Thermocatalytic conversion of fatty acids and their derivatives to aromatics over MoOx/HZSM-5 via tandem deoxygenation–aromatization
Received
4th December 2025
, Accepted 16th April 2026
First published on 29th April 2026
Abstract
Benzene, toluene and xylenes (BTX) serve as critical components in enhancing cold flow properties in high-performance fuels such as jet fuel, preventing fuel gelling and crystallization at low temperatures while maintaining combustion efficiency; however, their production remains largely reliant on conventional crude oil-derived processes. Alternate pathways such as catalytic pyrolysis, methanol-to-aromatics processes, etc. remain underexplored for sustainable synthesis of these aromatic hydrocarbons. This study presents a novel approach for producing aromatic hydrocarbons using MoOx/HZSM-5 catalyst in a continuous fixed-bed reactor. Feedstocks of interest such as oleic acid (OA), linoleic acid (LA), corn distiller's oil (CDO), and used cooking oil (UCO) were individually co-fed with water through the catalyst bed. A comprehensive design of experiments was employed to systematically elucidate the effect of critical reaction parameters such as temperature (350–375 °C), space time (1–4 h), and Mo(VI) loading on HZSM-5 (0–10 wt%). With no additional H2 requirement for the process, the maximized operating conditions generated a maximum yield of 85% aromatic hydrocarbons in the liquid product obtained. The major platform chemicals produced during the conversion were benzene, toluene, ethylbenzene, and xylenes (BTEX), which were characterized using GC-MS. Further evidence for the extent of deoxygenation of feedstocks and presence of aromatic compounds in the product was supported by IR and 1H-NMR analyses respectively. The catalysts synthesized were characterized using BET, XRD, IR, SEM, NH3 and XPS. This study positions MoOx/HZSM-5 as a promising catalyst enabling the efficient, H2-free production of BTX from renewable fatty acid feedstocks, advancing the development of sustainable routes to produce these platform chemicals.
1. Introduction
Benzene and its alkyl-substituted derivatives such as toluene, ethyl benzene, and xylenes (BTEX) serve as the fundamental building blocks for essential commodities like plastics, rubbers, pharmaceuticals, and cosmetics.1 The global BTEX market was reported around 129 million tonnes in 2022, and is projected to reach 181 million tonnes by 2031.2 Currently, the production of these aromatics is dominated by catalytic reforming of naphtha, a process inherently dependent on crude oil-derived feedstocks.3 However, crude oil and its constituents are subject to price volatility, which is driven by market demand and geopolitical factors. Beyond the inherent price volatility of crude oil, the substantial greenhouse gas footprint of fossil-based processes has created a growing impetus for the sustainable production of high-value aromatics from carbon-neutral sources.
Renewable feedstocks containing triglycerides and free fatty acids, commonly sourced from both edible and non-edible oils, have emerged as promising candidates for the sustainable production of bio-based aromatics. The source of these feedstocks includes used cooking oil (UCO) such as canola, corn, soybean, sunflower, and safflower oil etc. Corn Distiller's Oil (CDO) is a by-product of ethanol production,4 and is mainly comprised of triacylglycerols with (poly)unsaturated fatty acids; mainly oleic acid (OA) and linoleic acid (LA). These feedstocks contain oxygenated compounds, which can undergo deoxygenation, hydrotreating, and alkylation reactions to produce drop-in fuels, and can be blended with conventional jet fuel without any modifications.5 UCO has also been evaluated as a promising feedstock for the production of sustainable aviation fuels (SAFs).6,7 In this context, Singh et al.8 recently provided a dedicated review of deoxygenation pathways specifically for SAF production from UCO, documenting how catalyst choice and process parameters critically govern selectivity between decarboxylation, decarbonylation, and hydrodeoxygenation routes and their consequences for jet-range hydrocarbon yield. Moreover, a highly selective NiMo/Al2O3–SiO2 catalyst system demonstrated a UCO conversion of over 86% with 74.8% jet-range hydrocarbon selectivity in a fixed-bed reactor, confirming the growing interest in tuning supported metal catalysts for SAF applications from waste lipid feedstocks. However, this requires additional regulatory checks to ensure the standards of the biofuel produced are met.9 Aromatic hydrocarbons such as BTEX possess high octane number, which makes them suitable for blending with gasoline and jet fuel to improve anti-knocking properties as well as cold-flow properties.10 With the growing demand for reducing our dependency on conventional feedstocks, upgrading bio-based feedstocks for production of aromatics such as BTEX can be considered as an alternative to support the drive towards sustainability.
Catalytic pyrolysis has been explored as a potential thermocatalytic technology to produce BTEX from bio-based feedstocks. The conversion involves a two-stage sequence: (i) pyrolysis of the feedstock at elevated temperatures (>550 °C), and (ii) subsequent cyclization and aromatization of the pyrolyzed vapors in a fixed-bed reactor over a reforming catalyst, operated under relatively high H2 pressures (10–30 bar), to generate aromatics.11 Anellotech LLC has demonstrated this catalytic pyrolysis approach for production of drop-in aviation fuels.12 However, catalytic pyrolysis produces substantial amounts of unsaturated compounds such as alkenes as by-products, which are undesirable in the petrochemical complexes for isomerization and blending purposes in drop-in fuels.13 A recent study by Wang et al. on enhanced bio-BTX formation from glycerol co-fed with in situ produced toluene over H-ZSM-5/Al2O3 illustrated that even optimized catalytic pyrolysis approaches struggle to surpass peak BTX carbon yields of ∼30 C% and suffer from rapid catalyst deactivation, reinforcing the fundamental limitations of high-temperature pyrolysis routes for selective monoaromatic production.14 Similarly, Miller et al. demonstrated a sequential ketonization–aromatization pathway over HZSM-5 for converting wet-waste-derived volatile fatty acids to BTEX at 350–450 °C, yet highlighted that the co-production of substantial light alkane by-products at near-equal proportions remains an unresolved challenge for achieving high aromatics selectivity from oxygenated bio-feedstocks.15
Catalytic deoxygenation is generally preferred over catalytic pyrolysis, as it proceeds at lower operating temperatures and minimizes the generation of undesirable by-products such as alkenes. These advantages not only improve process efficiency but also enhance selectivity toward desired liquid aromatics, thereby offering a more practical and sustainable pathway for upgrading renewable fatty acid-derived feedstocks. Transition metal-based catalysts for deoxygenation of fatty acids and their derivatives have been investigated under H2 pressures of 20–30 bar. These include NiMo,16–18 CoMo17–21 and Pt/Pd,22–28 which rely mainly on external H2 pressures for facilitating deoxygenation chemistry.29 Murzin et al.28,30 were among the pioneers to investigate the catalytic activity of Pd supported on activated carbon and alumina (Al2O3). Metal loadings between 1–5% were studied in a batch system with temperatures in the range of 300–370 °C and pressure range of 2.5–10 MPa. The increased reaction rates of decarboxylation were found with activated carbon as compared to alumina, in part due to its higher surface area (920 m2 g−1). Higher metal loadings generated favourable decarboxylation chemistry, primarily because of the large number of active sites available for the reaction. However, regeneration of the Pt/C catalyst resulted in the structural changes of the activated carbon support after regeneration at 650 °C and therefore impacted its catalytic activity. Wu et al. investigated decarboxylation under inert atmosphere with Ni/C as the catalyst.31 It was reported that same extent of decarboxylation similar to hydrotreating condition was achieved at 430 °C.32 They also showed that though transition metal-based catalysts such as Ni(II) and Co(II) have greater catalytic activity towards decarboxylation, they tend to deactivate quickly in comparison to Pd and Pt. Furthermore, the liquid fraction was dominated by aliphatic hydrocarbons enriched in alkenes, with no evidence of aromatic hydrocarbons. The high hydrogen consumption associated with conventional hydrotreating routes has spurred growing interest in hydrogen-free deoxygenation strategies. In a recent 2025 study, Pacheco et al. investigated a H2-free fast cracking of oleic acid over acidic HZSM-5 and Y-zeolites under conditions analogous to the fluid catalytic cracking process, establishing that the balance between Brønsted and Lewis acid sites governs the selectivity between deoxygenation pathways and the distribution of aromatic versus aliphatic products.33 Notably, they reported that HZSM-5 yielded a higher aromatic fraction than HY under comparable conditions, attributing this to the former's unique pore architecture and stronger Brønsted acidity, consistent with the mechanistic importance of acid site density in directing cyclization chemistry. The acidity of the catalyst support also contributes to the product distribution, which was demonstrated by Hengst et al. in their research for producing drop-in jet fuel.34 Similar conclusions were derived from the recent works by Charpentier et al.,25,35–37 where comparison of catalytic activity of Ni(II) and Mo(IV) loaded on Al2O3 for production of green diesel was demonstrated. Savage et al.38 also performed extensive research on different metal supported systems for conversion of mono-unsaturated and polyunsaturated fatty acids into heptadecane, which can be used as a drop-in diesel product. The group utilized Pt on activated carbon in the presence of water (sub-critical and supercritical). Han et al.39 investigated the decarboxylation and saturation of micro-algae oil in the presence of Pd:Au catalyst system in a packed bed system, which resulted in better catalyst stability as compared to standalone Pd. However, elimination of oxygen from the feedstock without isomerization or improving the octane rating of the resulting product does not address the requirement for jet fuels, which typically contains 8–25% aromatics.40 High operation costs and limited ability towards cyclization and aromatization with metal-based catalysts draws attention towards exploring other alternatives.
Direct conversion of fatty acids and their derivatives into aromatics at comparatively low temperatures (300–380 °C) without external H2 pressure has been underexplored. A few studies have emphasized on valorizing these feedstocks while investigating different catalyst systems at elevated temperatures (>400 °C).41–44 In some of these findings, catalytic decarboxylation of vegetable oils was carried out at temperatures ranging from 400–470 °C, resulting in low aromatic yields. Recent efforts using hierarchical ZSM-5 architectures have attempted to address the dual challenge of improving accessibility to active sites while preserving shape-selective aromatization. Liu et al. synthesized hierarchical ZSM-5 nanosheets with interconnected mesopore networks for the catalytic cracking of oleic acid, demonstrating that a self-pillared nanosheet structure with a Brønsted-to-Lewis acid ratio optimized toward strongly acidic Brønsted sites achieved markedly higher yields of light olefins and aromatics relative to conventional microporous ZSM-5.45 Nonetheless, the aromatic yields remained modest in the absence of a metal co-function capable of activating the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds of polyunsaturated fatty acids at moderate temperatures, pointing to a clear gap that bifunctional metal–zeolite catalysts are better positioned to fill. In a particularly relevant and recent contribution, Charpentier et al. reported the tandem deoxygenation and aromatization of oleic acid under subcritical hydrothermal conditions over MoO3/HZSM-5 catalyst, demonstrating synergistic interplay between the molybdenum oxide phase and the Brønsted acid sites of HZSM-5, thus enabling direct conversion of a model fatty acid to BTEX without the need for external hydrogen in a single-pot batch system.46 A clear consensus therefore indicates that transition metal-based catalysts have been reported to facilitate both deoxygenation and cyclization of fatty acids and their derivatives.47–51
C bonds of polyunsaturated fatty acids at moderate temperatures, pointing to a clear gap that bifunctional metal–zeolite catalysts are better positioned to fill. In a particularly relevant and recent contribution, Charpentier et al. reported the tandem deoxygenation and aromatization of oleic acid under subcritical hydrothermal conditions over MoO3/HZSM-5 catalyst, demonstrating synergistic interplay between the molybdenum oxide phase and the Brønsted acid sites of HZSM-5, thus enabling direct conversion of a model fatty acid to BTEX without the need for external hydrogen in a single-pot batch system.46 A clear consensus therefore indicates that transition metal-based catalysts have been reported to facilitate both deoxygenation and cyclization of fatty acids and their derivatives.47–51
In this study, we explore a novel thermocatalytic process using MoOx/HZSM-5 catalyst to facilitate tandem deoxygenation–aromatization of (poly)unsaturated fatty acids and their derivatives to produce BTEX, without added H2 in a continuous tubular reactor. The continuous-flow configuration used here offers specific benefits for interpreting the tandem deoxygenation–aromatization chemistry. By operating under well-defined space times and collecting products at fixed time-on-stream intervals, we anticipate that transient changes in conversion, deoxygenation extent, and aromatic selectivity can be linked more directly to catalyst-state evolution than in batch experiments, where concentrations, temperature history, and water content co-evolve in a less controlled manner. From a process-design perspective, we expect that the continuous system demonstrated here could be more readily translated into modular or pilot-scale units for decentralized upgrading of lipid-derived feedstocks. This work investigates MoOx/HZSM-5 as a novel catalyst for the tandem deoxygenation and aromatization of bio-based feedstocks without external hydrogen pressure, addressing a gap in the current literature where this specific metal–zeolite combination has not been explored in continuous mode for these coupled reactions without H2. This research further evaluates and emphasizes on comprehending the correlation between the reaction parameters (temperature, space time, and Mo loading on HZSM-5) and the extent of tandem deoxygenation–aromatization in a fixed bed tubular reactor.
2. Experimental section
2.1 Materials
Granular HZSM-5 (CBV 3024E, CAS no. 1318-02-1) with SiO2/Al2O3 = 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 was purchased from Zeolyst International, USA. It was obtained in powder form, the particle size being in the range of 20–200 nm. Ammonium molybdate tetrahydrate (AMT) > 98% purity, CAS no. 12054-85-2, oleic acid (OA) > 99% purity, CAS no. 112-80-1 and linoleic acid (LA) > 99% purity, CAS no. 60-33-3 were obtained from Sigma-Aldrich, Canada. Corn Distiller's Oil (CDO) was obtained from Greenfield Global and used as is. Used cooking oil (UCO) was obtained from the author's home and filtered before use. Ultrapure water system (EASY pure LF, Mandel Scientific Co., USA) was used to prepare deionized water (18.2 MΩ) for catalyst impregnation steps.
:1 was purchased from Zeolyst International, USA. It was obtained in powder form, the particle size being in the range of 20–200 nm. Ammonium molybdate tetrahydrate (AMT) > 98% purity, CAS no. 12054-85-2, oleic acid (OA) > 99% purity, CAS no. 112-80-1 and linoleic acid (LA) > 99% purity, CAS no. 60-33-3 were obtained from Sigma-Aldrich, Canada. Corn Distiller's Oil (CDO) was obtained from Greenfield Global and used as is. Used cooking oil (UCO) was obtained from the author's home and filtered before use. Ultrapure water system (EASY pure LF, Mandel Scientific Co., USA) was used to prepare deionized water (18.2 MΩ) for catalyst impregnation steps.
2.2 Catalyst preparation
Catalysts with varying MoOx loadings (5.0 wt%, 7.5 wt% and 10.0 wt%) on the HZSM-5 substrate were prepared by incipient-wetness impregnation method.35 The ZSM-5, obtained in its ammonium cation form (NH4+ZSM-5), was converted into protonic form, i.e. HZSM-5 by calcination at 550 °C for 4 h.34 Before the deposition of metal precursor, the HZSM-5 was preheated at 150 °C for 1 h to ensure removal of any residual volatile organic impurities. For the impregnation steps, a solution containing a desired amount of AMT in deionized water was introduced on the HZSM-5 using a syringe pump (Harvard Apparatus, PHD Ultra). The resulting mixture was then dried in a vacuum oven at 80 °C for 12 h, then calcined by heating in a muffle furnace at a rate of 10 °C min−1 to a temperature of 650 °C and was maintained at 650 °C for 4 h. The catalyst samples prepared were stored in a desiccator at ambient conditions. Detailed discussion regarding the effect of calcination temperatures on various other physicochemical properties of the zeolite has been incorporated in the later sections.
2.3 Catalyst testing
The prepared catalysts with varied MoOx loadings were evaluated in a continuous fixed bed reaction system (Bench-Top Reaction System Junior)™ purchased from Autoclave Engineers LLC to understand the interplay of reaction parameters to promote deoxygenation and aromatization of the feedstocks. For the experiments and the parametric study with the feedstocks, a specific catalyst weight of 1.47 g was utilized. The experiments utilized a water-to-feed weight ratio of 4:1 (w/w). This specific ratio was selected based on established protocols from prior research conducted by the same group on the deoxygenation of fatty acids.25,52 These parameters were investigated in a continuous fixed-bed tubular reactor system at temperatures ranging from 350 to 375 °C to evaluate their correlation with the efficiency of the tandem reaction. A comprehensive design of experiments was employed where the space time was varied between 1 and 4 hours. The process schematic for the reaction system is provided in SI (Fig. S1). The feedstocks and water were introduced into the reaction system using a dual-syringe pump system (260 D Teledyne Isco LLC). The feed-water mixer and the tubular reactor were placed inside a heated oven maintained at about 200 °C. The reactor assembly consisted of a 5 mL tubular reactor (316 SS) and was equipped with a band heater to maintain desired reaction temperatures. The catalyst bed inside the tubular reactor was prepared such that the desired amount of catalyst was sandwiched between known weight of ball bearings and quartz wool at both ends. The desired space time was achieved by adjusting the flow rates of the feedstock and water through the catalyst bed using the following equation:| |
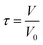 | (1) |
where V represents the volume of the catalyst bed, V0 represents the combined volumetric flow rates of water and feedstock, and τ represents the space time. The mixture of feed and water was introduced from the top of the tubular reactor, and the liquid product exiting from the bottom of the tubular reactor was introduced to a condensing gas–liquid separator maintained at 8.5 °C and was collected at regular intervals, which were quantified and characterized by the combination of GC-MS, IR, and 1H-NMR analyses. Errors in conversion, deoxygenation extent, and product yields were determined from replicate experiments, and the errors are presented as average ± average deviation. Each experiment was carried out in triplicates to confirm reproducibility, and the reported data represent the average of the individual runs, indicating good experimental repeatability. The gas samples were stored in airtight Tedlar gas bags (SKC Inc., PA) for GC-TCD analysis. A rigorous cleaning and preparation protocol was implemented to minimize cross-contamination and maintain integrity of the experimental results. At the end of each run, the spent catalysts were recovered, thoroughly washed with hexane to remove any residual organic products, dried at 120 °C overnight and analyzed via different catalyst characterization techniques.
2.4 Catalyst characterization
The Brunauer, Emmett and Teller (BET) method for determining specific surface area, BJH pore volume, and BJH pore diameter of the fresh and spent catalysts were analyzed by Tristar 3020. In this procedure, the catalyst samples were degassed overnight under N2 atmosphere at 200 °C and then subjected to N2 adsorption/desorption test. The X-ray diffraction (XRD) analysis was performed with a Bruker D2 Phaser powder diffractometer using Cu-Kα radiation (λ for Kα = 1.54059 Å) over 2θ ranging between 20–60°. The scanning electron microscopy (SEM) images were obtained with a model LEO1530 scanning electron microscope under different magnification levels. NH3-temperature programmed desorption (NH3-TPD) analysis was performed to characterize the acidity of the catalysts using a Micromeritics AutoChem II 2920 analyzer. Prior to analysis, samples were purged with high-purity helium (99.99%, 50 mL min−1) at 400 °C for 1 hour to remove adsorbed contaminants. Subsequently, the samples were cooled from 400 °C to 100 °C and saturated with ammonia by exposure to a flow of anhydrous 10% NH3 in helium (50 mL min−1) for 2 hours. Physiosorbed ammonia was then purged by flushing with helium (50 mL min−1) at 100 °C for 1 hour. NH3-TPD analysis was conducted over a temperature range from room temperature to 800 °C at a heating rate of 10 °C min−1. The amount of desorbed NH3 was quantified by integrating the area under the TPD curve, providing a direct measure of catalyst acidity. The X-ray photoelectron spectroscopy (XPS) analyses were carried out with a Kratos AXIS Supra X-ray photoelectron spectrometer using a monochromatic Al Kα source (15 mA, 15 kV). XPS was carried out by probing the surface of the sample to a depth of 7–10 nm, with detection limits ranging from 0.1–0.5 atomic% depending on the element. The instrument work function was calibrated to give a binding energy (BE) of 83.96 eV for the Au 4f7/2 line for metallic gold and the spectrometer dispersion was adjusted to give a BE of 932.62 eV for the Cu 2p3/2 line of metallic copper. The elemental composition of the prepared Mo/HZSM-5 catalysts was determined by inductively coupled plasma optical emission spectrometry (ICP-OES) using axial plasma viewing configuration. Catalyst samples were digested following EPA Method 3050B, and the resulting solutions were analyzed on an ICP-OES instrument in accordance with EPA Method 6010C. Analyses were performed by Lambton Scientific (a division of Technical Chemical Services Inc., Ontario, Canada), an accredited third-party analytical laboratory.
2.5 Product characterization
The gas chromatographic analysis of liquid products was performed with a Shimadzu 2014 gas chromatograph equipped with a 30 m × 0.25 mm × 0.25 µm DB-5 wax column (Agilent Technologies, Santa Clara, CA, USA), and a flame ionization detector (FID). The makeup gas, carrier gas, and ionization gas were, respectively, ultrapure air, helium, and hydrogen. In a typical analysis, 1 µL sample was injected with a split ratio of 10:1; carrier gas (helium) flow rate was 25 mL min−1; and the flow rate of H2 was 30 mL min−1 while that of ultrapure air was 400 mL min−1. The temperatures for injector and detector were set at 300 °C and 320 °C, respectively, and the following temperature program was applied after injection: 5 min hold at 50 °C; ramp at 5 °C min−1 to 150 °C; ramp at 10 °C min−1 to 300 °C; and a final hold at 300 °C for 2 min. Qualitative identification of compounds in the samples was based on comparison with retention times for aliphatic and aromatic hydrocarbons compounds in standards (Restek Inc). The GC instrument was calibrated using aromatic standard obtained from Restek (Certified Aromatics Mix, 5500 µg mL−1, P&T Methanol, 1 mL per ampul, catalog no. 30485) with known concentrations of individual components present. The calibration of the GC followed a series dilution methodology to establish a five-point calibration curve for each individual compound in the aromatic standard, which was in accordance with ASTM D5580 calculation procedure.53 The solvent used for preparing these standards was methanol, ensuring consistent dilution and accurate concentration measurements across the calibration range. Methanol was used as the solvent due to its compatibility with obtained standard for aromatic compounds and thus preventing interference and ensuring accurate standard preparation. Gaseous products formed during the reaction were collected in Tedlar gas bags and analyzed against a standard calibration mixture of gases (CO, CO2, N2, O2, H2, and gaseous hydrocarbons). The gas sample (1 mL) was filled into a gas tight syringe (Model 1001 LTN SYR, Cemented NDL, 22 ga, 2 in, point style 5) was injected with helium as the carrier gas. The gases were detected with a thermal conductivity detector (TCD). In GC-TCD analysis, the oven temperature was kept at 30 °C for 2 min followed by 15 °C min−1 ramp to 250 °C, held at 250 °C for 1 min. The temperature of both the injector and detector were maintained at 200 °C. 1H-NMR spectra of product hydrocarbon samples in deuterated chloroform (CDCl3) solvent were acquired with a Bruker 400 MHz spectrometer with referencing against the residual solvent signal (at δ 7.26 ppm) relative to tetramethylsilane at δ 0 ppm. All spectra were obtained under the same conditions (298 K, gas flow rate of 300 lph) and were processed with MestreNova software. Infrared spectra of the feed and the liquid products were acquired with a Nicolet 6700 Fourier Transform Infrared Spectrometer (Thermo Scientific) in the range of 600–4000 cm−1 with 64 scans at 8 cm−1 resolution. The extent of deoxygenation was calculated according to eqn (2) adapted from ASTM D3242,54 where peak area corresponding to absorbance band around 1710 cm−1 for carbonyl stretching mode characteristic of unconjugated carboxylic acids was calculated in feedstock and liquid product mixture.| |
 | (2) |
3. Results & discussions
3.1 Deoxygenation and aromatization studies with oleic acid
Preliminary experiments were conducted with mono-unsaturated fatty acid (OA) to understand the effect of varying MoOx loadings on HZSM-5 for deoxygenation and aromatization under the following reaction condition: weight of catalyst = 1.47 g, T = 370 °C, τ = 4 h, and water:feed (w/w) = 4:1. The selection of water:feed ratio was based on previous studies conducted by our research group addressing the deoxygenation of various fatty acids and their derivatives for production of green diesel.37 Fig. 1 represents the IR spectra of the liquid hydrocarbon products obtained under the above-mentioned reaction conditions. The reduction in the band intensity at 1710 cm−1 corresponding to the carbonyl group was observed in the liquid products; which was strongly influenced by MoOx loadings on HZSM-5; achieving 64% reduction in carbonyl band intensity using 5 wt% MoOx/HZSM-5, 99% reduction using 7.5 wt% MoOx/HZSM-5, and 90% reduction using 10 wt% MoOx/HZSM-5. Furthermore, when HZSM-5 was used without metal impregnation, the infrared spectra of the liquid products showed minimal changes in the carbonyl stretching vibration at 1710 cm−1 relative to oleic acid (OA), indicating that the carbonyl functional group remained unaltered under these conditions. The appearance of the significant CO stretching vibration at 1710 cm−1 can be primarily attributed to the inadequate catalytic activity of unloaded HZSM-5, resulting in incomplete deoxygenation. This finding was found consistent with deoxygenation studies done over Ni/MgO–Al2O3 catalyst by Liu et al.55 In their research, it was reported that metal loading improved the catalytic activity towards deoxygenation, however; increasing the metal loading after a threshold plateaued the catalytic activity due to agglomeration of NiO on the catalyst support. The IR spectroscopic analysis of the liquid products also revealed significant structural changes in comparison with OA, as evident by the diminished intensity of absorption bands corresponding to C–H stretching (2850–2950 cm−1) and bending (1460–1480 cm−1) vibrations of CH2 groups present in fatty acids (Fig. 1). Beyond the carbonyl region, the absorption bands associated with the aliphatic C–H stretching (2850–2950 cm−1) and bending (1460–1480 cm−1) vibrations also provide insight into the support's activity. In the case of liquid product obtained with pure HZSM-5 (0 wt% Mo), these bands demonstrated non-significant decrease in intensity compared to the raw oleic acid, suggesting partial transformation of aliphatics into cyclic compounds. In contrast, the Mo-promoted catalysts (5.0–10.0 wt% Mo) exhibit a much more dramatic suppression of these methylene vibrations. This near-disappearance of the aliphatic signature correlates with the high selectivity toward aromatics, indicating that the intermediate cracking products are rapidly undergoing cyclization and dehydrogenation over the bifunctional MoOx/HZSM-5 active sites. Furthermore, the appearance of weak vibrations in the 3000–3100 cm−1 (aromatic C–H stretch) and 1600–1650 cm−1 (aromatic CC ring stretch) regions for the Mo-loaded samples indicates towards transformation of the long-chain aliphatic backbone into desired cyclic aromatic structures. In all the remaining cases, the suppression of C–H stretching and bending vibrations corresponding to long methylene backbone in the product IR suggested a decrease in linear alkane chain content, attributed to C–C cleavage.
 |
| | Fig. 1 ATR-FTIR spectra of (a) oleic acid and liquid products obtained using (b) 10 wt% MoOx/HZSM-5, (c) 7.5 wt% MoOx/HZSM-5, (d) 5 wt% MoOx/HZSM-5 & (e) pure ZSM-5; reaction conditions: T = 370 °C, τ = 4 h & water:feed (w/w) = 4:1. | |
The evaluation of deoxygenation activity of varying MoOx/HZSM-5 loadings was continued for a time-on-stream (TOS) of 84 h through the fixed-bed reactor. The liquid product was collected after every 8 h from the start of the run and analyzed by IR to measure the extent of deoxygenation under identical operating conditions, as shown in Fig. 2. The gaseous products from each experimental run were collected in Tedlar gas bags and subsequently analyzed for CO and CO2 concentrations to validate and corroborate the results obtained from infrared (IR) spectroscopy. The normalized concentrations for CO and CO2 evolved during the experimental runs have been provided in Fig. S2. For the products collected at the same time intervals, notable reduction in catalytic activity towards decarboxylation of OA was observed with 5 wt% MoOx/HZSM-5 and HZSM-5 compared to 7.5 wt% MoOx/HZSM-5 and 10 wt% MoOx/HZSM-5. In case of 10 wt% MoOx/HZSM-5, agglomeration of MoOx in the zeolite channels could inhibit the reactant/product diffusion and therefore a potential reason for reduced deoxygenation activity, when compared to 7.5 wt% MoOx/HZSM-5.
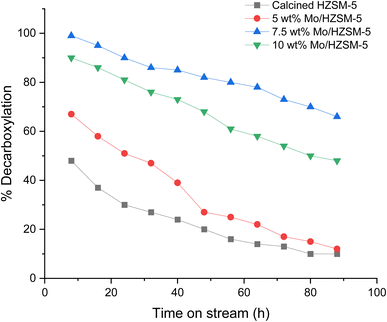 |
| | Fig. 2 Extent of decarboxylation of oleic acid. Reaction conditions: T = 370 °C, τ = 4 h, water/feed = 4:1. | |
3.1.1 Effect of temperature, space time and water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :feed ratio on deoxygenation. Parametric studies with OA were carried out with 7.5 wt% MoOx/HZSM-5 by varying temperature (350–375 °C); space time (1–4 h) and water:feed ratio (1:1–4:1) to investigate their synergistic effect on the extent of deoxygenation and conversion. Infrared (IR) analysis of the liquid products presented in Fig. 3 revealed that the extent of deoxygenation (calculated using eqn (2)) increased with reaction temperature. The extent of decarboxylation increased from 57% at 350 °C to 96% at 375 °C, which can be attributed to improved rates of decarboxylation due to increase in temperature. Notably, a near-complete deoxygenation (∼99.5%) was achieved at 360 °C. It is plausible that at higher temperatures, such as 375 °C and beyond, side reactions including coke formation, over-cracking, and condensation reactions become more prominent on MoOx/ZSM-5 catalysts. These side reactions lead to the deposition of carbonaceous species on active sites, blocking them and causing catalyst deactivation, which lowers the effective catalytic activity for decarboxylation. Furthermore, performing deoxygenation reactions with oleic acid at temperatures greater than 400 °C could result in trade-offs with product distribution and yields, as reported by Charpentier et al.56 In order to evaluate whether an increase in temperature above 375 °C would result in further improvement in deoxygenation, a control study was done at T = 390 °C. The total extent of deoxygenation obtained was 89.6% (Fig. S3) and was synonymous with those obtained at 370 °C, and therefore 360 °C was selected as the process temperature to evaluate other model compounds.
:feed ratio on deoxygenation. Parametric studies with OA were carried out with 7.5 wt% MoOx/HZSM-5 by varying temperature (350–375 °C); space time (1–4 h) and water:feed ratio (1:1–4:1) to investigate their synergistic effect on the extent of deoxygenation and conversion. Infrared (IR) analysis of the liquid products presented in Fig. 3 revealed that the extent of deoxygenation (calculated using eqn (2)) increased with reaction temperature. The extent of decarboxylation increased from 57% at 350 °C to 96% at 375 °C, which can be attributed to improved rates of decarboxylation due to increase in temperature. Notably, a near-complete deoxygenation (∼99.5%) was achieved at 360 °C. It is plausible that at higher temperatures, such as 375 °C and beyond, side reactions including coke formation, over-cracking, and condensation reactions become more prominent on MoOx/ZSM-5 catalysts. These side reactions lead to the deposition of carbonaceous species on active sites, blocking them and causing catalyst deactivation, which lowers the effective catalytic activity for decarboxylation. Furthermore, performing deoxygenation reactions with oleic acid at temperatures greater than 400 °C could result in trade-offs with product distribution and yields, as reported by Charpentier et al.56 In order to evaluate whether an increase in temperature above 375 °C would result in further improvement in deoxygenation, a control study was done at T = 390 °C. The total extent of deoxygenation obtained was 89.6% (Fig. S3) and was synonymous with those obtained at 370 °C, and therefore 360 °C was selected as the process temperature to evaluate other model compounds.
 |
| | Fig. 3 ATR-FTIR spectra of (a) oleic acid and liquid products formed at (b) 375 °C, (c) 370 °C, (d) 360 °C and (e) 350 °C; catalyst: 7.5 wt% MoOx/HZSM-5, τ = 4 h, water:feed = 4:1. | |
The effect of variation of space time and water:feed ratio on deoxygenation has been depicted in Fig. 4. Fig. 4(a) suggests that under the reaction conditions of 360 °C and 4:1 water:feed ratio, increasing space time from 1 to 4 hours substantially improved decarboxylation efficiency. Charpentier et al.56 have explained the correlation of longer space times with higher decarboxylation percentages, with 4 h yielding optimal results for deoxygenation of conversion yields. It can further be deduced from Fig. 4(b) that temperature significantly influences the deoxygenation of OA, with a marked increase from 350 °C to 360 °C and plateauing at higher temperatures. The decrease in deoxygenation of oleic acid (OA) at 370 °C compared to 360 °C is likely due to secondary reactions that dominate at higher temperatures, resulting in increased formation of light gases and coke rather than desired hydrocarbon products. Although temperature initially enhances catalyst activity and promotes deoxygenation up to a certain point, further increases lead to excessive decomposition, which lowers the yield of liquid products and can decrease deoxygenation efficiency. At temperatures greater than 400 °C, secondary reactions such as cracking, polymerization, and condensation are favored, leading to coke formation that blocks active sites on the catalyst. This hinders the access of oleic acid and intermediates, reducing deoxygenation efficiency.57
 |
| | Fig. 4 Extent of decarboxylation under different experimental conditions: (a) space time (τ); (b) temperature and (c) water:feed ratio. | |
This aligns with published findings showing that increasing temperature from 325 °C to 375 °C enhances decarboxylation from 55% to 92%.58 As shown in Fig. 4(c), increasing the water:feed ratio from 1:1 to 4:1 at 360 °C with 4 h space time progressively improved the decarboxylation efficiency. Earlier studies on optimization of reaction parameters have found that water:OA ratios between 1:1 and 4:1 significantly impact performance, with higher ratios generally yielding improved results.56 The literature reveals that water serves as an effective reaction medium, facilitating heat transfer and enabling more uniform temperature control throughout the reaction system, which helps achieve reproducible conversion results. Additionally, water promotes dispersion of the catalyst particles, which enhances interaction with OA, and plausibly would help in suppressing coking by providing a source of steam that helps prevent catalyst deactivation. The results obtained indicated that longer space times (4 h) and high water:feed (4:1) ratio showed a greater extent of deoxygenation of OA. Under preferred reaction conditions, the overall liquid yield during catalyst lifetime was found to be ca. 85% with OA. The liquid yield was calculated on a normalized mass basis of heptadecene present in the system, after complete decarboxylation of OA. Accordingly, the following conditions were selected as suitable conditions to achieve deoxygenation of poly(unsaturated) fatty acid: 7.5 wt% MoOx/HZSM-5, T = 360 °C, τ = 4 h, and water:feed = 4:1.
The IR spectra of the liquid products collected after the first 8 h of the experiments were analyzed to understand the changes in the functional groups during deoxygenation of OA, as depicted in Fig. 5. The prominent band at 1710 cm−1 corresponding to the CO stretching vibrations of carboxylic acid group present in OA (Fig. 5(a)) was significantly suppressed in the spectra shown in Fig. 5(e)–(g); therefore, suggesting the prominent effect of increasing severity of reaction parameters on deoxygenation. The fingerprint region below 1500 cm−1 reveals that the product consists of aromatic compounds, evident with IR signals around 1400–1460 cm−1 corresponding to vibrations associated with C–C stretching of aromatic ring. The C–H wag (900–675 cm−1) corresponding to aromatic ring was particularly notable in spectra (e) through (i). Absorbance bands corresponding to C–H stretching for aromatic compounds (3068 cm−1, 3028 cm−1) and C–C stretching attributable to the aromatic ring (1614 cm−1, 1506 cm−1 and 1465 cm−1) were observed. The IR results not only confirmed the catalytic activity of MoOx/HZSM-5 to promote deoxygenation but also indicated the potential of the catalyst to effect tandem deoxygenation and aromatization of OA.
 |
| | Fig. 5 IR spectra of (a) OA and the decarboxylated products formed at 360 °C under different conditions such as (b) τ = 1 h, water:feed = 4:1; (c) τ = 1 h, water:feed = 2:1; (d) τ = 1 h, water:feed = 1:1; (e) τ = 2 h, water:feed = 4:1; (f) τ = 2 h, water:feed = 2:1; (g) τ = 2 h, water:feed = 1:1; (h) τ = 4 h, water:feed = 2:1; (i) τ = 4 h, water:feed = 4:1. | |
3.1.2 GC-MS analysis of liquid products. The liquid products obtained through thermocatalytic deoxygenation of OA at different reaction temperature (350–375 °C) in the fixed bed tubular reactor using 7.5 wt% MoOx/HZSM-5 catalyst were also analyzed using GC-MS. The liquid products primarily included BTX (benzene, toluene, and xylenes), which contributed to more than ca. 65% of the total aromatics obtained. In maximized operating conditions, up to ca. 75% BTX were obtained. Besides, gas-phase components like short hydrocarbons (C1–C3) are formed in combination with low amounts of CO and CO2. Under maximized operating conditions, there was no trace of oleic acid in the liquid product, suggesting complete conversion. However, in cases where temperature was 350 °C, the catalyst activity dropped rapidly within the first 36 h, resulting in unconverted oleic acid in the liquid product. The remaining 10% product were gaseous products in the form of CO2, CO and C1–C4 small chain hydrocarbons. An overall mass balance for the reaction under optimized conditions is shown in Fig. S4. The product yields were based on the total liquid products obtained during each experimental runs and have been reported as % liquid yields. A quantitative comparison of the findings obtained in this study with existing state-of-the-art methods is presented in Table 1.
Table 1 State-of-the-art of production of aromatics from fatty acids and their derivatives
| Feedstock |
Reaction conditions, catalyst |
Maximum aromatic production |
Ref. |
| Peanut oil |
WHSV of 5.4 h−1, T: 450–550 °C, TOS: 30 min over H-ZSM-5 |
BTEX: 33 vol% |
58 |
| Palm oil |
WHSV of 5 h−1, T: 500 °C, TOS: 3 h over Zn/HZSM-5 |
BTEX: 65 wt% |
59 |
| Vegetable oil |
WHSV of 2.6 h−1, T: 550 °C, TOS > 6 h over FCC catalyst |
Average aromatic yield: 25 wt% with different feedstocks |
32 |
| Camelina oil |
WHSV of 0.6 h−1, T: 500 °C, TOS: 12 h over Zn/ZSM-5 |
BTX yield: 6.2 wt% |
60 |
| Soybean oil |
WHSV of 6.69 h−1, T: 460 °C, over Si-MCM-41 |
Aromatics: 25 wt% |
61 |
| Canola oil |
WHSV of 5 h−1, T: 400–500 °C, over SiO2 and similar |
BTX yield: 22.5 wt% |
62 |
| Rapeseed oil |
WHSV of 7.6 h−1, T: 550 °C, TOS: 3 h, over Ga and Zn/ZSM-5 |
Aromatics: 43% |
63 |
| Oleic acid |
WHSV of 0.25 h−1, T: 500 °C, TOS: 30 min over Al2O3, ZrO2, AC, MCM-41 |
BTX: 78% at 550 °C |
55 |
| Used cooking oil |
Batch reactor studies, catalyst: 4 mg, feed: 1 mg, T: 600 °C, over HZSM-5, reaction time: 4 h |
BTXNE: 58.6% |
64 |
| Vegetable oils |
WHSV of 0.1 h−1, reaction time: 180 min, T: 400 °C, over alkali treated HZSM-5 |
BTX: 28.4 wt% from coconut oil, 48 wt% from peanut oil |
59 |
| Palmitic acid |
Reaction time of 3 h, T: 400 °C, over zeolites Y, β |
BTX: 43.9 wt% |
26 |
| C18 fatty acids |
Reaction time: 180 min, T: 400 °C over HZSM-5 |
BTX: 7.6 wt% |
50 |
| Soybean oil |
Reaction time: 60 min, T: 430 °C, over HZSM-5 |
Aromatics: 21 wt% |
65 |
| Oleic acid |
Reaction time: 180 min, T: 400 °C, over HZSM-5 |
Aromatics: 47% |
11 |
| Oleic acid etc. |
Reaction time: 360 min, T: 360 °C, over Mo/HZSM-5 |
BTX > 75% |
Current work |
Furthermore, Fig. 6 presents the composition of various compounds at different reaction temperatures: 350 °C, 360 °C, 370 °C, and 375 °C. The compounds identified, as per the standard aromatic hydrocarbons, include benzene, toluene, xylenes, ethyl toluene, 1,3,5-trimethylbenzene, and isopropylbenzene. These results suggest that increasing the reaction temperature from 350 °C to 360 °C significantly enhanced the formation of aromatic compounds, while reducing non-aromatic byproducts. At 350 °C, the GC-MS analysis revealed that a substantial fraction of the liquid product remained non-aromatic, consisting predominantly of partially deoxygenated and unsaturated C18 derived intermediates, which is consistent with an incomplete progression of the tandem deoxygenation–aromatization network under these milder conditions. However, further increases in temperature (beyond 375 °C) begin to reduce the deoxygenation and liquid yields of the aromatics. We anticipate that the relatively lower extent of decarboxylation at 350 °C (≈57%) compared to 360–375 °C, as determined by IR, implies that a significant pool of long-chain olefinic and paraffinic species persists in the liquid phase and competes with cyclization pathways for consumption of reactive intermediates. Similar temperature-sensitive behavior has been reported for zeolite-catalyzed upgrading of C18 fatty acids and triglycerides, where operation near 325–350 °C favours the formation of long-chain alkanes/olefins and oxygenated residues, while higher temperatures are required to drive aromatic ring closure and subsequent dehydrogenation over Brønsted acid sites.25,66 It is worth noticing that the proportion of benzene in the product remained relatively low at different reaction temperatures. The major product in the reactions were toluene, ethylbenzene, m-xylene, and p-xylene followed by some minor products such as 1,3,5-trimethylbenzene and isopropyl benzene. The concentration of toluene significantly increased with increase in temperature, roughly from 18% at 350 °C and to about 35% at 360 °C and 375 °C, indicating that higher temperature favors the formation of toluene. Similar trends were observed for xylenes and ethylbenzene. This is in accordance with other relevant literatures, which demonstrated the role of temperature and catalyst concentration during the decarboxylation of fatty acids such as oleic acid, linoleic acid, stearic acid, palmitic acid, and linolenic acid.67 Yang et al.68 reported that an increase in temperature promotes α/β scission of the hydrocarbon backbone, which is directly correlated with a higher degree of aromatization and enhanced deoxygenation rates at temperatures greater than 360 °C found in our studies.
 |
| | Fig. 6 Distribution of aromatic hydrocarbons produced as a result of thermocatalytic deoxygenation followed by cyclization and aromatization of OA under different reaction temperatures. The products were analyzed using GC-MS. Reaction conditions: catalyst = 7.5 wt% MoOx/HZSM-5; water:feed ratio = 4:1; τ = 4 h. | |
3.1.3 1H-NMR analysis of deoxygenated products. As the functional group vibrations seen in IR spectra of deoxygenated liquid products strongly indicated the presence of aromatic hydrocarbons, the deoxygenated products were analyzed by 1H-NMR spectroscopy, and the relative abundance of protons associated with different chemical functionalities were assessed. Broadly, protons corresponding to methyl group of aliphatic hydrocarbons (resonating in 0.5–0.8 ppm region of 1H NMR), methylene (–CH2–) group of aliphatic hydrocarbons (resonating in 1.0–1.5 ppm), benzylic (Ar–CH2–) group of alkyl aromatic hydrocarbons (resonating in 2.2–2.7 ppm), vinylic (CC) groups (resonating in 4.5–6.5 ppm region) and aromatic (resonating in 6.5–8 ppm region) groups were taken into consideration. The integral ratios of the protons corresponding to these moieties/functional groups were calculated to assess the transformation of aliphatic moieties of oleic acid into aromatic moieties. A typical 1H-NMR spectrum of OA is presented in Fig. S5, where the 1H-NMR signals for different moieties/functional groups have been attributed as follows: protons attached to vinylic (CC) group resonated at 5.34 ppm; protons associated with methylene group (–CH2) resonated in 1.25–1.62 ppm region; and the protons associated with terminal methyl group (–CH3) resonated at around 0.87 ppm. The 1H-NMR spectrum of the deoxygenated product of OA obtained under maximized operating conditions is shown in Fig. 7. The integral ratio of protons associated with aromatic, benzylic, and aliphatic functionality/moiety in the product of OA is 2.5:2.78:1, which is commensurate of substantially aromatic compounds.
 |
| | Fig. 7 1H-NMR spectra of decarboxylated product (reaction conditions: T = 360 °C, water:feed ratio = 4:1; τ = 4 h). | |
A comparison of the integral ratios of protons resonating in aromatic (A), vinylic (B), benzylic/allylic (C), and methyl and methylene regions (D) in the 1H-NMR spectra of OA and its product are presented in Fig. S6. The deoxygenated product of OA contained high integral ratio for protons resonating in benzylic and allylic region of the 1H-NMR spectrum but had negligible vinylic protons; signifying that the protons resonating in the allylic and benzylic regions of the 1H NMR spectrum of the deoxygenated product were primarily benzylic protons (the protons in the alkyl substituent of benzene). It was also observed that the deoxygenated product had significantly lower integral ratio for protons resonating in methylene and methyl (–CH2– and CH3) region of the 1H-NMR spectrum, suggesting that the alkyl substituents were short-chain molecules.
Further analysis of splitting patterns and position of 1H NMR signals in Fig. 7 of deoxygenated product further highlighted distinctive triplet signal at δ = 0.97 ppm; attributed to the protons associated with terminal methyl groups of short-chain alkyl groups. Multiplets in δ = 6.9–7.3 ppm indicated the presence of substituted benzene, whereas singlets at δ = 7.37, 7.11, and 7.09 ppm indicated the presence of symmetrically substituted (2,4-disubstituted and 1,3,5-trisubstituted) benzene. Singlets at δ = 2.28, 2.32 and 2.37 ppm were indicative of methyl groups attached to benzene, suggesting methyl substituted benzene like toluene, xylenes, and mesitylene. Multiplets in δ = 2.24 to δ = 2.68 ppm connoted that some alkyl substituents contain two or more carbon atoms (e.g. ethyl, n-propyl, isopropyl, etc.). Comparison of the 1H-NMR of the deoxygenated product of OA with that of a simulated/predicted 1H-NMR spectrum for a mixture of aromatic hydrocarbons containing benzene, toluene, xylene, and ethylbenzene obtained by using an open access tool (https://nmrdb.org) indicates identical integral ratios and 1H-NMR signal splitting patterns; corroborating that the deoxygenated product of OA is the mixture of alkyl aromatic compounds where the alkyl groups are primarily methyl, ethyl, and propyl. The predicted 1H-NMR spectrum, for the mixture of aromatic hydrocarbons is shown in Fig. S7.
A comparison of the 1H-NMR spectrum of the deoxygenated product of OA obtained by using HZSM-5 and 7.5% MoOx/HZSM-5 as catalyst (demonstrated in Fig. 8) signifies the role of MoOx to promote tandem deoxygenation and aromatization of OA. The two products compared in Fig. 8 were obtained under identical reaction conditions (T = 360 °C, τ = 4 h, and water:feed ratio = 4:1). The 1H-NMR spectrum of the product using 7.5% MoOx/HZSM-5 as catalyst (Fig. 8(b)) signifies substantially aromatic compounds whereas the 1H-NMR of the product using HZSM-5 as catalyst is commensurate of open-chain hydrocarbons with small proportion of aromatic hydrocarbons (Fig. 8(a)). The relative abundance of protons in aromatic, benzylic/allylic, and aliphatic regions in the product of HZSM-5 catalyst is 0.09:0.37:1, suggesting that only limited tandem cyclization–aromatization of OA was achieved with HZSM-5 catalyst without Mo loading. On the contrary, the integral ratio of protons in aromatic, benzylic/allylic, and aliphatic regions in the product with 7.5 wt% MoOx/HZSM-5 was found to be 2.54:2.81:1, indicating that remarkable enhancement in aromatization. This is attributed to the presence of MoOx on HZSM-5, which promotes dehydrogenation of aliphatic hydrocarbons and subsequent cyclization to aromatics presumably via Brønsted acid sites within the catalyst channels.
 |
| | Fig. 8 1H-NMR spectra of the deoxygenated product of OA; deoxygenated with (a) HZSM-5 and (b) 7.5% MoOx/HZSM-5. | |
3.2 Tandem decarboxylation–aromatization of linoleic acid, used cooking oil & corn distiller's oil over 7.5 wt% MoOx/HZSM-5
The catalytic activity of 7.5 wt% MoOx/HZSM-5 was further evaluated for deoxygenation and aromatization of linoleic acid (LA), and real-world feedstocks including Used Cooking Oil (UCO) and Corn Distiller's Oil (CDO), which primarily comprise of LA. The free fatty acid fraction, predominantly oleic acid, constituted approximately 13% of the total lipid composition in both UCO and CDO. A detailed composition for UCO and CDO has been provided in Fig. S8. The preferred reaction parameters obtained from parametric evaluation using OA (T = 360 °C, τ = 4 h, water:feed ratio = 4:1, catalyst = 7.5 wt% MoOx/HZSM-5) were used for evaluation of deoxygenation, cyclization, and aromatization efficacy of the catalyst with different feedstocks.
3.2.1 Deoxygenation of LA, UCO and CDO: ATR-FTIR. The absorption bands corresponding to CO at 1710 cm−1 present in OA (vide supra) were shifted to 1743 cm−1, confirming the presence of esters (R–CO–OR) in UCO and CDO. In the IR spectra of the liquid products obtained from OA, UCO and CDO, remarkable reduction in the intensity of CO stretching signal is observed (Fig. 9(b), (d) and (f), respectively), which indicates the outstanding efficacy of 7.5 wt% MoOx/HZSM-5 catalyst for deoxygenation of fatty acids and fatty esters. Additionally, the IR band at 3005 cm−1 due to C–H stretching vibrations attributing to CC were significantly reduced and shifted to 3028 cm−1, corresponding to C–H aromatic stretching vibrations. C–H wag attributed to aromatic and alkyl aromatics was predominantly visible in the fingerprint region from 860 cm−1 and 770 cm−1, further validating the tandem deoxygenation–aromatization chemistry during the run.
 |
| | Fig. 9 ATR-FTIR spectra of feedstocks and their deoxygenated products under preferred reaction conditions: (a) LA, and (b) its deoxygenated product P-1; (c) UCO, and (d) deoxygenated product P-2; (e) CDO, and (f) its deoxygenated product P-3. | |
3.2.2 GC-MS analysis of liquid products. Fig. 10 highlights the major products identified and quantified in the products of LA, UCO and CDO under the mentioned reaction conditions, which, based on the results of parametric evaluation for conversion of OA, are considered to be appropriate for maximizing liquid yield and deoxygenation of the product. The major compounds in the liquid product of LA, UCO, and CDO consisted of benzene, toluene, m-xylene, p-xylene, ethyl toluene, and mesitylene. A representative chromatogram for the GC-MS analysis has been presented in Fig. S10. The total yield of aromatic hydrocarbons was 78%, which was substantially higher and achieved under lower temperature conditions than those reported with rapeseed oil (22.5–26 wt%),59,62 camelina seed oil (6 wt%),59,61 soybean oil (24–25 wt%)59,61 and palm oil (22 wt%).59 These results corroborate the initial identification of the chemical functionalities analyzed by 1H-NMR and FTIR (vide supra). In all cases, using LA, UCO, and CDO as feedstock, the combined mass fraction of benzene, toluene, and xylenes (BTX) was greater than ca. 60%. In Fig. 10, others fraction constituted a non-negligible portion of the liquid product, and we anticipate that this pool comprises a mixture of partially deoxygenated and re-functionalized species originating from the distinct lipid profiles of CDO, UCO, and the model fatty acid feedstocks. It can be speculated that for all three feeds, residual oxygenates such as esters, ketones, and short-chain carboxylic acids could persist when decarboxylation and decarbonylation pathways remain incomplete, especially for heavier or more sterically hindered components. In addition, we anticipate that oligomeric and lightly cracked species derived from C18 unsaturated chains (e.g., dimers, cyclic oligomers, and branched aliphatic structures) may populate this category, reflecting secondary condensation and oligomerization reactions on the zeolite acid sites.
 |
| | Fig. 10 GC-MS analyses of decarboxylated products obtained from (a) linoleic acid, (b) used cooking oil and (c) corn distiller's oil; reaction conditions: 7.5 wt% Mo/HZSM-5 = 1.47 g, T = 360 °C, τ = 4 h. | |
Liquid yields of ca. 82%, ca. 75% and ca. 61% were reported on mass basis for products obtained with LA, UCO and CDO. The reduction in the total liquid yield is attributed to the chemical structure of these feedstocks, which contain a bulky aliphatic backbone (–CH2–CH–CH2) attached with three fatty acids per mole. As water is used during the reaction, hydrolysis of these feedstocks results in formation of three moles of free fatty acids and glycerol per mole of feedstock. The glycerol formed doesn't take part in the deoxygenation and aromatization chemistry, and thus results in overall yield loss on mass basis. This finding was further supported by analyzing the feedstock and hydrolyzed product without deoxygenation and aromatization using 1H-NMR spectroscopy (Fig. 11). A substantial decrease in the integral area corresponding to the proton associated with the acyl group in the feedstock (splitted doublets between δ 4.3 and 4.35, followed by quintet at δ 4.15) was observed in the hydrolyzed feedstock, confirming the absence of aliphatic backbone and generation of glycerol as a side product. In addition, 3 moles of CO2 are also removed per mol of hydrolyzed fatty acid derivatives upon deoxygenation, leading to an overall mass reduction of ∼30%.
 |
| | Fig. 11 (a) 1HNMR of CDO, highlighting the proton signals corresponding to the acyl group present in the aliphatic backbone; (b) 1HNMR of CDO after hydrolysis; decrease in the integral area of protons associated with acyl group is visible. | |
The gaseous product obtained with these feedstocks were also analyzed using GC-TCD, which majorly comprised of CO, CO2, H2 and short chain alkenes (![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) C5) (Fig. S9). The production of gaseous products during decarboxylation and aromatization chemistry arose from distinct yet interconnected reaction pathways. Decarboxylation generated CO2 through the elimination of carboxyl groups (–COOH), while CO could have formed via secondary processes such as partial oxidation or thermal decomposition of intermediate carbonyl species. Concurrently, aromatization chemistry could be attributed to H2 evolution through dehydrogenation steps required to establish stable aromatic systems, and short-chain alkenes (C7) emerged as fragmentation products from catalytic cracking of larger molecules, side-chain elimination from aromatic intermediates, or β-elimination reactions. The coexistence of CO2, CO, H2, and light alkenes in the gas profile reflected the parallel progression of these processes, with decarboxylation liberating volatile carbon oxides and aromatization producing hydrogen and unsaturated hydrocarbons.
C5) (Fig. S9). The production of gaseous products during decarboxylation and aromatization chemistry arose from distinct yet interconnected reaction pathways. Decarboxylation generated CO2 through the elimination of carboxyl groups (–COOH), while CO could have formed via secondary processes such as partial oxidation or thermal decomposition of intermediate carbonyl species. Concurrently, aromatization chemistry could be attributed to H2 evolution through dehydrogenation steps required to establish stable aromatic systems, and short-chain alkenes (C7) emerged as fragmentation products from catalytic cracking of larger molecules, side-chain elimination from aromatic intermediates, or β-elimination reactions. The coexistence of CO2, CO, H2, and light alkenes in the gas profile reflected the parallel progression of these processes, with decarboxylation liberating volatile carbon oxides and aromatization producing hydrogen and unsaturated hydrocarbons.
3.2.3 1H-NMR analysis of the products obtained from LA, UCO and CDO. 1H-NMR of the liquid products obtained from LA, UCO, and CDO were analyzed for assessment of integral ratios of protons in different chemical environments (aromatic vs. benzylic/allylic vs. methyl and methylene). The relative integral values of protons attributable to aromatic (A), vinylic (B), benzylic and allylic (C), and methyl and methylene (D) regions were assessed, and are summarized in Table 2. In all cases, protons in vinylic region were negligible, and high integral value was observed for aromatic and benzylic protons as compared to aliphatic methyl and methylene protons; suggesting that the products in all cases constitute substantially aromatic compounds. The spectrum in each case exhibited a sharp singlet attributed to hydrogens resonating at δ 7.40 ppm, which confirmed the presence of benzene. Appearance of singlets corresponding to δ 7.37, 7.11, 7.09, 2.4, 2.36, 2.35 and 2.3 ppm followed by multiplets between δ 6.9 and δ 7.2 ppm suggest the presence of toluene, xylenes and similar alkyl substituted benzenes in the liquid mixture. The signals corresponding to vinyl protons in linoleic acid, used cooking oil and CDO at δ 5.34 ppm disappeared in the spectra of their respective decarboxylated products, which is shown in figure (Fig. S11). In control experiments with no metal loading on the HZSM-5 support, 1H-NMR signals corresponding to vinyl group present in the fatty acids/fatty acid moiety were noticed in the liquid product. This is attributable to compromised catalytic activity of HZSM-5 towards aromatization.
Table 2 Relative integral ratios of protons associated with different chemical environment (chemical shift) in the deoxygenated products obtained from different feedstocks
| Product of |
Integral value of protons resonating at different chemical shifts/regions of 1H-NMR spectrum |
| Aromatic (A) |
Vinylic (B) |
Benzylic & allylic (C) |
Methylene and methyl (D) |
| Linoleic acid |
4.42 |
Negligible |
3.75 |
1 |
| Used cooking oil |
0.99 |
Negligible |
0.99 |
1 |
| Corn distiller's oil |
3.04 |
Negligible |
2.96 |
1 |
3.3 Proposed reaction mechanism
Deoxygenation of fatty acids catalyzed by reducible oxides such as MoO3, which can be partially reduced to MoO2, proceeds through initial removal of the carboxyl group and subsequent CO and C–O bond cleavage.69–72 This holds valid for both saturated and unsaturated fatty acids. It is plausible that used cooking oil and corn distiller's oil after hydrolysis also undergo similar reactions, liberating CO2. The absence of long-chain alkanes or olefins in the liquid product indicates that MoOx/HZSM-5 not only facilitates deoxygenation but also actively promotes C–C bond fragmentation, olefin generation, cyclization, and aromatization reactions. Fig. 12 highlights a proposed mechanism pathway for tandem deoxygenation–aromatization of fatty acids and their derivatives, which plausibly follows a series of reaction steps.
 |
| | Fig. 12 Plausible mechanism for tandem decarboxylation–aromatization of fatty acids (oleic acid has been used as a model compound; (A) decarboxylation of the feedstock, (B) C–C bond scission forming an allylic radical, (C) formation of terminal alkene, (D) generation of cationic centers in the presence of Mo/HZSM-5, (E) alternate pathway for allylic radical to produce cis-alkene. | |
In the proposed mechanism scheme, Reaction A involves the catalytic decarboxylation/decarbonylation of fatty acids, whereby CO2 is released, and shorter-chain alkenes such as heptadec-8-ene are generated. This is accompanied by homolytic bond scission at T ≥ 360 °C, giving rise to highly reactive free radical intermediates. Reaction B corresponds to β-scission and isomerization of radical species. Alkyl radicals undergo fragmentation into olefins and smaller radical intermediates, while the unsaturated hydrocarbons generated undergo skeletal rearrangements consistent with Zaitsev's rule, leading to more stable alkene isomers. Reaction C denotes disproportionation and hydrogen abstraction processes, where radical intermediates are quenched to form equimolar amounts of an alkane and an alkene. These pathways further contribute to the olefinic pool available for downstream transformations. Reaction D highlights the crucial cyclization of unsaturated intermediates as radical species undergo 1–6 endo cyclization or 1–6 exo cyclization reactions. The resulting cyclic intermediates, upon subsequent abstraction and stabilization, yield cycloalkane structures. Reaction E represents the dehydrogenation of cycloalkanes to stable aromatic compounds, a transformation strongly facilitated by the MoOx/HZSM-5 system. The resulting products predominantly consist of alkyl-substituted aromatic hydrocarbons, including toluene and xylenes. Product characterization by GC-FID and 1H-NMR supports the predominance of BTX-type aromatics while simultaneously confirming the absence of oxygenated intermediates.
Additionally, the environment provided by MoOx/HZSM-5 promotes secondary transformations such as alkylbenzene disproportionation, whereby toluene is converted to xylenes and mesitylene under the prevailing reaction conditions, as mentioned in other studies with ZSM based catalyst.73 Toluene produced via pathways shown in Fig. S12 shows interaction with another toluene molecule to produce xylene (specifically para-xylene) and benzene. This is also supported with the GC-MS results obtained in the previous section, where m-xylene and p-xylene are formed in higher amounts as compared to o-xylene. Taken together, these results highlight the dual functionality of MoOx/HZSM-5 in promoting both deoxygenation and hydrocarbon skeletal rearrangement, providing experimental evidence that consolidates the mechanistic scheme in Fig. 12 and establishes aromatization as the dominant terminal reaction route.
4. Catalyst characterization
4.1 BET model for specific surface area and BJH model for pore size & volume
In order to obtain insights into the catalyst structure and any changes that might have occurred in the course of chemical reactions, fresh and spent MoOx/HZSM-5 were characterized by various analytical techniques. Table 3 presents the specific surface area, pore volume and pore size of different types of fresh as well as spent catalyst. A small increase in the specific surface area of the impregnated catalyst is observed, when compared to non-impregnated catalyst support. During catalyst preparation by impregnation method, once the HZSM-5 pores are filled with MoOx, it can be deduced that the residual MoOx species tend to agglomerate on the support surface, thereby increasing the surface area. This is an intriguing observation, which has also been reported by other studies.74 However, after the threshold is reached, a further increase in loading of the metal on the catalyst surface results in agglomeration, thereby not affecting the surface area. The pore volumes of the fresh catalyst were found higher than the reported catalyst systems with HZSM-5 doped with other transition metals evaluated.11
Table 3 Specific surface area of fresh and spent catalysts using BET model
| Catalyst |
S.S.A (m2 g−1) |
BJH pore volume (cm3 g−1) |
BJH pore size (Å) |
| Pure HZSM-5 (calcined) |
398.4 |
0.295 |
86.2 |
| Fresh catalyst (5% MoOx/HZSM-5) |
410.2 |
0.310 |
83.5 |
| Fresh catalyst (7.5% MoOx/HZSM-5) |
445.6 |
0.304 |
81.3 |
| Fresh catalyst (10% MoOx/HZSM-5) |
432.6 |
0.280 |
82.4 |
| Spent catalyst (5% MoOx/HZSM-5) |
378.3 |
0.278 |
79.7 |
| Spent catalyst (7.5% MoOx/HZSM-5) |
412.3 |
0.27 |
76.5 |
| Spent catalyst (10% MoOx/HZSM-5) |
401.2 |
0.256 |
74.9 |
4.2 Scanning electron microscopy (SEM)
Scanning electron microscopy (SEM) analysis of the MoOx/HZSM-5 catalysts were carried out to study the dispersion of MoOx species on the zeolite surface under varying loadings on HZSM-5. Similar morphologies of fresh and spent catalyst under preferred reaction condition were observed (Fig. 13). Spherical agglomerates of MoOx/HZSM-5 with less than 20 µm average particle size can be seen. Agglomeration of the catalyst is further attributed to provide secondary porosity to the catalyst surface, resulting in formation of mesopores.75 SEM analysis also suggested carbon-rod like/filamentous coke structures of carbon formed on the surface of spent MoOx/HZSM-5 (Fig. 13(d)), indicating coke formation during the reaction.
 |
| | Fig. 13 Scanning electron microscopy images of (a) H-ZSM5, (b) 7.5% MoOx/HZSM-5 & spent 7.5% MoOx/HZSM-5 catalyst (c and d). The formation of carbon-rod like structure in (d) suggest catalyst deactivation. | |
4.3 NH3-TPD
The total acidity of the calcined HZSM-5 and MoOx/HZSM-5 catalysts were investigated by NH3-TPD. The TPD profiles, shown in Fig. 14, reveal distinct features corresponding to the distribution of acidic sites for each catalyst. For calcined HZSM-5, the NH3-TPD trace exhibits two prominent desorption peaks: a low-temperature peak centered around 200 °C, attributed to NH3 desorption from weak acid sites, and a high-temperature peak near 400–450 °C, which arises from strong acid sites, located within the zeolite framework. The presence of both weak and strong acid sites in the parent zeolite was consistent with classic reports on ZSM-5 acidity profiles. The total number of weak and strong acid sites, peak temperatures and total acid amount are listed in Table 4.
 |
| | Fig. 14 NH3-TPD results of HZSM-5 parent and various loadings of MoOx/HZSM-5. | |
Table 4 NH3-TPD results of HZSM-5 parent and 7.5 wt% MoOx/HZSM-5
| Catalyst |
Total acid sites (mmol g−1) |
Peak temperature |
Distribution of different acid sites (mmol g−1) |
| Weak |
Strong |
Weak |
Strong |
| Calcined HZSM-5 |
0.33 |
210.5 |
433 |
0.312 |
0.017 |
| 5 wt% MoOx/HZSM-5 |
0.24 |
198.2 |
462.1 |
0.236 |
0.004 |
| 7.5 wt% MoOx/HZSM-5 |
0.29 |
194.9 |
456.7 |
0.287 |
0.003 |
| 10 wt% MoOx/HZSM-5 |
0.26 |
205.4 |
466.4 |
0.254 |
0.006 |
Upon impregnation with Mo, the NH3-TPD profile demonstrates a marked reduction in the intensity of the high-temperature peak, signifying a loss of strong acid sites. Incorporation of Mo into HZSM-5 results in a notable decrease in the total number of acid sites, with the most significant reduction observed at 5 wt% Mo loading. The persistence of weak acid sites associated with dispersed MoOx may also contribute to aromatization through facilitating hydride abstraction and carbenium ion stabilization, albeit less efficiently than strong acid sites. Furthermore, a reduction in overall acid site density, particularly of strong acid sites, carries the ancillary benefit of suppressing undesirable side reactions such as excessive cracking, coke precursor formation, and secondary oligomerization, all of which compete with the desired aromatization pathway and compromise catalyst longevity.52 At 7.5 wt% Mo loading, a partial restoration of total acid sites is observed compared to 5 wt%, suggesting an optimum Mo content where both acidity and acid strength are balanced. In Fig. S13, a NH3-TPD analysis for 2.5 wt% MoOx/HZSM-5 catalyst can be seen. It is visible that the reduction in the peak corresponding to the strong acid sites starts at a lower loading, suggesting that during impregnation and calcination, Mo species can potentially migrate into the zeolite channels and substitute the Brønsted protons and thereby reducing the number of accessible acid sites. Furthermore, the introduction of the Mo species on the zeolite can initiate and accelerate the dehydration of strong acid sites within the zeolite channels, resulting in lower strong acid sites. These trends align closely with literature findings reported by Bao et al. wherein progressive Mo loading leads to a partial replacement of protons by MoOx species, diminishing overall strong acid sites but enhancing weak/medium acid sites.76 In parallel, the area associated with weak acid sites remains similar, likely due to the generation of additional acidic centers resulting from Mo species dispersed in or on the surface of the zeolite.
4.4 Fourier transform infrared spectroscopy
Strong bands around 1030–1060 cm−1 are attributed to asymmetric stretching of X–O–X bonds, where X represents Si or Al in the tetrahedral framework. A shoulder around 1220 cm−1 is also observed for the same stretching. The band at 790 cm−1 represents symmetrical stretching vibrations of the X–O bonds in the Si(Al)O4 tetrahedron and is in accordance with studies reporting the crystalline structure of MoOx/HZSM-5.77 These characteristic bands confirmed the formation of MFI zeolite (HZSM-5) structure. Infrared spectroscopy of the fresh MoOx/HZSM-5 did not indicate any functional groups or impurities other than the lattice vibrations of the zeolites, which are a result of tetrahedron and bending vibrations (Fig. S14). In spent HZSM-5, the functional group corresponding to the residual product/unconverted reactants can be seen around 1707 cm−1 (CO stretching) and 2920 cm−1 (C–H stretching vibrations), suggested the deactivation of the catalyst due to pore blockage.
4.5 X-ray diffraction and impact of calcination temperature on physicochemical properties of zeolite
While differences in calcination temperature can, in principle, influence the physicochemical properties of HZSM-5 support, it has been well established in the literature that the critical threshold temperature for framework degradation and changes in physicochemical properties lies considerably above the temperature employed in the present work. Specifically, dealumination and a concomitant reduction in zeolite crystallinity become significant at calcination temperatures at or above 750 °C.78,79 X-Ray Diffraction (XRD) patterns of the fresh, spent MoOx/HZSM-5 and the substrate (HZSM-5) were investigated to study differences in the zeolite framework. As seen in Fig. 15, it is notable that XRD peaks located between 20° < 2θ < 25° were intrinsic zeolite channel inner structures,80 which were present in all catalysts evaluated. The peaks were observed at 2θ = 23.42°, 24.07°, 27.05°, 30.19° and 45.64°, which are the characteristic diffraction angle for HZSM-5. These findings show minimal changes due to the impregnation of MoOx in the HZSM-5 framework. The calcination temperature of 650 °C employed in the present study is well within the range commonly reported in the Mo/HZSM-5 literature and below the threshold at which structural degradation becomes problematic. Furthermore, studies examining the effect of calcination temperatures between 500 and 650 °C on Mo/HZSM-5 have demonstrated that catalytic performance and Mo dispersion are sensitive to temperature within this window, with 600–650 °C representing a regime of acceptable thermal treatment for catalyst preparation.79 The XRD signal characteristic of crystalline MoO3 which typically appears at 2θ values of approximately 33.5° was not observed, which indicates that the loaded MoOx was well dispersed on the surface during impregnation and calcination of the catalyst.77,80,81
 |
| | Fig. 15 X-ray diffraction patterns of MoOx/HZSM-5 with various metal loadings; (a) fresh HZSM-5, (b) 7.5 wt% fresh Mo/HZSM-5, (c) 7.5 wt% spent Mo/HZSM-5, (d) 10 wt% fresh Mo/HZSM-5, (e) 10 wt% spent Mo/HZSM-5, (f) 7.5 wt% regenerated Mo/HZSM-5 and (g) 10 wt% regenerated Mo/HZSM-5. | |
4.6 XPS analysis
The XPS analysis was performed to analyze the surface composition and electronic state of Mo(VI) in the fresh and spent catalyst under optimal catalyst loadings (7.5 wt%). The XPS survey spectra obtained consisted of characteristic peaks for O, C, Mo, Si and Al (Fig. 16). The XPS fitting was carried out with the FWHM kept constant across all samples. The presence of carbon can be attributed to residual impurities during sample preparation and coke formation in fresh and spent catalyst respectively. As seen in Fig. 17, the high-resolution XPS profile of Mo(VI) spectra for fresh and spent catalysts exhibited peaks attributed to Mo(VI) 3d transitions, which consisted of two spin orbit split contributions; 3d5/2 and 3d3/2, separated by 3 eV and intensity ratio of 3:2.
 |
| | Fig. 16 Survey spectrum collected from fresh 7.5% MoOx/HZSM-5 catalyst. | |
 |
| | Fig. 17 XPS of fresh catalyst (a) 7.5% MoOx/HZSM-5 and spent catalyst (b) 7.5% MoOx/HZSM-5370 °C, 4 h. | |
Mo(VI) 3d5/2 and Mo 3d3/2 components were located at 232.69 and 235.81 eV respectively. There is a visible shift towards higher binding energy in Mo(VI) 3d3/2, compared to the BE value of Mo(VI) 3d5/2 in bulk MoO3, which can be explained based on strong metal–support interaction. In case of the spent catalyst, the observable shift in the spin–orbit doublets likely reflect a lower oxidation state. However, it was evident from the XPS data that both fresh and spent catalyst samples contained significant fraction of Mo6+ species in the form of MoO3. To complement the surface-sensitive XPS analysis and obtain bulk elemental composition of the catalysts, inductively coupled plasma optical emission spectrometry (ICP-OES) was employed. The measured Mo concentrations for the 7.5 wt% and 10 wt% nominal loading catalysts were 74800 mg kg−1 (7.48 wt%) and 98700 mg kg−1 (9.87 wt%), respectively, confirming successful incorporation of molybdenum onto the HZSM-5 support via incipient wetness impregnation. (Fig. S15) The close agreement between theoretical and measured Mo loadings, combined with the predominance of Mo6+ species identified by XPS, is consistent with the formation of well-dispersed MoO3 surface domains rather than bulk-phase segregation.
5. Conclusions and future directives
The catalyst prepared by loading MoOx on H/ZSM-5 demonstrated remarkable activity towards tandem deoxygenation and aromatization of fatty acids and their derivatives. Systematic evaluation of the effects of catalyst type and reaction parameters demonstrated that the 7.5 wt% MoOx/HZSM-5 catalyst was effective in chemical transformation of fatty acids and triglycerides to aromatic hydrocarbons under subcritical hydrothermal conditions. Analysis of 1H-NMR spectra of the products indicated that benzene, toluene, and xylenes were the major compounds in the product, which was corroborated by GC-MS analysis; the BTX composition being about 65% in the product. The remarkable results in chemical transformation of triglycerides and fatty acids to aromatic platform compounds was achieved in continuous, flow-through reactor without the addition of external H2 for about 84 hours. The fundamental knowledge gained from these studies associated with monounsaturated fatty acids was applied to valorization of real-world bio-based feedstocks such as CDO and used cooking oil. With oleic acid and linoleic acid as the model compounds, the aromatic yields at T = 350 °C were severely affected, which suggests that higher temperatures are required to facilitate the tandem deoxygenation–aromatization. The highest proportion of monocyclic aromatics and BTEX in recovered liquid was 82% and 75%, respectively. With a decrease in the metal loading from 10% to 5%, significant reduction in the extent of decarboxylation and saturation was observed. These results direct future research and development to utilize these feedstocks towards production of bio-based aromatics. Furthermore, achieving higher selectivity in the production of BTX mixture would require addition catalyst modifications by varying the Si:Al ratio to alter the acidity of the solid acid support, followed by alternate methods for catalyst preparation. A life cycle assessment (LCA) with energy and mass balance, environmental impacts, etc. would result in a deeper understanding of the scalability of the process. The pathway described for production of BTX under given reaction conditions without hydrotreating, along with the achieved product yields during the process represents significant improvements over what has been reported in this research domain, thereby serving as a cornerstone for future research endeavours.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5se01600b.
References
- E. A. R. Zuiderveen, C. Caldeira, T. Vries, N. J. Schenk, M. A. J. Huijbregts and S. Sala, et al., Evaluating the Environmental Sustainability of Alternative Ways to Produce Benzene, Toluene, and Xylene, ACS Sustain. Chem. Eng., 2024, 12(13), 5092–5104, DOI:10.1021/acssuschemeng.3c06996.
- Global Benzene-Toluene-Xylene (BTX) Market Extends at a Healthy CAGR of 3.80% by 2031, https://straitsresearch.com/press-release/global-benzene-toluene-xylene-market-trends Search PubMed.
- H. Jiang, Z. Li, Y. Sun, S. Jiang and J. Tian, Optimization of the Countercurrent Continuous Reforming Process Based on Equation-Oriented Modeling and the SQP Algorithm, ACS Omega, 2022, 7(2), 1757–1771, DOI:10.1021/acsomega.1c04651.
- R. A. Moreau and M. E. Hums, Corn Oil and Distillers Corn Oil, in Bailey's Industrial Oil and Fat Products, Wiley, 2020, pp. 1–27, DOI:10.1002/047167849X.bio007.pub2.
- Q. Tian, K. Qiao, F. Zhou, K. Chen, T. Wang and J. Fu, et al., Direct Production of Aviation Fuel Range Hydrocarbons and Aromatics from Oleic Acid without an Added Hydrogen Donor, Energy Fuels, 2016, 30(9), 7291–7297, DOI:10.1021/acs.energyfuels.6b00978.
- M. Carrasco-Suárez, A. G. Romero-Izquierdo, C. Gutiérrez-Antonio, F. I. Gómez-Castro and S. Hernández, Production of renewable aviation fuel by waste cooking oil processing in a biorefinery scheme: intensification of the purification zone, Chem. Eng. Process., 2022, 181, 109103, DOI:10.1016/j.cep.2022.109103.
- B. H. H. Goh, C. T. Chong, Y. Ge, H. C. Ong, J. H. Ng and B. Tian, et al., Progress in utilisation of waste cooking oil for sustainable biodiesel and biojet fuel production, Energy Convers. Manage., 2020, 223, 113296, DOI:10.1016/J.ENCONMAN.2020.113296.
- R. K. Singh, D. Panda and S. Singh, Deoxygenation Pathways for Sustainable Aviation Fuel from Used Cooking Oil: A Review on Catalyst and Operating Parameters, J. Hazard., Toxic Radioact. Waste, 2024, 28(4), 1327, DOI:10.1061/JHTRBP.HZENG-1327.
- ASTM D6751-20A Specification for Biodiesel Fuel Blend Stock (B100) for Middle Distillate Fuels, ASTM International, DOI:10.1520/d6751-20a.
- T. Michler, B. Niethammer, C. Fuchs, O. Toedter, U. Arnold and T. Koch, et al., Further Development of Gasoline from the bioliq® Process with Focus on Particulate and Hydrocarbon Emissions, Fuels, 2023, 4(2), 205–220, DOI:10.3390/fuels4020013.
- S. He, F. G. H. Klein, T. S. Kramer, A. Chandel, Z. Tegudeer and A. Heeres, et al., Catalytic Conversion of Free Fatty Acids to Bio-Based Aromatics: A Model Investigation Using Oleic Acid and an H-ZSM-5/Al 2 O 3 Catalyst, ACS Sustain. Chem. Eng., 2021, 9(3), 1128–1141, DOI:10.1021/acssuschemeng.0c06181.
- Plastics Today NewsFeed, Anellotech demonstrates Plas-TCat process to make chem feed from plastic waste, Focus Catal., 2022, 2022(11), 6, DOI:10.1016/j.focat.2022.10.029.
- N. Belinskaya, E. Ivanchina, E. Ivashkina and G. Silko, Effect of Feed Composition Changing at Naphtha Catalytic Reforming Unit Due to Involvement of Gasoline Fraction Obtained by Diesel Fuels Hydrodewaxing into the Processing, Procedia Chem., 2014, 10, 267–270, DOI:10.1016/j.proche.2014.10.044.
- F. Wang, T. S. Kramer, B. Yan, L. Zhu, Y. Zhu and A. Heeres, et al., Enhanced Bio-BTX Formation by Catalytic Pyrolysis of Glycerol with In Situ Produced Toluene as the Cofeed, ACS Sustain. Chem. Eng., 2024, 12(15), 5731–5737, DOI:10.1021/acssuschemeng.4c00451.
- J. H. Miller, A. M. Al, J. Stunkel, A. J. Koehler, M. R. Wiatrowski and R. L. McCormick, et al., Catalytic upgrading of wet waste-derived carboxylic acids to sustainable aviation fuel and chemical feedstocks, EES Catal., 2024, 2(5), 1111–1125, 10.1039/D4EY00087K.
- A. Srifa, K. Faungnawakij, V. Itthibenchapong, N. Viriya-empikul, T. Charinpanitkul and S. Assabumrungrat, Production
of bio-hydrogenated diesel by catalytic hydrotreating of palm oil over NiMoS2/γ-Al2O3 catalyst, Bioresour. Technol., 2014, 158, 81–90, DOI:10.1016/j.biortech.2014.01.100.
- A. Afshar Taromi and S. Kaliaguine, Green diesel production via continuous hydrotreatment of triglycerides over mesostructured γ-alumina supported NiMo/CoMo catalysts, Fuel Process. Technol., 2018, 171, 20–30, DOI:10.1016/j.fuproc.2017.10.024.
- P. Arora, E. L. Grennfelt, L. Olsson and D. Creaser, Kinetic study of hydrodeoxygenation of stearic acid as model compound for renewable oils, Chem. Eng. J., 2019, 364, 376–389, DOI:10.1016/j.cej.2019.01.134.
- P. A. Nikulshin, V. A. Salnikov, A. N. Varakin and V. M. Kogan, The use of CoMoS catalysts supported on carbon-coated alumina for hydrodeoxygenation of guaiacol and oleic acid, Catal. Today, 2016, 271, 45–55, DOI:10.1016/j.cattod.2015.07.032.
- P. Arora, E. L. Grennfelt, L. Olsson and D. Creaser, Kinetic study of hydrodeoxygenation of stearic acid as model compound for renewable oils, Chem. Eng. J., 2019, 364, 376–389, DOI:10.1016/j.cej.2019.01.134.
- P. A. Nikulshin, V. A. Salnikov, A. N. Varakin and V. M. Kogan, The use of CoMoS catalysts supported on carbon-coated alumina for hydrodeoxygenation of guaiacol and oleic acid, Catal. Today, 2016, 271, 45–55, DOI:10.1016/j.cattod.2015.07.032.
- I. H. Choi, J. S. Lee, C. U. Kim, T. W. Kim, K. Y. Lee and K. R. Hwang, Production of bio-jet fuel range alkanes from catalytic deoxygenation of Jatropha fatty acids on a WOx/Pt/TiO2 catalyst, Fuel, 2018, 215, 675–685, DOI:10.1016/j.fuel.2017.11.094.
- K. W. Jeon, J. O. Shim, J. W. Cho, W. J. Jang, H. S. Na and H. M. Kim, et al., Synthesis and characterization of Pt-, Pd-, and Ru-promoted Ni–Ce0.6Zr0.4O2 catalysts for efficient biodiesel production by deoxygenation of oleic acid, Fuel, 2019, 236, 928–933, DOI:10.1016/j.fuel.2018.09.078.
- C. Ochoa-Hernández, Y. Yang, P. Pizarro, V. A. de la Peña O'Shea, J. M. Coronado and D. P. Serrano, Hydrocarbons production through hydrotreating of methyl esters over Ni and Co supported on SBA-15 and Al-SBA-15, Catal. Today, 2013, 210, 81–88, DOI:10.1016/j.cattod.2012.12.002.
- M. Z. Hossain, M. B. I. Chowdhury, A. K. Jhawar, W. Z. Xu, M. C. Biesinger and P. A. Charpentier, Continuous Hydrothermal Decarboxylation of Fatty Acids and Their Derivatives into Liquid Hydrocarbons Using Mo/Al 2 O 3 Catalyst, ACS Omega, 2018, 3(6), 7046–7060, DOI:10.1021/acsomega.8b00562.
- C. Miao, O. Marin-Flores, S. D. Davidson, T. Li, T. Dong and D. Gao, et al., Hydrothermal catalytic deoxygenation of palmitic acid over nickel catalyst, Fuel, 2016, 166, 302–308, DOI:10.1016/j.fuel.2015.10.120.
- K. W. Jeon, H. S. Na, Y. L. Lee, S. Y. Ahn, K. J. Kim and J. O. Shim, et al., Catalytic deoxygenation of oleic acid over a Ni-CeZrO2 catalyst, Fuel, 2019, 258, 116179, DOI:10.1016/j.fuel.2019.116179.
- K. Jeništová, I. Hachemi, P. Mäki-Arvela, N. Kumar, M. Peurla and L. Čapek, et al., Hydrodeoxygenation of stearic acid and tall oil fatty acids over Ni-alumina catalysts: influence of reaction parameters and kinetic modelling, Chem. Eng. J., 2017, 316, 401–409, DOI:10.1016/j.cej.2017.01.117.
- S. Fangkoch, S. Boonkum, S. Ratchahat, W. Koo-amornpattana, A. Eiad-Ua and W. Kiatkittipong, et al., Solvent-Free Hydrodeoxygenation of Triglycerides to Diesel-like Hydrocarbons over Pt-Decorated MoO 2 Catalysts, ACS Omega, 2020, 5(12), 6956–6966, DOI:10.1021/acsomega.0c00326.
- I. Hachemi and D. Y. Murzin, Kinetic modeling of fatty acid methyl esters and triglycerides hydrodeoxygenation over nickel and palladium catalysts, Chem. Eng. J., 2018, 334, 2201–2207, DOI:10.1016/j.cej.2017.11.153.
- W. Wang, Z. Qiao, K. Zhang, P. Liu, Y. Yang and K. Wu, Highly selective catalytic hydrodeoxygenation of C aromatic –OH in bio-oil to cycloalkanes on a Ce–Ni–W–B amorphous catalyst, RSC Adv., 2014, 4(70), 37288–37295, 10.1039/C4RA04364B.
- C. Zhao, T. Brück and J. A. Lercher, Catalytic deoxygenation of microalgae oil to green hydrocarbons, Green Chem., 2013, 15(7), 1720, 10.1039/c3gc40558c.
- J. G. A. Pacheco, J. F. Padilha, B. L. Santos, M. R. Santos, D. O. Liborio and L. A. M. Pontes, et al., Hydrogen-free deoxygenation of oleic acid on acidic and basic ZSM-5 and Y-zeolites: products for biofuel and reaction pathways, Catal. Today, 2025, 445, 115094, DOI:10.1016/j.cattod.2024.115094.
- K. Hengst, M. Arend, R. Pfützenreuter and W. F. Hoelderich, Deoxygenation and cracking of free fatty acids over acidic catalysts by single step conversion for the production of diesel fuel and fuel blends, Appl. Catal., B, 2015, 174–175, 383–394, DOI:10.1016/j.apcatb.2015.03.009.
- M. B. I. Chowdhury, M. M. Hossain and P. A. Charpentier, Effect of supercritical water gasification treatment on Ni/La2O3-Al2O3-based catalysts, Appl. Catal., A, 2011, 405(1–2), 84–92, DOI:10.1016/J.APCATA.2011.07.031.
- M. Z. Hossain, M. B. I. Chowdhury, A. K. Jhawar, W. Z. Xu, M. C. Biesinger and P. A. Charpentier, Continuous Hydrothermal Decarboxylation of Fatty Acids and Their Derivatives into Liquid Hydrocarbons Using Mo/Al 2 O 3 Catalyst, ACS Omega, 2018, 3(6), 7046–7060, DOI:10.1021/acsomega.8b00562.
- M. Z. Hossain, A. K. Jhawar, M. B. I. Chowdhury, W. Z. Xu, W. Wu and D. V. Hiscott, et al., Using Subcritical Water for Decarboxylation of Oleic Acid into Fuel-Range Hydrocarbons, Energy Fuels, 2017, 31(4), 4013–4023, DOI:10.1021/acs.energyfuels.6b03418.
- N. Mo and P. E. Savage, Hydrothermal Catalytic Cracking of Fatty Acids with HZSM-5, ACS Sustain. Chem. Eng., 2014, 2(1), 88–94, DOI:10.1021/sc400368n.
- J. G. Na, B. E. Yi, J. K. Han, Y. K. Oh, J. H. Park and T. S. Jung, et al., Deoxygenation of microalgal oil into hydrocarbon with precious metal catalysts: optimization of reaction conditions and supports, Energy, 2012, 47(1), 25–30, DOI:10.1016/j.energy.2012.07.004.
- I. Gilmore and D. Kettner, Bio-based Aromatics Give Pathway to Drop-In 100% SAF, 2023 Search PubMed.
- A. G. Dedov, A. S. Loktev, A. E. Gekhman, T. V. Kosakova, E. A. Isaeva and M. N. Kartasheva, et al., Direct conversion of fatty acid triglycerides into motor fuel components, Theor. Found. Chem. Eng., 2012, 46(5), 556–562, DOI:10.1134/S004057951205003X.
- R. F. Beims, V. Botton, L. Ender, D. R. Scharf, E. L. Simionatto and H. F. Meier, et al., Effect of degree of triglyceride unsaturation on aromatics content in bio-oil, Fuel, 2018, 217, 175–184, DOI:10.1016/j.fuel.2017.12.109.
- K. M. Doll, B. R. Moser and G. Knothe, Decarboxylation of oleic acid using iridium catalysis to form products of increased aromatic content compared to ruthenium systems, Int. J. Sustain. Eng., 2021, 14(6), 2018–2024, DOI:10.1080/19397038.2021.1978589.
- N. Mo, W. Tandar and P. E. Savage, Aromatics from saturated and unsaturated fatty acids via zeolite catalysis in supercritical water, J. Supercrit. Fluids, 2015, 102, 73–79, DOI:10.1016/j.supflu.2015.03.018.
- H. Liu, W. Luo, K. Wang, Y. Wang and H. Yuan, Hierarchical ZSM-5 nanosheets for production of light olefins and aromatics by catalytic cracking of oleic acid, Sustainable Energy Fuels, 2025, 9(1), 152–171, 10.1039/D4SE01167H.
- S. Dixit, A. K. Jhawar, B. Adhikari, M. Trygstad and P. A. Charpentier, Catalytic Upgrading of Oleic Acid to Aromatic Hydrocarbons by Tandem Deoxygenation and Aromatization over MoO 3/HZSM-5, ACS Sustain. Chem. Eng., 2025, 13(41), 17139–17150, DOI:10.1021/acssuschemeng.5c04263.
- T. Prasomsri, M. Shetty, K. Murugappan and Y. Román-Leshkov, Insights into the catalytic activity and surface modification of MoO 3 during the hydrodeoxygenation of lignin-derived model compounds into aromatic hydrocarbons under low hydrogen pressures, Energy Environ. Sci., 2014, 7(8), 2660–2669, 10.1039/C4EE00890A.
- J. Liang, T. Chen, J. Liu, Q. Zhang, W. Peng and Y. Li, et al., Chemoselective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over Ni/MoO2@Mo2CTx catalyst with extraordinary synergic effect, Chem. Eng. J., 2020, 391, 123472, DOI:10.1016/j.cej.2019.123472.
- R. Ding, Y. Wu, Y. Chen, J. Liang, J. Liu and M. Yang, Effective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over MoO 2/CNTs catalyst, Chem. Eng. Sci., 2015, 135, 517–525, DOI:10.1016/j.ces.2014.10.024.
- N. Krobkrong, V. Itthibenchapong, P. Khongpracha and K. Faungnawakij, Deoxygenation of oleic acid under an inert atmosphere using molybdenum oxide-based catalysts, Energy Convers. Manage., 2018, 167, 1–8, DOI:10.1016/j.enconman.2018.04.079.
- S. Janampelli and S. Darbha, Highly efficient Pt-MoOx/ZrO2 catalyst for green diesel production, Catal. Commun., 2019, 125, 70–76, DOI:10.1016/j.catcom.2019.03.027.
- D. B. Lukyanov and T. Vazhnova, Highly selective and stable alkylation of benzene with ethane into ethylbenzene over bifunctional PtH-MFI catalysts, J. Mol. Catal. A:Chem., 2008, 279(1), 128–132, DOI:10.1016/j.molcata.2007.10.016.
- Test Method for Determination of Benzene, Toluene, Ethylbenzene, p/m-Xylene, o-Xylene, C9 and Heavier Aromatics, and Total Aromatics in Finished Gasoline by Gas Chromatography, ASTM International, West Conshohocken, PA, 2021, DOI:10.1520/D5580-21.
- ASTM International, Standard Test Method for Acidity in Aviation Turbine Fuel, 2017, retrieved from https://compass.astm.org/document/?contentCode=ASTM%7CD3242-23%7Cen-US Search PubMed.
- F. Shi, H. Wang, Y. Chen, Y. Lu, D. Hou and C. Liu, et al., Green diesel-like hydrocarbon production by H2-free catalytic deoxygenation of oleic acid via Ni/MgO-Al2O3 catalysts: effect of the metal loading amount, J. Environ. Chem. Eng., 2023, 11(5), 110520, DOI:10.1016/J.JECE.2023.110520.
- M. Z. Hossain, M. B. I. Chowdhury, A. K. Jhawar, W. Z. Xu and P. A. Charpentier, Continuous low pressure decarboxylation of fatty acids to fuel-range hydrocarbons with in situ hydrogen production, Fuel, 2018, 212, 470–478, DOI:10.1016/j.fuel.2017.09.092.
- M. Arumugam, C. K. Goh, Z. Zainal, S. Triwahyono, A. F. Lee and K. Wilson, et al., Hierarchical HZSM-5 for Catalytic Cracking of Oleic Acid to Biofuels, Nanomaterials, 2021, 11(3), 747, DOI:10.3390/nano11030747.
- R. Hilten, R. Speir, J. Kastner and K. C. Das, Production of aromatic green gasoline additives via catalytic pyrolysis of acidulated peanut oil soap stock, Bioresour. Technol., 2011, 102(17), 8288–8294, DOI:10.1016/j.biortech.2011.06.049.
- P. Bielansky, A. Weinert, C. Schönberger and A. Reichhold, Catalytic conversion of vegetable oils in a continuous FCC pilot plant, Fuel Process. Technol., 2011, 92(12), 2305–2311, DOI:10.1016/j.fuproc.2011.07.021.
- X. Zhao, L. Wei, S. Cheng, Y. Huang, Y. Yu and J. Julson, Catalytic cracking of camelina oil for hydrocarbon biofuel over ZSM-5-Zn catalyst, Fuel Process. Technol., 2015, 139, 117–126, DOI:10.1016/j.fuproc.2015.07.033.
- Z. Zheng, J. Wang, Y. Wei, X. Liu, F. Yu and J. Ji, Effect of La-Fe/Si-MCM-41 catalysts and CaO additive on catalytic cracking of soybean oil for biofuel with low aromatics, J. Anal. Appl. Pyrolysis, 2019, 143, 104693, DOI:10.1016/j.jaap.2019.104693.
- R. O. Idem, S. P. R. Katikaneni and N. N. Bakhshi, Catalytic conversion of canola oil to fuels and chemicals: roles of catalyst acidity, basicity and shape selectivity on product distribution, Fuel Process. Technol., 1997, 51(1–2), 101–125, DOI:10.1016/S0378-3820(96)01085-5.
- R. Ramos, A. García, J. A. Botas and D. P. Serrano, Enhanced Production of Aromatic Hydrocarbons by Rapeseed Oil Conversion over Ga and Zn Modified ZSM-5 Catalysts, Ind. Eng. Chem. Res., 2016, 55(50), 12723–12732, DOI:10.1021/acs.iecr.6b03050.
- Z. Ullah, M. A. Bustam and Z. Man, Biodiesel production from waste cooking oil by acidic ionic liquid as a catalyst, Renewable Energy, 2015, 77, 521–526, DOI:10.1016/J.RENENE.2014.12.040.
- C. Kadrmas, M. Khambete, A. Kubátová, E. Kozliak and W. Seames, Optimizing the Production of Renewable Aromatics via Crop Oil Catalytic Cracking, Processes, 2015, 3(2), 222–234, DOI:10.3390/pr3020222.
- E. Santillan-Jimenez and M. Crocker, Catalytic deoxygenation of fatty acids and their derivatives to hydrocarbon fuels via decarboxylation/decarbonylation, J. Chem. Technol. Biotechnol., 2012, 87(8), 1041–1050, DOI:10.1002/jctb.3775.
- M. d. S. B. d. Silva, J. G. L. d. Araujo, J. C. C. V. Bento, A. M. d. Azevedo, C. R. O. Souto and A. S. D. d. Anjos, et al., Nickel-catalyzed reductive decarboxylation of fatty acids for drop-in biofuel production, RSC Adv., 2022, 12(43), 27889–27894, 10.1039/D2RA04057C.
- Y. Yang, Q. Wang, X. Zhang, L. Wang and G. Li, Hydrotreating of C18 fatty acids to hydrocarbons on sulphided NiW/SiO2–Al2O3, Fuel Process. Technol., 2013, 116, 165–174, DOI:10.1016/j.fuproc.2013.05.008.
- R. Ding, Y. Wu, Y. Chen, J. Liang, J. Liu and M. Yang, Effective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over MoO 2/CNTs catalyst, Chem. Eng. Sci., 2015, 135, 517–525, DOI:10.1016/j.ces.2014.10.024.
- J. Liang, T. Chen, J. Liu, Q. Zhang, W. Peng and Y. Li, et al., Chemoselective hydrodeoxygenation of palmitic acid to diesel-like hydrocarbons over Ni/MoO2@Mo2CTx catalyst with extraordinary synergic effect, Chem. Eng. J., 2020, 391, 123472, DOI:10.1016/j.cej.2019.123472.
- I. T. Ghampson, G. Pecchi, J. L. G. Fierro, A. Videla and N. Escalona, Catalytic hydrodeoxygenation of anisole over Re-MoO x/TiO 2 and Re-VO x/TiO 2 catalysts, Appl. Catal., B, 2017, 208, 60–74, DOI:10.1016/j.apcatb.2017.02.047.
- G. H. Liu, Z. M. Zong, Z. Q. Liu, F. J. Liu, Y. Y. Zhang and X. Y. Wei, Solvent-controlled selective hydrodeoxygenation of bio-derived guaiacol to arenes or phenols over a biochar supported Co-doped MoO2 catalyst, Fuel Process. Technol., 2018, 179, 114–123, DOI:10.1016/j.fuproc.2018.05.035.
- M. A. Uguina, J. L. Sotelo and D. P. Serrano, Roles of ZSM-5 modifier agents in selective toluene disproportionation, Can. J. Chem. Eng., 1993, 71(4), 558–563, DOI:10.1002/cjce.5450710408.
- H. Ronduda, M. Zybert, W. Patkowski, K. Sobczak, D. Moszyński and A. Albrecht, et al., On the effect of metal loading on the performance of Co catalysts supported on mixed MgO–La 2 O 3 oxides for ammonia synthesis, RSC Adv., 2022, 12(52), 33876–33888, 10.1039/D2RA06053A.
- B. Golman, Z. Ohashi, K. Shinohara, S. I. Fujita, M. Shimokawabe and M. Arai, NO Adsorption Characteristics of Agglomerated Zeolite Particles, J. Chem. Eng. Jpn., 2002, 35(6), 590–594, DOI:10.1252/jcej.35.590.
- D. Ma, W. Zhang, Y. Shu, X. Liu, Y. Xu and X. Bao, MAS NMR, ESR and TPD studies of Mo/HZSM-5 catalysts: evidence for the migration of molybdenum species into the zeolitic channels, Catal. Lett., 2000, 66(3), 155–160, DOI:10.1023/A:1019099607029.
- R. Li, S. Chong, N. Altaf, Y. Gao, B. Louis and Q. Wang, Synthesis of ZSM-5/Siliceous Zeolite Composites for Improvement of Hydrophobic
Adsorption of Volatile Organic Compounds, Front. Chem., 2019, 16, 7, DOI:10.3389/fchem.2019.00505.
- P. L. Tan, Y. L. Leung, S. Y. Lai and C. T. Au, The effect of calcination temperature on the catalytic performance of 2 wt.% Mo/HZSM-5 in methane aromatization, Appl. Catal., A, 2002, 228(1–2), 115–125, DOI:10.1016/S0926-860X(01)00955-3.
- C. M. Sinnathamb, Effect of Calcination on Mo-Modified Zeolite Catalyst, J. Appl. Sci., 2011, 11(8), 1336–1340, DOI:10.3923/jas.2011.1336.1340.
- B. Li, S. Li, N. Li, H. Chen, W. Zhang and X. Bao, et al., Structure and acidity of Mo/ZSM-5 synthesized by solid state reaction for methane dehydrogenation and aromatization, Microporous Mesoporous Mater., 2006, 88(1–3), 244–253, DOI:10.1016/J.MICROMESO.2005.09.016.
- B. Liu, L. France, C. Wu, Z. Jiang, V. L. Kuznetsov and H. A. Al-Megren, et al., Methanol-to-hydrocarbons conversion over MoO 3/H-ZSM-5 catalysts prepared via lower temperature calcination: a route to tailor the distribution and evolution of promoter Mo species, and their corresponding catalytic properties, Chem. Sci., 2015, 6(9), 5152–5163, 10.1039/C5SC01825K.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  a,
Anil Kumar Jhawarb,
Birendra Adhikarib,
Marcus Trygstadb,
Craig Piaceka and
Paul A. Charpentier*a
a,
Anil Kumar Jhawarb,
Birendra Adhikarib,
Marcus Trygstadb,
Craig Piaceka and
Paul A. Charpentier*a


















