
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrochemical lithiation of the layered superionic conductors AgCrSe2 and CuCrSe2
Seongbak
Moon
a,
Md Towhidur
Rahman
 c,
Noah P.
Holzapfel
a,
Alexandra
Zevalkink
b and
Veronica
Augustyn
*a
c,
Noah P.
Holzapfel
a,
Alexandra
Zevalkink
b and
Veronica
Augustyn
*a
aDepartment of Materials Science and Engineering, North Carolina State University, Raleigh, NC 27695, USA. E-mail: vaugust@ncsu.edu
bDepartment of Chemical Engineering and Materials Science, Michigan State University, East Lansing, MI 48824, USA
cDepartment of Mechanical Engineering, Michigan State University, East Lansing, MI 48824, USA
First published on 19th December 2025
Abstract
The layered transition metal chalcogenides MCrX2 (M = Ag, Cu; X = S, Se, Te) are of interest for energy storage because chemically Li-substituted analogs were reported as superionic Li+ conductors. The coexistence of fast ion transport and reducible transition metal and chalcogen elements suggests that this family may offer multifunctional capability for electrochemical storage. Here, we investigate the electrochemical reduction of AgCrSe2 and CuCrSe2 in non-aqueous Li- and Na-ion electrolytes using electrochemical measurements coupled with ex situ characterization (scanning electron microscopy, energy-dispersive X-ray spectroscopy, X-ray diffraction, and X-ray photoelectron spectroscopy). Both compounds delivered high initial specific capacities (∼560 mAh g−1), corresponding to 6.6 and 5.7 Li+/e− per formula unit for AgCrSe2 and CuCrSe2, respectively. We attribute this difference to distinct reduction pathways: (1) Li+ intercalation to form LiCrSe2 and extruded Ag or Cu, (2) conversion of LiCrSe2 to Li2Se, and (3) formation of an Ag–Li alloy at the lowest potential, operative only in AgCrSe2. Consistent with this proposed mechanism, step 3 was absent during reduction of AgCrSe2 in a Na-ion electrolyte since Ag does not alloy with Na. These results demonstrate the complex reduction chemistry of MCrX2 in the presence of alkali ions, providing insights into the use of MCrX2 materials as alkali ion superionic conductors or high capacity electrodes for lithium or sodium-ion type batteries.
1. Introduction
The ever-increasing demand for high energy density lithium-ion rechargeable batteries motivates the search for new types of high capacity electrode materials. On the anode side, this includes metallic anodes such as lithium metal, alloying-type anodes such as silicon, and a broad range of compounds including those based on transition metal chalcogenides that undergo conversion-type reaction mechanisms.1–3 Conversion-type electrodes achieve higher specific capacities than insertion-type electrodes because they can undergo a greater degree of charge transfer involving large microstructural transformations. Transition metal chalcogenides are one class of conversion-type electrodes, which upon lithiation, undergo multi-step reactions involving Li+ intercalation followed by conversion to metal nanoparticles and Li2X (X = S, Se, Te).4–7 While such reactions lead to high capacities they are often accompanied by poor electronic conductivity,8–10 significant volume changes,11–13 and dissolution14,15 of reaction intermediates. Nevertheless, their redox-active metal centers and tunable interlayer spacing render the materials as valuable model systems for understanding conversion mechanisms. Insights from transition metal chalcogenides provide a useful comparative framework for evaluating less-explored layered materials like MCrX2 (M = Ag, Cu; X = S, Se, Te). With M in the structure, we can anticipate additional reduction mechanisms possible through extrusion, as reported for Fe-doped CeO2.16High ionic conductivity is an important characteristic for solid-state electrolytes and mixed ionic-electronic electrode materials for battery applications. MCrX2 (M = transition metal and X = chalcogenide) is a class of layered materials that exhibit superionic conductivity of the M cation upon an order–disorder transition at elevated temperatures. This dynamic disorder in the cation sublattice not only enables fast ion transport but also suppresses lattice thermal conductivity, making these materials attractive for both solid-state electrolyte and thermoelectric applications.17–19 In addition to intrinsic M+ conductivity, the partial substitution of M+ with Li+ was reported in AgCrS2.20 The proposed lithiated Li0.31Ag0.69CrS2 had an ionic conductivity of 19.6 mS cm−1 at 298 K, greater than that of standard liquid lithium-ion battery electrolytes (∼10 mS cm−1). This result suggested the tantalizing possibility of Li+ solid state superionic conductors formed simply through partial ion exchange of Ag+ with Li+, although the method employed a strong chemical reducing agent, tert-butyllithium. This finding raised the question of the exact mechanism of Li+ substitution into AgCrS2 in reducing conditions. While the electrochemical behavior of conventional transition metal chalcogenides has been broadly characterized, prior studies on MCrX2 materials have mainly centered on their cation superionic conductivity rather than their reduction chemistry. In this context, examining the electrochemical reduction of AgCrSe2 and CuCrSe2 provides information that can be considered alongside what is known for other layered chalcogenide systems. To the best of our knowledge, there is no prior work on the chemical or electrochemical reduction mechanism of AgCrSe2 and related materials. Therefore, we decided to investigate the electrochemical behavior of this class of materials to determine whether Li+/Ag+ substitution is possible, as well as whether this class of materials exhibits promising electrochemical properties for non-aqueous rechargeable batteries.
In this work, we performed electrochemical reduction of AgCrSe2 and CuCrSe2 in non-aqueous lithium and sodium-ion electrolytes. We analyzed the electrochemical behavior together with a suite of ex situ characterization techniques, including scanning electron microscopy (SEM), energy-dispersive X-ray spectroscopy (EDS), X-ray diffraction (XRD), and X-ray photoelectron spectroscopy (XPS). AgCrSe2 had a maximum capacity of 560 mAh g−1 corresponding to the storage of 6.6 electrons and Li+ per AgCrSe2. We identified three sequential lithium-ion coupled electron transfer reduction mechanisms in AgCrSe2: (1) Li intercalation to form LiCrSe2 and with the exsolution of Ag0,nd (2) a conversion reaction to form Li2Se, and (3) an Ag–Li alloying reaction. For CuCrSe2 in Li+ and AgCrSe2 in a Na+ electrolyte, reaction (3) was absent due to the lack of possible Cu–Li and Ag–Na alloying. We found only limited Li+ substitution into AgCrSe2, and it was concomitant with the formation of Ag0 at the surface.
2. Experimental methods
2.1. Chemicals
All chemicals were used as received. Silver (Ag; shot, 99.99%), chromium (Cr; chips, 99.995%), n-methyl-2-pyrrolidone (NMP; anhydrous), lithium perchlorate (LiClO4; anhydrous, battery grade), propylene carbonate (PC; anhydrous), and 1.0 M lithium hexafluorophosphate in ethylene carbonate/dimethyl carbonate [LiPF6 in EC/DMC, 50/50 (v/v)] were purchased from Millipore-Sigma. Copper (Cu; powder, 99.9%), selenium (Se; shot, 99.999%), and acetylene black (100% compressed) were purchased from Fisher Scientific. Polyvinylidene fluoride (PVDF; Kynar HSV 900) was obtained from Arkema Inc. Cu foil (9 µm thick) was purchased from MTI Corp.2.2. Material synthesis
Polycrystalline AgCrSe2 and CuCrSe2 were synthesized by solid state reaction. The corresponding stoichiometric amounts of Ag (shot, Sigma-Aldrich, 99.99% purity), Cr (chips, Sigma-Aldrich, 99.995% purity), and Se (shot, Alfa-Aesar, 99.999% purity) were weighed in an argon-filled glovebox. For AgCrSe2, the weighted elements were sealed in a quartz ampoule under static vacuum, heated up to 1173 K at 1 K per minute rate for 24 hours, followed by slow cooling in 12 hours, and finally hand-ground by mortar and pestle into fine powder inside the glovebox. Phase purity of the obtained powder was confirmed with XRD. The synthesis of CuCrSe2 involved an initial ball-milling step, which is reported to help avoid formation of CuCr2Se4 as a secondary phase. The elemental precursors were sealed in stainless steel SPEX vials inside of an argon-filled glovebox. 10 g of powder were then ball milled with three 7/16″-stainless steel ball bearings for 3 hours using a ball mill (SPEX SamplePrep 8000D). After milling, approximately 93% of the initial load was recovered. The powder was vacuum sealed in a quartz ampoule, heated to 973 K, held for 4 hours and air quenched. Lastly, the sample was ground by mortar and pestle and sintered in a spark plasma sintering press at 873 K for 20 minutes to obtain a phase-pure consolidated sample, which was later crushed into fine powder for electrode preparation. AgCrSe2 and CuCrSe2 are moderately air sensitive and show oxidation after several hours to days of exposure. To minimize exposure, the samples were synthesized in an inert atmosphere and stored in an argon-filled glovebox.2.3. Electrode preparation
AgCrSe2 and CuCrSe2 powders were mixed with acetylene black and PVDF in a weight ratio of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 in NMP solvent. Electrodes were prepared by drop-casting the slurry onto a 1.13 cm2 area of Cu foil for an active material mass loading of ∼1.6 mg cm−2 for electrochemical characterization and ∼5.7 mg cm−2 for ex situ and operando electrochemical XRD characterization. All slurry mixing and drop-casting steps were carried out outside the glovebox. The electrodes were dried at 60 °C for 12 h in air followed by 120 °C for 12 h under vacuum prior to transfer into an argon-filled glovebox (MBraun Labstar Pro) for coin cell assembly.
:1:1 in NMP solvent. Electrodes were prepared by drop-casting the slurry onto a 1.13 cm2 area of Cu foil for an active material mass loading of ∼1.6 mg cm−2 for electrochemical characterization and ∼5.7 mg cm−2 for ex situ and operando electrochemical XRD characterization. All slurry mixing and drop-casting steps were carried out outside the glovebox. The electrodes were dried at 60 °C for 12 h in air followed by 120 °C for 12 h under vacuum prior to transfer into an argon-filled glovebox (MBraun Labstar Pro) for coin cell assembly.
2.4. Electrochemical characterization
Coin cells (CR2032, MTI Corp.) were fabricated inside an argon-filled glovebox (MBraun Labstar Pro) with Ar and H2O levels < 1 ppm. The cathode consisted of the AgCrSe2 or CuCrSe2 porous composite electrode, while a metallic Li chip was used as the anode. The electrolyte was 1.0 M LiPF6 in EC/DMC (50:50, v/v) or 1.0 M LiClO4 in PC. For the Na system, Na metal was used as the anode with 1.0 M NaClO4 in PC. A glass microfiber filter (GF/F, Whatman) was used as the separator. The coin cells were crimped with a pressure of 0.8 torr using a digital pressure controlled electric crimper (MSK-160E, MTI Corp.). Electrochemical characterization methods, including galvanostatic cycling with potential limitation (GCPL), galvanostatic intermittent titration technique (GITT), and electrochemical impedance spectroscopy (EIS), were conducted using a potentiostat (BioLogic VMP3). GCPL was performed with a constant applied current of 10 mA g−1 to different lower cut-off potentials: 2.1, 1.5, 0.8, and 0.1.
2.5. Physical characterization
Standard powder and ex situ powder XRD were performed using a Rigaku SmartLab X-ray diffractometer in Bragg–Brentano geometry with Cu Kα radiation (λ = 1.5406 Å). Samples for ex situ XRD were prepared in an argon-filled glovebox in a coin cell (MTI Corporation) with 8 µm Kapton film window (Spex CertiPrep). Scanning electron microscopy (SEM) measurements were performed on a Hitachi SU8700 field-emission scanning electron microscope equipped with Oxford energy dispersive X-ray spectrometer (EDS) detector. Ex situ samples were prepared in an argon-filled glovebox in a plastic sample holder sealed with Parafilm. During loading, samples were exposed to the ambient for approximately 2 minutes. SEM images and EDS mapping data were collected at 5 keV and 20 keV, respectively. X-ray photoelectron spectroscopy (XPS) measurements were conducted on a SPECS PHOIBOS 150. Pristine and ex situ electrodes on Cu foil were attached to the stainless steel sample holder in an argon-filled glovebox using carbon tape. During loading, samples were exposed to the ambient for approximately 1 minute.2.6. Operando electrochemical X-ray diffraction (EC-XRD)
Operando EC-XRD of AgCrSe2 was collected on a PANalytical X'Pert powder diffractometer (45 kV, 40 mA, sealed Cu tube, Kα1λ = 1.5406 Å, Kα2λ = 1.5444 Å) with a coin cell stage and X'Celerator position-sensitive detector in Bragg–Brentano geometry. Li-metal half-cells (CR2032) consisted of an anode casing, spring, two spacers, 8 mm Li disk, and glass fiber separator with 120 µL of 1 M LiClO4 in PC. Composite slurry electrodes (3.8 mg cm−2) were coated onto stainless steel mesh, dried at 120 °C under vacuum, and punched into 19 mm disks. The cathode casing had a 7 mm hole covered with 8 µm Kapton film. PXRD patterns were collected from 20°–43° 2θ (0.033° step, 7 min scan). Cells were discharged at 30 mA g−1 for one cycle.3. Discussion
We characterized the crystal structure and microstructure of the synthesized (Ag,Cu)CrSe2 with XRD and SEM, respectively. Rietveld refinement of the structural models based on the XRD data (Fig. 1a and b) confirmed that both AgCrSe2 and CuCrSe2 crystallize in a rhombohedral structure (space group R3m) without detectable secondary phases or impurities. The refined lattice parameters were a = 3.6760(1) Å and c = 19.3854(2) Å for CuCrSe2, and a = 3.6803(1) Å and c = 21.2170(7) Å for AgCrSe2. The larger c-axis of AgCrSe2 reflects the larger ionic radius of Ag+ compared to Cu+, leading to an expanded interlayer spacing. The low Rwp values (1.54% for CuCrSe2 and 1.90% for AgCrSe2) indicate excellent agreement between the observed and calculated patterns. Both structures exhibited high crystallinity, as indicated by the sharp diffraction peaks and low background noise. These results confirm the successful synthesis of the phase-pure layered chalcogenides, suitable for further studies on their electrochemical or transport properties.21 The morphology of the synthesized AgCrSe2 consists of non uniform particles with sizes on the order of tens of micrometers (Fig. 1c). A zoomed-in view of the side of a particle reveals the layered crystalline structure as indicated by the uniform striation features along the block facet (Fig. 1d). | ||
| Fig. 1 Rietveld refinement of the structural models based on the XRD data collected for (a) AgCrSe2 and (b) CuCrSe2, confirming phase-pure rhombohedral R3m structure with high crystallinity; insets show the corresponding refined crystal structures. (c) SEM image of AgCrSe2 particles, revealing non-uniform tens-µm morphology. (d) Magnified view of the region marked in (c), highlighting layered striated features consistent with the crystalline framework. | ||
3.1. Mechanism of electrochemical reduction
Next we considered the electrochemical behavior of the layered (Ag,Cu)CrSe2 in Li-ion and Na-ion non-aqueous electrolytes, respectively, for which there are no prior reports. These studies allowed us to determine the electrochemical behavior as a function of the transition metal (Ag+vs. Cu+) and charge-compensating cation in the electrolyte (Li+vs. Na+). The first galvanostatic discharge and charge cycles under the various conditions tested are shown in Fig. 2. First we consider the reduction of AgCrSe2 to 0.1 V in a Li+-containing electrolyte, which is associated with a specific capacity of 560 mAh g−1. Assuming that the electrochemical reduction is accompanied by a Li+ cation (Li+-coupled electron transfer), this corresponds to the transfer of 6.6 Li+, e− per AgCrSe2. Upon oxidation, the capacity is 427 mAh g−1 (5.1 Li+, e− per AgCrSe2), indicating that ∼76% of the charge is reversible. Based on the number of plateau regions in the discharge profile and corresponding number of peaks in the differential capacity (dQ/dV) plot, we propose a three-step mechanism (regions I–III, labeled in Fig. 2a) for the electrochemical reduction of AgCrSe2 in a non-aqueous Li+ electrolyte.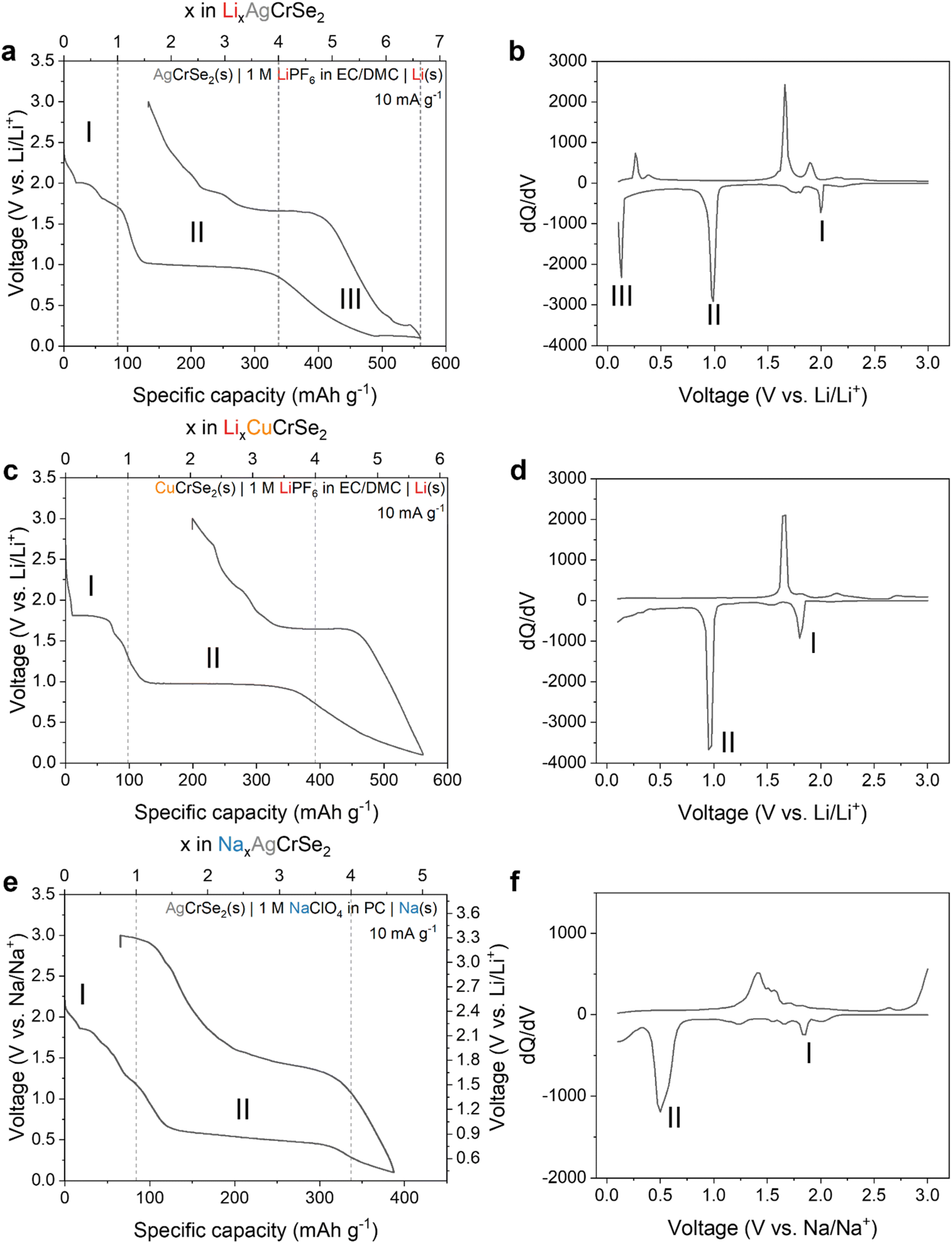 | ||
| Fig. 2 Galvanostatic profiles at 10 mA g−1 and corresponding differential capacity plots. (a and b) Lithiation of AgCrSe2 shows three reduction regions (∼2.0, 1.0, 0.13 V) that suggest stepwise processes. (c and d) Lithiation of CuCrSe2 displays two main regions, lacking the low-voltage feature seen in AgCrSe2. (e and f) Sodiation of AgCrSe2 shows two plateaus (∼2.15, 1.0 V), consistent with the absence of a low-voltage alloying step. | ||
Region I is associated with a one electron reduction process consisting of multiple shoulder plateaus around 2.0 V, which may include Ag+ extrusion and Li+ insertion processes that cannot be clearly separated in the voltage profile. Region II is associated with a reduction step requiring the transfer of three electrons and a flat voltage profile near 1.0 V, while region III is associated with the remaining 2.6 electrons and a sloping voltage profile followed by a third plateau above 0.1 V. The three distinct reduction steps can also be identified from the differential capacity (dQ/dV) plot (Fig. 2b), corresponding to three distinct reduction peaks at 2.0 V, 1.0 V, and 0.13 V. To help us develop a mechanism for the electrochemical reduction of AgCrSe2 with Li+, we evaluated the reversibility of the electrochemical reactions by using galvanostatic cycling with decreasing cathodic potential cutoffs (Fig. S1, and Table 1). We quantified the reversibility in terms of the degree of electron transfer (coulombic efficiency) and voltage (voltage hysteresis) during reduction and oxidation. Between 3.0 and 2.1 V (region I), AgCrSe2 shows reversible electrochemical behavior as characterized by high average coulombic efficiency (99.8%) and negligible voltage hysteresis. Expanding the range to 3.0–1.5 V completes the voltage plateau at 2.0 V (region I), but leads to lower coulombic efficiency (98.3%) and a greater voltage hysteresis of ∼0.2 V. Lowering the potential further to 0.8 V leads to the appearance of the voltage plateau at 1.0 V (region II), with a coulombic efficiency of 89.1% and a voltage hysteresis of 0.7 V. Finally, further decreasing it to 0.1 V (region III) leads to the appearance of a sloping region from 1.0 to 0.13 V, followed by an additional plateau at 0.13 V, with a coulombic efficiency of 96.6% and a voltage hysteresis of approximately 0.2 V. Based on these features, we propose that the electrochemical reduction of AgCrSe2 in a Li+-containing non-aqueous electrolyte proceeds as follows:
| AgCrSe2 + Li+ + e− → Ag + LiCrSe2 | (1) |
| LiCrSe2 + 3Li+ + 3e− → Cr + 2Li2Se | (2) |
| Ag + 2.6Li+ + 2.6e− → Li2.6Ag | (3) |
| Voltage range (V) | Curve shape | Voltage hysteresis (V) | Coulombic efficiency (%) |
|---|---|---|---|
| 3.0–2.1 | Sloping | Negligible | 99.8 |
| 3.0–1.5 | Plateau | 0.2 | 98.3 |
| 3.0–0.8 | Plateau | 0.7 | 89.1 |
| 3.0–0.1 | Plateau | 0.2 | 96.6 |
Evidence supporting this proposed mechanism is presented in the following sections. The Li–Ag alloying reaction occurs at approximately 0.28 V.22
To help evaluate the proposed mechanism for AgCrSe2 lithiation, we performed electrochemical reduction of CuCrSe2 in a non-aqueous Li+ electrolyte (Fig. 2c). The first cycle reduction capacity was 562 mAh g−1, which corresponds to approximately 5.7 e−, Li+ per CuCrSe2, which is almost 1 electron less than the capacity obtained for the Ag-analog. We hypothesized that, since Step I involves replacing the monovalent metal ions (Ag+ and Cu+) with Li+, there would be no change in the degree of lithiation per formula unit. This holds even though the first voltage plateau for CuCrSe2 appears at 1.8 V, approximately 0.2 V lower than that for AgCrSe2. This is in line with the lower standard reduction potential of Cu+vs. Ag+.23 The plateau at 1.0 V is the same as in AgCrSe2. Since it likely corresponds to the conversion of LiCrSe2 to Li2Se and Cr (step II), these reactions are the same in CuCrSe2 and AgCrSe2, and so the voltage plateau is unchanged. Reduction of AgCrSe2 to 0.13 V, just before the third plateau, delivers 484 mAh g−1 (5.7 Li+ per AgCrSe2), compared to 562 mAh g−1 (5.7 Li+ per CuCrSe2). This indicates that the same amount of Li+ per (Ag or Cu)CrSe2 participates in the reaction before the assumed Li–Ag alloying. This reinforces our assumption that step III includes the Li–Ag alloying reaction. Finally, the smaller discharge capacity and absence of the third reaction plateau in CuCrSe2 may be attributed to the fact that Cu does not alloy with Li, unlike Ag. This is clearly shown in Fig. 2d by the absence of the third reduction peak.
Next we investigated the role of the charge compensating cation in the electrolyte by performing electrochemical characterization in a non-aqueous Na+-containing electrolyte. Reduction of AgCrSe2 under these conditions (Fig. 2e) shows two primary voltage plateaus, at 2.15 and 1.0 V vs. Na/Na+ (1.85 and 0.7 V vs. Li/Li+). The loss of the third and lowest potential plateau is attributed to the inability of Na to alloy with Ag.24 The second voltage plateau, starting near 1.0 V, is likely related to the following conversion reaction:
| NaCrSe2 + 3Na+ + 3e− → 2Na2Se + Cr | (4) |
3.2. Morphological and compositional changes during lithiation
Our proposed mechanism, involving metal exsolution, conversion, and alloying, should lead to significant changes in the microstructure, crystal structure, and surface composition of the electrodes. To that end, we performed ex situ SEM, EDS, XRD, and XPS characterization of AgCrSe2 at different states of charge. Porous AgCrSe2 electrodes were discharged at 10 mA g−1 to 2.1 V, 1.5 V, 0.8 V, and 0.1 V, followed by chronoamperometry at each potential for 12 hours to ensure completion of the electrochemical reaction. The ex situ SEM images in Fig. 3 demonstrate the expected morphological changes during lithiation. Pristine AgCrSe2 has a layered microstructure. By 2.1 V, small particles appeared on the surface of the material while at 1.5 V, the surface became extensively covered by these particles that grew and accumulated, indicating progressive surface reconstruction during lithiation. Further reduction to 0.8 V led to growth of the overlayer, creating a rugged surface, and by 0.1 V, these valleys and lower surfaces were filled, resulting in a flatter appearance. These microstructural changes indicate a progressive transformation of the electrode material, with surface particle formation and growth likely driven by Ag extrusion and Li+ insertion. The associated microstructural evolution, including phase transitions and possible rearrangement of the AgCrSe2 framework, remains to be clarified, particularly whether Ag de-insertion and Li+ insertion occur simultaneously or in distinct steps. | ||
| Fig. 3 Ex situ SEM images of AgCrSe2 electrodes during electrochemical reduction in a non-aqueous Li+ electrolyte to different lower cutoff voltages: (a) pristine, (b) 2.1 V, (c) 1.5 V, (d) 0.8 V, and (e) 0.1 V, showing progressive surface reconstruction with the emergence and growth of Ag nanoparticles consistent with the proposed extrusion–conversion–alloying mechanism. | ||
We performed ex situ energy dispersive X-ray spectroscopy (EDS) to analyze the distribution of Ag, Cr, and Se. All three elements were evenly distributed throughout the material in pristine AgCrSe2 (Fig. S2). Upon discharge to 2.1 V, we found regions with high Ag intensity and the absence of Cr and Se. At 1.5 V, these intensity differences became more pronounced, indicating further changes in elemental distribution. At 0.8 V and 0.1 V, scattered Ag intensity spots were observed, with lower Cr and Se intensities at those spots. The redistribution of Ag further indicates an exsolution process occurring in AgCrSe2. This could proceed via a nucleation and growth mechanism, whereby Ag clusters form and coalesce into distinct, localized spots, separating from the lattice (Fig. S2d and e). In contrast, Cr does not form a distinct phase, which may explain why it is not observed in EDS. This is possibly due to the limited diffusion of Cr atoms, residual Cr–Se interactions, or amorphous phase formation.
Fig. S2 and Table S1 show the EDS oxygen mapping results of the ex situ samples at each cutoff voltage after approximately two minutes of air exposure. The signal intensity increases with deeper lithiation, likely due to the higher reactivity of more reduced samples under ambient conditions. This trend is quantitatively confirmed by the atomic percentage data, which show an increase in the oxygen to carbon ratio (O:C) from 0.03 in the pristine sample to 0.29 in the 0.1 V sample.
3.3. Structural evolution during lithiation
We performed ex situ X-ray diffraction (XRD) of the AgCrSe2 porous electrode at various cutoff voltages using an airtight cell with a Kapton window to prevent air exposure, since the reduced electrode is air sensitive (Fig. 4). The XRD pattern of pristine AgCrSe2 powder matched well with the previously reported pattern.25,26 In the porous electrode on a Cu foil substrate, Cu peaks dominated but the AgCrSe2 peaks remained visible. At 2.1 V, new peaks corresponding to LiCrSe2 appeared around 32° and 40° 2θ. The ex situ sample, marked as 2.1 V was discharged to 2.1 V and subsequently held at 2.1 V by chronoamperometry for about 4 hours. Based on the specific capacity, this corresponds to Li0.21AgCrSe2 (Fig. S3). In comparison with the operando XRD (Fig. S5), which still shows AgCrSe2 reflections at 2.1 V, the ex situ XRD indicates more complete LiCrSe2 formation. This difference arises from the chronoamperometric hold, which allows further reaction progress toward equilibrium. Therefore, the electrochemical lithiation of AgCrSe2 can be viewed as an intermediate process occurring at different potentials, while the discrepancy between ex situ and operando results reflects kinetic limitations during continuous discharge. The XRD pattern at 2.1 V does not show a shift of the (003) peak, as reported by Peng et al.20 Instead, the AgCrSe2 reflections disappear and LiCrSe2 reflections appear. This suggests that phenomena such as Li insertion into the AgCrSe2 lattice to form a LixAg1−xCrSe2 solid solution do not occur. Rather, this reaction appears to be limited to AgCrS2 or to chemical reduction conditions. Upon further reduction to 1.5 V, the LiCrSe2 peaks intensified, and metallic Ag peaks emerged. This supports the interpretation that the first potential plateau corresponds to the insertion of one lithium per formula unit, involving Ag extrusion and Li intercalation into the interlayer. | ||
| Fig. 4 Ex situ XRD of AgCrSe2 electrodes during the first discharge in 1 M LiClO4 in PC. Electrodes were stopped at selected voltages (pristine, 2.1, 1.5, 0.8, 0.1 V). (a) Galvanostatic profile from OCV to 0.1 V with cutoff points indicated. (b) XRD patterns of electrodes collected at these voltages in the 10–80° 2θ range, showing the sequential appearance and disappearance of diffraction peaks as a function of discharge potential. | ||
At 0.8 V, the LiCrSe2 peaks disappeared, while peaks for Li2Se appeared, signaling the proposed conversion reaction. However, there is no clear evidence of Cr metal formation. This could be due to (1) the Cr-metal peaks overlapping with the Ag-metal peaks, making it difficult to observe, and/or (2) insufficient Cr growth to be detected by XRD, which aligns with the EDS results (Fig. S2). At 0.1 V, the Li2Se peaks persisted with slightly increased intensity, while the metallic Ag peaks shifted to higher diffraction angles, consistent with Li–Ag alloy formation due to the incorporation of smaller Li atoms into Ag.
Peng et al. reported that chemical lithiation of AgCrS2, a compositionally and structurally related layered chalcogenide with the same R3m space group symmetry.20 The proposed mechanism suggests only partial Ag-extrusion and Li-intercalation leading to the formation of a (Li–Ag)CrS2 phase as indicated by changes in interlayer spacing from XRD measurements. However, in this study, the interlayer (006) peak at 25° remains unchanged in both ex situ and operando XRD (Fig. S5), indicating that electrochemical lithiation of AgCrSe2 does not result in the formation of a (Li–Ag)CrSe2 phase. Instead of a peak shift, which would indicate a solid solution phase, the operando XRD results show a gradual evolution: initially, the AgCrSe2 peak at 41° is present, followed by the coexistence of peaks at 41° (AgCrSe2) and 40° (LiCrSe2), and finally, only the 40° (LiCrSe2) peak remains until reaction II completes. In addition, the peak at 38° exhibits broadening and a slight increase in intensity, suggesting a phase transition. Since both AgCrSe2 and metallic Ag have reflections at this position, the broadening may indicate the formation of nanosized Ag particles, consistent with a conversion reaction.
3.4. Surface chemistry changes during lithiation
Ex situ XRD confirms the formation of LiCrSe2, Ag, and Li2Se during the lithiation process. Due to the similar XRD peak positions of Cr and Li–Ag alloy with Ag, these phases are difficult to confirm by XRD alone. To verify their presence and finalize the lithiation mechanism, X-ray photoelectron spectroscopy (XPS) was conducted. XPS calibration was performed using the C–C single bond peak, set at 285 eV (Fig. S6a). The pristine C 1s spectrum was deconvoluted into five peaks: C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C (283.8 eV), C–C (285 eV), C–O (286.4 eV), CO (289.4 eV), and C–F2 (290.3 eV). The C–F2 peak originates from the PVDF binder used in the composite slurry electrode, while the other peaks arise from both acetylene black and the PVDF backbone.27 In ex situ samples, the CC peak weakens, while CO and C–O peaks intensify, suggesting the introduction of an electrolyte on the surface, covering the acetylene black. The pristine O 1s spectrum includes peaks for O–C (533.1 eV), OC (531.9 eV), and SeO2 (530.4 eV).28 For ex situ samples, with the addition of carbonate electrolyte and ClO4− salt, the peak at 533.1 eV is significantly enhanced. The introduction of the ClO4− anion is confirmed by the chlorine 2p spectrum at 208.5 eV (2p3/2) and 210.0 eV (2p1/2) in Fig. S6c. In addition, a novel peak below 530 eV could be attributed to the formation of metal oxides (e.g., Ag2O, Li2O) when the highly reduced ex situ samples were exposed to ambient air, as also confirmed by the EDS oxygen maps (Fig. S4). The O–C and OC peaks are confirmed from the C 1s spectrum, and the SeO2 peak is identified in the Se 3d spectrum (Fig. 5c).
C (283.8 eV), C–C (285 eV), C–O (286.4 eV), CO (289.4 eV), and C–F2 (290.3 eV). The C–F2 peak originates from the PVDF binder used in the composite slurry electrode, while the other peaks arise from both acetylene black and the PVDF backbone.27 In ex situ samples, the CC peak weakens, while CO and C–O peaks intensify, suggesting the introduction of an electrolyte on the surface, covering the acetylene black. The pristine O 1s spectrum includes peaks for O–C (533.1 eV), OC (531.9 eV), and SeO2 (530.4 eV).28 For ex situ samples, with the addition of carbonate electrolyte and ClO4− salt, the peak at 533.1 eV is significantly enhanced. The introduction of the ClO4− anion is confirmed by the chlorine 2p spectrum at 208.5 eV (2p3/2) and 210.0 eV (2p1/2) in Fig. S6c. In addition, a novel peak below 530 eV could be attributed to the formation of metal oxides (e.g., Ag2O, Li2O) when the highly reduced ex situ samples were exposed to ambient air, as also confirmed by the EDS oxygen maps (Fig. S4). The O–C and OC peaks are confirmed from the C 1s spectrum, and the SeO2 peak is identified in the Se 3d spectrum (Fig. 5c).
 | ||
| Fig. 5 Ex situ XPS core spectra obtained from AgCrSe2 electrodes discharged to different cutoff voltages (pristine, 2.1, 1.5, 0.8, and 0.1 V) in 1 M LiClO4 in PC: (a) Cr 2p, (b) Ag 3d, and (c) Se 3d and Li 1s. The spectra reveal progressive reduction of Cr3+, the emergence and loss of Ag signals, and shifts in Se and Li peaks reflecting changes in bonding environments during lithiation. | ||
Fig. 5 shows the evolution of the chemical states and bonding environments of the elements composing the active electrode materials upon lithiation. Pristine chromium 2p spectrum consists of peaks at 2p1/2 (596.1 eV) and 2p3/2 (576.8 eV). The Cr 2p3/2 peak at 576.8 eV originates from the Cr3+ ion in AgCrSe2 and this matches with other Cr3+-containing materials such as Cr(III) oxide (575.7 eV) and Cr(III) hydroxide (577.7 eV).29 The peak position remains unchanged when reduced to 2.1 V, indicating that reaction I is not yet complete. At 1.5 V, where reaction II is considered to have progressed, both 2p1/2 and 2p3/2 shift to a lower binding energy of 576.1 eV indicating some degree of Cr3+ reduction. When the entire reduction process is finished, the peak is centered at 575.9 eV. Given that metallic chromium (Cr(0)) is located at a lower binding energy than Cr3+, the result suggests the reduction of Cr3+ to Cr0. This behavior aligns with the conversion reaction (LiCrSe2 + 3Li+ + 3e− → Cr + 2Li2Se), as confirmed by XRD.
Ag 3d peaks appear at 373.6 eV (3d3/2) and 367.6 eV (3d5/2). Upon reduction to 2.1 V and 1.5 V, a subtle increase in binding energy is observed. Since Ag0 generally exhibits a higher binding energy than Ag+, this shift suggests the reduction of Ag+ to Ag metal. At 0.8 V and 0.1 V, no Ag-related peaks are detected.30 This absence is likely due to a decrease in Ag atomic %, which may result from the oxidation of Ag or the removal of Ag particles during ex situ sample preparation.
The pristine Se 3d spectrum exhibits peaks corresponding to SeO2 (58.3 eV) and AgCrSe2 (53.6 eV). The Se 3d spectrum is not deconvoluted into 3d3/2 and 3d5/2 components because the typical spin–orbit splitting of Se 3d is only 0.86 eV,31 which is significantly smaller than the 4.7 eV binding energy difference arising from different bonding environments. After lithium introduction, a distinct Li 1s peak appears at 55.7 eV which matches with other Li+-containing species such as LiO. The peak shift of Se 3d to lower energy indicates a change in the Se bonding environment even at a low state of lithiation (2.1 V). This suggests that modifications in AgCrSe2 begin occurring during the initial stage of the lithiation reaction. The Li 1s peak shifts to lower binding energies, indicating a transition from ionic lithium to metallic lithium during Ag–Li alloy formation, well before the theoretical lithium deposition potential.
3.5. Li ion diffusion behavior during lithiation
Peng et al. report an increase in the ionic conductivity of LixAg1−xCrS2 with Li content x, reaching a maximum conductivity at x = 0.31.20 Ionic conductivity (σ) and the diffusion coefficient (D) are directly related by the Nernst–Einstein equation: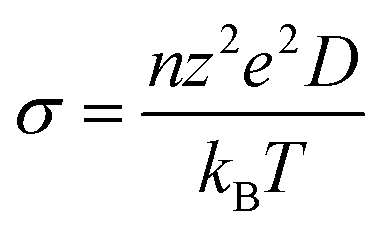 | (5) |
We obtained the diffusion coefficient using two methods: galvanostatic intermittent titration technique (GITT) and electrochemical impedance spectroscopy (EIS). GITT was employed to calculate the apparent diffusion coefficient during lithiation and delithiation. Unlike the trend reported by Peng et al., the diffusion coefficient fluctuates but generally decreases in region I (3.0–1.5 V), where lithiation induces a phase transition from AgCrSe2 to LiCrSe2 (Fig. S7).20
We also determined the diffusion coefficient using EIS with the method described in Ref. 32, based on the Warburg constant (σW). σW was obtained from the slope of the real part of the impedance (Z′) plotted against the inverse square root of frequency. The diffusion coefficient is then calculated using the equation:
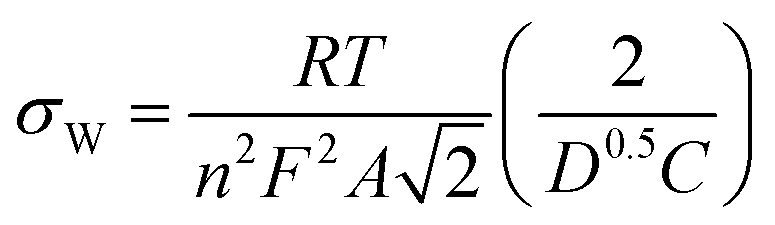 | (6) |
4. Conclusions
In this study, we employed electrochemical characterization and ex situ characterization to investigate the electrochemical lithiation mechanism of layered AgCrSe2 and CuCrSe2. The results reveal a three-step process involving the total transfer of ∼6.6 Li+ per AgCrSe2 and 5.7 Li+ per CuCrSe2. In the first step, Ag extrusion from AgCrSe2 is combined with Li+ intercalation to yield LiCrSe2 decorated with Ag nanoparticles. The second step is the conversion of LiCrSe2 into Li2Se and Cr. In the third and final step, the extruded Ag reacts with Li+ at approximately 0.13 V to form a Li–Ag alloy. CuCrSe2 follows only the first two steps due to the absence of Li–Cu alloying. In both materials, electrochemical reduction is accompanied by significant microstructural reconstruction, Ag redistribution, and changes in Se bonding environments. Measurements of Li-ion diffusivity indicate a general decrease in diffusion coefficient during Ag–Li exchange. This behavior is in contrast to the concept of room-temperature superionic conductivity achieved through the ‘pillar effect’ reported in (Ag–Li)CrS2, suggesting that the substitution of Se for S fundamentally alters the transport mechanism. These findings provide the electrochemical reduction mechanisms of the MCrX2 family of materials in the presence of alkali ions and offer guidance for the design of layered transition-metal chalcogenides as high-energy-density lithium-ion battery electrodes or electrolytes.Conflicts of interest
There are no conflicts to declare.Data availability
Data from this work is openly available at https://doi.org/10.5281/zenodo.17244893.Supplementary information: supplementary figures and tables including additional electrochemical data, ex situ SEM/EDS characterization, operando XRD patterns, XPS spectra, impedance and GITT-based kinetic analyses, and Rietveld refinement results. See DOI: https://doi.org/10.1039/d5ta08061d.
Acknowledgements
This material is based upon work supported by the National Science Foundation under Grants No. DMREF-2119377 (S. M., N. P. H. and V. A.) and DMREF-2118463 (M. T. R. and A. Z.). This work was performed in part at the Analytical Instrumentation Facility (AIF) at North Carolina State University, which is supported by the State of North Carolina and the National Science Foundation (award number ECCS-2025064). The AIF is a member of the North Carolina Research Triangle Nanotechnology Network (RTNN), a site in the National Nanotechnology Coordinated Infrastructure (NNCI).References
- J. W. Choi and D. Aurbach, Nat. Rev. Mater., 2016, 1, 16013 CrossRef CAS.
- X. Zuo, J. Zhu, P. Müller-Buschbaum and Y.-J. Cheng, Nano Energy, 2017, 31, 113–143 CrossRef CAS.
- T. Wang, S. Chen, H. Pang, H. Xue and Y. Yu, Adv. Sci., 2016, 3, 1600289 Search PubMed.
- W. Choi, Y. S. Choi, H. Kim, J. Yoon, Y. Kwon, T. Kim, J.-H. Ryu, J. H. Lee, W. Lee, J. Huh, J. M. Kim and W.-S. Yoon, Chem. Mater., 2021, 33, 1935–1945 CrossRef CAS.
- Y.-Z. Wang, X.-Y. Shan, D.-W. Wang, Z.-H. Sun, H.-M. Cheng and F. Li, Joule, 2018, 2, 1278–1286 CrossRef CAS.
- X. Li, J. Zhi, Y. Xie, Y. Huang and H. Li, RSC Adv., 2015, 5, 85857–85863 Search PubMed.
- R. Bhandavat, L. David and G. Singh, J. Phys. Chem. Lett., 2012, 3, 1523–1530 CrossRef CAS PubMed.
- F. Han, J. Yue, X. Fan, T. Gao, C. Luo, Z. Ma, L. Suo and C. Wang, Nano Lett., 2016, 16, 4521–4527 CrossRef CAS PubMed.
- D. Su, D. Zhou, C. Wang and G. Wang, Adv. Funct. Mater., 2018, 28, 1800154 CrossRef.
- G. Xiang, X. Yan, X. Xiong, Y. Zong, S. Aziz, Z. Zhu and Y. Wu, Mater. Today Energy, 2025, 51, 101894 CrossRef CAS.
- P. Yu, S. Sun, C. Sun, C. Zeng, Z. Hua, N. Ahmad, R. Shao and W. Yang, Adv. Funct. Mater., 2024, 34, 2306939 CrossRef CAS.
- Y. Fujita, K. Münch, T. Asakura, K. Motohashi, A. Sakuda, J. Janek and A. Hayashi, Chem. Mater., 2024, 36, 7533–7540 CrossRef CAS.
- F. Wu, J. T. Lee, Y. Xiao and G. Yushin, Nano Energy, 2016, 27, 238–246 CrossRef CAS.
- J. H. Um, A. Jin, X. Huang, J. Seok, S. S. Park, J. Moon, M. Kim, S. H. Kim, H. S. Kim, S.-P. Cho, H. D. Abruña and S.-H. Yu, Energy Environ. Sci., 2022, 15, 1493–1502 RSC.
- D. You, W. Yang, Y. Liang, C. Yang, Y. Yu, Z. Zhu, X. Li, Y. Zhang and Y. Zhang, Adv. Funct. Mater., 2025, 35, 2421900 CrossRef CAS.
- Y. Ma, Y. Ma, G. Giuli, H. Euchner, A. Groß, G. O. Lepore, F. d'Acapito, D. Geiger, J. Biskupek, U. Kaiser and H. M. Schütz, Adv. Energy Mater., 2020, 10, 2000783 CrossRef CAS.
- J. Ding, M. Towhidur Rahman, C. Mao, J. L. Niedziela, D. Bansal, A. F. May, D. L. Abernathy, Y. Ren, A. Zevalkink and O. Delaire, et al. , Phys. Rev. Mater., 2025, 9, 035402 CrossRef CAS.
- M. T. Rahman, N. P. Holzapfel, K. Ciesielski, W. Guetari, E. Toberer, V. Augustyn and A. Zevalkink, Chem. Mater., 2025, 37, 6718–6726 CrossRef CAS PubMed.
- M. T. Rahman, K. Ciesielski, J. Pelkey, A. K. M. A. Shawon, E. Toberer and A. Zevalkink, J. Phys.: Energy, 2025, 7, 035016 CAS.
- J. Peng, Y. Liu, Y. Pan, J. Wu, Y. Su, Y. Guo, X. Wu, C. Wu and Y. Xie, J. Am. Chem. Soc., 2020, 142, 18645–18651 CrossRef CAS PubMed.
- D. Wu, S. Huang, D. Feng, B. Li, Y. Chen, J. Zhang and J. He, Phys. Chem. Chem. Phys., 2016, 18, 23872–23878 RSC.
- A. Khan, M. Orbay, N. Dupré and E. Gautron and others, Energy Storage Mater., 2024, 70, 103431 CrossRef.
- D. A. Skoog, F. J. Holler and S. R. Crouch, Principles of Instrumental Analysis, Cengage Learning, Boston, 7th edn, 2018, pp. 914–915 Search PubMed.
- A. D. Pelton, Bull. Alloy Phase Diagrams, 1986, 7, 133–136 CrossRef CAS.
- P. F. Bongers, C. F. Van Bruggen, J. Koopstra, W. P. F. A. M. Omloo, G. A. Wiegers and F. Jellinek, J. Phys. Chem. Solids, 1968, 29, 977–984 CrossRef CAS.
- B. Li, H. Wang, Y. Kawakita, Q. Zhang, M. Feygenson, H. L. Yu, D. Wu, K. Ohara, T. Kikuchi, K. Shibata, T. Yamada, X. K. Ning, Y. Chen, J. Q. He, D. Vaknin, R. Q. Wu, K. Nakajima and M. G. Kanatzidis, Nat. Mater., 2018, 17, 226–230 CrossRef CAS PubMed.
- P. Viswanath and M. Yoshimura, SN Appl. Sci., 2019, 1, 1519 CrossRef.
- R. Ciceo-Lucacel, T. Radu, O. Ponta and V. Simon, Mater. Sci. Eng., C, 2014, 39, 61–66 CrossRef CAS PubMed.
- M. C. Biesinger, B. P. Payne, A. P. Grosvenor, L. W. M. Lau, A. R. Gerson and R. S. C. Smart, Appl. Surf. Sci., 2011, 257, 2717–2730 CrossRef CAS.
- C. D. Wagner, NIST Technical Note 1289: the NIST X-Ray Photoelectron Spectroscopy (XPS) Database, National Institute of Standards and Technology, Washington, DC, 1991 Search PubMed.
- J. F. Moulder, W. F. Stickle, P. E. Sobol and K. D. Bomben, Handbook of X-Ray Photoelectron Spectroscopy, Perkin-Elmer Corporation, Eden Prairie, MN, 1992 Search PubMed.
- R. Vedalakshmi, S. Velu, H.-W. Song and N. Palaniswamy, Corros. Sci., 2009, 51, 1299–1307 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |