
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSulfide electrolyte additive enables multi-ionic transfer pathways in alkaline iron redox
Divakar
Arumugam
a,
A. M.
Milinda Abeykoon
 b,
Gihan
Kwon
b,
Cheng-Hung
Lin
b,
Lihua
Zhang
c and
Xiaowei
Teng
*c
b,
Gihan
Kwon
b,
Cheng-Hung
Lin
b,
Lihua
Zhang
c and
Xiaowei
Teng
*c
aDepartment of Chemical Engineering, Worcester Polytechnic Institute, Worcester, Massachusetts 01609, USA
bNational Synchrotron Light Source II, Brookhaven National Laboratory, Upton, New York 11973, USA
cCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA. E-mail: xteng@wpi.edu
First published on 22nd December 2025
Abstract
Traditional alkaline iron (Fe) batteries rely on the conversion reaction between Fe and Fe(OH)2 but suffer from hydrogen generation upon Fe formation on charging. Fe2+/Fe3+ redox is a promising anode reaction for alkaline Fe batteries, as it alleviates the formation of hydrogen gas during charging. However, achieving complete Fe2+/Fe3+ redox with a one-electron transfer reaction is challenging due to the formation of electrochemically inert Fe3O4 materials. Here, we demonstrate that an alkaline sulfide-containing electrolyte facilitates the reversible multi-ion transfer and transport pathways within (and beyond) the Fe2+/Fe3+ redox system, including Fe(OH)2/Fe3O4 conversion, intercalation of hydrosulfide into layered Fe(OH)2, and hydrogen deposition, mediated by hydroxyl ions, hydrosulfide anions, and protons, respectively. This multi-ionic charge storage mechanism delivers a compelling discharge capacity of up to 330 mA h g−1, as determined by chronopotentiometry measurements at a current density of 0.1 A g−1, exceeding the theoretical iron redox capacity solely relying on Fe2+/Fe3+ redox (∼299 mA h g−1). Our study will highlight the potential of alkaline iron redox as a green anode reaction for various aqueous energy storage systems by utilizing a scalable and low-concentration hydrosulfide electrolyte additive.
Introduction
The intermittent nature of wind and solar renewable energy necessitates the development of energy storage technologies that can bridge the mismatch between energy generation and demand.1–3 Long-duration energy storage (LDES) can address the intermittency of renewable energy sources and ensure grid resilience and stability, especially for critical infrastructure such as data centres, hospitals, and campuses that require uninterrupted power. LDES requires scalable, reliable, and cost-effective stationary energy storage systems. Rechargeable non-aqueous lithium-ion batteries (LIBs) have been extensively deployed in portable electronics and electric vehicles.4 However, their implementation in LDES is limited by concerns over cost, safety, and reliance on critical materials such as lithium, cobalt, and graphite.5,6 In contrast, aqueous metal-ion batteries offer a safer and more sustainable alternative by utilizing non-flammable electrolytes and earth-abundant building materials.7,8 Among potential candidates, Fe is the fourth most abundant element in the Earth's crust. It stands out for its multiple oxidation states, widespread availability, low cost, and environmental compatibility. Additionally, the United States alone generates over 15 million tons of unrecycled scrap iron annually, mainly in the form of Fe(OH)3 and FeOOH.9,10 Alkaline iron-based battery systems, such as Fe–Ni, Fe–air, and Fe–S batteries, present a promising opportunity to upcycle scrap iron into high-value materials for LDES applications.11–15The primary technical challenge of conventional alkaline iron redox reactions has been the hydrogen evolution reaction (HER; H2O → H2) during charging since the invention of the Fe–Ni alkaline battery in 1901, where the Fe/Fe(OH)2 couple served as the primary anode reaction.16 According to the iron–water Pourbaix diagram, the reduction potential for the HER is close to that for the Fe(OH)2/Fe reduction reaction. Our previous study demonstrated that the formation of metallic Fe significantly accelerates the HER,17 as Fe is a more active HER catalyst than Fe(OH)2 with nearly two orders of magnitude higher HER exchange current.18 The parasitic HER consumes nearly the entire electrical energy to reduce water instead of Fe(OH)2, thereby significantly lowering the coulombic efficiency.17 A potential strategy to alleviate the HER involves replacing the Fe0/Fe2+ redox couple with Fe2+/Fe3+, which operates at a higher potential range and, thus, is less prone to hydrogen generation. However, this pathway typically involves the formation of a spinel-type Fe3O4 intermediate, a close-packed phase that is electrochemically stable and has a high energy barrier for further redox transformations. The accumulation of Fe3O4 over repeated cycles impairs the coulombic efficiency, requiring high overpotentials for its conversion to Fe(OH)2 during the charging process, thereby reducing the overall energy efficiency of the iron redox.19,20
Another technical challenge of iron redox reactions is the sluggish conversion of Fe2+ to Fe3+ during the discharging. Various strategies have been explored to improve the Fe2+/Fe3+ redox. For instance, the formation of anion-intercalated layered double hydroxide (LDH) intermediate phases—commonly referred to as “green rust” (GR), with the general formula [Fe1−x2+Fex3+(OH)2]X+[(An−)x/n]X− (A = SO42−, CO32−, and Cl−), which facilitates the Fe2+/Fe3+ conversion.21–23 According to the Pourbaix diagram, these GR phases serve as intermediates bridging the Fe(OH)2 and FeOOH phases. The intercalation of anions during the Fe2+ → Fe3+ transition suppresses Fe3O4 accumulation, thereby enhancing the reversibility of the redox cycle. Additionally, electrolyte engineering, such as the use of additives like sodium silicate, has been demonstrated to selectively modulate iron redox chemistry.24 Sodium silicate can strengthen the hydrogen-bonding network in the electrolyte, thereby reducing the water activity and suppressing the hydrogen evolution reaction (HER).25 Furthermore, it interacts with iron hydroxide surfaces to restrict water access to the electrode interface, promoting the Fe(OH)2 → FeOOH conversion over undesired side reactions. Despite these advances, key fundamental questions remain unanswered: can the electrolyte environment be further tailored to modulate electrode–electrolyte interactions and activate the otherwise inert Fe3O4 redox chemistry?
Sulfide salts have been identified as effective additives, either incorporated into electrodes (e.g., Bi2S3) or introduced directly into the electrolyte (e.g., K2S or Na2S),19,26,27 mitigating the HER in alkaline Fe batteries. Although the underlying mechanisms remain under investigation, it has been proposed that S2− ions interact with metallic Fe to form an insoluble Fe–S interphase on the electrode surface, which increases the overpotential for the HER. In this work, we report, for the first time, that Na2S additives play multifaceted roles in enhancing Fe redox chemistry, particularly in the Fe2+/Fe3+ redox regime. Specifically, we find that during discharge the HS− anions intercalate into the interlayer region of Fe(OH)2 to form a green rust (GR) phase, diverting from the Fe(OH)2/Fe3O4 redox pathway and enhancing the overall Fe2+ → Fe3+ conversion, and vice vera during the charge process, where GR is reduced to Fe(OH)2via HS− anion de-intercalation. Moreover, we also found that Na2S additive enables the deep charging process to enhance the Fe3O4 → Fe(OH)2 conversion and hydrogen deposition on the Fe(OH)2 phase. The resulting multi-ionic transport mechanism, facilitated by HS− anions, comprises Fe(OH)2/Fe3O4 conversion, intercalation of hydrosulfide into layered Fe(OH)2, and hydrogen deposition, collectively contributing to the overall discharge capacity of up to 330 mA h g−1, exceeding the theoretical iron redox capacity solely relying on Fe2+/Fe3+ redox (∼299 mA h g−1), along with excellent cycling stability.
Experimental section
Materials and equipment
The goethite (α-FeOOH) material was synthesized by dissolving ferrous sulfate heptahydrate (FeSO4·7H2O, 2.085 g) and ferric sulfate hydrate (Fe2(SO4)3·H2O, 0.66 g) in 50 mL of deionized (DI) water until the solution turned clear brown. 0.6 g of NaOH was dissolved in 50 mL of DI water and then added to the solution, which was stirred for 24 hours under an air overflow. The product was washed and dried overnight at 80 °C and ground for material and electrochemical characterization studies.High-resolution transmission electron microscopy (TEM) and High-Angle Annular Dark-Field (HAADF) imaging were conducted at the Centre for Functional Nanomaterials in Brookhaven National Laboratory (BNL) using an FEI Talos F200X scanning transmission electron microscope.
Operando X-ray diffraction (XRD) and X-ray absorption spectroscopy (XAS) measurements were carried out at the 28-ID-1 and 6-BM beamlines at the National Synchrotron Light Source-II at BNL.
Electrochemical studies
Chronopotentiometry (CP) measurements were conducted in an Arbin BT-G battery cycler (S/N: 177907) using a three-electrode half-cell system. The three-electrode cell was a home-made acrylic cell with an O-ring sealing assembled using the working electrode, which was prepared by drop-casting the ink slurry containing the active material on a 5% Teflon-coated Toray carbon paper (Fuel Cell Earth) of an area of 1 cm−2 to give an active material loading of 1.4 mg cm−2, a platinum wire counter electrode, and a silver/silver chloride reference electrode filled with 3 M KCl solution. The ink slurry was prepared by mixing 70% FeOOH and 30% carbon black in a mixture of 800 µL ethanol and 200 µL DI water at a concentration of 10 mg mL−1, and a 5 wt% Nafion 117 solution was used as the binder at a concentration of 30 µL mL−1 solvent. The electrolyte consisted of sodium hydroxide, sodium sulfide, and 5000 ppm sodium silicate (Na2SiO3). All electrolytes were degassed by purging with argon gas for at least 2 hours before each measurement.The CP measurements during the operando experiments were conducted using a CH Instruments 660D/E electrochemical workstation. 0.01 M NaOH/5000 ppm Na2SiO3/12 mM Na2S was used as the electrolyte for XRD and XAS measurements. The electrodes were prepared, and the cells were assembled following the same procedure as mentioned above. Operando XRD and XAS measurements were conducted at a current density of 0.1 A g−1 and 0.2 A g−1, respectively.
Galvanostatic intermittent titration technique (GITT) measurements were conducted in an Arbin BT-G battery cycler (S/N: 177907) using a three-electrode half-cell system. The electrode was first stabilized at each potential step in CP measurements. At each step, the switching current (0.1 A g−1) was applied for 10 minutes, followed by a 20 minute relaxation. The diffusion coefficient at each pulse current during CP testing was calculated based on eqn (1), where τ is the duration of the current pulse (s); nm is the number of moles; Vm is the molar volume of the electrode (cm3 mol−1); S is the electrode and electrolyte contact area (cm2); ΔEs is the steady-state voltage change during the constant current pulse without the iR drop (concentration polarization); ΔEt is the voltage for each current pulse.
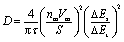 | (1) |
Electrochemical Impedance Spectroscopy (EIS) measurements were conducted with a CH Instruments 660D/E electrochemical workstation in a three-electrode cell with a frequency range of 0.1 MHz to 0.001 Hz. CP measurements were conducted at 0.1 A g−1, and EIS measurements were conducted at various potentials. In the NaOH/Na2SiO3/Na2S electrolyte, the selected potentials (with respect to Hg/HgO) for the EIS measurements were −0.7 V (C1), −0.9 V (C2), and −1.1 V (C3) during charging and −0.85 V (D1), −0.56 V (D2), and −0.2 V (D3) during discharging. In the NaOH/Na2SiO3 electrolyte, the selected potentials were −0.9 V (C1) and −1.1 V (C2) during charging and −0.56 V (D1) during discharging.
Results and discussion
Electrochemical analysis of Fe redox
The starting electrode material, α-FeOOH, was synthesized via a co-precipitation method by mixing ferric sulfate with NaOH, followed by thermal treatment in air. X-ray diffraction (XRD) confirmed the formation of the pure-phase α-FeOOH (Fig. S1). Transmission electron microscopy (TEM) revealed highly crystalline particles with dimensions of approximately 10–100 nm (Fig. S2). Notably, during the initial electrochemical formation cycle, the α-FeOOH precursor was reduced to Fe3O4. Consistent with previous studies,25,28 the electrodes stabilized into a reversible Fe2+/Fe3+ redox couple in the NaOH electrolyte over subsequent charge–discharge cycles.Fig. 1a shows the CP plots of Fe2+/Fe3+ redox in 0.01 M NaOH/5000 ppm sodium silicate electrolyte with and without the 12 mM Na2S additive. The low-concentration sodium silicate was used because it can strengthen the hydrogen-bonding network in mild alkaline solutions, thereby reducing water activity, and can also interact with the Fe(OH)2 surface to minimize the HER.25 The charge (reduction) profiles in both electrolytes show a similar two-stage behavior, where the first plateau at ∼−0.9 V relates to the Fe3O4 → Fe(OH)2 conversion (confirmed by operando XRD, discussed later), while the second plateau near −1.1 V is primarily attributed to the HER with minor contribution of continuous formation of Fe(OH)2, as shown in our previous study.17 However, the discharge profiles in both electrolytes are markedly different. In NaOH/Na2SiO3/Na2S electrolyte, CP exhibits three distinct voltage plateaus at approximately −0.9 V, −0.6 V, and −0.3 V, also clearly reflected in the dQ/dV (1st order derivative of electrical charge over potential) plots (Fig. 1b). These multiple discharge plateaus indicate complex and sequential charge storage mechanisms associated with the Fe2+ → Fe3+ redox, leading to an exceptional discharge capacity of 330 mA h g−1 and exceeding the theoretical capacity of Fe(OH)2 (299 mA h g−1) based on a single-electron Fe2+/Fe3+ transfer. Comparable redox features were also observed at current densities of 0.1, 0.2, and 0.5 A g−1 (Fig. S3). As the current density increased, Fe(OH)2/Fe3O4 redox in NaOH/Na2SiO3/Na2S electrolytes showed capacity values ranging from 330 to 281 and to 276 mA h g−1. This indicates the robustness of the Fe2+/Fe3+ redox behaviors across different rates, assisted by the Na2S electrolyte additive. The electrolyte used in this study was 0.01 M NaOH with 12 mM Na2S, indicating that hydrosulfide ions (HS−) are the predominant species in the solution. It is known that HS− can be oxidized to polysulfides (SX2−) and sulfite (SO32−) ions in alkaline solutions with pH values ranging from 7 to 13 using different electrode materials, including Pt, Au, and IrO2, at high anodic potentials.29–31 Therefore, the observed high discharge capacity is unlikely due to the electrochemical oxidation of hydrosulfide species within the applied potential range.
 | ||
| Fig. 1 (a) CP curves and (b) corresponding dQ/dV plots of Fe2+/Fe3+ redox obtained in NaOH/Na2SiO3 and NaOH/Na2SiO3/Na2S electrolytes at a current density of 0.1 A g−1. CP measurements of 300 charge–discharge cycles at 0.2 A g−1 in (c) NaOH/Na2SiO3 and (d) NaOH/Na2SiO3/Na2S electrolytes. | ||
The long-term cycling performances of Fe2+/Fe3+ redox are shown in Fig. 1c and d. In the NaOH/Na2SiO3/Na2S electrolyte system, Fe2+/Fe3+ redox shows excellent cycling performance with a discharge capacity of 174 mA h g−1 at a current density of 0.2 A g−1 and a capacity retention of 80.6% after 300 charge–discharge cycles, much better than that in NaOH/Na2SiO3 electrolyte (118 mA h g−1). The effect of Na2S concentration on Fe redox was also investigated, as shown in Fig. S4. As the concentration of Na2S increases, the discharge capacity initially rises proportionally, indicating an enhanced discharge capacity. However, with further increases in Na2S, the growth of discharge capacity gradually diminishes, suggesting that low concentrations of Na2S effectively improve Fe redox, where additional increases in Na2S no longer contribute significantly to changes in Fe redox.
Operando XRD and XAS analyses
To elucidate the unexpected high discharge capacity beyond the theoretical Fe2+/Fe3+ redox capacity in the NaOH/Na2SiO3/Na2S electrolyte system and its underlying charge storage mechanism, operando XRD was conducted under CP cycling at a current density of 0.1 A g−1 in a three-electrode half-cell. Fig. 2 shows the XRD contour map, corresponding unfolded CP profiles, and the evolution of crystalline iron phases in molar fractions (relative to the total number of Fe atoms), derived from Rietveld refinement. The complete set of XRD patterns is presented in Fig. S5. | ||
| Fig. 2 The molar fraction of phases, based on total Fe atoms, was calculated by Rietveld refinement. Contour plots were obtained from operando XRD measurements acquired simultaneously with CP measurements in the NaOH/Na2SiO3/Na2S electrolyte at a current density of 0.1 A g−1. | ||
We first examine the charging (reduction) process, during which Fe3O4 and Fe(OH)2 were the only crystalline phases identified. As the applied potential decreased from −0.92 V (point a) to −1.08 V (point b), the phase fraction of Fe3O4 remained constant. The conversion of Fe3O4 to Fe(OH)2 commenced at −1.08 V. Between −1.08 V and −1.14 V (point c), Fe(OH)2 rapidly formed, with its phase fraction increasing from 0% to 62.7%. Unexpectedly, the potential then increased from −1.14 V to −1.10 V and plateaued at −1.10 V (point d) for the remainder of the charging process. Under constant cathodic current, one would expect the external potential to continue decreasing as the reduction process proceeds. This anomalous increase in potential suggests the occurrence of the HER. It is likely that upon reaching a critical accumulation of Fe(OH)2 (e.g., 62.7% in this study), trace amount of metallic Fe forms on the Fe(OH)2 surface, since −1.14 V exceeds the theoretical reduction potential of the Fe(OH)2 → Fe0 conversion (approximately −0.90 V vs. Hg/HgO). While the quantity of metallic Fe may be too low to be detected by XRD, it may still be sufficient to catalyze the HER. The observed shift in potential from −1.14 V to −1.10 V can thus be interpreted as a transition to a regime where the HER dominates the cathodic current. This is consistent with the notion that metallic Fe, being a much more active HER catalyst than Fe(OH)2, lowers the overpotential required to sustain the 0.1 A g−1 cathodic current. Notably, during the charging process, operando XRD and Rietveld refinement did not detect FeS or FeS2 formation, largely due to the low concentration of the Na2S solution. However, it is still possible that a thin layer of amorphous FeS or FeS2 is formed on the Fe(OH)2 surface to inhibit the HER, which warrants further study.
In contrast to the charging process, the discharging process reveals the presence of Fe3O4, Fe(OH)2, and green rust (GR) crystalline phases, as identified by Rietveld refinement. During the initial discharge stage from −1.04 V to −0.67 V (point e), Fe(OH)2 remains largely unreacted. A distinct discharge plateau centered at approximately −0.87 V corresponds to a capacity of ∼60 mA h g−1. This feature is likely associated with the oxidation of deposited hydrogen, which will be discussed in more detail along with operando XAS analysis in Fig. 3. As the potential increases from −0.67 V (point e) to −0.37 V (point f), Fe3O4 forms progressively via the oxidation of Fe(OH)2, eventually reaching a maximum phase fraction of 89%, suggesting that Fe(OH)2 → Fe3O4 conversion is the primary driver for the Fe2+ → Fe3+ redox. Uniquely, a new set of diffraction peaks appear at 2θ values of 8.1°, 16.2°, and 24.3° when the potential reaches −0.37 V (point f). These peaks are, respectively, indexed to the (001), (002), and (003) planes of hydrosulfide-intercalated green rust (GR) with a nominal composition of [Fe1−x2+Fex3+(OH)2]x+[(HS−)x]x−, formed via the intercalation of HS− anions between the Fe–O molecular layers of Fe(OH)2. The insertion of hydrosulfide ions is accompanied by partial oxidation of Fe2+ to Fe3+, contributing approximately 65 mA h g−1 to the total discharge capacity. Meanwhile, the Fe–O layered framework remains intact, preserving structural integrity during charge transfer. Unlike the conventional conversion mechanism involving Fe(OH)2 → Fe3O4, this intercalation-based redox reaction, Fe(OH)2 → [Fe1−x2+Fex3+(OH)2]x+[(HS−)x]x−, proceeds without a reconstructive phase transition. As a result, the potential–capacity profile during this process exhibits a sloped curve rather than a well-defined plateau, which is characteristic of intercalation reactions.
 | ||
| Fig. 3 Operando XAS measurements in the NaOH/Na2SiO3/Na2S electrolyte. (a) CP measurement conducted at 0.2 A g−1, contour plots showing the evolution of (b) a white-line shape and 1/2 µ(E) in energy space, (c) Fe–O and Fe–Fe bond distances in R-space and (d) evolution of Fe valence during the charging process. | ||
A critical question arises regarding the observed one electron transfer per Fe atom (corresponding to a capacity of ∼300 mA h g−1) in the NaOH/Na2SiO3/Na2S electrolyte, particularly since XRD analyses indicate that the Fe2+ → Fe3+ redox process yields Fe3O4 and GR as discharge products because combining both would not exceed one electron transfer per Fe atom. To address this discrepancy, synchrotron-based X-ray absorption spectroscopy (XAS) was employed to probe the valence states of iron in selected electrochemical states spanning from −0.95 V to −1.2 V (Fig. S6).
By fitting the energy position at half-absorption intensity [½µ(E)], we tracked the evolution of Fe valence across different potentials. Fig. 3 shows a continuous decrease in the Fe oxidation state from −0.95 V (point a′) to −1.11 V (point b′), which is consistent with the reduction of Fe3O4 to Fe(OH)2. However, in contrast to the XAS data, XRD patterns during this range show no observable change in the Fe3O4 crystalline phase fraction, which remains at 100%. This apparent discrepancy is attributed to the fundamental differences in probing mechanisms: XAS captures the average oxidation state of all iron species, including both crystalline and amorphous phases, whereas XRD exclusively detects Bragg reflections from long-range ordered crystalline materials. The combined results imply that the reduction of Fe3O4 initially produces amorphous Fe(OH)2, which is invisible to XRD but contributes to the valence change detected by XAS. Further evidence for amorphous phase formation is provided by the white-line contour plots of the K-edge spectra (Fig. 3b), which exhibit a progressive shift to lower energies, accompanied by a decline in the intensity of Fe–O and Fe–Fe coordination features (Fig. 3c). While the exact structural evolution remains uncertain, the emergence of amorphous domains suggests that Fe-ion relocation is accompanied by lattice disordering. This amorphization may enhance ionic diffusion pathways and facilitate the Fe3O4 → Fe(OH)2 conversion, thereby contributing to the observed high capacity. Operando XRD/XAS and CP measurements collectively indicated that the formation of amorphous and crystalline Fe(OH)2 is highly potential-dependent, and CP results suggest that the amorphous phase corresponds to approximately 48% of the total Fe(OH)2.
A similar discrepancy between XRD and XAS is observed in the potential range between −1.16 V and −1.12 V (points c′ → d′), where XAS indicates a continued decrease in the average Fe valence, while XRD reveals minimal changes in the crystalline phase, which remains dominated by Fe(OH)2. This divergence is likely due to the deposition of hydrogen atoms in Fe(OH)2. The phenomenon of simultaneous proton reduction and hydrogen atom deposition and insertion, without the formation of molecular hydrogen gas, is considered a particularly promising charge carrier due to its unique characteristics, as highlighted in recent studies.32,33 Support for this hypothesis comes from the electrochemical behaviors of the Fe(OH)2 electrode during the subsequent discharge (oxidation) cycle. A distinct potential plateau appears at −0.87 V, corresponding to a discharge capacity of 60 mA h g−1. This plateau is not associated with the Fe(OH)2 → Fe3O4 conversion, which occurs at a higher potential (−0.60 V), and is therefore attributed to the deposited H oxidation. The presence of deposited H atoms contributes an additional charge storage mechanism beyond Fe2+/Fe3+ redox, explaining the observed capacity (330 mA h g−1) exceeding the theoretical value for a single-electron Fe2+/Fe3+ redox process (299 mA h g−1). Notably, the hydrogen deposition/removal, Fe(OH)2/Fe3O4 conversion, and hydrosulfide intercalation/deintercalation processes, each involving multiple ionic charge carriers such as protons, hydroxide ions, and hydrosulfide anions, exhibit pronounced potentiodynamic behaviors. Therefore, their quantitative contributions to the overall charge-storage process can be estimated from the discharge capacities obtained from the CP measurements, which yield 88, 182, and 60 mA h g−1, corresponding to approximately 26.7%, 55.1%, and 18.2% of the total storage capacity, respectively. Collectively, all three charge-storage mechanisms contribute to a total capacity of 330 mA h g−1.
Ionic transport study
The ionic transport behaviors during Fe2+/Fe3+ redox in NaOH/Na2SiO3 and NaOH/Na2SiO3/Na2S electrolytes were determined by galvanostatic intermittent titration technique (GITT) measurements (Fig. S7), further supporting the reported multi-ion storage mechanisms. GITT measurement during a current pulse is schematically shown in Fig. 4a and b. When the current reduction is switched on, a steep potential decrease appears (IR drop, E0 → E1). Subsequently, the steady decrease in potential (E1 → E2) indicates the development of concentration polarization. After that, switching off the reduction current is accompanied by a potential increase, along with the second IR drop (E2 → E3), followed by a relaxation transient with a slow potential increase (E3 → E4), related to the balancing of ion concentrations to reach the next quasi-equilibrium state. The diffusion coefficients during CP measurements were calculated using eqn (1), as detailed in the Experimental section. | ||
| Fig. 4 GITT measurements of Fe2+/Fe3+ redox. (a) The schematics of GITT measurements and (b) one representable GITT segment obtained experimentally. The GITT curve and the evolution of diffusion coefficients from (c) charging and (d) discharging processes in NaOH/Na2SiO3 and NaOH/Na2SiO3/Na2S electrolytes. | ||
Fig. 4c shows the evolution of the diffusion coefficient during the charging process in NaOH/Na2SiO3 and NaOH/Na2SiO3/Na2S electrolytes. In the Na2S-containing electrolyte, the initial charging process from 0 to 100 mA h g−1 exhibits fast kinetics, with diffusion coefficients of the order of 10−10 cm2 s−1. This suggests a de-intercalation of hydrosulfide ions from the interlayer regions of the GR host electrode, accompanied by facile charge transfer and minimal structural change. In the sequential charging stage from 100 to 225 mA h g−1, corresponding to the GR → Fe3O4 → Fe(OH)2 conversion process, the diffusion coefficient shows a U-shaped curve, a sharp decrease followed by a rapid increase with an overall decreased diffusion coefficient, congruent with sluggish kinetics of the conversion reaction involving phase transition of the electrode material. The first decrease in the diffusion coefficient of the U-shaped profile may be due to the creation of energy barriers resulting from structural reorganization during the transition from deintercalation to conversion. In contrast, the subsequent increase in diffusion coefficient can be attributed to the stabilization of the Fe(OH)2 phase, which facilitates improved percolation for ionic transport. In the final charging stage from 225 to 400 mA h g−1, corresponding to the Fe(OH)2 → Fe and HER, showing a relatively constant diffusion coefficient in the lowest value range from 10−13 and 10−14 cm2 s−1. While the HER is a relatively fast charge transfer process, the low diffusion coefficient suggests that the bubble accumulation on the electrode surface significantly increases the ionic transport barrier. The evolution of the diffusion coefficient in the NaOH/Na2SiO3 electrolyte is simpler than that in NaOH/Na2SiO3/Na2S electrolyte, where the initial slow decrease from 0 to 150 mA h g−1 is consistent with the Fe3O4 → Fe(OH)2 conversion, and the sequential sharp decrease from 150 to 350 mA h g−1 might be due to the H2 formation.
Fig. 4d illustrates the evolution of the diffusion coefficient during the discharging process, where the Fe2+/Fe3+ redox in Na2S-containing electrolyte exhibits a complex trend similar to that observed during the charging process. During the initial discharge from 0 to 60 mA h g−1, diffusion coefficients show high values ranging from 10−10 and 10−11 cm2 s−1, congruent with fast kinetics of oxidation of deposited H in an alkaline solution. During the following discharging from 60 to 180 mA h g−1, a similar U-shaped trend appears, consistent with the transition from the deposited H oxidation into Fe(OH)2 → Fe3O4 conversion process. The last discharge stage from 180 to 275 mA h g−1 is accompanied by the decreasing diffusion coefficient, which is related to the formation of the GR phase via the insertion of hydrosulfide into Fe(OH)2. Notably, this hydrosulfide intercalation process shows much lower diffusion coefficients (10−13 and 10−11 cm2 s−1) than the deintercalation process (∼10−10 cm2 s−1) in Fig. 4c, possibly because the incoming hydrosulfide anion faces strengthened electrostatic repulsion from previously intercalated anions, slowing down the insertion process, reducing the apparent diffusion coefficient. It is worth noting that a low-alkalinity electrolyte (e.g., 0.01 M NaOH) is used in this study to promote hydrosulfide intercalation and deintercalation. Our previous studies showed that a low-alkalinity solution favours anion intercalation in Fe(OH)2 layered materials during discharge, forming an anion-intercalated green rust phase and mitigating the Fe(OH)2/Fe3O4 conversion reaction.21
To elucidate how the sulfide additive influences the charge-transfer kinetics of the iron redox processes, electrochemical impedance spectroscopy (EIS) was conducted at selected charging (C1–C3) and discharging (D1–D3) potentials during the CP measurements (Fig. 5). The charge-transfer resistance (Rct) extracted from the Nyquist plots shows trends consistent with those from the GITT analysis. In the NaOH electrolyte containing sulfide, the Rct values progressively decreased from 80.2 Ω to 52.8 Ω and 39.3 Ω as the charging potential shifted from C1 (−0.7 V) to C2 (−0.9 V) and C3 (−1.1 V), corresponding to HS− deintercalation, Fe3O4/Fe(OH)2 conversion, and the onset of the HER, respectively. Conversely, during discharge, the Rct values increased from 36.2 Ω to 60.4 Ω and 94.7 Ω as the potential increased from D1 (−0.85 V) to D2 (−0.56 V) and D3 (−0.2 V), reflecting deposited-hydrogen removal, Fe(OH)2/Fe3O4 conversion, and HS− intercalation, respectively.
 | ||
| Fig. 5 CP measurements during (a and b) charging and (c and d) discharging in (a, c and e) NaOH/Na2SiO3/Na2S and (b, d and f) NaOH/Na2SiO3 electrolytes. EIS measurements at selected potentials in (e) NaOH/Na2SiO3/Na2S and (f) NaOH/Na2SiO3 electrolytes. | ||
In contrast, significantly higher Rct values were observed in NaOH electrolyte without the sulfide additive for both charging and discharging processes involving the Fe3O4 redox couple, indicating that sulfide promotes faster charge-transfer kinetics, consistent with GITT results. Notably, in additive-free NaOH, the HER became much more vigorous during charging, leading to severe H2 bubble accumulation on the electrode surface, and preventing the acquisition of reliable EIS spectra at highly negative potentials.
Conclusions
In this study, we reveal how sulfide-containing alkaline electrolytes fundamentally alter the Fe2+/Fe3+ redox system by enabling multiple ion-transfer pathways. Using operando X-ray diffraction and X-ray absorption spectroscopy, combined with galvanostatic intermittent titration, we demonstrate three complementary charge storage mechanisms: (i) Fe(OH)2/Fe3O4 conversion, (ii) hydrosulfide intercalation into layered Fe(OH)2, and (iii) hydrogen deposition on Fe(OH)2 surfaces. These hydroxide-, hydrosulfide-, and proton-mediated processes collectively deliver a reversible capacity of up to 330 mA h g−1, exceeding the theoretical limit of single-electron Fe2+/Fe3+ redox. Our findings demonstrate that the resulting multi-ionic mechanism provides faster kinetics, reduced polarization, and significantly improved cycling stability compared to conventional alkaline Fe systems. Significantly, this strategy relies on low-concentration sulfide additives in dilute alkaline solutions, ensuring compatibility with scalable and cost-effective electrolyte formulations. By uncovering these intertwined redox pathways, this work establishes a new design principle for revitalizing iron chemistry in aqueous batteries. Our ongoing work includes full-cell configurations at high areal density and more concentrated alkaline solutions to assess the rate performance, cycling stability, and system-level applicability of the iron redox chemistry. We believe our reported studies will lead to a practical route to upgrade abundant iron resources—including waste Fe oxides—into high-performance, sustainable anodes for LDES applications.Author contributions
X. T. conceived the research idea, designed the experiments, and supervised the project. D. A. performed material synthesis and characterization, as well as electrochemical experimental measurements. D. A., X. T., M. A., G. K., and C.·H. L. performed operando XRD measurements. D. A. performed X-ray analysis. X. T. and D. A. wrote the manuscript. All the authors contributed to the editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the main text and/or the supplementary information (SI). Supplementary information: XRD and TEM analysis of the as-made electrode materials, electrochemical measurements at different current densities and sulfide additive concentrations, and complete in situ XRD and XAS data. See DOI: https://doi.org/10.1039/d5ta08086j.Acknowledgements
This research received financial support from the National Science Foundation under the Award Number 2222928 (D. A. and X. T.). This research used the FEI Talos 200X at the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility at Brookhaven National Laboratory under Contract No. DE-SC0012704. This research used 28-ID-1 and 6-BM beamlines of the National Synchrotron Light Source II, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. We thank Dr Bruce Ravel for assisting with the XAS experiments at beamline 6-BM at BNL-NSLS II.References
- B. Dunn, H. Kamath and J. M. Tarascon, Electrical Energy Storage for the Grid: A Battery of Choices, Science, 2011, 334(6058), 928–935 Search PubMed.
- M. Pasta, C. D. Wessells, R. A. Huggins and Y. Cui, A high-rate and long cycle life aqueous electrolyte battery for grid-scale energy storage, Nat. Commun., 2012, 3, 1149 CrossRef.
- J. Ma, Y. Li, N. S. Grundish, J. B. Goodenough, Y. Chen, L. Guo, Z. Peng, X. Qi, F. Yang, L. Qie, C.-A. Wang, B. Huang, Z. Huang, L. Chen, D. Su, G. Wang, X. Peng, Z. Chen, J. Yang, S. He, X. Zhang, H. Yu, C. Fu, M. Jiang, W. Deng, C.-F. Sun, Q. Pan, Y. Tang, X. Li, X. Ji, F. Wan, Z. Niu, F. Lian, C. Wang, G. G. Wallace, M. Fan, Q. Meng, S. Xin, Y.-G. Guo and L.-J. Wan, The 2021 battery technology roadmap, J. Phys. D: Appl. Phys., 2021, 54(18), 183001 Search PubMed.
- M. S. Whittingham, Lithium batteries and cathode materials, Chem. Rev., 2004, 104(10), 4271–4301 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Challenges for Rechargeable Li Batteries, Chem. Mater., 2010, 22(3), 587–603 CrossRef CAS.
- B. Scrosati and J. Garche, Lithium batteries: status, prospects and future, J. Power Sources, 2010, 195(9), 2419–2430 CrossRef CAS.
- Y. Sui and X. Ji, Electrolyte Interphases in Aqueous Batteries, Angew. Chem., Int. Ed., 2024, 63(2), e202312585 CrossRef CAS PubMed.
- Y. Liang and Y. Yao, Designing modern aqueous batteries, Nat. Rev. Mater., 2023, 8(2), 109–122 Search PubMed.
- B. Björkman and C. Samuelsson, Chapter 6 – Recycling of Steel, in Handbook of Recycling, ed. Worrell, E. and Reuter, M. A., Elsevier, 2014, pp. 65–83 Search PubMed.
- R. U. Ayres, Metals recycling: economic and environmental implications, Resour., Conserv. Recycl., 1997, 21(3), 145–173 CrossRef.
- R. D. McKerracher, C. Ponce de Leon, R. G. A. Wills, A. A. Shah and F. C. Walsh, A Review of the Iron–Air Secondary Battery for Energy Storage, ChemPlusChem, 2015, 80(2), 323–335 CrossRef CAS.
- N. Chawla, Recent advances in air-battery chemistries, Mater. Today Chem., 2019, 12, 324–331 CrossRef CAS.
- J. M. E. Abarro, J. N. L. Gavan, D. E. D. Loresca, M. A. A. Ortega, E. A. Esparcia and J. A. D. R. Paraggua, A Tale of Nickel–Iron Batteries: Its Resurgence in the Age of Modern Batteries, Batteries, 2023, 9(7), 383 CrossRef CAS.
- J. Islam, R. Anwar, M. Shareef, H. M. Zabed, J. N. Sahu, X. Qi, M. U. Khandaker, A. Ragauskas, I. Boukhris, M. R. Rahman and F. Islam Chowdhury, Rechargeable metal–metal alkaline batteries: recent advances, current issues and future research strategies, J. Power Sources, 2023, 563, 232777 Search PubMed.
- Y. Yang, H. Yang, X. Wang, Y. Bai and C. Wu, Multivalent metal–sulfur batteries for green and cost-effective energy storage: Current status and challenges, J. Energy Chem., 2022, 64, 144–165 CrossRef CAS.
- R. Saltat, Nickel-iron battery system safety, Final report; Eagle-Picher Industries, Inc., Joplin, MO (USA), Electronics Div., 1984. https://www.osti.gov/servlets/purl/6758087.
- D. Arumugam, T. Zhou, S. N. Jagadeesan, L. Zhang, R. T. Pidathala, A. M. M. Abeykoon, G. Kwon, D. Olds, B. Narayanan and X. Teng, Electrochemical Reduction Pathways from Goethite to Green Iron in Alkaline Solution with Silicate Additive, ACS Sustain. Chem. Eng., 2025, 13(7), 2633–2640 CrossRef CAS PubMed.
- S. Trasatti, Work function, electronegativity, and electrochemical behaviour of metals: III. Electrolytic hydrogen evolution in acid solutions, J. Electroanal. Chem. Interfacial Electrochem., 1972, 39(1), 163–184 Search PubMed.
- A. K. Manohar, C. Yang and S. R. Narayanan, The Role of Sulfide Additives in Achieving Long Cycle Life Rechargeable Iron Electrodes in Alkaline Batteries, J. Electrochem. Soc., 2015, 162(9), A1864–A1872 CrossRef CAS.
- S. R. Narayanan, G. K. S. Prakash, A. Manohar, B. Yang, S. Malkhandi and A. Kindler, Materials challenges and technical approaches for realizing inexpensive and robust iron-air batteries for large-scale energy storage, Solid State Ionics, 2012, 216, 105–109 Search PubMed.
- F. Guo, S. N. Jagadeesan, R. T. Pidathala, S. Kim, X. Shan, N. A. Deskins, A. M. M. Abeykoon, G. Kwon, D. Olds, B. Narayanan and X. Teng, Revitalizing Iron Redox by Anion-Insertion-Assisted Ferro- and Ferri-Hydroxides Conversion at Low Alkalinity, J. Am. Chem. Soc., 2022, 144(27), 11938–11942 CrossRef CAS PubMed.
- S. N. Jagadeesan, G. D. Barbosa, F. Guo, L. Zhang, A. M. M. Abeykoon, G. Kwon, D. Olds, C. H. Turner and X. Teng, Chloride Insertion Enhances the Electrochemical Oxidation of Iron Hydroxide Double-Layer Hydroxide into Oxyhydroxide in Alkaline Iron Batteries, Chem. Mater., 2023, 35(16), 6517–6526 Search PubMed.
- M. Usman, J. M. Byrne, A. Chaudhary, S. Orsetti, K. Hanna, C. Ruby, A. Kappler and S. B. Haderlein, Magnetite and Green Rust: Synthesis, Properties, and Environmental Applications of Mixed-Valent Iron Minerals, Chem. Rev., 2018, 118(7), 3251–3304 CrossRef CAS PubMed.
- E. De Ketelaere, D. Moed, K. Verbeken and T. Depover, A comprehensive electrochemical evaluation of the corrosion inhibition by sodium silicate on carbon steel, Electrochim. Acta, 2024, 507, 145053 CrossRef CAS.
- S. N. Jagadeesan, F. Guo, R. T. Pidathala, A. M. M. Abeykoon, G. Kwon, D. Olds, B. Narayanan and X. Teng, Unlocking High Capacity and Reversible Alkaline Iron Redox Using Silicate-Sodium Hydroxide Hybrid Electrolytes, ChemSusChem, 2024, 17(19), e202400050 CrossRef CAS.
- H. Kitamura, L. Zhao, B. T. Hang, S. Okada and J.-i. Yamaki, Effect of Charge Current Density on Electrochemical Performance of Fe/C Electrodes in Alkaline Solutions, J. Electrochem. Soc., 2012, 159(6), A720–A724 CrossRef CAS.
- H. Weinrich, M. Gehring, H. Tempel, H. Kungl and R.-A. Eichel, Electrode thickness-dependent formation of porous iron electrodes for secondary alkaline iron-air batteries, Electrochim. Acta, 2019, 314, 61–71 CrossRef CAS.
- D. Arumugam, T. Zhou, S. N. Jagadeesan, R. T. Pidathala, L. Zhang, A. M. M. Abeykoon, G. Kwon, D. Olds, B. Narayanan and X. Teng, Electrochemical Reduction Pathways from Goethite to Green Iron in Alkaline Solution with Silicate Additive, ACS Sustain. Chem. Eng., 2025, 13(7), 2633–2640 CrossRef CAS PubMed.
- B. Miller and A. Chen, Oscillatory instabilities during the electrochemical oxidation of sulfide on a Pt electrodeJ, Electroanal. Chem., 2006, 588(2), 314–323 CrossRef CAS.
- A. N. Buckley, I. C. Hamilton and R. Woods, An investigation of the sulphur(−II)/sulphur(0) system on bold electrodes, J. Electroanal. Chem. Interfacial Electrochem., 1987, 216(1), 213–227 CrossRef CAS.
- B. Miller and A. Chen, Effect of concentration and temperature on electrochemical oscillations during sulfide oxidation on Ti/Ta2O5−IrO2 electrodes, Electrochim. Acta, 2005, 50(11), 2203–2212 CrossRef CAS.
- N. P. Holzapfel, V. Augustyn and V. Balland, Fundamentals of Proton-Insertion Coupled Electron Transfer (PICET) in Metal Oxides for Aqueous Batteries, ACS Energy Lett., 2025, 10(3), 1143–1164 CrossRef CAS.
- H. Li, M. Abdelgaid, J. R. Paudel, N. P. Holzapfel, V. Augustyn, J. R. McKone, G. Mpourmpakis and E. J. Crumlin, Operando Unveiling of Hydrogen Spillover Mechanisms on Tungsten Oxide Surfaces, J. Am. Chem. Soc., 2025, 147(8), 6472–6479 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |