
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Catalytic hydrodeoxygenation of black soldier fly larval lipids and co-processing with vacuum gas oil into biofuel intermediates
Jon
Selimi
 *ab,
Tove A.
Kristensen
ab,
Ziyauddin S.
Qureshi
cd,
Christian P.
Hulteberg
ab and
Omar Y.
Abdelaziz
*ce
*ab,
Tove A.
Kristensen
ab,
Ziyauddin S.
Qureshi
cd,
Christian P.
Hulteberg
ab and
Omar Y.
Abdelaziz
*ce
aDivision of Chemical Engineering, Department of Process and Life Science Engineering, Lund University, Naturvetarvägen 14, SE-221 00 Lund, Sweden. E-mail: jon.selimi@ple.lth.se
bHulteberg Chemistry & Engineering AB, Krusegatan 32, SE-212 25 Malmö, Sweden
cInterdisciplinary Research Center for Refining & Advanced Chemicals, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
dDepartment of Chemistry, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia
eDepartment of Chemical Engineering, King Fahd University of Petroleum & Minerals, Dhahran 31261, Saudi Arabia. E-mail: omar.abdelaziz@kfupm.edu.sa
First published on 15th December 2025
Abstract
The aviation industry's decarbonization requires sustainable aviation fuel (SAF) feedstocks that do not compete with food resources. In this context, black soldier fly larvae (BSFL) lipids represent a non-food, second-generation feedstock with strong potential for drop-in SAF production. This work presents an integrated evaluation of BSFL-derived lipids hydroprocessed over a commercial Ce/La-doped NiMo/Al2O3 catalyst across batch, continuous, and co-processing modes. In batch hydrodeoxygenation (HDO), oxygen was consistently reduced to below the analytical limit of detection, with oil yields averaging 66.3 wt% and reaching a maximum of 72.2 wt%. Maximum kerosene- and diesel-range yields were 37.8 wt% and 29.0 wt%, respectively. Pressure was the dominant factor affecting yields, with temperature–pressure interactions being most significant, while stirring improved performance under mass-transfer-limited conditions. Continuous fixed-bed HDO runs showed that efficient catalyst wetting was achieved at LHSV 0.5 h−1 and H2/oil ≥800 mL mL−1, conditions under which selectivity shifted toward HDO rather than decarboxylation/decarbonylation (deCOx). Co-processing BSFL lipids with vacuum gas oil enhanced hydrogen availability, promoted HDO over deCOx pathways, and yielded high kerosene- and diesel-range fractions, demonstrating the potential for integration of insect-derived lipids into existing refinery infrastructure.
1 Introduction
Since the start of the industrial revolution, energy use in the transportation sector has increased rapidly each year, particularly in commercial passenger and freight air transport.1,2 The demand for aviation fuel is estimated to increase in the next decades, making the need to produce sustainable aviation fuel (SAF) even more necessary to lower the carbon output rate in the atmosphere.3,4 In the short term, global air transport is expected to increase by approximately 5% annually until 2030, according to the European Commission.5 Governmental bodies, particularly the EU, are planning to enforce stricter policies to include SAF as drop-in options for aviation applications. The aim is to reduce reliance on fossil fuels to lower environmental impact from aviation transport. According to the ReFuelEU Aviation initiative, aviation fuel provided by fuel suppliers is required to contain at least 2% SAF starting in 2025 and gradually increase to 70% in 2050.6 Moreover, federal government agencies in the USA have launched a SAF Grand Challenge through a Memorandum of Understanding, as an initiative to accelerate the development of new technologies to boost production of SAF. They have set an interim target of 3 billion gallons per year by 2030, with the goal of reaching 35 billion gallons annually by 2050.7 These regulations will require SAF produced from various sources of biomass to maintain supply in line with future demand.A potential approach to replace a portion of the petroleum oil used today is sourcing it from organic material, such as biomass, found on or near the Earth's surface, rather than extracting it from deep underground where fossil oil is found. A promising source for this is second- and third-generation feedstocks, which include organic food waste, animal biomass, wood, agricultural residues, and algae.8–10 Using these non-edible feedstocks offers the advantage of avoiding competition with food production. Processing them through various methods results in an oil to be obtained for further treatment. Oils produced from biomass typically contain a higher proportion of heteroatoms, with a significant focus on oxygen.11–13 Removing the oxygen is crucial for producing a fuel that is suitable for engines used in aviation.14,15 One technology for achieving this is hydroprocessing, which enables the upgrading of renewable feedstocks into liquid fuel intermediates with improved fuel properties. Specifically, hydrodeoxygenation (HDO) is essential for converting oxygen-rich fats and oils into hydrocarbons, thereby making the product more comparable to conventional petroleum fuels.16–19 In this pathway, oxygen is removed as H2O, preserving carbon number while consuming H2. On sulfided NiMo sites, unsaturated chains first hydrogenate, followed by C–O bond scission through direct HDO or through decarboxylation/decarbonylation (deCOx),20 which release CO2 and CO, respectively, reducing H2 consumption while decreasing carbon number. Selectivity between HDO and deCOx depends on operating conditions. Hydrogenation proceeds at lower temperatures, while extensive oxygen removal typically requires higher temperatures,20 which increases the risk of secondary reactions such as hydrocracking. Higher hydrogen partial pressure favors HDO and suppresses deCOx. Moderate decreases in space velocity in continuous operation can favor HDO by increasing deoxygenation. The products from this process are intermediates that typically undergo further refining, such as hydrocracking and hydroisomerization, to meet aviation fuel specifications.
Building on this potential, one promising approach is utilizing lipids derived from black soldier fly larvae (Hermetia illucens, BSFL) as a feedstock. Currently, BSFL-derived fat is primarily used as a nutritional supplement in livestock feed.21 However, significant investments in insect farms, where protein meal is the primary product and animal fat a by-product, highlight the potential for increased availability of this resource. For instance, the Danish ENORM Biofactory, recently expanded their facility, aiming for an annual processing capacity of approximately 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 500 tonnes of larvae.22 Notably, BSFL lipid feedstock is relatively well-defined prior to upgrading,23 simplifying downstream processing. Unlike biodiesel production from vegetable oils and animal fats, HDO offers an alternative route that produces fuels chemically similar to petroleum-based fuels, thereby improving miscibility and compatibility.17 Given the expanding capacity, there is significant potential to utilize the production of fats for jet fuel intermediate generation through HDO. Hence, it is essential to advance the understanding of this upgrading process by studying the behavior of key process parameters and exploring the integration of such feedstocks into existing infrastructure, for instance, through co-processing with fossil-based oil. This improves the efficiency and economic viability of lipid oil hydroprocessing while also diversifying the feedstock portfolio for SAF production.
500 tonnes of larvae.22 Notably, BSFL lipid feedstock is relatively well-defined prior to upgrading,23 simplifying downstream processing. Unlike biodiesel production from vegetable oils and animal fats, HDO offers an alternative route that produces fuels chemically similar to petroleum-based fuels, thereby improving miscibility and compatibility.17 Given the expanding capacity, there is significant potential to utilize the production of fats for jet fuel intermediate generation through HDO. Hence, it is essential to advance the understanding of this upgrading process by studying the behavior of key process parameters and exploring the integration of such feedstocks into existing infrastructure, for instance, through co-processing with fossil-based oil. This improves the efficiency and economic viability of lipid oil hydroprocessing while also diversifying the feedstock portfolio for SAF production.
This research addresses this need by identifying conditions for achieving complete deoxygenation and high oil yield during BSFL lipid feedstock upgrading through a multi-dimensional study. It aims to investigate the influence of process parameters on lipid feedstock hydroprocessing in a batch system, understanding mass transfer phenomena relevant to scale-up in continuous flow systems, and conduct an initial co-processing study with vacuum gas oil (VGO) in a batch system. This study provides valuable insights into the hydroprocessing of BSFL lipid feedstock, supporting the development of efficient, scalable, and industry-relevant technologies that advance the production of SAF.
2 Experimental
2.1 Materials
The lipid wax used in this work consisted mainly of triglycerides, with the fatty acid profile and free fatty acid (FFA) content detailed in Table 1, provided by the lipid manufacturer ENORM Biofactory (Flemming, Denmark). VGO was provided by Shell and had a sulfur content of 1.9% and a viscosity of 11.8 mm2 s−1. Sulfur was introduced into the feed and also contributed to catalyst activation via the addition of dimethyl disulfide (DMDS) (≥99.0%, Sigma Aldrich). Hydrogen and nitrogen gases were supplied by Strandmöllen. During the continuous experiments, decalin (98.0%, Sigma Aldrich) acted as the solvent.| Fatty acid | Concentration (wt%) |
|---|---|
| C10:0 |
0.8 |
| C12:0 |
47.2 |
| C14:0 |
11.1 |
| C16:0 |
14.3 |
| C16:1 |
3.7 |
| C18:0 |
1.9 |
| C18:1 |
9.8 |
| C18:2 |
9.2 |
| C18:3 |
1.9 |
| FFA (oleic acid) | 1.5 |
2.2 Catalyst synthesis and activation
Commercial NiMo/Al2O3 catalysts were synthesized in-house by incipient wetness impregnation to the commercial specification (Hulteberg Chemistry and Engineering AB, Sweden). The catalysts used in the experiments were doped with 1% Ce and 1% La, and had a bimetallic composition of 3.5% Ni (quality level 100, CAS number 13478-00-7, Sigma Aldrich) and 8% Mo (quality level 300, CAS number 12054-85-2, Sigma Aldrich) supported on Al2O3. In accordance with a previous study, the same catalyst formulation and preparation conditions were applied, adding Ce and La to the Al2O3 support to stabilize the structure and tune acidity, which has been shown to influence HDO selectivity and reduce carbon formation during hydrotreating.24 The particle sizes were 0.09–0.18 mm for the batch runs and 0.85–2 mm for the continuous runs. Catalyst particle sizes were selected to suit each reactor mode. For the autoclave experiments, smaller particles were chosen to avoid attrition of larger particles during stirring. Smaller particles also provide greater external surface area, shorter diffusion paths inside the particle, and remain suspended at lower stirring rates. Together these effects improve contact between phases and reduce temperature gradients, at the cost of requiring more thorough filtration of the product oil after the experimental run. For the continuous runs, the particle size was chosen to be large enough to limit pressure drop, yet small enough to avoid channeling, limit hot spots, ensure adequate wetting, and enhance removal of exothermic heat generated within the catalyst particles. To ensure this and to limit wall effects, a ratio between inner reactor diameter (D) and particle diameter (dp) of D/dp ≥ 8–10 was maintained.25 Prior to use, the catalyst intended for batch runs was sulfided using DMDS at a DMDS/catalyst mass ratio of 3.4/4.5 (g g−1) under hydrogen pressure. The reactor was initially flushed three times with 5 bar of N2, followed by the same procedure with H2. Finally, the autoclave reactor was pressurized to 20 bar with H2 at room temperature and heated to 400 °C, holding this temperature for 4 hours. The catalyst for continuous runs was sulfided in a tube reactor under 1% H2S in H2 at 400 °C and 8 bar for 4 hours.2.3 Catalyst characterization
Nickel, molybdenum, and sulfur content of the sulfided catalysts were measured by inductively coupled plasma sector field mass spectrometry (ICP-SFMS). Samples were prepared by digestion in accordance with laboratory SOP S-PA16-HB for solid matrices, and analyses followed SS-EN ISO 17294-2:2023 and US EPA Method 200.8 (Revision 5.4, 1994).26 Textural properties of the catalysts were determined by adsorption–desorption experiments on a Micromeritics ASAP 2020 instrument using N2 gas at −196 °C. Prior to measurements, the catalysts were degassed under vacuum for 3 h at 350 °C. The surface area and porosity data were determined based on the Brunauer–Emmett–Teller (BET) and Barrett–Joyner–Halenda methods, respectively. Morphological examination of the catalysts was conducted on an Inovenso's benchtop scanning electron microscope (SEM) – IEM 11 field emission scanning electron microscope operating at 20 kV, and elemental analysis was performed using the energy-dispersive X-ray spectroscopy (EDS) detector coupled to the SEM. Thermogravimetric analysis (TGA) of the catalyst samples was carried out using a Mettler Toledo TGA/DSC2 system. About 25 mg of the fresh or spent catalyst sample was heated at a rate of 10 °C min−1 from room temperature to 550 °C under air flow at the rate of 50 mL min−1.2.4 Hydrodeoxygenation in batch mode
Before each run, the reactor was filled with 100 mL of hydrocarbon feedstock, either lipid wax or a mixture of lipid wax and VGO, along with 5 or 10 wt% sulfided catalyst and DMDS at a mass ratio of 0.4 (g g−1) relative to the catalyst weight. The reactor was flushed three times with N2, before repeating the same procedure with H2. It was pressurized to the desired pressure at ambient temperature with H2. Stirring and heating were initiated simultaneously, with stirring set to either 400 or 800 rpm to ensure proper mixing. Heating was set to a controlled rate of 10 °C min−1 until the target temperature was reached. Experimental parameters for each test in the lipid HDO parametric study are presented in Table 2. For co-processing with VGO, all tests used a stirring rate of 800 rpm, an initial H2 pressure of 70 bar at room temperature, 5% catalyst loading, and temperature at 420 °C. The lipid content in the co-processing feed varied at 0 wt% (X1), 10 wt% (X2), 20 wt% (X3), and 100 wt% (X4). Once the target temperature was reached, it was maintained for 4 hours. After the reaction, heating and stirring were stopped, and the autoclave reactor was allowed to cool to room temperature. Gas sampling was performed using a steel sample cylinder for analysis. The organic and aqueous phases were separated by decantation, the catalyst was recovered by filtration (1–2 µ m) in a fume hood, and the recovered catalyst and separated phases were subsequently weighed.
| Sample | Temperature (°C) | Initial pressure at RT (bara) | Catalyst loading (wt%) | Stirring rate (rpm) |
|---|---|---|---|---|
| a B7 and B10 were each repeated for a total of 4 and 2 runs, respectively, to improve the 2FI model fit in design of experiments (DoE). RT = room temperature. | ||||
| B1 | 350 | 30 | 5 | 400 |
| B2 | 390 | 30 | 5 | 400 |
| B3 | 370 | 30 | 5 | 800 |
| B4 | 390 | 30 | 10 | 400 |
| B5 | 350 | 30 | 10 | 800 |
| B6 | 370 | 70 | 5 | 400 |
| B7 | 390 | 70 | 5 | 800 |
| B8 | 350 | 70 | 5 | 800 |
| B9 | 350 | 70 | 10 | 400 |
| B10 | 370 | 30 | 10 | 800 |
| B11 | 390 | 70 | 10 | 800 |
| B12 | 370 | 70 | 10 | 400 |
As part of the evaluation method, oil yield Yoil for the batch runs was calculated gravimetrically with the following equation:
 | (1) |
Furthermore, to specifically assess the HDO performance, the degree of deoxygenation (DoD) was calculated as follows:
 | (2) |
2.5 Hydrodeoxygenation in continuous mode
The reactor system consisted of a preheater packed with α-alumina beads (4.76 mm) to ensure uniform heat distribution and flow prior to the reactor. The fixed-bed reactor was a 15 cm stainless steel tube with an inner diameter of 15.75 mm. A total of 10 mL of catalyst was used, diluted with inert α-alumina (0.65–0.85 mm) at a volume ratio of 1:1 to improve heat transfer and mixing. Quartz wool was added at the top and bottom of the reactor to keep the catalyst bed stable. Feed oil was pumped using an LC-10AD HPLC pump (Shimadzu), and hydrogen gas flow and system pressure were controlled by mass flow controllers, with the system operated in a downflow configuration. The reactor system was also equipped with a condenser to improve gas–liquid product separation.
Decalin (20 wt%) was used as a solvent to enable continuous feeding of the lipid feedstock, which is otherwise in wax form at room temperature. To maintain the catalyst sulfided and active during the run, 1 wt% DMDS was added to the feed. The process parameters varied across experiments included liquid hourly space velocities (LHSV) of 0.5, 1, and 2 h−1, and hydrogen-to-oil ratios (H2/oil) of 400, 800, and 1200 mL mL−1. H2/oil is defined as the volumetric flow of hydrogen (at standard conditions) divided by the volumetric flow of liquid feed at the pump, reported as mL mL−1. All experiments were conducted at a constant temperature of 350 °C and 50 bar H2 pressure.
To obtain a representative liquid product sample, the system was first allowed to reach the process condition setpoints and stabilize for 2 h, during which the sample collection tube was emptied. The experiment then continued for an additional hour, after which a liquid product sample was collected for analysis.
2.6 Product analysis
The boiling point ranges and carbon number distributions of the hydrotreated oil samples were determined by gas chromatography following ASTM D7213. The analysis was conducted using a SCION 456 gas chromatograph (GC) from SCION Instruments. The samples were dissolved in 90% CS2 (ACS reagent, ≥99.9%, Sigma Aldrich) before analysis, and the GC data were processed using Eclipse 3.0.2.0 software.Oxygen in oil samples was determined based on ASTM D5622, which involves reductive pyrolysis. In this method, a sample of the oil is pyrolyzed in a high-temperature furnace (950–1300 °C) with metallized carbon. Oxygen-containing compounds in the sample are converted into carbon monoxide, which is detected and quantified to determine the total oxygen content in the sample. The oxygen is then reported as mass percentage of the fuel. The accuracy of the analytical instrument is such that it can confidently measure oxygen levels at or above 0.5 wt%.
The hydrotreated oil samples were analyzed using a GC-VUV system from VUV Analytics equipped with a VGA-101 detector, in accordance with ASTM D8071. This method utilizes vacuum ultraviolet (VUV) spectroscopy to generate a PIONA report, providing detailed information on the paraffin, iso-paraffin, olefin, naphthene, and aromatic content of the samples.
The gas composition of the reaction gas at the end of the test run was analyzed using ASTM D1946 and ASTM D2163 with a gas chromatograph, the SCION Rapid Refinery Gas Analyzer from SCION Instruments. This method determines the concentrations of hydrogen, permanent gases, and light hydrocarbons, generating a detailed profile of the reaction gas composition.
The elemental analysis (C, H, N, S) was performed by using a vario MICRO cube instrument (Elementar Analysensystem GmbH, Langenselbold, Germany) equipped with a thermal conductivity detector (TCD). Helium (20 mL min−1) and oxygen (10 mL min−1) were used during the experiment and a column temperature of 1100 °C was applied.
The calorific value was measured by using a liquid calorimeter system IKA C5000 (IKA, Staufen, Germany). The samples were measured according to DIN 51900. The calorimeter consists of a sealed, thick-walled steel vessel (calorimetric bomb) in which a precisely weighed quantity (1 g) of the sample is burnt in oxygen at a pressure of 30–35 bar after ignition by an electric glow wire. During combustion, the bomb is immersed in a measured quantity of water, which absorbs the combustion heat of the burnt sample and causes a measurable increase in temperature, from which the amount of heat released can be determined. Additionally, density measurements were conducted with an Eraspec FTIR analyzer (Eralytics) in full compliance with ASTM D4052.
The cold flow properties were measured by using a OptiMPP (Mini Cloud & Pour Point Tester, PAC, Lauda, Germany). For the measurements 0.5 mL of sample were used for each test and measured twice according to ASTM D7689 (Cloud Point) and ASTM D7346 (no-flow point and pour point) in slow test mode.
3 Results and discussion
3.1 Catalyst properties
The measured Ni and Mo contents of the fresh catalysts after sulfidation are close to the intended synthesis levels (Table 3). Taken together with the sulfidation protocol, the Ni, Mo, and S contents are consistent with formation of the sulfided NiMoS phase on Al2O3-supported NiMo catalysts under standard sulfidation conditions.28| Sample | Ni (wt%) | Mo (wt%) | S (wt%) |
|---|---|---|---|
| B0 | 3.85 | 7.94 | 9.60 |
| C0 | 4.67 | 9.39 | 7.22 |
Fresh catalyst (B0) and spent catalysts (B7 and B10) from batch lipid HDO experiments were characterized by SEM-EDS. SEM images were examined for sample homogeneity and surface morphology changes. No significant differences were observed between fresh and spent catalysts, suggesting that reaction conditions had minimal impact on surface structure. Most catalyst granulates showed relatively flat surfaces in each sample, with some displaying a coarser texture, indicating generally uniform morphology with minor variations. A representative comparison of B0 and B7 is shown in Fig. 1.
 | ||
| Fig. 1 SEM images of (a) B0 and (b) B7. | ||
In addition to morphological analysis, EDS was conducted to obtain the surface elemental composition of the fresh B0 and spent catalysts B7 and B10 (Table 4). In line with the alumina support, oxygen and aluminum were the dominant elements across all samples. The analysis also verified the presence of the added active metals, molybdenum and nickel, while sulfur levels (7.49 wt% in B0) confirmed effective sulfidation of the fresh catalyst and remained comparable in the spent catalysts (11.31 wt% in B7 and 5.82 wt% in B10), indicating that sulfidation was maintained throughout the experiments. Molybdenum and nickel remained at significant levels, indicating limited metal loss under the conditions tested. Although EDS is used as compositional evidence only and is not used to assign the active phase, the applied sulfidation procedure is expected to form NiMoS under the stated conditions. Calcium was detected in B7 (0.08 wt%) and is likely derived from the feedstock, as reported by Selimi et al., who noted that ash components can contribute to catalyst deactivation.23 Iron was detected in both spent samples, despite only trace levels being reported in the same feedstock, suggesting that Fe contamination likely originated from the reactor system itself, possibly through leaching or wear of metallic components during operation.
| Element | B0 (wt%) | B7 (wt%) | B10 (wt%) |
|---|---|---|---|
| O | 54.66 | 47.16 | 52.01 |
| Al | 24.99 | 20.50 | 33.46 |
| S | 7.49 | 11.31 | 5.82 |
| Mo | 7.26 | 12.83 | 5.36 |
| Ni | 5.44 | 7.14 | 3.00 |
| Fe | — | 0.98 | 0.34 |
| Ca | — | 0.08 | — |
| Zr | 0.16 | — | — |
N2 physisorption measurements on the same catalysts showed that the BET surface area decreased from 57.7 m2 g−1 in B0 to 26.9 m2 g−1 in B7 and 44.0 m2g−1 in B10 (Table 5), indicating a reduction in available surface area after reaction. Corresponding decreases in total pore volume and average pore diameter suggest partial pore blockage or structural changes, likely due to coke deposition or accumulation of heavy species during the HDO process. The fresh catalyst C0 used in the continuous reactions exhibited a higher surface area of 98.5 m2 g−1 than its counterpart used in the batch reactions. Although C0 exhibits higher surface area and larger pores, the active phase and catalyst formulation are otherwise nearly identical, differing in the type of alumina used as the support.
| Sample | S BET (m2 g−1) | V Pore (cm3 g−1) | d (nm) | Total mass loss (mg) |
|---|---|---|---|---|
| a S BET values for B0 represent the mean ± standard deviation from three BET measurements. Other samples were measured once. | ||||
| B0 | 57.73 ± 0.23 | 0.233 ± 0.001 | 12.74 ± 0.04 | 1.07 |
| B7 | 26.90 | 0.088 | 7.33 | 1.28 |
| B10 | 44.00 | 0.127 | 8.46 | 1.23 |
| C0 | 98.50 | 0.639 | 20.45 | — |
TGA was performed on the same selected batch catalysts. Mass loss was evaluated from 100 °C to the final temperature to remove moisture and physisorbed species. In this range, the used catalysts B7 and B10 showed 14.95–19.62 wt% higher mass loss compared to the fresh catalyst B0 (Table 5), indicating the formation of carbonaceous residues or coke during reaction. However, because mass loss can include sulfur loss and impurity decomposition, results are presented as total mass loss. Notably, both B7 and B10 exhibited coke deposition despite being tested under different conditions of temperature, pressure, and catalyst loading, suggesting that coke formation occurs consistently across the range of tested conditions. The fresh catalyst B0 also showed some mass loss, likely due to desorption of residual surface species or decomposition of synthesis-related compounds. Characterization after TGA was not performed, therefore the extent of coke removal and any changes to the catalyst surface cannot be confirmed.
Overall, the batch catalysts showed favorable initial properties, with high surface area and accessible active metals indicating good catalytic potential. After reaction, structural and compositional changes, particularly the reduction in surface area and pore volume with limited evidence of sintering, indicate catalyst deactivation over time. The observed coke deposition, evidenced by greater mass loss in TGA for used catalysts B7 and B10 relative to B0, correlates with reductions measured by N2 physisorption and supports pore blocking as a primary deactivation mechanism. Compared with the fresh sample, the used catalysts show higher total mass loss in TGA and lower BET surface area, pore volume, and pore diameter. These trends are consistent with deposit-induced pore blocking rather than irreversible support collapse.
In this context, the differences observed in EDS surface composition reflect the surface sensitive nature of the technique and are attributed to varying extents of coke deposition, sulfur redistribution, and partial coverage of active metal sites during batch operation. In line with these observations, the differences in physisorption properties are consistent with progressive pore blockage and textural modification during catalyst deactivation in the batch system. The spent catalysts are described by surface composition and textural characterization, with bulk elemental analysis recommended for future work.
3.2 Parameter effects in batch mode
To investigate the effects of process parameters on the hydrotreating of lipids in batch mode with respect to yield and oil properties, a series of experiments were conducted using a fractional factorial design, varying temperature, pressure, stirring rate, and catalyst loading.| Source | SoS | df | MS | F-value | p-value | Significance |
|---|---|---|---|---|---|---|
| a SoS = sum of squares, df = degrees of freedom, MS = mean squares, F-value = ratio of mean squares, and p-value = probability. | ||||||
| Model | 957.10 | 8 | 119.64 | 71.14 | <0.0001 | Significant |
| A-temperature | 140.92 | 1 | 140.92 | 83.79 | <0.0001 | |
| B-pressure | 380.88 | 1 | 380.88 | 226.48 | <0.0001 | |
| C-stirring rate | 108.74 | 1 | 108.74 | 64.66 | <0.0001 | |
| D-catalyst loading | 4.02 | 1 | 4.02 | 2.39 | 0.1662 | |
| AB | 96.80 | 1 | 96.80 | 57.56 | 0.0001 | |
| AC | 34.62 | 1 | 34.62 | 20.59 | 0.0027 | |
| AD | 30.38 | 1 | 30.38 | 18.06 | 0.0038 | |
| BC | 36.03 | 1 | 36.03 | 21.42 | 0.0024 | |
| Residual | 11.77 | 7 | 1.68 | |||
| Lack of fit | 9.15 | 3 | 3.05 | 4.66 | 0.0857 | Not significant |
The ANOVA demonstrates that the model is statistically significant, as indicated by a high F-value (71.14) and a very low p-value (<0.0001), while the Lack of fit test resulted in an insignificant F-value (4.66) and p-value (0.0857). This confirms that the 2FI model adequately represents the relationships between experimental variables and oil yield, capturing the underlying trends within the studied process conditions.
Among the individual process parameters, temperature (A), pressure (B), and stirring rate (C) each showed a significant linear effect, with pressure (B) having the strongest influence (F-value = 226.48). Catalyst loading (D), however, showed no significant linear impact (p-value = 0.1662), indicating a minimal effect on yield. For interaction effects, four terms were statistically significant: temperature–pressure (AB), temperature–stirring rate (AC), temperature–catalyst loading (AD), and pressure–stirring rate (BC). The AB interaction had the strongest effect (F-value = 57.56), emphasizing the importance of maintaining both high temperature and pressure to enhance yield. The AC, AD, and BC interactions also showed significant effects with F-values of 21.42, 20.59, and 18.06, respectively. The regression equation in terms of coded factors (eqn (3)) quantifies the relative influence of each factor and interaction on the response, consistent with the ANOVA results.
| Yield = 65.22 + 4.68A + 6.04B + 3.21C + 0.6215D − 4.62AB − 2.76AC − 2.01AD − 2.20BC | (3) |
The robustness and reliability of the developed 2FI model was supported by the statistical summary presented in Table 7. The high coefficient of determination (R2 = 0.9878) and adjusted R2 (0.9740) indicate that the model effectively describes the variation in oil yield. Although the predicted R2 (0.7918) is slightly lower, it remains in reasonable agreement with the adjusted R2, with a difference of less than 0.2, suggesting the model has reasonable predictive capability. The low coefficient of variation (C.V. = 1.96%) shows satisfactory model precision and reliability compared to the mean response (66.30). The adequate precision value of 34.19, which is well above the signal-to-noise threshold of 4, demonstrates that the model produces a strong signal.
| Std. dev | 1.30 | R 2 | 0.9878 |
| Mean | 66.30 | Adjusted R2 | 0.9740 |
| C.V. % | 1.96 | Predicted R2 | 0.7918 |
| Adeq. precision | 34.1877 |
Model diagnostics were further assessed by plotting the predicted oil yields against the experimental results, and the residuals against the run order. The predicted values closely match the experimentally observed values by clustering near the diagonal line, which implies good agreement and high model accuracy, as shown in Fig. 2. Examining the residuals against the run order, it is evident that the residuals are randomly scattered around zero without any apparent pattern, and that all values are found within the acceptable limits ±4.76 (Fig. 3). The absence of any clear trend or outliers suggests minimal influence from the order of experimentation or other systematic effects.
 | ||
| Fig. 2 Comparison between predicted oil yield values and those obtained experimentally. | ||
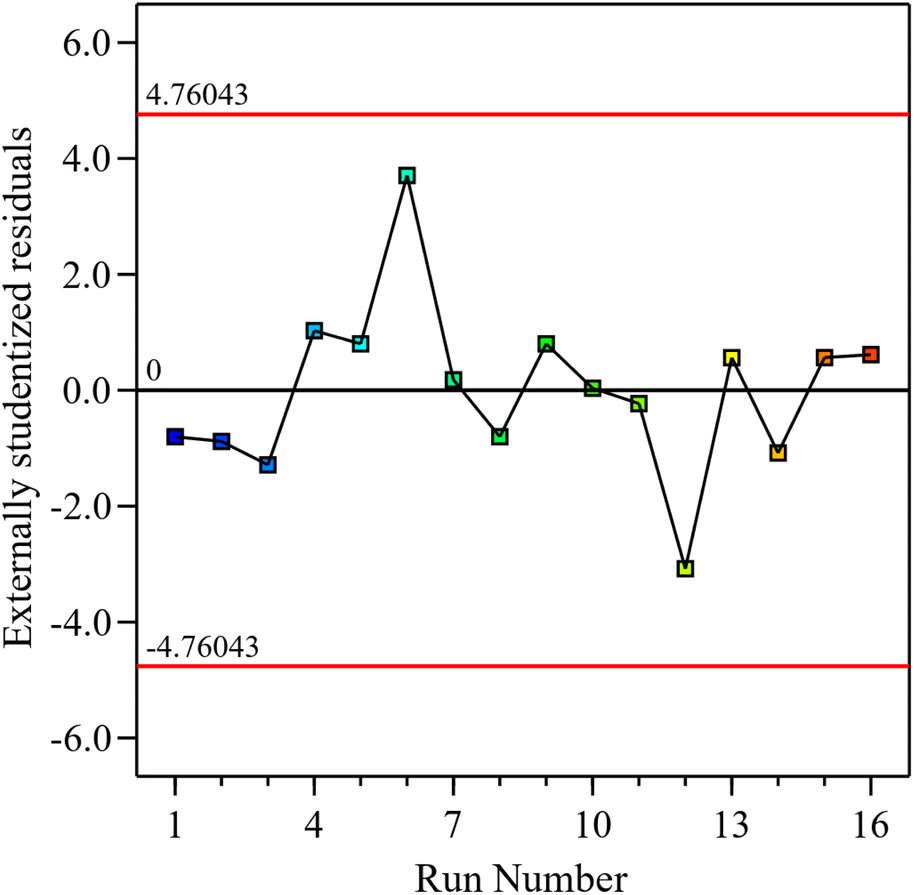 | ||
| Fig. 3 Residuals plotted against experimental run order. | ||
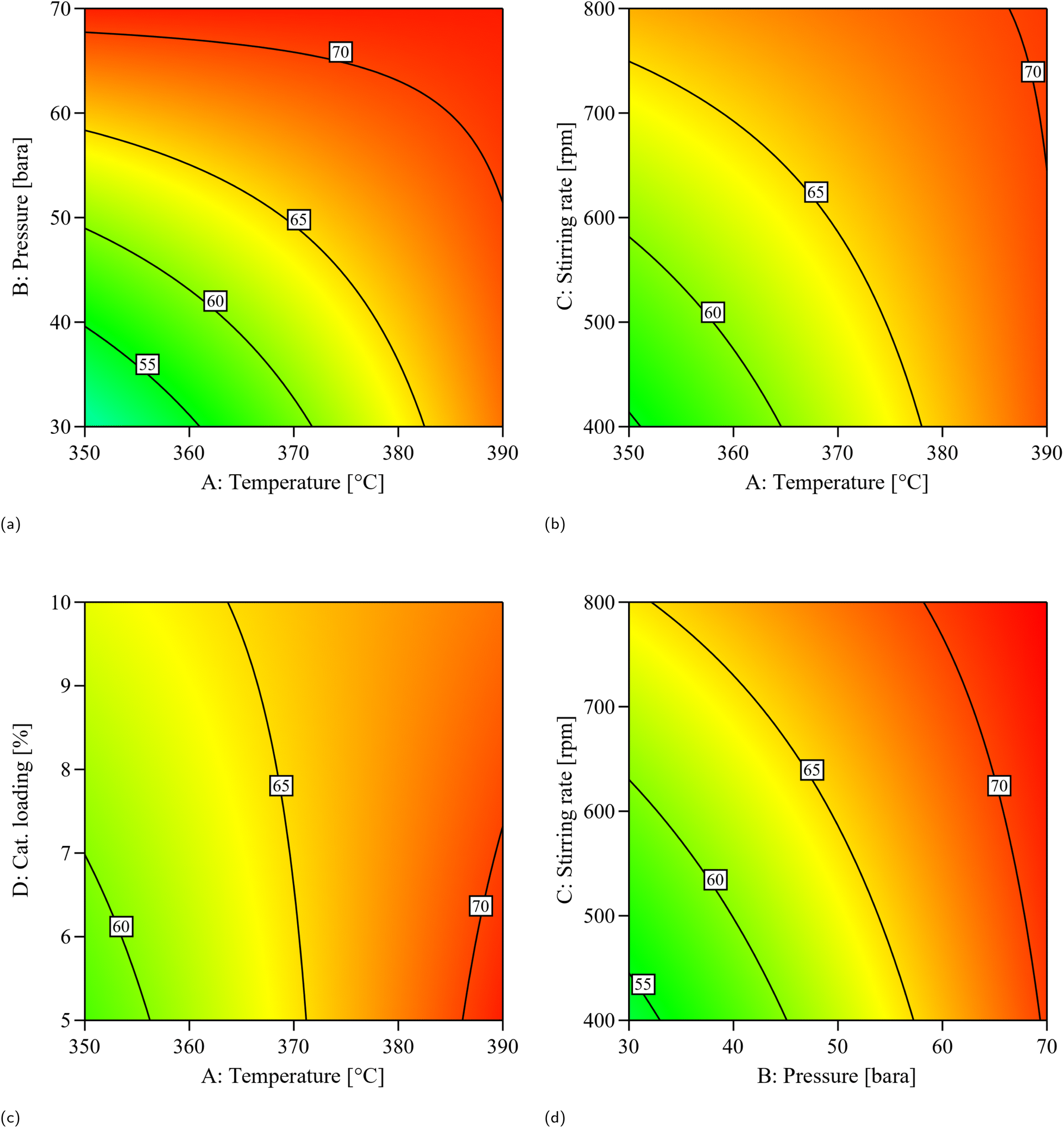 | ||
| Fig. 4 Contour plots illustrating oil yield response to key variable combinations: (a) pressure vs. temperature, (b) stirring rate vs. temperature, (c) catalyst loading vs. temperature, and (d) stirring rate vs. pressure. | ||
 | ||
| Fig. 5 Image of raw, untreated lipid feedstock (left) and hydrotreated lipid oil (right), highlighting the increase in clarity. | ||
In addition, a range of properties commonly used to assess fuel quality was obtained for the samples (Table 8). While the product samples are not considered finished fuel products, evaluating these properties still provides valuable insight into their fuel potential and the impact of process conditions. It can be observed that the properties overall substantially improved compared to the feedstock, approaching characteristics more typical of aviation fuel. The hydrogen content and higher heating value (HHV) showed relatively minor variation among the samples across the different conditions. Examining the results in more detail, pressure generally appeared to have the greatest impact, enhancing the HHV, with the highest values (47.2–47.4 kJ g−1) observed at an initial pressure of 70 bar. It also had a noticeable effect on density, flash point, and cold-flow properties. In high DoD samples, increasing the temperature resulted in minor changes, with the most evident trend being an improvement in cold-flow properties accompanied by a reduction in flash point. Varying the catalyst loading improved cold-flow properties, as shown by the cloud point of B3 (−5 °C) compared to B10 (−8.9 °C). This suggests that the additional active sites present during the reaction increase the occurrence of secondary reactions such as isomerization and cracking, which in turn enhance cold-flow properties. Stirring rate also influenced product properties, mainly under conditions where mass transfer limitations are more significant. At 30 bar, increasing stirring from 400 to 800 rpm generally resulted in higher flash points (51–68 °C) and lower cloud points (down to −8.9 °C). At 70 bar, however, flash point and cold-flow properties were similar across stirring rates. These results suggest improved hydrogen-to-oil mass transfer at lower pressure, where reactions are more limited by stirring.
| Sample | Hydrogen (wt%) | HHV (kJ g−1) | Density (kg m−3) | Flash point (°C) | Cloud point (°C) | Pour point (°C) |
|---|---|---|---|---|---|---|
| a All density measurements were performed at temperatures between 27.0 °C and 28.6 °C. | ||||||
| Untreated lipids | 11.1 | 38.3 | — | 200.3 | 17.5 | 12 |
| B1 | 13.4 | 43.5 | 803.0 | 73.9 | 16.6 | 6 |
| B2 | 13.5 | 46.2 | 769.3 | 44.3 | 1.4 | −12 |
| B3 | 13.4 | 46.5 | 770.9 | 61.0 | −5 | −9 |
| B4 | 13.1 | 46.1 | 774.0 | 45.0 | −0.92 | −12 |
| B5 | 13.9 | 46.0 | 781.6 | 68.0 | 2.5 | −9 |
| B6 | 14.3 | 47.2 | 757.5 | 69.3 | −3.5 | −3 |
| B7 | 14.0 | 47.4 | 755.5 | 69.3 | −3.2 | −3 |
| B8 | 13.9 | 47.3 | 755.1 | 71.1 | −1.6 | −3 |
| B9 | 13.9 | 47.2 | 757.4 | 68.0 | −2.4 | −3 |
| B10 | 13.2 | 46.5 | 777.3 | 51.0 | −8.9 | −9 |
| B11 | 14.0 | 47.3 | 755.9 | 68.5 | −4.6 | −3 |
| B12 | 14.4 | 46.1 | 757.4 | 69.7 | −3.3 | −3 |
| Untreated VGO | 11.6 | 44.5 | — | >330 | 39 | 36 |
| X1 | 12.3 | 45.2 | — | 34.7 | 35.0 | 33 |
| X2 | 11.7 | 45.4 | — | 24.3 | 31.1 | 27 |
| X3 | 11.9 | 45.4 | — | 29.1 | 32.0 | 30 |
| X4 | 13.9 | 47.1 | — | 22.5 | −8.0 | −9 |
Across conditions, the liquid products were dominated by paraffinic hydrocarbons, typically 75–90 wt%, with B1 as a clear outlier at 46 wt% paraffin and 22 wt% olefins (Table S2). Outside B1, olefins were generally at or below about 11 wt%. Naphthenes and aromatics remained minor in most cases, usually at or below about 5 wt%. Moving to higher initial pressure increased liquid yields to about 68–72 wt%, while the liquids in those cases remained strongly paraffinic with very low olefin fractions (Table S1). Furthermore, all experiments delivered high yields of kerosene- and diesel-range fractions, with maximum of 37.8 wt% for jet fuel (180–250 °C) and 29.0 wt% for diesel (250–350 °C),33 as detailed in Table S4.
The gas phase was primarily composed of CO2 with CH4, propane, and smaller amounts of CO, ethane, and trace C4–C5 hydrocarbons (Table S5). CH4 and propane were consistent secondary components, and CH4 was toward the higher end in the lowest liquid-yield case B1. Comparing lower-pressure runs with otherwise similar settings showed a modest shift toward propene in the gas and a higher overall olefin share in the liquid. This coincided with lower initial hydrogen availability in the liquid phase, whereas higher-pressure cases tended to show lower olefin content and more paraffinic liquids.
3.3 Effect of LHSV and H2/oil in continuous mode
Hydrotreating of lipid oil dissolved in decalin (20 wt%) was conducted in a continuous fixed-bed reactor. Within this system, the influence of LHSV and H2/oil ratio on catalyst wetting was evaluated through three indicator product ratios (Fig. 6). These experiments were conducted as single runs per condition to map operating trends. | ||
| Fig. 6 Effect of LHSV on the (a) even-to-odd carbon number ratio, (b) CO2/CO ratio, and (c) propane/COx ratio. Each line represents a different H2/oil volume ratio (400, 800, 1200). | ||
The ratios may reflect local hydrogen availability in the oil phase at the catalyst surface and could indicate liquid film coverage and conditions favoring optimal hydrogen utilization. The even-to-odd carbon number ratio compares the concentration of hydrocarbons with even-numbered carbon chains to those with odd-numbered chains. As all fatty acid chains in the triglyceride feedstock are even-numbered in terms of carbon number (Table 1), the presence of even-numbered products suggests HDO, while odd-numbered products indicate deCOx reactions. At the lowest LHSV (0.5 h−1), the even-to-odd carbon number ratio increased from 0.80 to 0.95 with increasing H2/oil ratio, from 400 to 1200 mL mL−1 (Fig. 6a). This suggests that higher H2/oil favors HDO under low LHSV conditions.34 However, at an intermediate LHSV of 1 h−1, the ratio declined across all gas rates, indicating reduced catalyst wetting that became more apparent with higher hydrogen flow. A similar trend was observed at 2 h−1, except for a slight reversal at an H2/oil ratio of 1200 mL mL−1. Moreover, the CO2/CO ratio was lower at 0.5 h−1 LHSV for H2/oil ≤800 mL mL−1, indicating more HDO and decarbonylation activity (Fig. 6b). In contrast, other LHSV conditions showed less favorable results, except for a partial recovery at 2 h−1 LHSV with H2/oil at 1200 mL mL−1. Finally, the highest propane/COx ratio, which compares triglyceride backbone cleavage with oxygen removal via deCOx reactions, was observed at 0.5 h−1 LHSV and 800H2/oil (Fig. 6c). At LHSV values of 1 h−1 and above, the ratio consistently remained around 0.9–1.0, regardless of H2/oil, indicating that LHSV was the dominant limiting factor. Lower LHSV increases liquid residence time and can improve catalyst wetting and hydrogen availability, which favors HDO relative to deCOx.35 This effect applies while liquid distribution remains adequate, and very low LHSV may instead lead to insufficient wetting. It is important to note that these three ratios probe different aspects of the oxygen-removal chemistry. The even-to-odd and propane/COx ratios reflect the balance between HDO and deCOx and therefore show similar qualitative trends. In contrast, the CO2/CO ratio distinguishes decarboxylation from decarbonylation, so its variation with LHSV can differ. Taken together, the optimal conditions for efficient wetting appear to be at a 0.5 h−1 LHSV and an H2/oil ratio ≥800 mL mL−1. Under these conditions, the indicator ratios suggest efficient catalyst wetting, combining favorable liquid film coverage with sufficient hydrogen at the catalyst surface, thereby enhancing HDO and suppressing deCOx pathways.
3.4 Performance of co-processing with vacuum gas oil
In an initial study, BSFL lipid feedstock was co-processed with oxygen-free VGO in a batch autoclave reactor to assess the HDO performance and product oil quality. The feed used in the experiments contained varying lipid content, ranging from 0 to 100 wt%. All experiments were conducted under the same operating conditions: 420 °C, 70 bar, a stirring rate of 800 rpm, and a catalyst loading of 5 wt%. This temperature was selected as lower values led to a more waxy product due to VGO, while 420 °C produced a liquid product that improved product recovery and handling. This exploratory series was conducted as single runs per condition.The mass of water produced in each experiment was compared to the corresponding lipid mass in the feed, Fig. 7. Normalization performed on a lipid content basis was intended to evaluate how HDO efficiency is affected by increasing lipid content. The level ranged from 3.6–4.2 wt% for feed lipid content of 10 and 20 wt%, without showing any clear trend. However, at a feed lipid content of 100 wt%, the experiment resulted in a significantly lower water yield of 1.8 wt%.
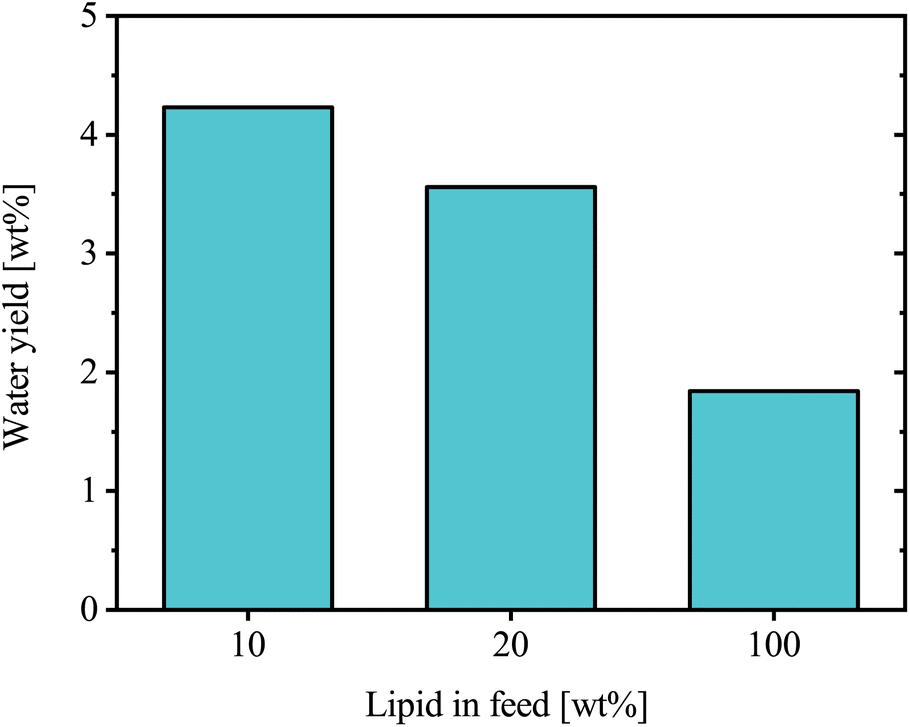 | ||
| Fig. 7 Water yield obtained at different lipid concentrations in the feed. | ||
The concentration of CO2 and CO were measured to understand oxygen removal by deCOx reactions. The gas species were calculated relative to propane as the triglyceride-backbone reference, normalizing for VGO in the gas data. While some propane may further crack into smaller hydrocarbons, this provides a more direct approach of comparison. There was a steady increase in CO2/propane from 0.2% to 4.9% over the 10–100 wt% lipid content range (Fig. 8). CO/propane, however, did not follow the same trend, as further illustrated by the CO2 selectivity relative to gas-phase oxygenates.
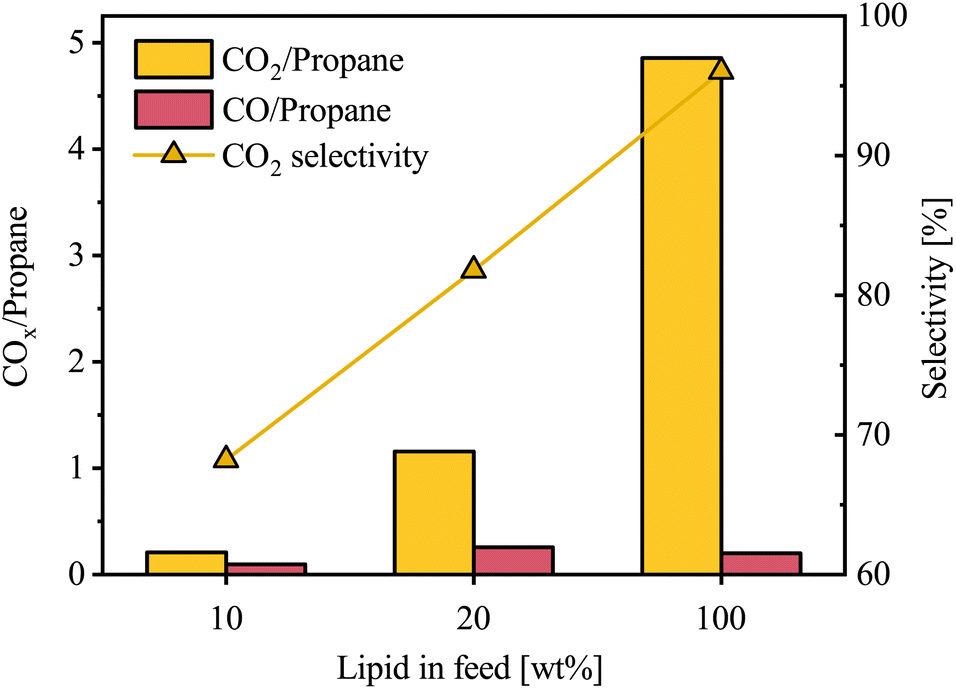 | ||
| Fig. 8 COx/propane ratio and CO2 selectivity at different lipid concentrations in the feed. | ||
It is evident that with increasing lipid content, oxygen removal reactions shift progressively from HDO toward deCOx. Since the autoclave reactor was charged with the same number of moles of hydrogen between runs, increasing lipid content in the feed mixture raised the oxygen load and therefore lowered the H2/O ratio, which favors deCOx over HDO. By the same reasoning, the deCOx reactions also shifts toward decarboxylation, because decarbonylation requires net H2, albeit less than HDO, whereas decarboxylation does not. The larger amount of oxygenated compounds in the feed has to compete for the same amount of hydrogen, leading to greater CO2 formation. It is also likely that the VGO in the feed facilitates and enhances HDO reactions, primarily because it provides dilution effects that mitigate the instability of oxygenates and improves hydrogen solubility, thereby enhancing hydrogen mass transfer.36 VGO also contains naphthenic and aromatic species that can act as hydrogen-donor solvents under hydrotreating conditions, transferring hydrogen to reactive oxygenates and helping suppress condensation and coke.37–39 Another reason could be attributed to water formed during HDO, which increases with higher lipid oxygen content and can partially oxidize the sulfided NiMoS phase, thereby inhibiting HDO activity.40
Additionally, the oil products were analyzed by simulated distillation to determine yield distributions across fuel-range boiling-point cuts: gasoline 50–180 °C, jet fuel 180–250 °C, diesel 250–350 °C, heavy fuel 350 °C–FBP. Hydrotreated products from VGO mainly occupy fractions similar to naphtha/gasoline, while those from BSFL lipid feedstock are mainly found in the diesel range, Fig. 9. Another noteworthy detail is that higher yields of heavy fractions (350 °C–FBP) from mixtures of VGO and lipids, are also observed. This could be due to synergy effects from both feedstocks, facilitating stabilization of intermediate products and minimizing overcracking.
 | ||
| Fig. 9 Simulated distillation yields for hydrotreated products obtained from feeds of VGO, BSFL lipids, and their blends. Bars are colored by lipid in feed (wt%) as indicated in the legend. Increasing lipid in the feed shifts products toward the diesel range. Distillation temperature regions used are gasoline 50–180 °C, jet fuel 180–250 °C, diesel 250–350 °C, heavy fuel 350 °C–FBP. FBP = final boiling point. | ||
Similar liquid-property tests to those used in the lipid-batch experiments were also performed on the co-processed product samples, as shown in Table 8. The tests showed that increasing the BSFL lipid fraction led to an HHV increase from 45.2 to 47.1 kJ g−1 and marginal improvements in cloud and pour points for the mixed-feed samples, while flash points remained low overall. The enhanced HHV is likely due to increased saturation as indicated by the hydrogen content. Sample X1, however, does not follow this trend and is considered an outlier. As noted in Section 3.2.3, these samples show system behavior and should not be regarded as final fuel products.
Overall, batch co-processing of the BSFL lipid feedstock with a petroleum feed such as VGO under hydrotreating conditions effectively removed oxygen via both HDO and deCOx reactions, increasing saturation, energy density and improving cold-flow properties. The improved diesel-range yield of lipid-derived products offers potential for conversion into SAF via hydrocracking and hydroisomerization.
4 Conclusion
BSFL-derived lipids were hydroprocessed with a commercial Ce/La doped NiMo/Al2O3 catalyst to produce biofuel intermediates for subsequent conversion to SAF across three parallel studies comprising parametric batch experiments, continuous trickle bed runs, and batch mode VGO co-processing runs. Across the lipid HDO batch series, deoxygenation to oxygen concentrations at or below the analytical limit of detection was consistently achieved, indicating essentially complete deoxygenation under the tested parameters. Moreover, oil yields remained high, averaging 66.3 wt% with a maximum of 72.2 wt% (Table S1). The 2FI model identified pressure as the dominant factor influencing oil yield, with the temperature–pressure interaction exhibiting the greatest effect. Stirring had a stronger positive influence on yield under mass-transfer-limited conditions, up to a pressure of 60 bar.The co-processing of BSFL lipid feedstock with VGO notably improved hydrogen availability, promoting HDO over deCOx pathways. This suggests that existing refinery streams could actively enhance the upgrading of bio-based feedstocks, improving energy density while efficiently removing oxygen and producing high yields of kerosene- and diesel-range fractions. This combined effect presents a strong case for integrating lipids derived from insects as drop-in components within existing petroleum refinery infrastructure, potentially lowering barriers to the implementation of new sustainable feedstocks.
In the continuous fixed-bed runs, efficient catalyst wetting was achieved at LHSV 0.5 h−1 with H2/oil ≥800 mL mL−1. Under these conditions, the indicator product ratios consistently pointed toward enhanced HDO selectivity and suppression of deCOx pathways, supporting this operating window for scale-up in industrial continuous-flow systems.
To build upon these findings, future work should address long-term catalyst stability and recyclability. Since oxygen removal was consistently successful, efforts should focus on minimizing carbon formation and improving oil retention. The measurable pore blockage observed after catalyst use indicates that coke is the primary factor limiting catalyst lifetime, so regeneration and reuse should be assessed with multi-cycle tests in batch and extended time-on-stream in continuous operation with oxidative cleaning and resulfiding between cycles. Reducing coking will require continued development of optimized catalysts and studies of catalyst-bed configurations that use multiple catalyst layers with differing reactivity. Additionally, future work should include optimizing downstream hydroisomerization to meet jet-fuel specifications and conducting techno-economic and life-cycle assessments (LCA) to evaluate commercial viability. LCAs should account for feedstock type and origin, including production method, regional conditions, and logistics.
Author contributions
Conceptualization, J. S., C. P. H. and O. Y. A.; methodology, J. S.; software, J. S. and O. Y. A.; validation, J. S. and C. P. H.; formal analysis, J. S., T. A. K., Z. S. Q. and O. Y. A.; investigation, J. S.; resources, J. S, C. P. H. and O. Y. A.; data curation, J. S.; writing – original draft preparation, J. S.; writing – review and editing, J. S., Z. S. Q., T. A. K., C. P. H. and O. Y. A.; visualization, J. S. and O. Y. A.; supervision, C. P. H. and O. Y. A.; project administration, J. S. and O. Y. A.; funding acquisition, C. P. H. and O. Y. A. All authors read and approved the final manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: the dataset comprises oil yields from batch HDO experiments used to generate the statistical model. See DOI: https://doi.org/10.1039/d5se01232e.Acknowledgements
Funding for this research was provided by the European Union's Horizon 2020 research and innovation programme under grant agreement number 101007130, and the Swedish Energy Agency under the project “Eco-efficient biorefinery for competitive production of green renewable shipping fuels (ECO-FORCE FUELS)”, reference number 2022-201046. O.Y.A. acknowledges the support provided by the Deanship of Research at King Fahd University of Petroleum & Minerals under the Early Career Research Grant (Project No. EC251006). The authors would like to express their sincere appreciation to the colleagues at Hulteberg Chemistry & Engineering AB for fruitful discussions. We are also grateful to Dr Fanny Langschwager at Rostock University for providing the VGO and for performing the heating value and cold flow characteristics analysis, and Dr Katharina Groβmann at the German Aerospace Center (DLR) for elemental analysis. Jane L. Sam and Toke M. Schou at Enorm Biofactory are acknowledged for supplying the lipid material used in this study. Z. S. Q. and O. Y. A. gratefully acknowledge the support provided by the Interdisciplinary Research Center for Refining & Advanced Chemicals at KFUPM.References
- U.S. Energy Information Administration, World Jet Fuel Consumption, https://www.eia.gov/international/data/world, accessed 2025-08-13.
-
Statistical Office of the European Communities, Energy, Transport and Environment Statistics - 2020 Edition, Publications Office of the European Union, Luxembourg, 2020 Search PubMed
.
- Clean Skies for Tomorrow: Sustainable Aviation Fuels as a Pathway to Net-Zero Aviation, McKinsey & Company, Inc, https://www.weforum.org/publications/clean-skies-for-tomorrow-sustainable-aviation-fuels-as-a-pathway-to-net-zero-aviation/Publisher Search PubMed.
- European Environment Agency and European Union Aviation Safety Agency, European Aviation Environmental Report 2022, Publications Office of the European Union, 2023 Search PubMed.
- Mobility and Transport, Air, 2024, https://transport.ec.europa.eu/transport-modes/air_en, accessed 2024-09-13.
- European Commission, European Green Deal: new law agreed to cut aviation emissions by promoting sustainable aviation fuels, 2023, https://ec.europa.eu/commission/presscorner/detail/en/ip_23_2389, accessed 2024-11-17.
- Sustainable Aviation Fuel Grand Challenge, https://www.energy.gov/eere/bioenergy/sustainable-aviation-fuel-grand-challenge, accessed 2024-09-13.
- M. R. L. Leal, M. V. Galdos, F. V. Scarpare, J. E. Seabra, A. Walter and C. O. Oliveira, Biomass Bioenergy, 2013, 53, 11–19 CrossRef
- S. Karatzos, J. S. van Dyk, J. D. McMillan and J. Saddler, Biofuels, Bioprod. Biorefin., 2017, 11, 344–362 CrossRef
-
G. Dragone, B. D. Fernandes, A. A. Vicente and J. A. Teixeira, Current Research, Technology and Education Topics in Applied Microbiology and Microbial Biotechnology, Formatex Research Center, Badajoz, Spain, 2010, vol. 2, pp. 1355–1366 Search PubMed
- J. P. Diebold, A review of the chemical and physical mechanisms of the storage stability of fast pyrolysis bio-oils, http://www.osti.gov/servlets/purl/753818/, accessed 2024-09-13.
- T. M. Dabros, M. Z. Stummann, M. Høj, P. A. Jensen, J.-D. Grunwaldt, J. Gabrielsen, P. M. Mortensen and A. D. Jensen, Prog. Energy Combust. Sci., 2018, 68, 268–309 CrossRef
- M. A. Peters, C. T. Alves and J. A. Onwudili, Energies, 2023, 16, 6100 CrossRef
- A. T. Hoang, M. Tabatabaei and M. Aghbashlo, Energy Sources, Part A, 2020, 42, 2923–2943 CrossRef
- D02 Committee, Specification for Aviation Turbine Fuel Containing Synthesized Hydrocarbons, http://www.astm.org/cgi-bin/resolver.cgi?D7566-24D, accessed 2025-08-14.
- E. Furimsky, Appl. Catal., A, 2000, 199, 147–190 CrossRef
- G. Huber and A. Corma, Angew. Chem., Int. Ed., 2007, 46, 7184–7201 CrossRef PubMed
- P. Zhu, O. Y. Abdelaziz, C. P. Hulteberg and A. Riisager, Curr. Opin. Green Sustainable Chem., 2020, 21, 16–21 CrossRef
- S. Khan, A. N. Kay Lup, K. M. Qureshi, F. Abnisa, W. M. A. Wan Daud and M. F. A. Patah, J. Anal. Appl. Pyrolysis, 2019, 140, 1–24 CrossRef
- D. Kubička, M. Bejblová and J. Vlk, Top. Catal., 2010, 53, 168–178 CrossRef
- Enorm - larvae for green development of the agricultural and food industry, https://enormbiofactory.com/, accessed 2024-12-04.
- Opening of the Largest Insect Factory in Northern Europe, https://www.vilofoss.com/News/Opening-of-the-Largest-Insect-Factory-in-Northern-Europe, accessed 2024-12-04.
- J. Selimi, F. Langschwager, P. Tunå, C. P. Hulteberg and O. Y. Abdelaziz, Biomass Convers. Biorefin., 2025, 15, 5523–5531 CrossRef
- T. A. Kristensen, C. P. Hulteberg, R. L. Wallenberg, O. Y. Abdelaziz and S. Blomberg, Energy Fuels, 2024, 38, 9827–9835 CrossRef CAS PubMed
-
C. J. Geankoplis, A. A. Hersel and D. H. Lepek, Transport Processes and Separation Process Principles, Prentice Hall, Boston, MA, 2018 Search PubMed
-
U. E. P. Agency, Method 200.8: Determination of Trace Elements in Waters and Wastes by Inductively Coupled Plasma-Mass Spectrometry, U.s. environmental protection agency technical report, 1994 Search PubMed
-
Stat-Ease, Inc., Design-Expert® Software, Stat-Ease, Inc., Minneapolis, MN, USA, 2021 Search PubMed
-
H. Topsøe, B. S. Clausen and F. E. Massoth, Catalysis: Science and Technology, Springer Berlin Heidelberg, Berlin, Heidelberg, 1996, pp. 1–269 Search PubMed
- J. Blažek, D. Toullis, P. Straka, M. Staš and P. Šimáček, Catalysts, 2021, 11, 1093 CrossRef
- R. Sotelo-Boyás, Y. Liu and T. Minowa, Ind. Eng. Chem. Res., 2011, 50, 2791–2799 CrossRef
- S. Bezergianni, A. Dimitriadis, A. Kalogianni and P. A. Pilavachi, Bioresour. Technol., 2010, 101, 6651–6656 CrossRef CAS
- A. Afshar Taromi and S. Kaliaguine, Fuel Process. Technol., 2018, 171, 20–30 CrossRef CAS
- S. K. Tanneru and P. H. Steele, Fuel, 2014, 133, 326–331 CrossRef CAS
- A. A. Hebish, N. W. M. Zulkifli, N. M. Julkapli and M. F. A. Patah, Appl. Catal. O: Open, 2024, 190, 206926 CAS
- K. Treusch, N. Schwaiger, K. Schlackl, R. Nagl, A. Rollett, M. Schadler, B. Hammerschlag, J. Ausserleitner, A. Huber, P. Pucher and M. Siebenhofer, React. Chem. Eng., 2018, 3, 258–266 RSC
- M. Al-Sabawi and J. Chen, Energy Fuels, 2012, 26, 5373–5399 CrossRef CAS
- D. S. Stratiev, I. K. Shishkova, R. K. Dinkov, I. P. Petrov, I. V. Kolev, D. Yordanov, S. Sotirov, E. Sotirova, V. Atanassova, S. Ribagin, K. Atanassov, D. D. Stratiev, S. Nenov, L. Todorova-Yankova and K. Zlatanov, Resources, 2021, 10, 71 CrossRef
- L. Alemán-Vázquez, J. L. Cano-Domínguez and J. García-Gutiérrez, Procedia Eng., 2012, 42, 532–539 CrossRef
- F. Zhao, T. Xu, G. Zhu, K. Wang, X. Xu and L. Liu, Sustainable Energy Fuels, 2022, 6, 1866–1890 RSC
- T. M. H. Dabros, A. Gaur, D. G. Pintos, P. Sprenger, M. Høj, T. W. Hansen, F. Studt, J. Gabrielsen, J.-D. Grunwaldt and A. D. Jensen, Appl. Catal., A, 2018, 551, 106–121 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2026 |