
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of superionic conduction in K2NiF4-type Ba–Li oxyhydrides from a first-principles molecular dynamics study
Jun
Haruyama
 *a,
Fumitaka
Takeiri
ab,
Genki
Kobayashi
ac and
Osamu
Sugino
d
*a,
Fumitaka
Takeiri
ab,
Genki
Kobayashi
ac and
Osamu
Sugino
d
aSolid State Chemistry Laboratory, Pioneering Research Institute (PRI), RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan. E-mail: jun.haruyama@riken.jp
bDepartment of Chemistry, Kindai University, 3-4-1 Kowakae, Higashiosaka, Osaka 577-8502, Japan
cDepartment of Electrical Engineering and Bioscience, School of Advanced Science and Engineering, Waseda University, 3-4-1 Okubo, Shinjuku-ku, Tokyo 169-8555, Japan
dThe Institute for Solid State Physics, The University of Tokyo, 5-1-5 Kashiwanoha, Kashiwa, Chiba 277-8581, Japan
First published on 7th January 2026
Abstract
Hydride-ion (H−) conductors have attracted considerable attention as charge carriers for hydrogen transport applications. To understand the microscopic H− dynamics and elucidate the superionic conduction characteristics, we investigated K2NiF4-type Ba–Li oxyhydrides, Ba1·75LiH2·7O0.9 (BLHO). Three microscopic structural models were constructed, Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29, in which the H and O atoms at the apical sites were ordered in two ways (β- and δ-phase-like). By using first-principles electronic structure calculations, the constructed models showed band gaps of 2.2–2.7 eV, occupied H-1s orbitals, and Bader charges of Ba1.37+, Li0.85+, H0.73−, and O1.45−, which are consistent with those of pure H− conductors and electrochemical H− carriers observed in previous studies. Using first-principles molecular dynamics (FPMD) simulations, the H− diffusion coefficients of the β- and δ-like models were in good agreement with those reported in previous quasi-elastic neutron scattering analyses. The activation energies of the ab-plane (0.52 and 0.55 eV in the β and δ models, respectively) and c-axis components (0.78 eV in the δ model) from FPMD simulations were found to be related to migration barriers of distinct H− transfer pathways. Furthermore, the Lindemann index of H exceeded the threshold for a solid–liquid transition at temperatures above 700 K, suggesting sublattice melting of the H− equatorial sites. Finally, above the sublattice melting temperature (T ≥ 700 K), the van Hove correlation function showed that the peak growth at the correlation hole position increased for a few picoseconds, and rearrangement indicator analysis showed that H− hopping events tend to form a large cluster, indicating cooperativity in the H− diffusion.
1. Introduction
Energy and chemical conversion technologies based on hydrogen transport have become one of the solutions for realizing environmental sustainability.1 Hydride ions (H−), which possess a strong reducing ability due to their redox potential of approximately 0.36 V vs. Li+/Li,2 have been proposed as alternative charge carriers to protons (H+) for electrochemical applications.3,4 Recently, intensive efforts have been devoted to exploring their potential for the development of novel electrochemical devices.5–7 Initial studies on H− conductors primarily focused on alkaline-earth metal hydrides.8–10 Significant developments occurred in 2016 with the realization of pure H− conduction in oxyhydrides, which opened new directions in this field of research. The first example, La–Sr–Li oxyhydride (La2–x–ySrx+yLiH1+x+yO3–y) with a K2NiF4-type structure, features a framework in which hydride and oxide ions are ordered, thereby forming H− diffusion pathways within the oxide-based lattice.3 In this compound, tuning the La/Sr ratio allows for control over the H/O ratio and concentration of anion vacancies (V), enabling the modulation of H− conductivity. The discovery of La–Sr–Li oxyhydrides stimulated the exploration of H− conductors, particularly mixed-anion compounds such as oxyhydrides.11–16 Among the various compounds reported to date, Ba1.75LiH2.7O0.9 (BLHO), which adopts the same K2NiF4-type structure as the La–Sr–Li oxyhydride, is of particular interest because of its H− superionic conductivity that emerges upon undergoing a phase transition at approximately 300 °C.17–19The ionic conductivity of BLHO was determined by electrochemical impedance spectroscopy (EIS) measurements in a wide temperature range (200 °C ≤ T ≤ 350 °C, T is temperature).17 It exhibited an ionic conductivity of 10−5–10−7 S cm−1 with an activation energy of approximately 103 kJ mol−1 at low temperatures (T ≤ 300 °C), a drastic increase from 10−5 to 10−2 S cm−1 across 300 °C < T ≤ 315 °C, and a nearly temperature-independent value at high temperatures (315 °C < T). This conductivity behavior of BLHO is almost the same as that of type I superionic conductors. Usually, superionic conductors can be categorized by their phase transition behaviors; type I involves a discontinuous conductivity jump with a structural (first-order) phase transition, whereas type II shows continuous conductivity change with gradual defect accumulation (higher-order phase transition).20,21 The representative AgI shows a conductivity jump of four orders of magnitude at the α–β transition temperature, and the superionic conductivity of the high-temperature α-AgI with the quasi-molten cationic sublattice is characterized by low activation energies (0.1 eV).22–24 Therefore, understanding the relationship between the ionic conductivity, crystal structures, and H− dynamics in BLHO from a solid-state ionic perspective is important.
However, from a crystal structure viewpoint, the phase transition behavior of BLHO differs from that of AgI. BLHO adopts a K2NiF4-type structure in which perovskite and rock-salt layers are alternately stacked along the c-axis. In this structure, the equatorial sites bridging LiX4 planes, where X = H− and V, are either occupied by H− anions (Heq) or empty (VH). The apical sites are, on the other hand, occupied either by H− (Hap) or O2− anions (Oap). The occupied and empty Ba sites are denoted as Ba and VBa, respectively. These sites (Ba/VBa, the Heq/VH, Hap/Oap, and Ba/VBa) are illustrated in Fig. 1(a). Under ambient pressure, the stable composition is represented as (Ba1.75V0.25)Li(H1.6V0.4)eq(H1.1O0.9)ap, which composition was confirmed by neutron diffraction analysis.17 While the composition of BLHO includes large amounts of intrinsic H and Ba vacancies, no significant change in composition was observed during the β–γ–δ phase transitions. As shown in Fig. 1(a)–(c), there are different anion configurations depending on temperature, defined as the β-phase (T ≤ 300 °C), γ-phase (300 °C ≤ T ≤ 350 °C), and δ-phase (350 °C ≤ T) from the low-temperature side.17 The β–γ and γ–δ phase transitions with increasing temperature can be interpreted as the order/disorder transitions of Heq/VH (and also Ba/VBa) and Hap/Oap sites, respectively, accompanied by a crystal symmetry change from the space groups Pnm21 through Pnma to I4/mmm. The behavior, where superionic conductivity emerges through changes in the ordering of carrier ions while maintaining the basic framework structure, is akin to that of type II superionic conductors such as β-PbF2. They exhibit a gradual transition from an insulating to a conducting state25 with a second-order phase transition.20,26,27 Regardless of type I or II, superionic conduction can be characterized in terms of sublattice melting, which involves the disordering of one of the ion sublattices. In the case of BLHO, the equatorial LiX4 plane can be interpreted as an H− diffusion layer; thus, the disordered arrangement of Heq/VH at the equatorial sites implies the occurrence of sublattice melting.
 | ||
| Fig. 1 Crystal structures of (a) β-, (b) γ-, and (c) δ-phase BLHO (side and top views). Ba/VBa, Li, Heq/VH, and Hap/Oap sites are represented by green/white, light blue, blue/white, and blue/red spheres, respectively. Blue octahedra and black rectangles represent the LiX6 unit and periodic unit cell, respectively. | ||
In the previous study, an indicative analysis of the H− dynamics in BLHO was conducted using quasi-elastic neutron scattering (QENS).17 From the momentum transfer dependence of the QENS signal at 315 °C, the self-diffusion coefficient of hydrogen (DH) estimated based on a jump-diffusion model was 3–4 × 10−7 cm2 s−1. Considering the H− concentration in BLHO, this value is consistent with the ionic conductivity obtained from EIS, providing evidence that the high conductivity of BLHO arises from fast H− diffusion. In contrast, the QENS signal at 275 °C, prior to the conductivity jump and β–γ transition, showed a comparable level of H− diffusivity to that at 315 °C, suggesting that only local H− motion was accelerated in the β phase, whereas the structural phase transition to the γ phase enabled long-range H− diffusion that directly contributed to ionic conductivity. These results suggest that the conduction mechanism of H− changes from the typical jump diffusion model through the β–γ transition; however, they do not detect sublattice melting that characterizes the superionic state.
Therefore, in this study, we performed first-principles molecular dynamics (FPMD) simulations to investigate the relationship between H− diffusion and BLHO sublattice melting. Since X-ray diffraction experiments provide only information on the average crystal structure, we first constructed several microscopic structural models. The structural models satisfying periodic boundary conditions consists of approximately 200 atoms. Next, we verified the structural models constructed for each phase by comparing their H− diffusion coefficients and activation energies with those from EIS and QENS data. Then, sub-nanosecond-scale FPMD calculations revealed behavior interpretable as H− sublattice melting, and rapid H− diffusion accompanied by collective motion. Comparison with previous studies on typical superionic conductors (PbF2 and AgI) suggests that the large H− conductivity of BLHO is likely due to sublattice melting.
2. Methods
2.1 Models
Before conducting the FPMD simulations, we carefully examined several microscopic structural models of the system. Due to the non-stoichiometric nature of BLHO and the disordered arrangement of ions and vacancies, it is necessary to construct a microscopic supercell model. However, this model cannot be determined uniquely. Among the possible models, an appropriate one can be selected based on the reproducibility of measured quantities such as electronic structure and diffusion properties.First, considering the pure H− conducting behavior, the BLHO models should be bandgap semiconductors, which require a charge neutrality condition with respect to Ba2+, Li+, H−, and O2− ions.
As a model that reproduces the experimental composition ratio as faithfully as possible, we selected the β-type 2 × 4 × 1 model shown in Fig. 2(a) and S1(a)–(c). In this model, the H atoms and O atoms at the vertex are relatively neatly aligned along the b axis. Using this as a reference, we created three variants by adding a small number of Ba, H, and O atoms, that is, based on this model: Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29. The composition ratios and numbers of Heq and Ba vacancies of the three models are listed in Table S1 of the SI. The configurations and cell-optimized structures are shown in Fig. S2(a)–(c). In all models, the Li–O and counter side Li–Hap distances varied between 1.85–2.43 and 1.93–3.41 Å, respectively, resulting in the formation of LiH3O tetrahedra and free Hap atoms.
 | ||
| Fig. 2 Crystal structures of (a) β-like Ba56Li32H88O28 and (b) δ-like Ba14Li8H22O7. Ba, Li, H, and O atoms are depicted as green, light blue, blue, and red spheres, respectively. VH and VBa are depicted as small and big white spheres, respectively. (c) PDOSs of the δ-like Ba14Li8H22O7 model. The black, green, light blue, blue, and red lines represent the total, Ba, Li, H, and O contributions, respectively. The dotted line indicates the Fermi level located at the middle of the bandgap. | ||
Next, we moved on to constructing the δ-like model. Fig. 2(b) and S3(a)–(c) show the reference model of Ba14Li8H22O7 (Ba1·75LiH2·75O0.875). In this model, the H and O atoms at the apical sites are alternately aligned along both the a- and b-axes of the unit cell. Based on this δ-like 2 × 2 × 1 model, we constructed three composition-adjusted models in a 4 × 4 × 1 supercell: Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29, whose configurational and cell-optimized structures are shown in Fig. S4(a)–(c). Similar to the β-like models, the Li–O and counter side Li–Hap distances varied between 1.81–1.93 and 2.73–3.69 Å, respectively, resulting in the formation of LiH3O tetrahedra. The large variation of distances might be related to the observed large B factors of Li atoms in BLHO, BLi = 1.90 (RT), 6.1 (314 °C), and 6.4 (368 °C) Å2, as shown in ref. 17. As expected from the experimental phases, the optimized energies of the δ-like models are higher than those of the β-like models by approximately 0.12–0.16 eV per Li. In this study, while the local structures and vacancy concentration of constructed models do not contradict high-precision neutron diffraction analysis,17 it is not possible to verify their thermodynamic characteristics such as vacancy formation energy. Therefore, verification from this perspective remains a topic for future research.
The optimized cell parameters and calculated bandgap energies are listed in Table S2 in the SI. Slight compositional changes do not significantly affect the calculated values. Although the cell parameters are slightly overestimated compared with those of the β-phase, which is typical for standard density functional theory (DFT) calculations. All models show electronic bandgap energies of 2.2–2.7 eV. Considering that DFT calculations with semilocal exchange–correlation functionals tend to underestimate the bandgap value, the calculated values suggest the semiconducting or insulating electronic conductivity of BLHO. Additionally, we confirmed with spin-polarized calculations that the total magnetic moments of all the relaxed structures are negligibly small.
Fig. 2(c) shows the projected densities of states (PDOSs) of the δ-like Ba14Li8H22O7 model, which shows typical properties in BLHO structure models. The H-1s and O-2p orbitals mainly contribute to the valence band, and the Li-2s orbitals contribute to the conduction band. Regardless of the bonding and antibonding states, all orbitals with a significant H-1s component are occupied, which can be interpreted as an H− like character. This valence band property is almost the same as that of K2NiF4-type La–Sr–Li and Ba–Sc oxyhydrides.28–31
To discuss the effective charge distributions in BLHO, we assigned an electronic charge density to each atom using the Bader charge analysis.32 Positive and negative ions are easily distinguished from the Bader charges, as listed in Table 1. These values are close to the formal ionic charges (Ba2+, Li+, H−, and O2−), and the fractional parts of the charge value represent a small amount of covalent character. In addition, the Bader charges of Li, H, and O in BLHO are similar to those in La–Sr–Li oxyhydrides,28,29 indicating that the ionic and bonding characteristics of K2NiF4-type oxyhydrides are common.
| Atom | Charge [e] |
|---|---|
| Ba | +1.37 |
| Li | +0.85 |
| H | −0.73 |
| O | −1.45 |
Based on the above preparative calculations, we adopted the simplest models, β- and δ-like Ba56Li32H88O28, as the initial structures used in the FPMD simulations. Namely, we conducted long term FPMD simulations using two possible configurations that mimic the β and δ crystal structures. Because the crystallographic difference between the β and γ phases can be attributed to the partial occupancy ratios of the equatorial anion sites, we consider that the phase transition from β to γ can be included automatically in the FPMD simulations at high temperatures, even though the initial structures started from β-like models. In addition, there are numerous possible configurations for three partial occupancy sites: Heq/VH, Ba/VBa and Hap/Oap. For the partial occupancy effect at the Heq/VH site, H− hopping between Heq/VH sites frequently occurred at higher temperatures and a suitable statistical ensemble can be accounted for to some context. While for the Ba/VBa and Hap/Oap sites, this is not the case owing to the limited duration of the FPMD simulations, necessitating further investigation in future studies.
2.2 Computational details
The β- and δ-like models as shown in Fig. 2(a) and (b), respectively, were constructed from the refined structural parameters of β- and δ-BLH O.17 The VESTA package was used to visualize the crystal structures and atom coordinates in this study.33To obtain optimized atomic configurations and cell parameters, band-gap energies, and PDOSs, the Quantum ESPRESSO (QE) code,34,35 which uses a plane-wave basis within the ultrasoft pseudopotential framework,36,37 was used to perform spin-unpolarized DFT calculations. It was confirmed that spin-polarized calculations, performed by starting from various initial spin configurations, yielded the same non-spin states as obtained from spin-unpolarized calculations. This indicates that the H− property is maintained throughout the nudged elastic band (NEB) calculations. The electronic configurations were 1s1 for H, 1s22s1 for Li with nonlinear core correction (NLCC),38 2s22p4 for O with NLCC, and 5s25p66s2 for Ba with NLCC; the pseudopotentials of H.pbe-rrkjus_psl.1.0.0.UPF, Li.pbe-sl-rrkjus_psl.1.0.0.UPF, O.pbe-n-rrkjus_psl.1.0.0.UPF, and Ba.pbe-spn-rrkjus_psl.1.0.0.UPF from the QE pseudopotential database were used.39,40 All DFT calculations were performed using the Perdew–Burke–Ernzerhof exchange–correlation functional.41 The cutoff energies were set to 40/320 Ry (1 Ry = 13.606 eV) for the wave functions/augmented charge. Only for the cell optimization procedure, they increased to 60/480 Ry to ensure convergence. Converged k-point samplings of 4 × 4 × 2 and 2 × 2 × 2 were adopted for the 2 × 2 × 1 and 4 × 4 × 1 cells, respectively. The atomic positions and cell parameters were relaxed until the residual forces and stresses were less than 10−3 Ry bohr−1 (1 bohr = 0.52918 Å) and 0.5 kbar (1 bar = 100 Pa), respectively. For the Bader charge analysis, we used a program distributed by the Henkelman group.42,43
The NEB method,44,45 which can determine the minimum energy path between two stable points, was used to evaluate the activation energies of H− migration. The NEB method was applied to a discretized path using eight images. The start- and end-point configurations were constructed by replacing the H atom at the target site. All atoms in the start and end images were relaxed in advance of NEB calculations, whereas those in the intermediate images were relaxed until their forces converged within 0.05 eV Å−1. The electronic structure calculation conditions were the same as those used in the QE calculations. The cell parameters were fixed at the experimental values. All NEB calculations were performed using a representative δ-like model of Ba56Li32H88O28.
For FPMD simulations, we used the Vienna Ab initio Simulation Package (VASP),46–48 a plane-wave basis within the projector-augmented-wave method49,50 with a cutoff energy of 400 eV for the wave functions. A single Γ-centered k-point and a time step of 2 fs were adopted in spin-unpolarized DFT calculations. In hydrogen containing materials, a shorter timestep (1 fs) is typically chosen to describe rapid H vibrations, such as the OH stretching mode (frequency of 3000 cm−1). In hydride ion conductors, however, a longer timescale of 2 fs can be adopted29 because the maximum phonon frequency is significantly lower than the OH frequencies (e.g., approximately 1600 cm−1 in La–Li oxyhydride28). The relaxed β- and δ-like structures were assigned an initial temperature of 300 K according to the Boltzmann distribution and then heated to the desired temperatures in the NVT ensemble with a Nose–Hoover thermostat.51,52 FPMD simulations were conducted for approximately 200 ps under thermal equilibrium conditions at T = 500, 700, 900, and 1100 K. The self-diffusion coefficients, Lindemann indices, and van Hove time correlation functions were then calculated from the atomic trajectories obtained from the FPMD simulations.
The self-diffusion coefficient of H atoms was calculated using the mean square displacement (MSD) over interval time Δt;29,53
 | (1) |
The Lindemann index δL,54–56 which measures the atomic vibrational amplitude, was calculated as follows:
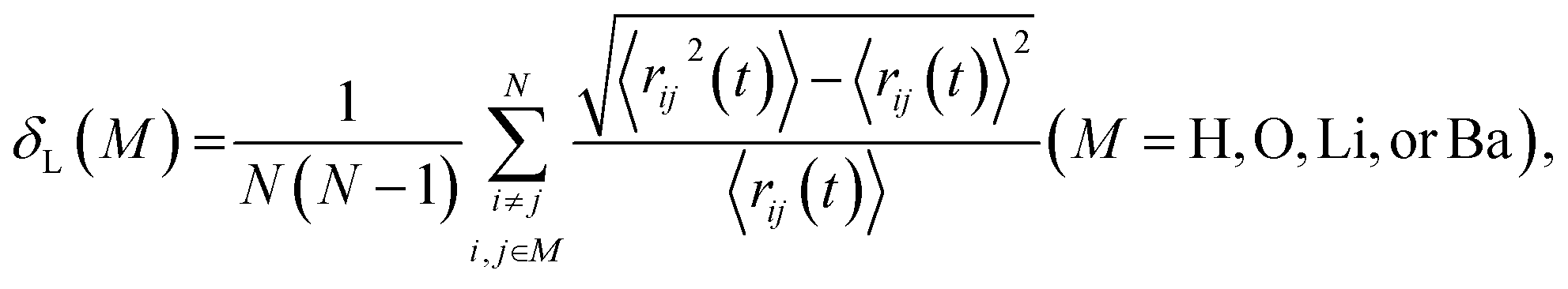 | (2) |
| rij(t) = |ri(t) − rj(t)|, | (3) |
The distinct part of van Hove correlation function Gd,57–60 which describes the time–space correlation between selected atoms, was calculated as a function of radial coordinate r and time distance Δt:53
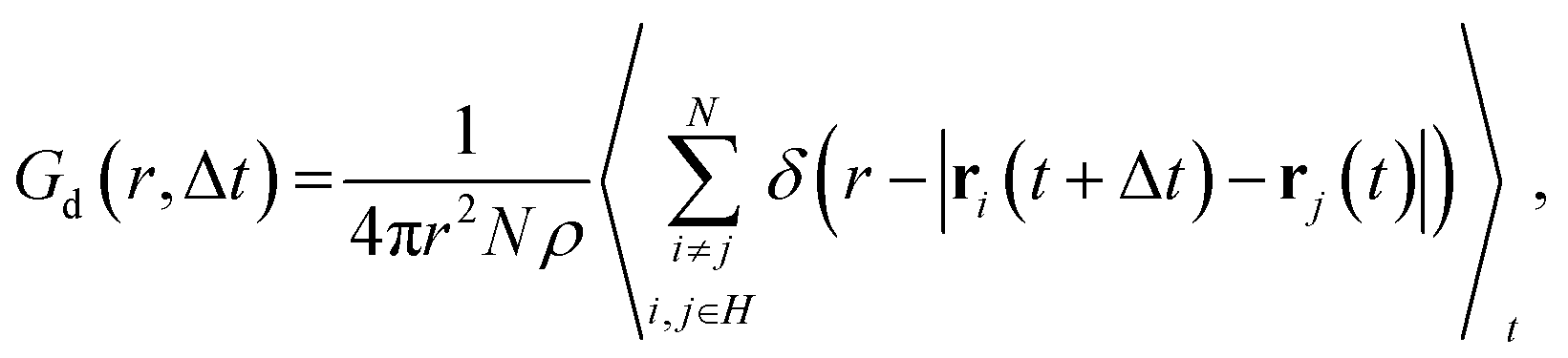 | (4) |
We analyzed the number of hopping events and their cluster sizes using Ri,61 (rearrangement index), an indicator of net atomic displacement. This index is characterized by a typical reconfiguration time interval, Δt:
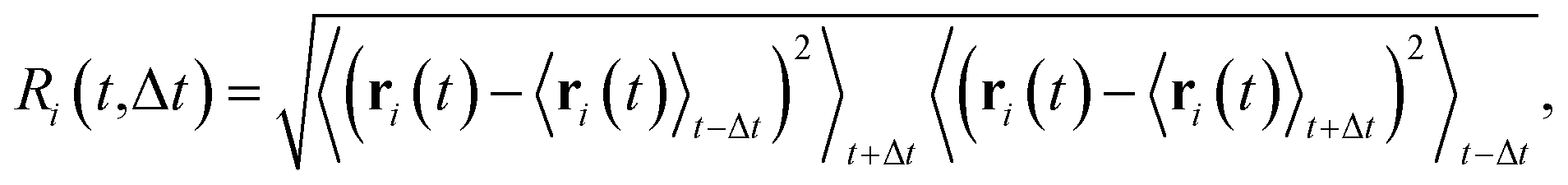 | (5) |
3. Results
Fig. S5 shows the calculated MSDs of H obtained from temperature-controlled FPMD simulations. At the lowest temperature of 500 K, sufficient H-hopping events were not observed during the simulation, and the values of D at 500 K contain errors. At higher temperatures, the diffusion process in the ab plane corresponds to H hopping among the equatorial sites, which is consistent with the experimental results of the crystal structure analysis. Moreover, at 1100 K, the δ-like model showed that diffusion in the c-direction is comparable to that in the ab-plane, as shown in Fig. S5(h), although BLHO is expected to melt in this high-temperature region.Fig. 3 shows the diffusion coefficients Dab and Dc obtained from the slopes of the MSD data. It should be noted that the simulation length did not reach the diffusion regime where the MSD is clearly linear with respect to time, and consequently, the diffusion barrier could not be accurately estimated. However, it is possible to compare qualitatively the differences arising from the phase variations. In the high-temperature region (700 K ≤ T ≤ 1100 K), the Dab values of the β model are almost the same as those of the δ model, indicating no significant difference between the β and δ phases regarding ab-plane diffusion and conduction. The Dc values are distinctly lower for the β-like model than for the δ-like model at temperatures above 900 K. This suggests that the alternating arrangement of the apical H and O atoms hinders c-axis diffusion. Furthermore, the interpolation of Dab of the δ model reproduces the reported value (D = 3–4 × 10−7 cm2 s−1 at 315 °C) from QENS analysis,17 and that of the β model is also consistent with the previous value. This agreement strongly suggests that the FPMD simulations of the constructed microscopic models is reliable for discussing the sublattice melting as well as the dynamical aspects of H− diffusion in BLHO.
 | ||
| Fig. 3 Diffusion coefficients D of H as a function of inverse temperature. Blue and orange represent β- and δ-like models, respectively. Solid and open circles represent the D of theab-plane and c-axis components, respectively. Dashed lines represent the corresponding activation energies of the ab-components in the high-temperature region (700 K ≤ T ≤ 1100 K). The cross mark represents the D value obtained from QENS analysis.17 | ||
The estimated activation energies Ea from Dab in the high-temperature region (700 K ≤ T ≤ 1100 K) are 0.52 and 0.55 eV for the β- and δ-models, respectively. These values differ from those obtained from experimental ionic conductivity: Ea = 1.1 eV (β phase) and 0.0 eV (γ phase). The comparison indicates that the rate-determining process of macroscopic conductivity is not the ab-plane diffusion of H− but is rather related to the ionic diffusion of H− and O2− along the c-direction, their diffusion at crystallite interfaces, or structural transition. In the low temperature region, c-direction diffusion is one of the possible rate-determining steps. The reliable large-scale simulations should be conducted in the future to obtain the activation energy of c-direction diffusion quantitatively.
The activation energies of H− diffusion from the FPMD simulations are associated with elementary processes by NEB calculations. We should note that, in a disordered system, it is necessary to perform numerous NEB calculations for various configurations to obtain the average activation energy. Hence, the NEB results can be regarded as the rough estimation of the activation energies and supporting data for FPMD calculations. Fig. 4(a) and (b) show the energy profiles along the ab-plane and c-axis H migration pathways, respectively, assuming VH-hopping processes within the δ-like model. We obtained several activation energies depending on the local bonding environment for ab-plane H migration or H hopping between the equatorial sites. For example, in the H1 migration path, the initial Li1–H1 bond length is large (LiX4 like) and rather unstable, which resulted in a slightly higher energy at the start point than that at the end. The H2 migration path, hopping between LiX5 sites, showed a high activation energy of approximately 0.4 eV. Finally, the energy range between the minimum (end point of the H1 path) and maximum (transition state of the H2 path) NEB images was 0.50 eV, which was consistent with the activation energy of 0.55 eV obtained from Dab of the δ-model.
 | ||
| Fig. 4 Energy profiles along the (a) ab-plane and (b) c-axis for H migration. Solid and open circles represent the relative energies from the NEB calculations, and solid lines are guides for the eye, the colors of which correspond to the migration paths indicated by the colored arrows in the subset structures. In the purple and green migration paths, the forward H atom moves to the next vacancy before the backward H atom starts moving. | ||
For c-axis diffusion or H hopping between the apical and equatorial sites, we obtained several activation energies that depended on the local bonding environment. In general, the relative energy of the VH at the apical site was approximately 0.2–0.3 eV higher than that at the equatorial site. Occasionally, high-energy configurations occurred, such as the end points of the H3 and H4 paths, which indicated that Li polyhedra constructed only from H−, that is, LiH4 and LiH6, were quite unstable. Finally, the energy range between the minimum and maximum energy configurations was 0.82 eV, which was consistent with the activation energy of 0.78 eV obtained from Dc of the δ-model in the high-temperature region (700 K ≤ T ≤ 1100 K).
4. Discussion
In this section, we discuss whether sublattice melting, the disordering of one of the ion sublattices, occurs in BLHO. This phenomenon is usually characterized by a peak in the specific heat;20 however, the available FPMD data for temperatures are rather sparse. Alternatively, we use the Lindemann index δL as an indicator of liquid-like behavior. Furthermore, we discuss the behavior of two-body correlations and multi-ion concerted diffusion of H− in terms of the van Hove correlation function and hopping cluster analysis.Fig. 5 shows the δL of each atom in BLHO obtained from the FPMD simulations. At a low temperature of 500 K, there is no significant difference between the β and δ models, and all values are lower than the critical threshold of the solid–liquid transition. At an intermediate temperature of 700 K, δL(H) exceeded the threshold for both models, suggesting sublattice melting at the anion equatorial sites of Heq/VH. It should be noted that only the equatorial site contribution, δL(Heq), is larger than the plotted values because the calculated δL(H) also includes fluctuations at the apical sites (Hap). Because the critical temperature exceeding the threshold of the solid–liquid transition is consistent with the β–γ transition temperature (T ≈ 300 °C), we consider the results of δL(H) as theoretical suggestions for sublattice melting; BLHO is one of the superionic conductors.
 | ||
| Fig. 5 Lindemann indices δL as a function of temperature T. Solid and open symbols represent the β- and δ-like models, respectively. Green, light blue, blue, and red colors represent the Ba, Li, H, and O atoms, respectively. The critical threshold of the solid–liquid transition (=0.1) is indicated by the black dash-dotted line. | ||
At a high temperature of 900 K, δL(H) significantly increased, and that of the δ model exceeded 0.2. The δL(H) difference between the β and δ models can be attributed to c-axis diffusion. The δL(Li) values are also comparable to the solid–liquid threshold. At the highest temperature of 1100 K, all the δL values of the δ model exceeded the threshold, indicating that BLHO cannot maintain a solid-state crystal structure, that is, a typical solid–liquid transition occurs below this temperature. Finally, it should be noted that the indication of sublattice melting is qualitative. Toward the quantitative analysis, it is desirable to obtain the dynamical property associated with the occurrence of sublattice melting, as discussed in the last paragraphs.
Several studies suggest that sublattice melting affects ion diffusion through significant coupling with anharmonic phonons and parent lattice vibrations.64,65 Besides the thermodynamic analysis using the Lindemann index, we briefly discuss the dynamic properties related to the sublattice melting of H− from here. Fig. 6(a) shows the radial distribution function g(r) of the δ model at the four controlled temperatures. At the lowest temperature of 500 K, the first peak position of 2.86 Å can be assigned to the first nearest-neighbor distance between equatorial sites, d1(Heq–Heq) ≈ 2.8 Å, and that between equatorial and apical sites, d1(Heq–Hap) ≈ 3.0 Å. The shoulder peak at approximately 4.0 Å corresponds to the second nearest-neighbor distance between equatorial sites, d2(Heq–Heq) ≈ 4.0 Å, and the first nearest-neighbor distance between apical sites, d1(Hap–Hap) ≈ 3.8 Å. The second peak at 5.02 Å corresponds to the second nearest-neighbor distance between the equatorial and apical sites, d2(Heq–Hap) ≈ 5.0 Å. At increased temperatures of 700–1100 K, the separation between the first and shoulder peaks becomes ambiguous. This continuous distribution of H− is consistent with the sublattice melting obtained from the Lindemann index.
 | ||
| Fig. 6 (a) Radial distribution function g(r) and (b) van Hove correlation function Gd (r, Δt = 10 ps) of the δ-like model. Magenta, green, blue, and orange lines represent those obtained at T = 500, 700, 900, and 1100 K, respectively. | ||
Fig. 6(b) shows the van Hove time-correlation function Gd with the interval time Δt set to 10 ps (the systematic Gd plots of the β and δ models with gradually changing interval times are shown in Fig. S6(a–h)). At all temperatures, the correlation holes at r = 0 quickly annihilated and peak growth continued over time intervals of several tens of picoseconds, which can be interpreted as VH created by a certain H-hopping event being immediately occupied by another H-hopping event. This is strong evidence for concerted-like hopping migration, as discussed in a previous study.53 The analysis reveals that most H− migrate in a highly concerted fashion and multiple ions hop simultaneously into their nearest sites within a few picoseconds. Finally, at all temperatures, the peak growth continued even at the maximum time of the FPMD simulations.
H atoms exhibit a string-like MD trajectory, which was analyzed using the rearrangement indicator following Mohn et al.61 In this study, we selected the reconfiguration time interval Δt = 0.3 ps, which was estimated from the MD trajectory as the timescale for site-to-site hopping of H− ions. It was confirmed that the resulting behavior is not sensitive to the choice of Δt (see Fig. S7), which suggests that the collective motion can be reliably captured by our analysis. All the strings appearing in the entire MD trajectory at T = 700 K are shown in Fig. 7(a), as typically found in supercooled liquids62 and glasses.63 The number of strings constituting a cluster, or the cluster size, is now analyzed. Fig. 7(b) and S7(a–d) show the distribution of the number of hopping events that occur forming a cluster of size n. As the temperature increases, hopping tends to occur forming a larger cluster, which is significantly above the sublattice melting temperature (T ≥ 700 K). This indicates increased cooperativity in H diffusion. In addition, the interlayer H migrations tend to occur through the Ba vacancy, VBa, indicating the important role played by the VBa configuration. Note that only one configuration, as appeared in the supercell model was examined in this study, but sampling the configuration would be an important target for future study. Next, we analyzed the correlation between H-movement in the c-axis direction and the magnitude of Li-displacement. The correlation is appreciable although not so strong, as shown in Fig. S8(a–c), which indicates the contribution of Li atoms in the collective H diffusion. Analysis of the MD trajectory has thus revealed that diffusion in BLHO is characterized by the correlated motion of H atoms accompanying the displacement of Li atoms. We consider that a large number of VH (and VBa) is an essential factor to induce the dynamical ion-lattice correlation as well as sublattice melting. It should be noted that the reason why BLHO can introduce such a large number of vacancies is not known in terms of solid-state chemistry; therefore, the thermodynamic stability of BLHO and defect properties should be clarified in the future.
 | ||
| Fig. 7 (a) Visualization of H− diffusion at T = 700 K. Blue strings represent the H atom trajectories with cluster sizes ≥3. Orange strings and red spheres represent the Ba atom trajectories and Ba vacancies, respectively. (b) Number of hopping events plotted against the cluster size involved in the hop. Green, blue, red, and black symbols represent the values obtained at T = 500, 700, 900, and 1100 K, respectively. | ||
5. Conclusions
We investigated the H− conduction mechanism and anion sublattice melting in BLHO using first-principles electronic structure calculations and molecular dynamics simulations. First, we examined several microscopic structure models, such as Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29, where the H and O atoms at the apical sites were ordered by β- and δ-phase like structures. The constructed models showed bandgaps of 2.2–2.7 eV, occupied H-1s orbitals, and Bader charges close to their formal ionic charges (Ba2+, Li+, H−, and O2−), consistent with the pure H− conduction and electrochemical H− carriers observed in experiments. Second, using the constructed β- and δ-like models of Ba56Li32H88O28, FPMD simulations were performed for several hundred picoseconds under temperature-controlled conditions at T = 500, 700, 900, and 1100 K. The obtained Dab of H was in good agreement with that obtained from the QENS measurements, indicating that the local-scale H− dynamics were well reproduced. The activation energies of Dab (0.52–0.55 eV in both models) and Dc (0.78 eV in the δ model) were associated with individual H− migration paths, with energy barriers of 0.50 and 0.82 eV for H− hopping between equatorial sites and between apical and equatorial sites, respectively. Furthermore, the Lindemann index of H exceeded the threshold for a solid–liquid transition at T ≥ 700 K, suggesting sublattice melting of the H− equatorial sites. Above the sublattice melting temperature, the van Hove correlation function and rearrangement indicator analysis showed that the peak growth at the correlation hole position increased for several tens of picoseconds and H− hopping events tended to form a large cluster, respectively, indicating cooperativity in H− diffusion.Author contributions
Jun Haruyama: conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, software, validation, visualization, writing – original draft. Fumitaka Takeiri: conceptualization, writing – review & editing. Genki Kobayashi: conceptualization, supervision, writing – review & editing. Osamu Sugino: conceptualization, formal analysis, funding acquisition, methodology, resources, software, supervision, validation, visualization, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
Data for this article, including VASP and QE inputs and trajectory analysis programs are available at the ISSP data repository, https://isspns-gitlab.issp.u-tokyo.ac.jp/j-haruyama/BLHO_DFT-MD. The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: crystal structures of the β-like Ba56Li32H88O28 model. Table of the number of atoms, composition ratios, and numbers of Heq and Ba vacancies in the composition change models. Optimized structures of β-like Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29 models. Crystal structure of the δ-like Ba14Li8H22O7 model. Optimized structures of δ-like Ba56Li32H88O28, Ba57Li32H90O28, and Ba58Li32H90O29 models. Table of the mimicked phases, number of atoms, optimized lattice constants, and calculated energy gaps of the composition change models. Mean square displacements of H obtained from the β- and δ-like models at controlled temperatures. van Hove correlation function as a function of the radial coordinate obtained from the β- and δ-like models at controlled temperatures. The number of hopping events as a function of cluster size and rearrangement time interval. Correlation between Li and H motions. See DOI: https://doi.org/10.1039/d5ta08063k.Acknowledgements
This work was supported in part by JSPS as Grants in Aid for Scientific Research on Innovative Areas “Hydrogenomics” (Grant Numbers JP18H05516 and JP18H05519), JSPS KAKENHI (Grant Numbers JP20K15376, JP24K08241, JP24H00390, JP24H02204, and JP25H01986), and the JST FOREST Program (Grant Number JPMJFR213H). The calculations were performed using the facilities of the Supercomputer Center at The Institute for Solid State Physics (ISSP), The University of Tokyo, and the Supercomputer HOKUSAI Bigwaterfall2 (HBW2) system in the Information Systems Division, RIKEN. We would like to thank Editage (https://www.editage.jp) for English language editing.References
- T. Norby, Solid-State Protonic Conductors: Principles, Properties, Progress and Prospects, Solid State Ionics, 1999, 125, 1–11 Search PubMed.
- H. Ito, Y. Hasegawa and Y. Ito, Li–H2 cells with Molten Alkali Chlorides Electrolyte, J. Appl. Electrochem., 2005, 35, 507–512 Search PubMed.
- G. Kobayashi, Y. Hinuma, S. Matsuoka, A. Watanabe, M. Iqbal, M. Hirayama, M. Yonemura, T. Kamiyama, I. Tanaka and R. Kanno, Pure H– Conduction in Oxyhydrides, Science, 2016, 351, 1314–1317 CrossRef CAS PubMed.
- S. Yamaguchi, Large, Soft, and Polarizable Hydride Ions Sneak around in an Oxyhydride, Science, 2016, 351, 1262–1263 Search PubMed.
- Y. Izumi, F. Takeiri, K. Okamoto, T. Saito, T. Kamiyama, A. Kuwabara and G. Kobayashi, Electropositive Metal Doping into Lanthanum Hydride for H− Conducting Solid Electrolyte Use at Room Temperature, Adv. Energy Mater., 2023, 13, 2301993 Search PubMed.
- J. Cui, R. Zou, W. Zhang, H. Wen, J. Liu, S. Wang, S. Liu, H. Chen, W. Liu, X. Ju, W. Wang, T. Gan, J. Li, J. Guo, T. He, H. Cao and P. Chen, A room temperature rechargeable all-solid-state hydride ion battery, Nature, 2025, 646, 338–342 Search PubMed.
- T. Hirose, N. Matsui, T. Itoh, Y. Hinuma, K. Ikeda, K. Gotoh, G. Jiang, K. Suzuki, M. Hirayama and R. Kanno, High-capacity, reversible hydrogen storage using H–-conducting solid electrolytes, Science, 2025, 389, 1252–1255 Search PubMed.
- M. C. Verbraeken, E. Suard and J. T. S. Irvine, Structural and electrical properties of calcium and strontium hydrides, J. Mater. Chem., 2009, 19, 2766–2770 Search PubMed.
- M. C. Verbraeken, C. Cheung, E. Suard and J. T. S. Irvine, High H– Ionic Conductivity in Barium Hydride, Nat. Mater., 2015, 14, 95–100 CrossRef CAS PubMed.
- G. J. Irvine, R. I. Smith, M. O. Jones and J. T. S. Irvine, Order–disorder and ionic conductivity in calcium nitride-hydride, Nat. Comm., 2023, 14, 4389 Search PubMed.
- A. Watanabe, G. Kobayashi, N. Matsui, M. Yonemura, A. Kubota, K. Suzuki, M. Hirayama and R. Kanno, Ambient Pressure Synthesis and H– Conductivity of LaSrLiH2O2, Electrochemistry, 2017, 85, 88–92 CrossRef CAS.
- Ø. S. Fjellvåg, J. Armstrong, W. A. Sławiński and A. O. Sjåstad, Thermal and Structural Aspects of the Hydride-Conducting Oxyhydride La2LiHO3 Obtained via a Halide Flux Method, Inorg. Chem., 2017, 56, 11123–11128 Search PubMed.
- Y. Iwasaki, N. Matsui, K. Suzuki, Y. Hinuma, M. Yonemura, G. Kobayashi, M. Hirayama, I. Tanaka and R. Kanno, Synthesis, Crystal Structure, and Ionic Conductivity of Hydride Ion-Conducting Ln2LiHO3 (Ln = La, Pr, Nd) Oxyhydrides, J. Mater. Chem. A, 2018, 6, 23457–23463 Search PubMed.
- N. Matsui, G. Kobayashi, K. Suzuki, A. Watanabe, A. Kubota, Y. Iwasaki, M. Hirayama and R. Kanno, Ambient Pressure Synthesis of La2LiHO3 as a Solid Electrolyte for a Hydrogen Electrochemical Cell, J. Am. Ceram. Soc., 2019, 102, 3228–3235 Search PubMed.
- N. Matsui, Y. Hinuma, Y. Iwasaki, K. Suzuki, J. Guangzhong, H. Nawaz, Y. Imai, M. Yonemura, M. Hirayama, G. Kobayashi and R. Kanno, R. The Effect of Cation Size on Hydride-Ion Conduction in LnSrLiH2O2 (Ln = La, Pr, Nd, Sm, Gd) Oxyhydrides, J. Mater. Chem. A, 2020, 8, 24685–24694 RSC.
- Y. Sasahara, T. Hirose, M. Yoshimoto, N. Matsui, S. Kobayashi, H. Ubukata, F. Takeiri, K. Suzuki, M. Hirayama, K. Nishio, R. Shimizu, R. Kanno, G. Kobayashi and T. Hitosugi, High H– Conductivities along the ab-Planes of La2LiHO3 Epitaxial Thin Films, Cryst. Growth Des., 2023, 23, 7103–7108 Search PubMed.
- F. Takeiri, A. Watanabe, K. Okamoto, D. Bresser, S. Lyonnard, B. Frick, A. Ali, Y. Imai, M. Nishikawa, M. Yonemura, T. Saito, K. Ikeda, T. Otomo, T. Kamiyama, R. Kanno and G. Kobayashi, Hydride-Ion-Conducting K2NiF4-Type Ba–Li Oxyhydride Solid Electrolyte, Nat. Mater., 2022, 21, 325–330 Search PubMed.
- K. Okamoto, F. Takeiri, Y. Imai, M. Yonemura, T. Saito, K. Ikeda, T. Otomo, T. Kamiyama and G. Kobayashi, Stabilization of a High H–-Conducting Phase via K Doping of Ba–Li Oxyhydride, J. Mater. Chem. A, 2022, 10, 23023–23027 RSC.
- K. Okamoto, F. Takeiri, Y. Imai, M. Yonemura, T. Saito, K. Ikeda, T. Otomo, T. Kamiyama and G. Kobayashi, Impact of Na Concentration on the Phase Transition Behavior and H– Conductivities in the Ba–Li–Na–H–O Oxyhydride System, Adv. Sci., 2023, 10, 2203541 Search PubMed.
- J. B. Boyce and B. A. Huberman, Superionic Conductors: Transitions, Structures, Dynamics, Phys. Reports, 1979, 51, 189–265 Search PubMed.
- O. Yamamoto, Solid State Ionics: A Japan Perspective, Sci. Technol. Adv. Mater., 2017, 18, 504–527 Search PubMed.
- J.-S. Lee, S. Adams and J. Maier, Defect Chemistry and Transport Characteristics of β-AgI, J. Phys. Chem. Solids, 2000, 61, 1607–1622 Search PubMed.
- Y.-G. Guo, J.-S. Lee, Y.-S. Hu and J. Maier, AgI Nanoplates in Unusual 7H/9R Structures, J. Electrochem. Soc., 2007, 154, K51–K60 CrossRef CAS.
- V. V. Tomaev, Y. S. Tver’yanovich and M. D. Bal’makov, Mechanical Modification of β-AgI Nanocrystals, Crystallgraphy Rep., 2012, 57, 948–954 CrossRef CAS.
- C. E. Derrington and M. O'Keeffe, Anion Conductivity and Disorder in Lead Fluoride, Nat. Phys. Sci., 1973, 246, 44–46 Search PubMed.
- C. E. Derrington, A. Navrotsky and M. O'Keeffe, High Temperature Heat Content and Diffuse Transition of Lead Fluoride, Solid State Comm., 1976, 18, 47–49 CrossRef CAS.
- L. M. Volodkovich, G. S. Petrov, R. A. Vecher and A. A. Vecher, Heat Capacity and Enthalpy of Phase Transitions of α- and β-Modifications of Lead Fluoride, Thermochim. Acta, 1985, 88, 497–500 Search PubMed.
- Ø. S. Fjellvåg, J. Armstrong, P. Vajeeston and A. O. Sjåstad, New Insights into Hydride Bonding, Dynamics, and Migration in La2LiHO3 Oxyhydride, J. Phys. Chem. Lett., 2018, 9, 353–358 CrossRef PubMed.
- Q. Bai, X. He, Y. Zhu and Y. Mo, First-Principles Study of Oxyhydride H– Ion Conductors: Toward Facile Anion Conduction in Oxide-Based Materials, ACS Appl. Energy Mater., 2018, 1, 1626–1634 Search PubMed.
- X. Liu, T. S. Bjørheim and R. Haugsrud, Formation of Defects and their Effects on Hydride Ion Transport Properties in a Series of K2NiF4-Type Oxyhydrides, J. Mater. Chem. A, 2018, 6, 1454–1461 RSC.
- A. Kuwabara, F. Takeiri, H. Nawaz and G. Kobayashi, First-Principles Calculations of Point Defect Formation and Anion Diffusion Mechanisms in the Oxyhydride Ba2ScHO3, ChemRxiv, 2020 DOI:10.26434/chemrxiv.12121254.v2.
- R. F. W. Bader, in Atoms in Molecules: A Quantum Theory, Oxford University Press, 1990 Search PubMed.
- K. Momma and F. Izumi, VESTA: A Three-Dimensional Visualization System for Electronic and Structural Analysis, J. Appl. Crystallogr., 2011, 44, 1272–1276 Search PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni and I. Dabo, et al., QUANTUM ESPRESSO: A Modular and Open-Source Software Project for Quantum Simulations of Materials, J. Phys.: Condens. Matter, 2009, 21, 395502 Search PubMed.
- P. Giannozzi, O. Andreussi, T. Brumme, O. Bunau, M. B. Nardelli, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli and M. Cococcioni, et al., Advanced Capabilities for Materials Modeling with QUANTUM ESPRESSO, J. Phys.: Condens. Matter, 2017, 29, 465901 Search PubMed.
- D. Vanderbilt, Soft Self-Consistent Pseudopotentials in a Generalized Eigenvalue Formalism, Phys. Rev. B, 1990, 41, 7892–7895 CrossRef PubMed.
- A. M. Rappe, K. M. Rabe, E. Kaxiras and J. D. Joannopoulos, Optimized Pseudopotentials, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 1227–1230 Search PubMed.
- S. G. Louie, S. Froyen and M. L. Cohen, Nonlinear Ionic Pseudopotentials in Spin-Density-Functional Calculations, Phys. Rev. B: Condens. Matter Mater. Phys., 1982, 26, 1738–1742 Search PubMed.
- A. Dal Corso, Pseudopotentials periodic table: From H to Pu, Comput. Mater. Sci., 2014, 95, 337–350 Search PubMed.
- Quantum ESPRESSO Pseudopotential Data Base, http://www.quantum-espresso.org/pseudopotentials, accessed: September 1, 2025 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- G. Henkelman, A. Arnaldsson and H. Jónsson, A Fast and Robust Algorithm for Bader Decomposition of Charge Density, Comput. Mater. Sci., 2006, 36, 354–360 Search PubMed.
- W. Tang, W. Sanville and G. Henkelman, A Grid-Based Bader Analysis Algorithm without Lattice Bias, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- G. Mills, H. Jónsson and G. K. Schenter, Reversible Work Transition State Theory: Application to Dissociative Adsorption of Hydrogen, Surf. Sci., 1995, 324, 305–337 Search PubMed.
- G. Henkelman, B. P. Uberuaga and H. Jónsson, A Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths, J. Chem. Phys., 2000, 113, 9901 Search PubMed.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Liquid Metals, Phys. Rev. B, 1993, 47, 558(R)–561(R) Search PubMed.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Efficiency of Ab Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mat. Sci., 1996, 6, 15–50 CrossRef CAS.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef CAS.
- S. Nosé, A Unified Formulation of the Constant Temperature Molecular Dynamics Methods, J. Chem. Phys., 1984, 81, 511–519 Search PubMed.
- W. G. Hoover, Canonical Dynamics: Equilibrium Phase-Space Distributions, Phys. Rev. A, 1985, 31, 1695 Search PubMed.
- X. He, Y. Zhu and Y. Mo, Origin of Fast Ion Diffusion in Super-Ionic Conductors, Nat. Comm., 2017, 8, 15893 CrossRef CAS PubMed.
- F. A. Lindemann, The Calculation of Molecular Vibration Frequencies, Phys. Z, 1910, 11, 609–612 Search PubMed.
- N. P. Gupta, On the Lindemann Law of Melting of Solids, Solid State Comm., 1973, 13, 69–71 CrossRef CAS.
- Y. Zhou, M. Karplus, K. D. Ball and R. Stephen Berry, The Distance Fluctuation Criterion for Melting: Comparison of Square-Well and Morse Potential Models for Clusters and Homopolymers, J. Chem. Phys., 2002, 116, 2323–2329 CrossRef CAS.
- L. van Hove, Correlations in Space and Time and Born Approximation Scattering in Systems of Interacting Particles, Phys. Rev., 1954, 95, 249–262 CrossRef CAS.
- W. Kob and H. C. Andersen, Testing Mode-Coupling Theory for a Supercooled Binary Lennard-Jones Mixture: The van Hove Correlation Function, Phys. Rev. E, 1995, 51, 4626–4641 Search PubMed.
- T. Aihara Jr, Y. Kawazoe and T. Masumoto, Molecular Dynamics Study of Structural Relaxation and Dynamical Correlations in Amorphous and Liquid Zr67Ni33 Alloys, Mater. Trans. JIM, 1995, 36, 835–841 Search PubMed.
- S. Banerjee, R. Chahal, A. S. Ivanov, S. Roy, V. S. Bryantsev, Y. Shinohara and S. T. Lam, Ab Initio Simulated Van Hove Correlation Function for Time-Resolved Local Dynamics in Molten MgCl2, J. Mol. Liquids, 2024, 412, 125821 Search PubMed.
- C. E. Mohn, M. Krynski, W. Kob and N. L. Allan, Cooperative Excitations in Superionic PbF2, Phil. Trans. R. Soc. A, 2021, 377, 20190455 Search PubMed.
- R. Candelier, O. Dauchot and G. Biroli, Building Blocks of Dynamical Heterogeneities in Dense Granular Media, Phys. Rev. Lett., 2009, 102, 088001 CrossRef CAS PubMed.
- R. Candelier, A. Widmer-Cooper, J. K. Kummerfeld, O. Dauchot, G. Biroli, P. Harrowell and D. R. Reichman, Spatiotemporal Hierarchy of Relaxation Events, Dynamical Heterogeneities, and Structural Reorganization in a Supercooled Liquid, Phys. Rev. Lett., 2010, 105, 135702 CrossRef CAS PubMed.
- J. L. Niedziela, D. Bansal, A. F. May, J. Ding, T. Lanigan-Atkins, G. Ehlers, D. L. Abernathy, A. Said and O. Delaire, Selective breakdown of phonon quasiparticles across superionic transition in CuCrSe2, Nat. Phys., 2019, 15, 73–78 Search PubMed.
- S. Kumar, M. K. Gupta, P. Goel, R. Mittal, O. Delaire, A. Thamizhavel, S. Rols and S. L. Chaplot, Solidlike to liquidlike behavior of Cu diffusion in superionic Cu2X (X = S, Se): An inelastic neutron scattering and ab initio molecular dynamics investigation, Phys. Rev. Mater., 2022, 6, 055403 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |