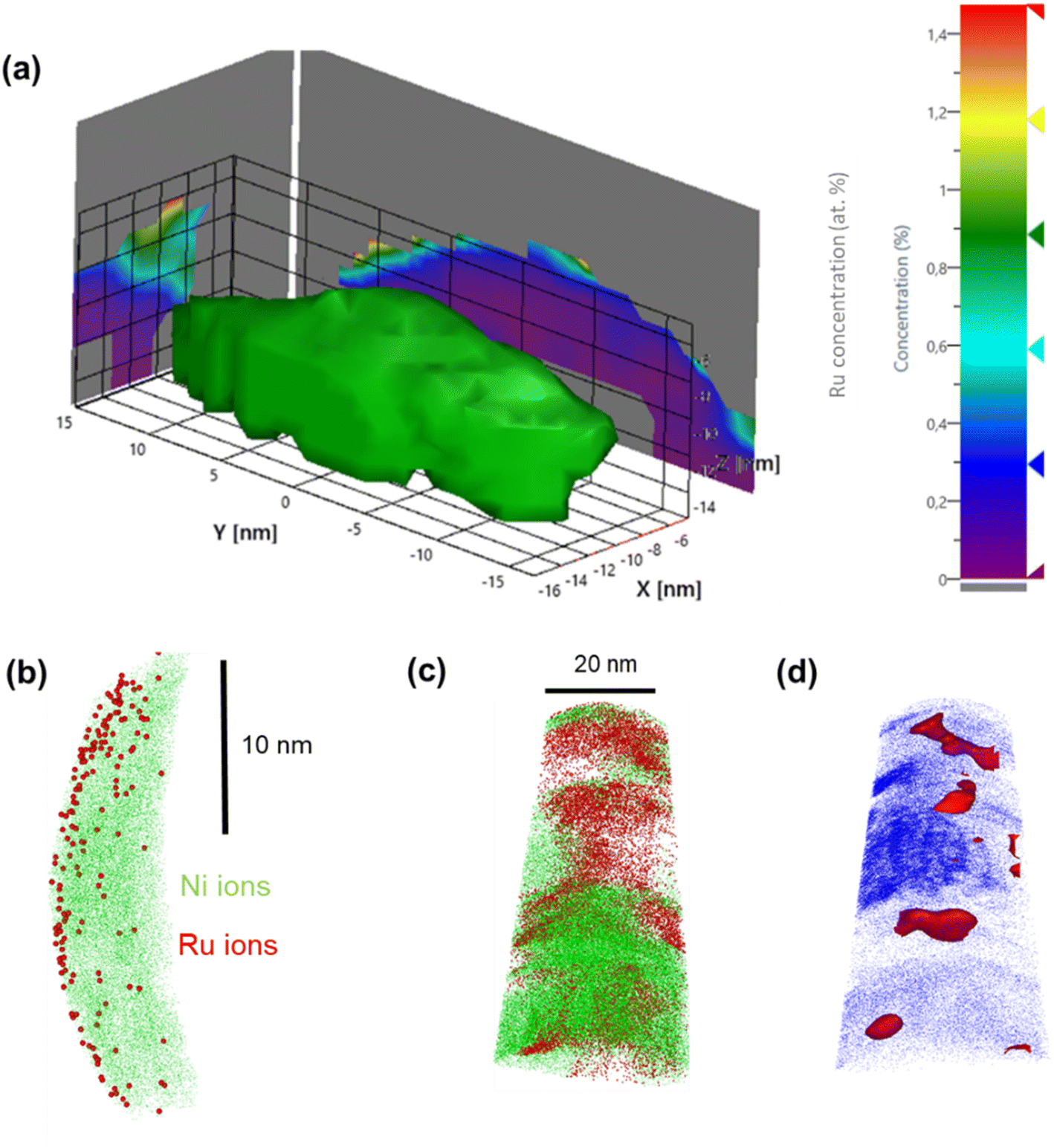 | ||
| Fig. 3 APT reconstruction of a Ru(L)@Ni-NW segment: Ni iso-concentration surface delineating the 3-D NW segment structure along with the 2-D Ru concentration map projected on the x-y plane (a). 2-D Ni and Ru ion distribution; Ru ions are localized at the surface of the Ni-NW (b). APT reconstruction of a Ru(H)@Ni-NW segment: Ni (green) and Ru (red) ion distribution at the 3.2% Ru iso-concentration surface (red) (c) and along the APT specimen delineated by the Co embedding matrix (blue) (d). | ||
XPS analysis in the Ru 3d5/2 region gave evidence of the presence of Ru in two different phases, namely metallic Ru (Ru(0)) and RuO2. Indeed, the intense peak can be deconvoluted into one component in the range of 279.7–280.3 eV (Ru(0)) and another one at 280.8 eV (RuO2) (Fig. 2b).55 Actually, and as expected after air exposure, the RuO2 contribution can be attributed to the formation of a RuO2 layer over the Ru(0) NPs.43,56 Interestingly, as shown in Fig. 2b, the Ru(0) component in Ru(L)@Ni-NW (279.6 eV) appears at lower binding energies than for Ru(H)@Ni-NW (279.9 eV) and related Ru NPs reported by our group,43,56,57 pointing at an effective interaction (electron transfer from Ni to Ru) between the two metals in the low Ru content system, analogously as described for NiRu alloys, where the electron donation from Ni to Ru provokes a negative shift (decrease in energy) of the Ru 3d5/2 peak.53
In contrast, APT measurements of Ru(H)@Ni-NW showed a different Ru distribution along the Ni-NW surface. This is highlighted in Fig. 3c, where the higher concentration of Ru appears as Ru agglomerates throughout the Ni-NW nanomaterial. An iso-concentration surface of the detected Ru ions (Fig. 3d) suggests that Ru is present in the form of NPs ranging from 5 to 10 nm in width, in concordance with the calculation of the crystallite sizes from PXRD analysis (Fig. S3b). The 3D reconstruction and Ru morphology seen in Fig. 3c are reminiscent of the results obtained by Rivas et al.,60 who studied the embedding of Ru NPs in a Co matrix using APT. This result further confirms that there is a certain Ru concentration threshold during synthesis above which Ru NPs are formed rather than single atoms or small clusters at the surface of Ni-NWs.
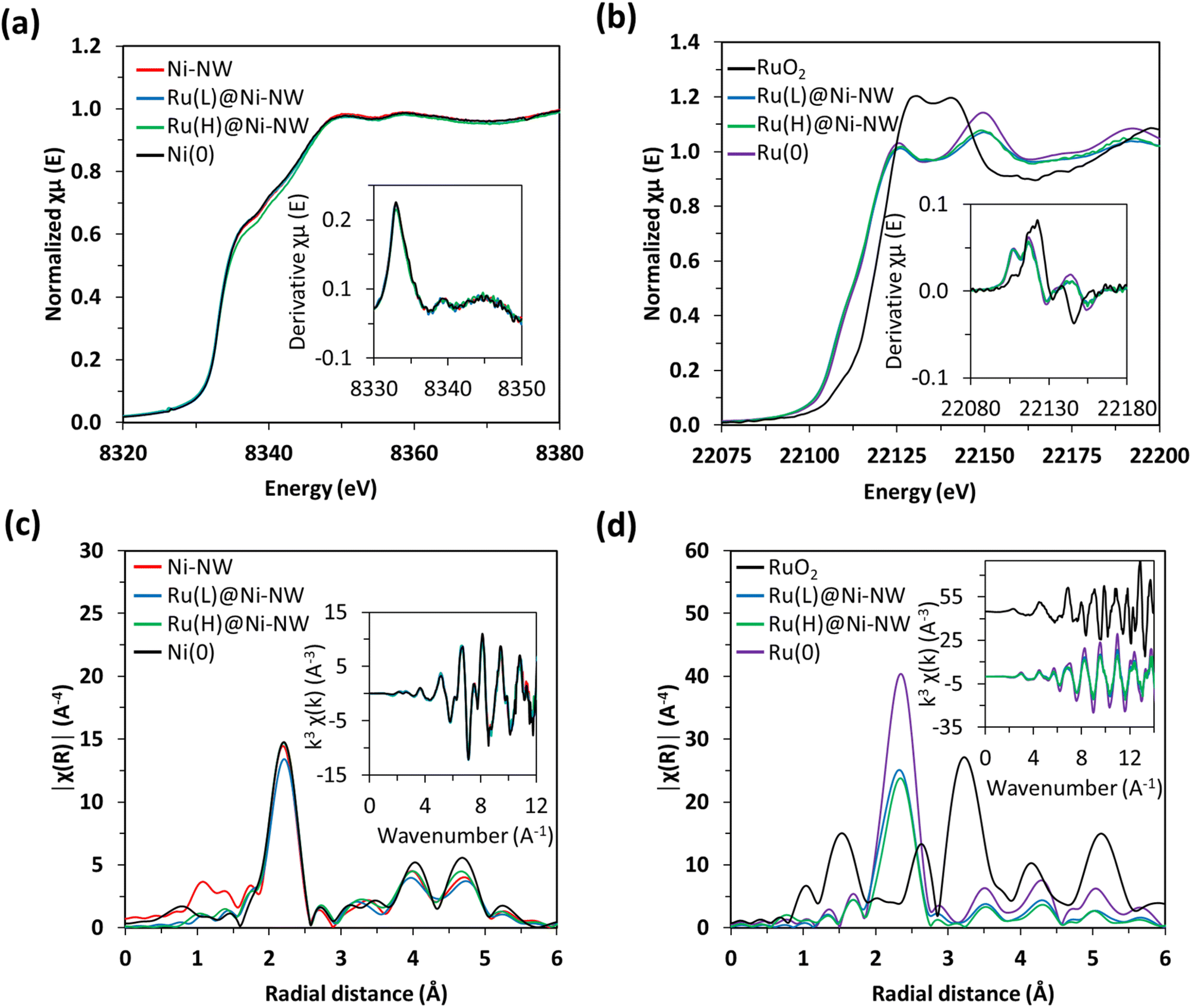 | ||
| Fig. 4 Normalized Ni K-edge XANES for Ni-NW, Ru(L)@Ni-NW and Ru(H)@Ni-NW non-passivated nanomaterials and for the Ni(0) reference. Inset: the derivative of the Ni K-edge (a). Normalized Ru K-edge XANES for as-synthesized RuO2, Ru(L)@Ni-NW and Ru(H)@Ni-NW, and the Ru(0) reference. Inset: the derivative of the Ru K-edge (b). Fourier transforms of k3-weighted Ni EXAFS. Inset: EXAFS region k3χ(k) for Ni systems together with a Ni(0) reference (c). Fourier transforms of k3-weighted Ru EXAFS. Inset: EXAFS region k3χ(k) for Ni systems together with Ru(0) and RuO2 references (d). | ||
Similar findings were observed for the Ru signals in the Ru(L)@Ni-NW and Ru(H)@Ni-NW nanomaterials, where the XANES spectra and their derivatives are overlapped with the spectrum of metallic Ru(0) foil (Fig. 4b). The comparison with the RuO2 reference spectrum shows that the presence of Ru(IV) in the whole Ru-incorporating Ni-NW materials is negligible, as expected for non-passivated nanomaterials. Furthermore, the R-space spectra showed the distinctive feature of Ru(0) attributed to the short Ru–Ru distance of 2.68 Å (Fig. 4d, S6 and Table S2). In fact, the EXAFS spectra perfectly overlap with that of Ru(0), suggesting the presence of small clusters or nanoparticles with well-resolved Ru–Ru scattering patterns rather than the Ru–Ni interactions expected for single-atom species. Focusing on the Ru(L)@Ni-NW nanomaterial, for which the presence of NPs was discarded in TEM images, this observation points to the presence of small (sub-nanometric) Ru clusters, not observable in low magnification electron microscopy images.
3.3. Electrocatalytic performance towards the OER in alkaline media
The catalytic activity of the passivated Ni-NWs, Ru(L)@Ni-NW and Ru(H)@Ni-NW nanomaterials was studied towards the OER at pH 14. First, 10 consecutive cyclic voltammetry (CV) cycles were performed from 1.05 V to 1.75 V vs. RHE in an Fe-free (see the Experimental section for further details) 1 M NaOH solution (Fig. 5). Second, the same electrodes were tested in an Fe-containing (non-purified) 1 M NaOH solution by performing another 10 consecutive CV cycles (Fig. 5).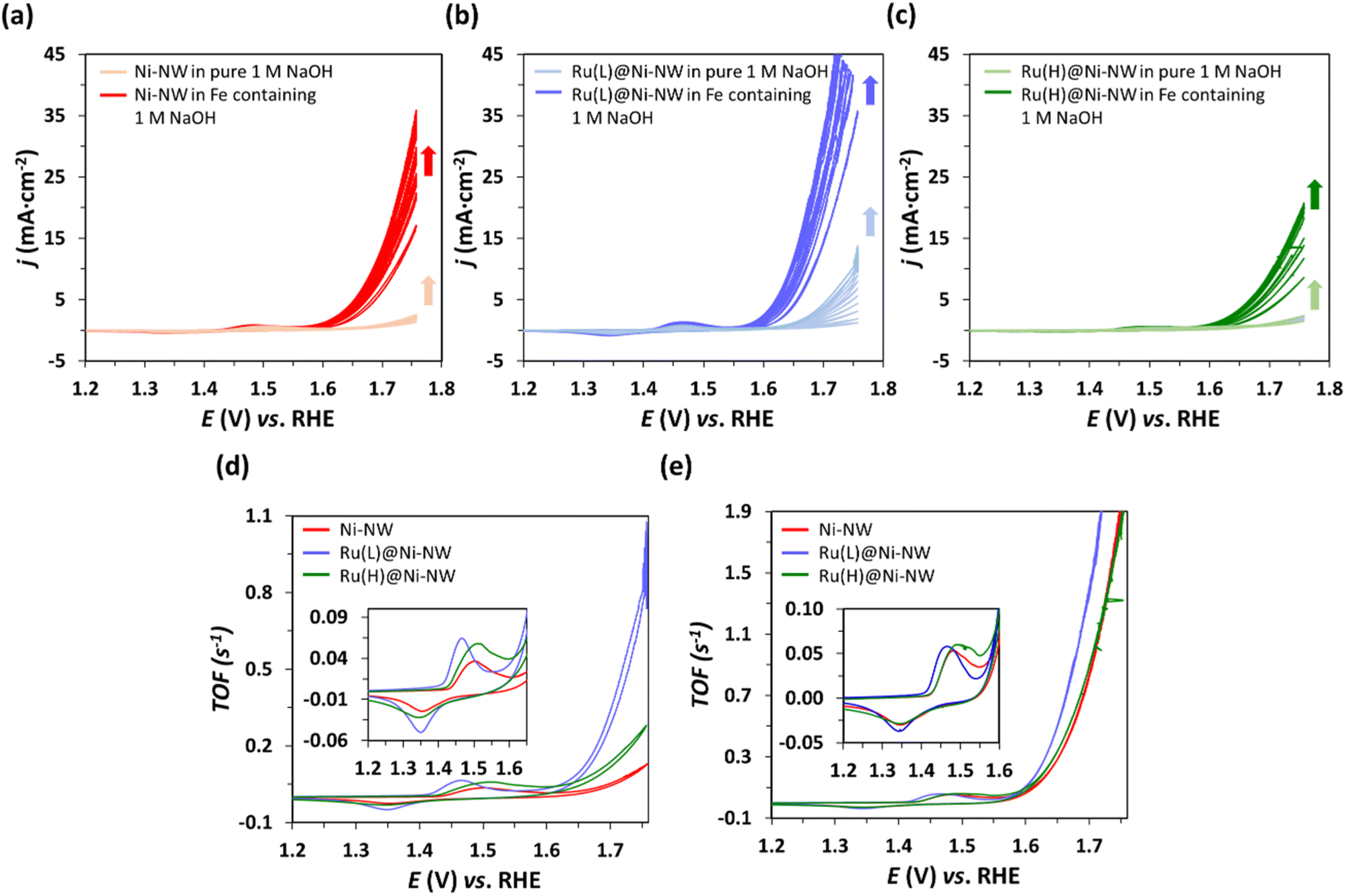 | ||
| Fig. 5 Consecutive CV cycles performed at pH 14 in Fe-free (purified) 1 M NaOH (light colours) and, afterwards, in Fe-containing (non-purified) 1 M NaOH (dark colours) to assess the OER electrocatalytic activity of Ni-NWs (a), Ru(L)@Ni-NW (b) and Ru(H)@Ni-NW (c). Comparison of the last CV normalized by the calculated TOF (see the Experimental section for TOF calculation) of the different electrocatalysts in Fe-free (d) and Fe-containing (e) electrolyte. | ||
As shown in Fig. 5, when scanned anodically up to 1.75 V vs. RHE, all electrodes showed one anodic peak in the oxidative forward scan prior to a sharp current increase assigned to the electrocatalytic OER. According to literature data,61,62 the first faradaic process observed in the voltammograms (Eap = ca. 1.5 V vs. RHE) could be attributed to the oxidation of Ni(II) to Ni(III), following eqn (1). The cathodic wave in the backward scan corresponds to the respective inverse reduction process of the NiO(OH) species.
| Ni(OH)2 + OH− ↔ NiO(OH) + H2O + e− | (4) |
The magnitude of this Ni(II)/Ni(III) redox wave (eqn (4)) is indicative of the number of Ni redox electroactive centers present in each electrode, and it has been used to estimate the TOF (s−1) evolution vs. E, considering Ni as the active species63 (Fig. 5d and e, see the Experimental section for further details). This enables the direct comparison of the electrochemical properties of the three nanomaterials studied despite the potential variation in the deposited amount of each electrocatalyst.
As can be observed in Fig. 5a–c there is an increase in j after 10 progressive CV cycles for all electrodes under both Fe-containing and Fe-free conditions. Interestingly, even when purified electrolyte (Fe-free) is employed, the three electrocatalysts activate, which may be due to their hydration and the Ni(0) oxidation.64 It is important to highlight that, under these Fe-free conditions, the electrocatalyst presenting higher activation and superior performance is Ru(L)@Ni-NW (Fig. 5d), indicating that the presence of the sub-nanometric Ru clusters is beneficial for the intrinsic catalytic properties of the Ni nanomaterial. These results agree with the superior performance of Ru(L)@Ni-NM in the Fe-free electrolyte. When the same electrodes were then submerged in an Fe-containing (non-purified) electrolyte and 10 additional consecutive CV cycles were performed, the activity of the electrodes increased more than before, mostly due to Fe incorporation into the Ni structure, a common phenomenon observed in Ni-based electrocatalysts under OER conditions in alkaline media.13 Due to the observed activation process, the overpotential needed to achieve the standard benchmarking current density of 10 mA cm−2 (η10) has been extracted from the 10th CV cycle to compare the samples, the obtained values being 454 mV, 414 mV and 476 mV for Ni-NWs, Ru(L)@Ni-NW and Ru(H)@Ni-NW, respectively. These results highlight that the incorporation of 0.43 % wt of Ru in the Ni-NWs led to a better catalyst for the OER, but a higher loading of Ru (3.2 % wt) was not beneficial, as will be further discussed. To study any possible effect of the Ru incorporation on the ability of the nanomaterials to incorporate Fe, the Ni-NW, Ru(L)@Ni-NW and Ru(H)@Ni-NW nanomaterials were analysed by APT after Fe incorporation. The results (Fig. S7–S9) show the presence of similar amounts of Fe on the surface of the three nanomaterials, thus indicating that the observed differences in OER performance are not caused by the existence of either a higher or lower percentage of this element. These results agree with the superior performance of Ru(L)@Ni-NW in the Fe-free electrolyte.
The long-term stability has been evaluated for the best performing system Ru(L)@Ni-NW at 10 mA cm−2 for more than 19 h, showing no decrease in activity (SI Fig. S10 and S11). In addition, its stability at higher current densities (50 mA cm−2) has also been assessed for 1 hour (Fig. S12 and S13), showing a slight increase in the overpotentials, pointing out the remarkable robustness of this nanomaterial under these demanding conditions.
Interestingly, the non-faradaic redox wave (eqn (4)) appearing before the catalytic process presented a shift in potential depending on the Ru content (insets in Fig. 5d and e). Ni-NWs and Ru(H)@Ni-NW presented their oxidation wave at 1.48 V vs. RHE in Fe-containing electrolyte and at 1.50 V vs. RHE in purified electrolyte. On the other hand, the Ni(II)/Ni(III) oxidation wave of Ru(L)@Ni-NW appears at 1.46 V vs. RHE in both electrolytes. This points that the Ru(L)@Ni-NW nanomaterial with 0.43 wt% Ru stabilizes higher oxidation states on the Ni-NWs.
3.4. XPS analysis after OER electrocatalysis
The evolution of the electrocatalysts during the activation process discussed above was analysed by XPS on the Ru(L)@Ni-NW and Ru(H)@Ni-NW electrodes after the first 5 CV cycles. As can be observed in Fig. 6a and d, initially both electrodes presented Ru(0) and RuO2 characteristic signals, as described in Section 3.2.3. However, after only 5 CV cycles at pH 14 from the open circuit potential up to 1.75 V vs. RHE, the Ru 3d5/2 XPS signal disappeared from the Ru(H)@Ni-NW electrode (Fig. 6e), whilst it was still present in the Ru(L)@Ni-NW electrode (Fig. 6b). In the case of Ru(L)@Ni-NW, this signal remained even after 100 CV cycles under the same conditions (Fig. 6c). It is worth noticing that the ruthenium oxidation state of the Ru(L)@Ni-NW electrodes shifted from Ru(0) to RuO2 as more CV cycles were run (Fig. 6a–c). The same oxidation tendency was found for the Ni metal centers (Fig. S14), where the signal of Ni(0) almost disappeared after only 5 CV cycles, indicating significant surface oxidation under operative conditions.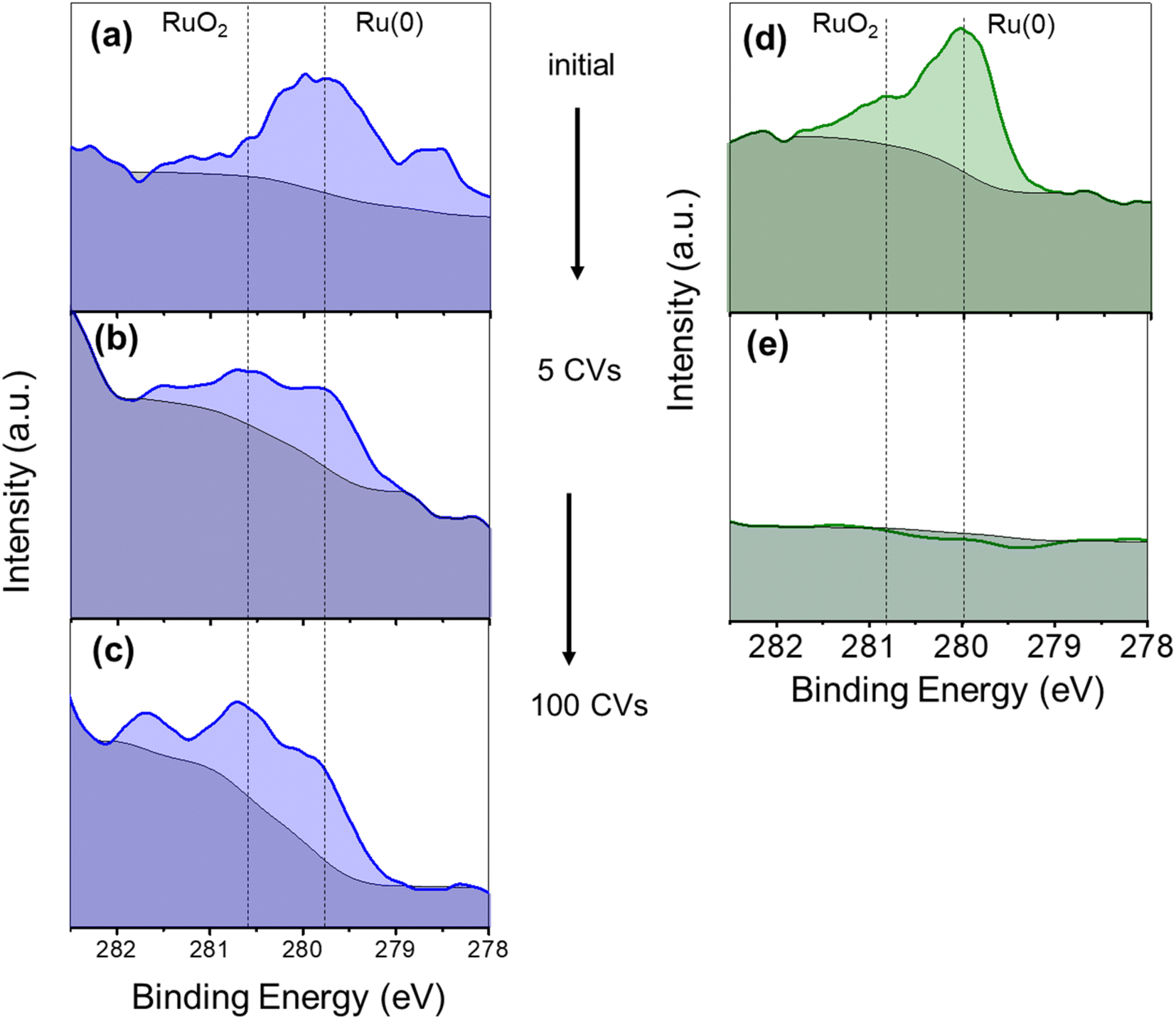 | ||
| Fig. 6 Ru 3d5/2 XPS spectra (279.7–280.3 eV) on FTO electrodes of as prepared Ru(L)@Ni-NW (a), after 5 CV cycles (b) and after 100 CV cycles (c), and of as prepared Ru(H)@Ni-NW (d) after 5 CV cycles (e) at pH 14 in Fe-containing 1 M NaOH. | ||
These experiments suggest a lack of stability under catalytic conditions for Ru(H)@Ni-NW, which contains Ru NPs of ca. 8 nm in size, in contrast to Ru(L)@Ni-NW, where the Ru is dispersed as small (sub-nanometric) clusters. This difference in stability between the Ru NPs and the sub-nanometric clusters can explain why the OER activity of Ru(H)@Ni-NW is similar to that of bare Ni-NWs, which do not contain Ru (Fig. 5).
To distinguish between potential and non-potential driven dissolution of the Ru NPs in Ru(H)@Ni-NW, an electrode containing this nanomaterial was immersed overnight into the non-purified 1 M NaOH electrolyte and its Ru 3d5/2 XPS spectrum was recorded. As shown in Fig. S15, the Ru XPS signal was still clearly visible after the long-time immersion, indicating the presence of the metal, which points to a potential driven process. The stability of RuO2 anodes at high applied potentials was previously studied in the literature, concluding that the oxidation of RuO2 leads to the formation of soluble RuO4, RuO4− or RuO42− species.65–67
3.5. UV-vis spectroelectrochemistry studies
In situ UV-vis spectroelectrochemistry has been used to track the changes and the accumulation of different species along a range of potentials for the three Ni-based nanomaterials.68 The objective was to track the oxidation process from Ni(OH)2 to NiO(OH) (eqn (4)) in Ni-NWs, Ru(L)@Ni-NW and Ru(H)@Ni-NW using pH 14 purified (Fe-free) and non-purified (Fe-containing) electrolytes after activation (10 consecutive CV cycles arriving at catalytic conditions, see Section 3.3 above and the Experimental section for further details).The difference in absorbance (ΔO.D.) when applying a potential below and above the potentials where the Ni oxidative wave appears is shown in Fig. 7 for the purified electrolyte and in Fig. S10 for the Fe-containing one. The spectral features for Ni-NWs and Ru(H)@Ni-NW are similar, with a broad band peak centered at 450 nm characteristic of the NiO(OH) species according to the literature (Fig. 7a and c, S16a and c).16,63 The observation of a similar electronic structure between activated Ni-NWs and Ni(H)@Ni-NW is in accordance with the post-electrocatalysis XPS results described above, demonstrating the loss of ruthenium after activation of the Ru(H)@Ni-NW nanomaterial. On the other hand, Ru(L)@Ni-NW presented a very different spectral pattern, with increasing absorption towards the NIR, peaking at 650 nm (Fig. 7b and S16b). This different spectral shape suggests that the presence of the small Ru clusters may change the electronic structure of the Ru(L)@Ni-NW nanomaterial.41,43
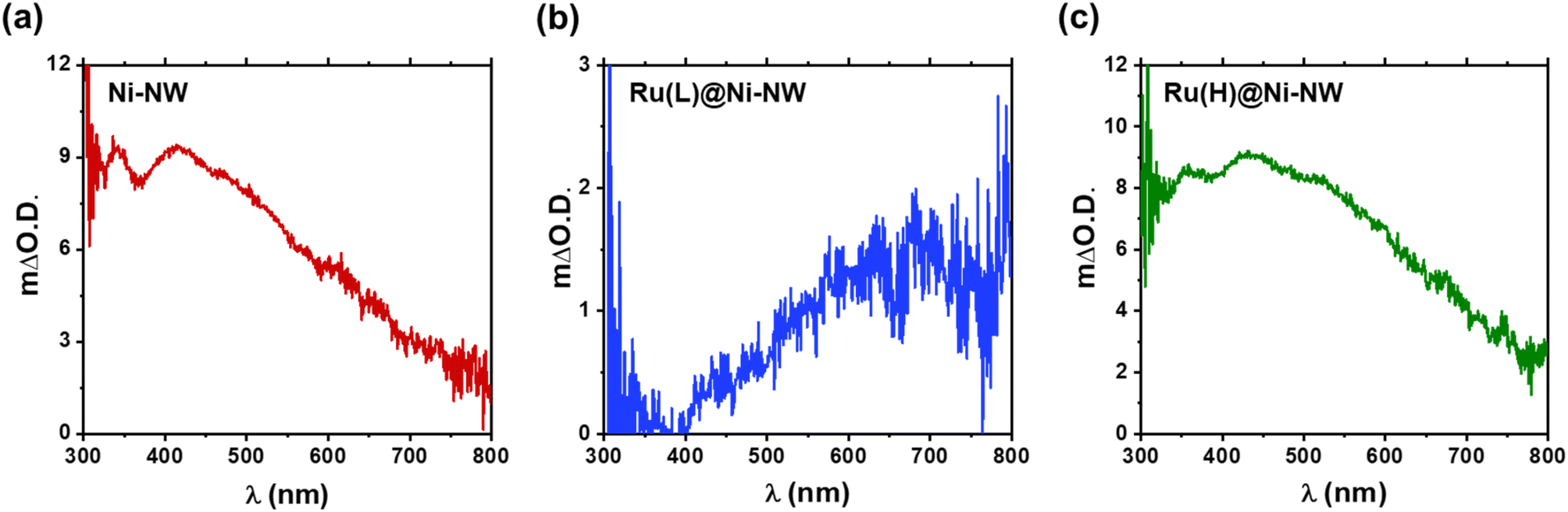 | ||
| Fig. 7 ΔO.D. UV-vis absorption spectra of the species accumulated after the Ni(II)/Ni(III) redox wave before the catalytic region in pure 1 M NaOH and after activation (10 CV cycles up to catalytic potentials to stabilize the systems). Ni-NWs (a), Ru(L)@Ni-NW (b) and Ru(H)@Ni-NW (c). | ||
3.6. DFT calculations
With the aim of analyzing the influence of Ru-based sub-nanometric clusters on the Ni(OH)2 to NiO(OH) oxidation process (eqn (4)), DFT (PBE-U-D3) calculations on the three model systems have been performed, as shown in Fig. 8. The models are based on either a Ru10, Ru17O15 or Ru17O27 motif supported on a (6 × 6) Ni(OH)2 monolayer supercell of the (001) surface in accordance with the sub-nanometric Ru clusters present on the Ru(L)@Ni-NW nanomaterial with consideration of different oxidation states of Ru (0, 1.8, and 3.2, respectively).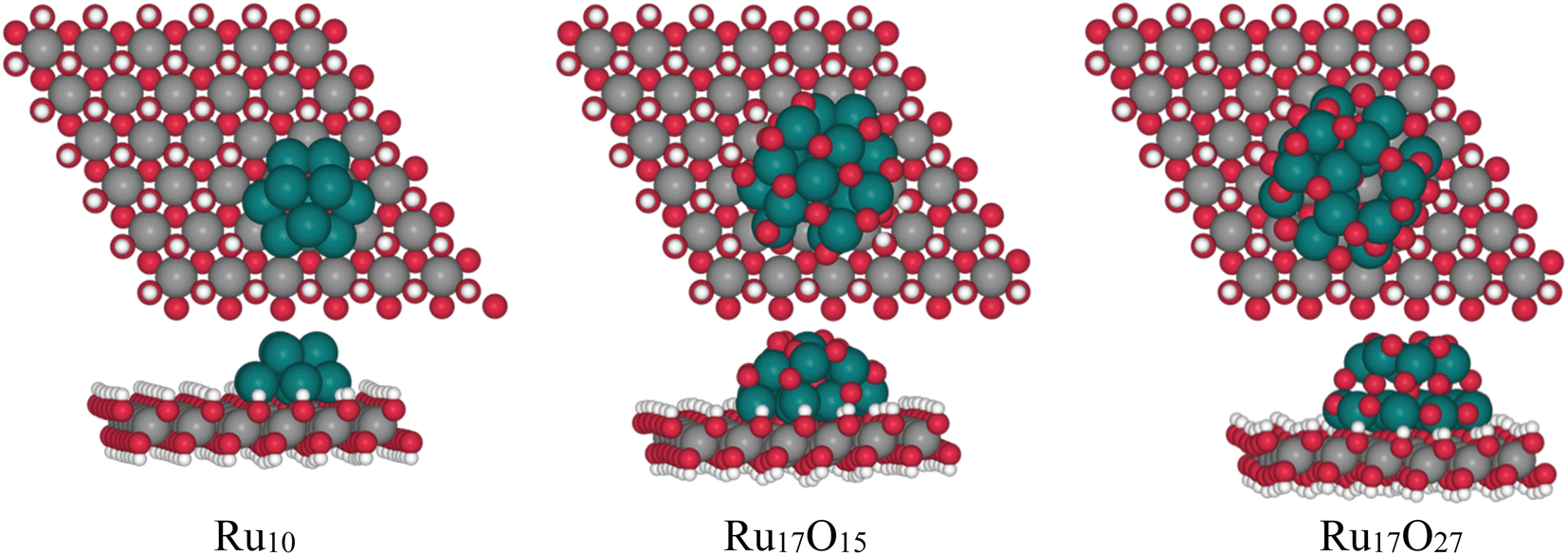 | ||
| Fig. 8 Optimized structures of Ru10, Ru17O15 and Ru17O27 clusters over a Ni(OH2) (100) layer. | ||
For each model, the exposed H atoms of Ni(OH)2 have been classified as a function of the distance and position with respect to the Ru cluster (atoms in blue shades in Fig. 9). The energy cost of the proton coupled electron transfer (PCET) step associated with the transformation of *OH into *O + 1H+ + 1e−, assuming the computational standard hydrogen electrode approach, has been computed for all of them.48 The nomenclature of the different OH groups was implemented in relation to the distance to the closest Ru atom of each cluster in a way that, within each model, A describes the OH group that is further away from the Ru cluster and E denotes the site that is the closest.
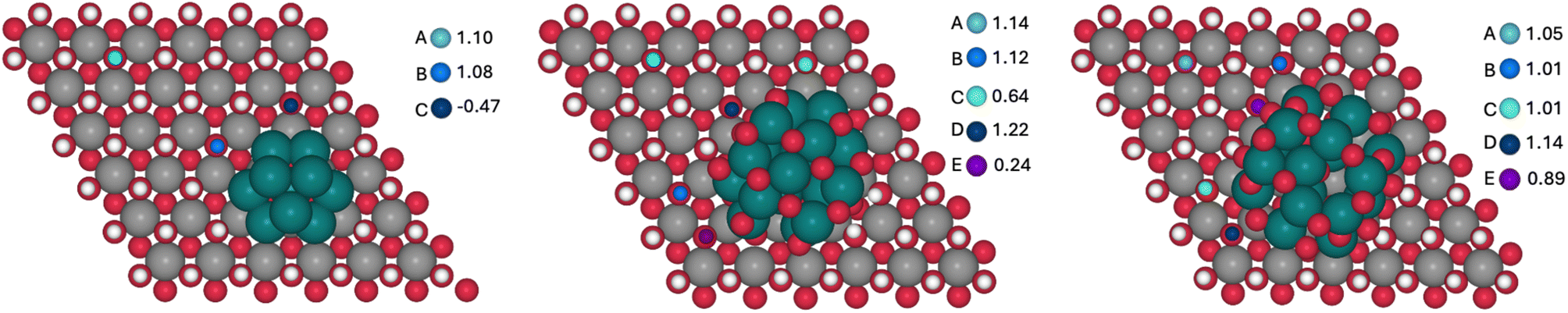 | ||
| Fig. 9 OH sites considered for the Ni(OH)2 oxidation in Ru10 (left), Ru17O15 (center) and Ru17O27 (right) model clusters and the corresponding computed potentials in V vs. RHE. | ||
The computed potentials at which PCET takes place vary from −0.47 to 1.10 V, from 0.24 to 1.22 V and from 0.89 to 1.14 V for Ru10, Ru17O15 and Ru17O27, respectively (Fig. 9). In general, the sites that are closer to the ruthenium cluster are oxidized at lower potentials than the ones that are further away. The only exceptions are the D sites of the Ru17O15 and Ru17O27 clusters. In these cases, the atom of the cluster that is closer to the nickel site is an oxygen (H⋯O = 2.3 Å). This favours a weak hydrogen bond interaction with a H atom of the initial Ni(OH)2 surface, thus stabilizing its structure and making its oxidation marginally more challenging. For the Ru cluster with lower O content, several surface sites are oxidized very easily (C of Ru10 and C and E of Ru17O15). This is associated with a large reorganization of the Ni nanomaterial in which the oxygen that loses the proton is transferred to the Ru cluster, oxidizing it. This suggests that Ru in Ru10 and Ru17O15 would be more easily oxidized than Ni(OH)2, in agreement with the experimental evidence that under catalytic conditions almost all Ru(0) is transformed into RuO2 in Ru(L)@Ni-NW (Fig. 6, left). Consequently, comparison with experiments should be made with the Ru17O27 model. Remarkably, even in this case, the absolute values seem to be underestimated by almost 0.4 V when compared to the experimental results. However, DFT simulations usually reproduce relative energies of equivalent processes better than absolute values, and thus the focus is on the differences between sites. The oxidation of A, which is essentially not influenced by the presence of the Ru cluster (the shortest H⋯Ru distance is 5.6 Å), is computed to occur at 1.05 V. For sites B/C and E, that are closer to the RuOX cluster than A, the potential required to oxidize the nickel centre decreases by 0.04 V and 0.16 V, respectively. In fact, the closer the site is to the cluster, the lower is the potential required to oxidize the nickel center. Remarkably, the substitution of one Ni surface center by Fe has little effect on the Ni2+/Ni3+ oxidation process (Fig. S17). Regardless of the considered H site, the oxidation of Ni becomes more demanding by 0.05–0.08 V. Moreover, the H sites closer to the Ru17O27 cluster are more easily oxidized than those that are far apart from the NPs. The only exception is the closest H site to Fe that implies the oxidation of iron. Overall, calculations suggest that the presence of the RuOX clusters on Ni(OH)2 tend to favour nickel oxidation, regardless of the presence or absence of Fe, with a few exceptions related to very particular local environments. This agrees with the experimental data and further supports the relevance of the sub-nanometric Ru clusters incorporated on Ni-NWs to improve the OER electrocatalytic performance.
4. Summary
In this work we have prepared and characterized four different Ni-based electrocatalysts, Ni-NWs, Ru(L)@Ni-NW, Ru(H)@Ni-NW and Ru(HH)@Ni-NW. After passivation in open air, the pristine Ni-NWs can be considered a core–shell structure where a thin layer of Ni(OH)2 covering the Ni(0) core, as evidenced by the surface sensitive XPS technique, whilst PXRD could only detect the presence of Ni(0).The pristine Ni-NWs were modified with three different concentrations (0.43 % wt, 3.2 % wt and 6.4 % wt) of ruthenium, leading to Ru(L)@Ni-NW, Ru(H)@Ni-NW and Ru(HH)@Ni-NW, respectively. Both Ru(H)@Ni-NW and Ru(HH)@Ni-NW contain Ru NPs of ca. 8 nm on the Ni-NW surface. Therefore, only Ru(H)@Ni-NW was further studied. The Ru present in Ru(H)@Ni-NW was found to be unstable under catalytic OER conditions (Scheme 2). The similar ΔO.D. spectra obtained for both the bare Ni-NW and the Ru(H)@Ni-NW nanomaterials and the absence of the Ru signal in the post-catalysis (after 5 CV cycles) XPS spectrum of Ru(H)@Ni-NW evidence the absence of Ru shortly after the OER turnover starts. According to the literature, this can be attributed to the formation of soluble RuO4, RuO4− or RuO42− species, which was reported under similar oxidative conditions.65–67
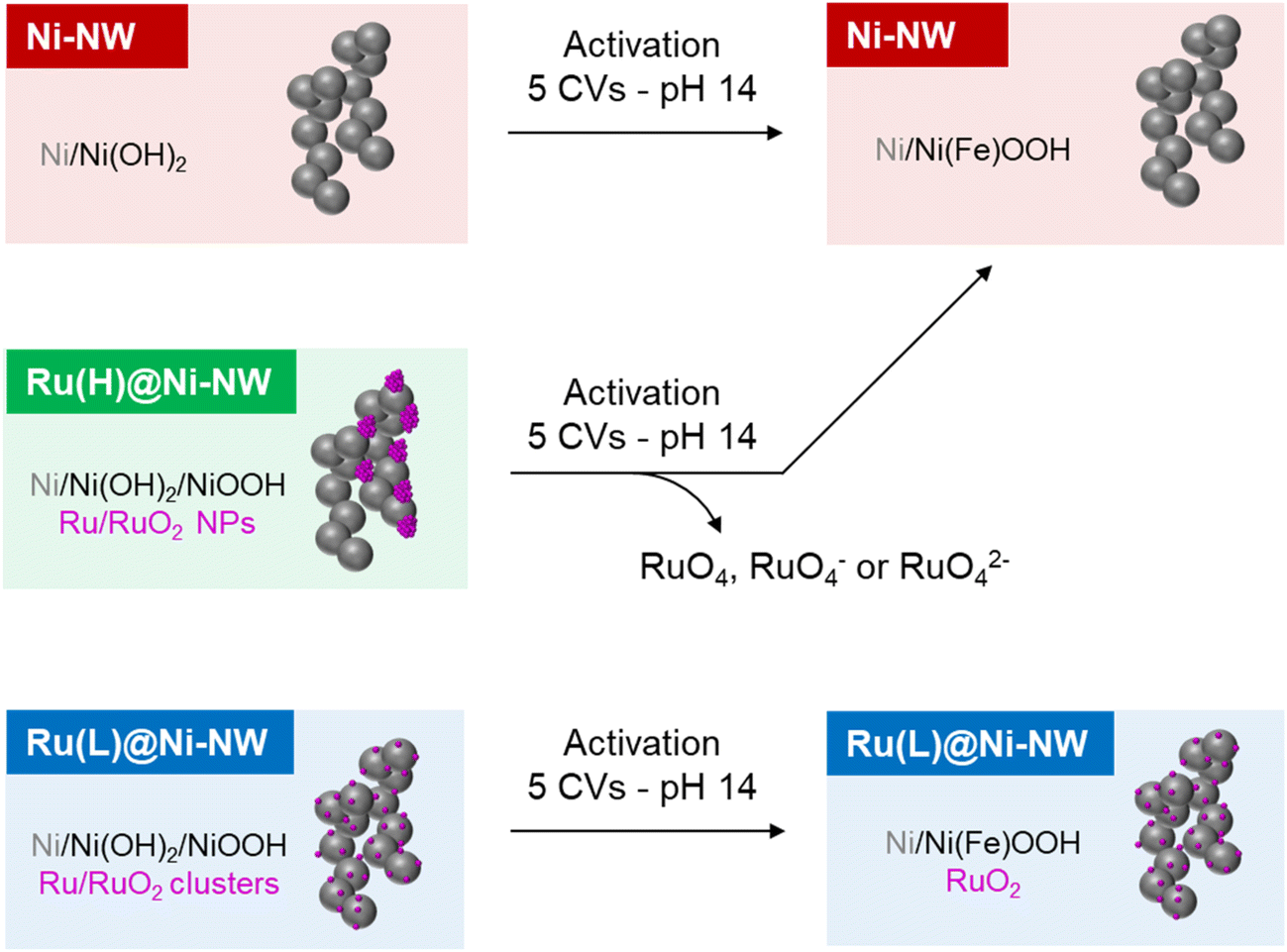 | ||
| Scheme 2 Schematic representation of the main processes described in this work. | ||
On the other hand, Ru(L)@Ni-NW displays highly dispersed Ru all over the Ni-NW surface, as observed by EDX mapping and APT measurements (Scheme 2). This Ru is in the form of sub-nanometric clusters, as indicated by the presence of Ru–Ru interactions in the EXAFS results and the impossibility to observe them by HAADF-STEM due to their small size. The fine interaction between the Ru clusters and the Ni-NW surface directly impacts (i) the longer stability (>100 CV cycles) of the incorporated Ru at the Ni-NW surface under OER turnover conditions (Fig. 6) and (ii) the stabilization of high Ni oxidation states, which have been found to be more stable in the presence of Ru, as indicated by the shift in the Ni(II)/Ni(III) (eqn (4)) redox wave to lower potentials. The latter is further supported by spectroelectrochemical experiments (Fig. 7) that suggest a change in the electronic structure of Ru(L)@Ni-NW compared to its Ru-free Ni-NW counterpart. The earlier accumulation of higher oxidation states observed by electrochemical experiments for this Ru cluster-incorporating nanomaterial was also supported by DFT calculations, where the oxidation of Ni sites gradually increases as they approach the incorporated Ru. Interestingly, these observations are not dependent on the presence or absence of Fe in the nanomaterials. Thus, the Ru-induced stabilization of higher oxidation states in Ru(L)@Ni-NW is the main reason behind the enhanced OER performance observed in this nanomaterial.41
5. Conclusions
This work demonstrates that small variations in the concentration of incorporated Ru in a Ni(OH)2/NiO(OH) electrocatalyst significantly impact its OER performance. This effect arises from differences in the structure and distribution of Ru atoms at the surface of the pristine Ni nanomaterial (Ni-NWs). At a high Ru concentration (Ru(H)@Ni-NW), Ru forms 8 nm NPs on the Ni-NW surface. Under catalytic oxidative conditions, the Ru NPs leach from the Ni-NW nanomaterial, as indicated by the absence of Ru-related peaks in post-reaction XPS analysis. Consequently, the activity of the Ru(H)@Ni-NW electrocatalyst becomes comparable to that of bare Ni-NWs. In contrast, at a low Ru concentration (Ru(L)@Ni-NW), Ru forms well-dispersed sub-nanometric clusters, facilitating a strong interaction between the Ru and Ni atoms. This interaction stabilizes higher Ni oxidation states, as evidenced by electrochemical experiments and DFT calculations, and provokes changes in the electronic structure of the Ru(L)@Ni-NW nanomaterial, which shows enhanced OER performance. These data point out the importance of controlling the morphology of this incorporated Ru onto Ni-based electrocatalysts, which influences the interaction between the two metals and critically determines both the system stability and kinetics in the OER. Therefore, this work will contribute to paving the way to a rational design of efficient Ru-incorporating Ni-based OER electrocatalysts.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5ta08099a.Acknowledgements
M. G. S. acknowledges funding from the HORIZON-MSCA-2021-PF-01 A-Trusol project No. 101063820. L. F. thanks MICIU/AEI/10.13039/501100011033 and FEDER, EU (PID2021-128197NA-I00) and El FSE invierte en tu futuro (RYC2018-025394-I Fellowship). X. S. and J. G.-A. thank MINECO/FEDER (PID2019-104171RB-I00) and MICIU (PID2023-146787OB-I00) for financial support. L. R. S. and X. S. M. (PID2023-151738NB-I00) thank MICIU for financial support. AGAUR is gratefully acknowledged for financial support (CLIMA2023-00036). CNRS, University of Toulouse Paul Sabatier and the Centre de Microcaractérisation R. Castaing (UAR 3623) are acknowledged for funding and access to scientific platforms. ANR PRECINANOMAT (project no. ANR-17-CE06-0017-01) is gratefully acknowledged for financial support (L. P.’s salary). AGM is thankful for Grant RYC2021 – 033479. XAS experiments were performed at the CLAESS beamline at the ALBA Synchrotron under proposal No. 2021095409 with the collaboration of ALBA staff. We especially acknowledge G. Gorni (CLAESS at ALBA) for his collaboration during the XAS experiments. AGM is thankful for Grant RYC2021 – 033479. C. S. acknowledges financial support from the Federal Ministry for Economic Affairs and Climate Action (BMWi) based on a decision taken by the German Bundestag.References
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- S. Yin Tee, K. Yin Win, W. Siang Teo, L.-D. Koh, S. Liu, C. Peng Teng, M.-Y. Han, S. Y. Tee, K. Y. Win, L. Koh, S. Liu, C. P. Teng, M. Han and W. S. Teo, Adv. Sci., 2017, 4, 1600337 CrossRef PubMed.
- Z. Abdin, A. Zafaranloo, A. Rafiee, W. Mérida, W. Lipiński and K. R. Khalilpour, Renewable Sustainable Energy Rev., 2020, 120, 109620 CrossRef CAS.
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CrossRef CAS PubMed.
- C. R. Lhermitte and K. Sivula, ACS Catal., 2019, 9, 2007–2017 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- D. Friebel, M. W. Louie, M. Bajdich, K. E. Sanwald, Y. Cai, A. M. Wise, M. J. Cheng, D. Sokaras, T. C. Weng, R. Alonso-Mori, R. C. Davis, J. R. Bargar, J. K. Nørskov, A. Nilsson and A. T. Bell, J. Am. Chem. Soc., 2015, 137, 1305–1313 CrossRef CAS PubMed.
- S. Chen, Z. Kang, X. Zhang, J. Xie, H. Wang, W. Shao, X. Zheng, W. Yan, B. Pan and Y. Xie, ACS Cent. Sci., 2017, 3, 1221–1227 CrossRef CAS PubMed.
- M. A. Oliver-Tolentino, J. Vázquez-Samperio, A. Manzo-Robledo, R. D. G. González-Huerta, J. L. Flores-Moreno, D. Ramírez-Rosales and A. Guzmán-Vargas, J. Phys. Chem. C, 2014, 118, 22432–22438 CrossRef CAS.
- N. Li, D. K. Bediako, R. G. Hadt, D. Hayes, T. J. Kempa, F. Von Cube, D. C. Bell, L. X. Chen and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1486–1491 CrossRef CAS PubMed.
- H. Xiao, H. Shin and W. A. Goddard, Proc. Natl. Acad. Sci. U.S.A., 2018, 115, 5872–5877 CrossRef CAS PubMed.
- N. Clament Sagaya Selvam, S. J. Kwak, G. H. Choi, M. J. Oh, H. Kim, W. S. Yoon, W. B. Lee and P. J. Yoo, ACS Energy Lett., 2021, 6, 4345–4354 CrossRef CAS.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253–17261 CrossRef CAS PubMed.
- S. Anantharaj, S. Kundu and S. Noda, Nano Energy, 2021, 80, 105514 CrossRef CAS.
- M. Gorlin, J. F. De Araujo, H. Schmies, D. Bernsmeier, S. Dresp, M. Gliech, Z. Jusys, P. Chernev, R. Kraehnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2017, 139, 2070–2082 CrossRef PubMed.
- B. J. Trześniewski, O. Diaz-Morales, D. A. Vermaas, A. Longo, W. Bras, M. T. M. Koper and W. A. Smith, J. Am. Chem. Soc., 2015, 137, 15112–15121 CrossRef PubMed.
- M. Görlin, P. Chernev, J. F. De Araújo, T. Reier, S. Dresp, B. Paul, R. Krähnert, H. Dau and P. Strasser, J. Am. Chem. Soc., 2016, 138, 5603–5614 CrossRef PubMed.
- G. Młynarek, M. Paszkiewicz and A. Radniecka, J. Appl. Electrochem., 1984, 14, 145–149 CrossRef.
- E. S. Da Silva, A. Macili, R. Bofill, J. García-Antón, X. Sala and L. Francàs, Chem.–Eur. J., 2024, 30, e202302251 CrossRef CAS PubMed.
- H. Zhang, Y. Lv, C. Chen, C. Lv, X. Wu, J. Guo and D. Jia, Appl. Catal., B, 2021, 298, 120611 CrossRef CAS.
- P. Zhai, M. Xia, Y. Wu, G. Zhang, J. Gao, B. Zhang, S. Cao, Y. Zhang, Z. Li, Z. Fan, C. Wang, X. Zhang, J. T. Miller, L. Sun and J. Hou, Nat. Commun., 2021, 12, 1–11 Search PubMed.
- M. Li, H. Wang, W. Zhu, W. Li, C. Wang, X. Lu, M. Li, W. Zhu, W. Li, C. Wang, X. G. Lu Alan MacDiarmid and H. Wang, Adv. Sci., 2020, 7, 1901833 CrossRef CAS PubMed.
- Z. Liu, M. Zha, Q. Wang, G. Hu and L. Feng, Chem. Commun., 2020, 56, 2352–2355 RSC.
- D. Kim, S. Park, J. Choi, Y. Piao, L. Yoon, S. Lee, D. Kim, L. Y. S. Lee, S. Park, Y. Piao and J. Choi, Small, 2024, 20, 2304822 CrossRef CAS PubMed.
- J. Liu, Y. Zheng, Y. Jiao, Z. Wang, Z. Lu, A. Vasileff and S. Z. Qiao, Small, 2018, 14, 1704073 CrossRef PubMed.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- Q. Sun, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2003, 67, 205424 CrossRef.
- Y. D. Kim, A. P. Seitsonen, S. Wendt, J. Wang, C. Fan, K. Jacobi, H. Over and G. Ertl, J. Phys. Chem. B, 2001, 105, 3752–3758 CrossRef CAS.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Collière, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374–3401 RSC.
- X. P. Fu, L. Peres, J. Esvan, C. Amiens, K. Philippot and N. Yan, Nanoscale, 2021, 13, 8931–8939 RSC.
- S. H. Kim, P. W. Kang, O. O. Park, J. B. Seol, J. P. Ahn, J. Y. Lee and P. P. Choi, Ultramicroscopy, 2018, 190, 30–38 CrossRef CAS PubMed.
- B. Ravel and M. Newville, J. Synchrotron Rad., 2005, 12, 537–541 CrossRef CAS PubMed.
- D. González, B. Camino, J. Heras-Domingo, A. Rimola, L. Rodríguez-Santiago, X. Solans-Monfort and M. Sodupe, J. Phys. Chem. C, 2020, 124, 1227–1237 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef CAS PubMed.
- S. L. Dudarev, G. A. Botton, S. Y. Savrasov, C. J. Humphreys and A. P. Sutton, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 1505–1509 CrossRef CAS.
- Y. F. Li and A. Selloni, ACS Catal., 2014, 4, 1148–1153 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 Search PubMed.
- N. Romero, D. A. Fenoll, L. Gil, S. Campos, J. Creus, G. Martí, J. Heras-Domingo, V. Collière, C. A. Mesa, S. Giménez, L. Francàs, L. Rodríguez-Santiago, X. Solans-Monfort, M. Sodupe, R. Bofill, K. Philippot, J. García-Antón and X. Sala, Inorg. Chem. Front., 2023, 10, 5885–5896 RSC.
- D. A. Fenoll, M. Sodupe and X. Solans-Monfort, Catal. Today, 2024, 442, 114908 CrossRef CAS.
- D. González, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, J. Catal., 2022, 412, 78–86 CrossRef.
- D. González, M. Sodupe, L. Rodríguez-Santiago and X. Solans-Monfort, Nanoscale, 2021, 13, 14480–14489 RSC.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106, DOI:10.1063/1.4865107/1003843.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- A. L. Patterson, Phys. Rev., 1939, 56, 978 CrossRef CAS.
- M. C. Biesinger, B. P. Payne, L. W. M. Lau, A. Gerson and R. S. C. Smart, Surf. Interface Anal., 2009, 41, 324–332 CrossRef CAS.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- L. Zhu, T. Zheng, J. Zheng, C. Yu, N. Zhang, Q. Zhou, W. Zhang and B. H. Chen, CrystEngComm, 2018, 20, 113–121 RSC.
- Y. Zhou, Y. Liu, H. Tang and B. L. Lin, J. Mater. Chem. A, 2023, 11, 10720–10726 RSC.
- S. A. Sreenavya, B. T. Baskaran, G. V. Ganesh, S. D. Sharma, N. Kulal and S. A. Sakthivel, RSC Adv., 2018, 8, 25248–25257 RSC.
- D. J. Morgan, Surf. Interface Anal., 2015, 47, 1072–1079 CrossRef CAS.
- J. Creus, S. Drouet, S. Suriñach, P. Lecante, V. Collière, R. Poteau, K. Philippot, J. García-Antón and X. Sala, ACS Catal., 2018, 8, 11094–11102 CrossRef CAS.
- J. Creus, L. Mallón, N. Romero, R. Bofill, A. Moya, J. L. G. Fierro, R. Mas-Ballesté, X. Sala, K. Philippot and J. García-Antón, Eur. J. Inorg. Chem., 2019, 12, 2071–2077 CrossRef.
- E. A. Marquis and F. Vurpillot, Microsc. Microanal., 2008, 14, 561–570 CrossRef CAS PubMed.
- B. Gault, M. P. Moody, J. M. Cairney and S. P. Ringer, Mater. Today, 2012, 15, 378–386 CrossRef CAS.
- N. A. R. Rivas, A. G. Manjón, M. Vega-Paredes, S. H. Kim, B. Gault, H. Jun, C. Jung, V. Berova, K. Hengge, T. Jurzinsky and C. Scheu, Ultramicroscopy, 2023, 254, 113831 CrossRef CAS PubMed.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- Y. Gao, H. Li and G. Yang, Cryst. Growth Des., 2015, 15, 4475–4483 CrossRef CAS.
- L. Francàs, S. Corby, S. Selim, D. Lee, C. A. Mesa, R. Godin, E. Pastor, I. E. L. Stephens, K. S. Choi and J. R. Durrant, Nat. Commun., 2019, 10, 1–10 Search PubMed.
- S. Corby, M.-G. Tecedor, S. Tengeler, C. Steinert, B. Moss, C. A. Mesa, H. F. Heiba, A. A. Wilson, B. Kaiser, W. Jaegermann, L. Francàs, S. Gimenez and J. R. Durrant, Sustainable Energy Fuels, 2020, 4, 5024–5030 RSC.
- F. Hess, Curr. Opin. Electrochem., 2023, 41, 101349 CrossRef CAS.
- W. Li, C. Wang and X. Lu, Nano Lett., 2024, 24, 11779–11792 CrossRef CAS PubMed.
- G. Chen, Y. Zhu, S. She, Z. Lin, H. Sun and H. Huang, InfoMat, 2024, e12609 CrossRef CAS.
- C. A. Mesa, E. Pastor and L. Francàs, Curr. Opin. Electrochem., 2022, 35, 101098 CrossRef CAS.
Footnotes |
| † Equal contribution. |
| ‡ Current address: Catalan Institute of Nanoscience and Nanotechnology (ICN2), CSIC and BIST, Campus UAB, Bellaterra, 08193 Barcelona, Catalonia, Spain. |
| This journal is © The Royal Society of Chemistry 2026 |