DOI:
10.1039/D5SD00155B
(Paper)
Sens. Diagn., 2026,
5, 337-347
π-Spacer modulation unlocking tunable photophysics and organelle-specificity in 2,6 disubstituted BODIPY-cyanostilbene fluorophores
Received
19th August 2025
, Accepted 30th November 2025
First published on 3rd December 2025
Abstract
Simultaneous visualization of two distinct organelles using a single fluorescent probe offers powerful opportunities for unravelling cellular signalling pathways and inter-organelle interactions. However, this goal remains challenging due to the complex microenvironment of subcellular compartments and the absence of robust design principles. In this study, we designed and synthesized two new donor–acceptor (D–A) BODIPY-cyanostilbene fluorophores, functionalized at the 2,6-positions with tailored π-spacers: phenyl (compound 7) and thiophene (compound 8) units. Strategic π-spacer engineering enabled precise control over both optical behavior and organelle-targeting selectivity. The thiophene analogue (compound 8) exhibited a remarkable Stokes shift (∼112 nm) and far-red/NIR emission (600–800 nm), overcoming two key limitations of conventional BODIPYs, particularly visible-range emission (490–510 nm) that restricts deep-tissue imaging, and narrow Stokes shifts that lead to spectral overlap and self-quenching. In contrast, the phenyl analogue (compound 7) emitted at 570 nm with a moderate 35 nm Stokes shift, primarily due to the extended π-conjugation framework. The photophysical properties were supported by DFT calculations (B3LYP/6-31G(d), Gaussian 09W). Both probes showed excellent photostability, minimal cytotoxicity, high chemical and pH stability, and non-interference from competitive species, making them highly suitable for cellular studies. Interestingly, both probes achieved dual-targeting of lipid droplets (LDs) and lysosomes, enabling real-time monitoring of dynamic changes in these organelles. This work highlights π-spacer engineering as a powerful strategy to unlock far-red/NIR dual-organelle probes, paving the way for advanced organelle-targeting imaging, and early-stage disease diagnostics.
1. Introduction
Fluorescence-based imaging using fluorescent probes has become a powerful platform for early disease diagnosis and surgical navigation, owing to its ability to offer real-time, non-invasive visualization of molecular interactions with high specificity and spatial resolution.1,2 However, the clinical translation of many fluorescent biomarkers, including several commercially available probes, remains limited due to their low signal-to-background ratios, poor biocompatibility, low fluorescence quantum yields, restricted structural tunability, short-wavelength emission, and inadequate photo, chemical, and thermal stability.3–5 Such drawbacks hinder precise drug delivery to target sites, reduce therapeutic efficacy, necessitating higher doses and intensify the risk of damage to healthy tissues during surgery. Among several classes of organic fluorophores, BODIPY (4,4′-difluoro-4-bora-3a,4a-diaza-s-indacene) dyes have attracted great attention due to their excellent structural versatility, high fluorescence quantum yields, excellent photostability, sharp emission profiles, and tunable optical properties.6–8 Consequently, they have been widely utilized in diverse research domains, including photosensitizers, lasers, optoelectronics, chemosensors, and bioimaging applications.9–16 Yet, conventional BODIPY derivatives suffer from two major limitations: emission predominantly in the visible range (490–510 nm), which restricts deep-tissue imaging, and a typically narrow Stokes shift. The rigidity of the BODIPY framework suppresses non-radiative decay, favoring locally excited (LE) states over charge-transfer (CT) interactions, which yields a minimal Stokes shift.6d,7a As a result, spectral overlap and self-quenching significantly reduce their effectiveness in biological imaging applications.
To overcome these drawbacks, extensive research has focused on molecular engineering strategies aimed at extending absorption and emission toward longer wavelengths, particularly into the near-infrared (NIR-I/NIR-II) regions, while achieving larger Stokes shifts. This facilitates deeper tissue penetration, reduced scattering, and minimal interference from biological autofluorescence, all of which are crucial for accurate and efficient fluorescence bioimaging.17 Among various approaches, the design of donor–acceptor (D–A), D–π–A, or D–A–D systems has proven especially effective. This strategy involves the incorporation of electron-donating and electron-withdrawing substituents, typically aromatic or heteroaromatic rings at different positions within the BODIPY framework.18 Such structural modulations enhance intramolecular charge transfer (ICT) interactions, leading to enlarged Stokes shifts, tunable excitation/emission characteristics through π-conjugation extension, and the ability to tailor organelle specificity through rational molecular design.19–23
While most studies have focused on substitutions at the meso, 1,7; 3,5; or 1,3,5,7-positions of the BODIPY core, only a limited number have explored donor substitution at the 2,6-positions, a promising yet unexplored strategy for improving BODIPY photophysical performance. Building on our previous work on lipid droplet (LD)-targeting BODIPY-cyanostilbene probes, where meso-substitution yielded minimal influence on optical behavior, we extended this concept by developing D–A conjugated BODIPY-cyanostilbene fluorophores featuring π-spacer substitution at the 2,6-positions.24,25 By introducing phenyl or thiophene spacers, we effectively tuned the photophysical characteristics and organelle-targeting behavior of these probes. As anticipated, the stronger electron-donating nature of the thiophene unit enhanced ICT effects, resulting in a remarkable red shift and a substantially increased Stokes shift. Notably, the thiophene-based derivative (compound 8) achieved a large Stokes shift (∼112 nm) and far-red/NIR emission (600–800 nm), ideal for bioimaging, while the phenyl analogue (compound 7) emitted at 570 nm with a Stokes shift of 35 nm. Both probes exhibited excellent stability, high photostability, and negligible cytotoxicity. They effectively enabled dual-targeting of LDs and lysosomes, organelles with distinct yet interrelated roles in cellular homeostasis.26,27 Additionally, their dual functionality enables tracking of dynamic changes in these organelles, providing valuable insight into organelle behavior under various physiological conditions.28 Overall, these findings underscore the role of π-spacer engineering in tuning BODIPY photophysics and biological performance, offering valuable design principles for next-generation far-red/NIR dual-organelle probes with potential applications in early disease diagnosis, mechanistic cell biology, and targeted theranostics.
2. Results and discussion
2.1 Synthesis and spectroscopic analysis
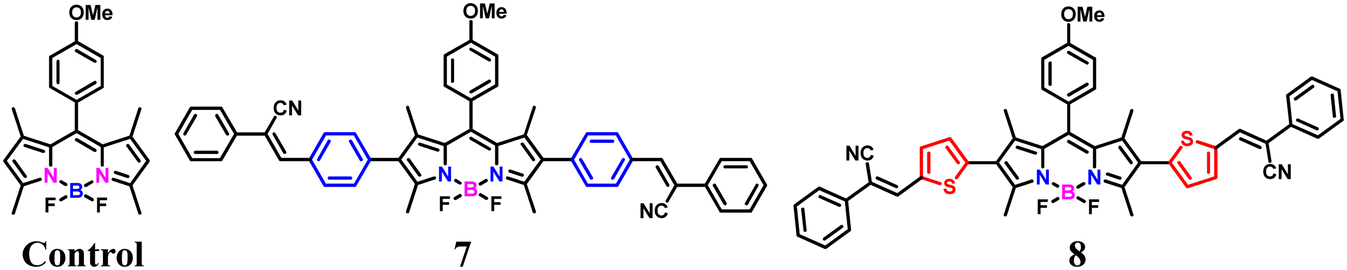
In the present work, we developed two target fluorophores (7 and 8) by fine tuning the 2,6 positions of the BODIPY core by introducing different π spacer units especially phenyl and thienyl moieties and tried to investigate the effect of different donor moieties on the optical characteristics of our target fluorophores. The synthetic protocol followed to synthesize the target fluorophores is outlined in Scheme 1 and the detailed synthetic procedures are provided in the experimental section (SI). Initially, a Knoevenagel condensation reaction between benzyl cyanide and 4-bromo benzaldehyde as well as 5-bromothiophene-2-carbaldehyde resulted in intermediates 1 and 3 in excellent yields.24,25,29 Further, they underwent a palladium-catalyzed Suzuki–Miyaura cross-coupling reaction yielding compounds 2 and 4 in moderate yields. The BODIPY core (compound 5) was synthesized according to previous literature reports, i.e. anisaldehyde was subjected to a trifluoroacetic acid (TFA)-catalyzed condensation reaction with 2,4 dimethyl pyrroles resulting in a dipyrromethane intermediate, followed by oxidation with DDQ (2,3-dichloro-5,6-dicyano-1,4-benzoquinone) and complexation with BF3·Et2O in dichloromethane (DCM), resulting in the formation of the stable compound 5.30 Next, compound 5 was reacted with N-iodo succinimide (NIS) yielding 2,6-diiodo BODIPY (compound 6).31 Finally, compound 6 underwent palladium-catalyzed Suzuki–Miyaura cross-coupling reactions with the intermediates 2 and 4, resulting in the synthesis of our target 2,6-di-substituted BODIPY fluorophores 7 and 8. The newly synthesized compounds were well characterized using various analytical techniques such as 1H NMR, 13C NMR, 11B NMR, and 19F NMR spectroscopy, as well as high-resolution mass spectrometry (HRMS).
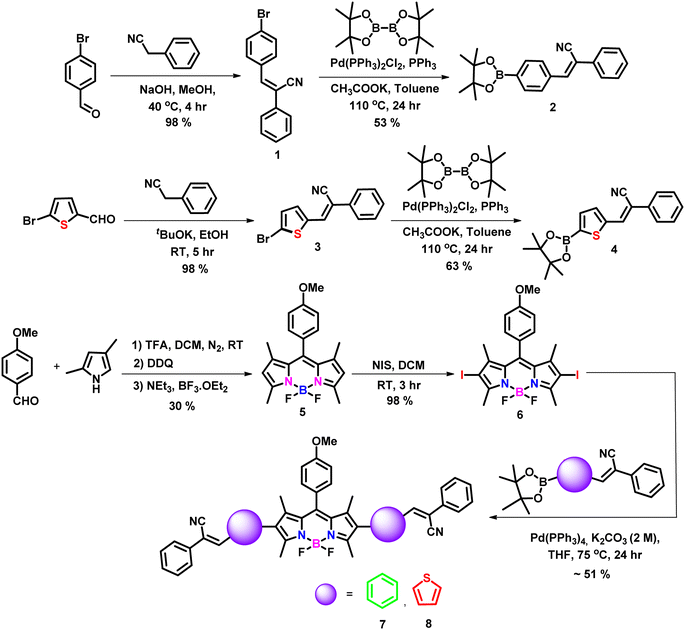 |
| | Scheme 1 Synthetic scheme adopted for the target molecules. | |
2.2 Photophysical studies
The photophysical properties of the target fluorophores (Chart 1) were initially studied by using UV-vis and fluorescence spectroscopic techniques in a DCM solvent system and the corresponding spectra are shown in Fig. 1. The summary of the spectral data is included in Table 1. The UV-vis absorption spectra (10 μM) of the control and compounds 7 and 8 exhibit two prominent bands in which the lower energy band corresponds to the S0 → S1 transition of the BODIPY core, while the higher energy band is in the range 300 to 460 nm. The maximum absorption of the control and compounds 7 and 8 were at 500 nm, 535 nm, and 543 nm, respectively. Compared to the unsubstituted BODIPY (control), the absorption maxima (λmax) of our target fluorophores are red shifted, i.e. probe 7 exhibits a 35 nm red shift while probe 8 exhibits 43 nm. This might be due to the extended π-conjugation framework of the target fluorophores. In order to further understand the optical properties, we studied the emission characteristics of our target molecules by exciting at 530 nm in DCM solvent (1.6 μM) as shown in Fig. 1 and S16. For the control, a typical small Stokes shift (11 nm) was observed, with the excitation and emission maxima at 485 and 512 nm, respectively. Meanwhile, compound 7, i.e. the phenyl-substituted derivative, exhibited emission maxima at 570 nm with a Stokes shift of 35 nm. This might be due to the efficient conjugation between the phenyl group and the BODIPY core.27 Interestingly, a much larger Stokes shift nearly 112 nm was observed for compound 8 with the thiophene–cyanostilbene functionalization at the 2,6-position of the BODIPY core, with emission maxima at 655 nm. The large Stokes shift observed in compound 8 may result from a de novo mechanism, as reported previously for BODIPY fluorophores with thienyl substituents at the 2,6- or 3,5-positions.32,33 Upon photoexcitation, the thienyl substituted derivatives likely experience a substantial geometric relaxation in the S1 excited state following vertical excitation from the ground S0 state, leading to a smaller emission energy and thus a larger Stokes shift.33 In contrast, 8-thienyl substitution often results in a smaller Stokes shift due to the rigidity of the BODIPY core, which suppresses non-radiative decay and favors locally excited (LE) transitions over charge-transfer (CT) states.34 Weak electronic coupling between the meso substituent and the BODIPY core further limits spectral shifts, underscoring the critical role of substituent position. Additionally, strong electron-donating thienyl groups can enhance intramolecular charge transfer (ICT) through π-conjugation extension, leading to both bathochromic emission and a large Stokes shift.35 Upon photoexcitation to the Franck–Condon state, structural relaxation and solvation favor electron transfer from donor to acceptor regions, reducing the HOMO–LUMO energy gap and resulting in longer-wavelength emission.18,21,35 The low-lying CT state is also more sensitive to environmental polarity, which can account for the reduced fluorescence quantum yield observed for compound 8 compared to the phenyl analogue (compound 7), as shown in Table 1. This is consistent with previous reports showing that greater molecular flexibility and enhanced ICT promote non-radiative decay and decrease fluorescence efficiency in the case of thienyl derivatives.32,33 The increased dipole moment in the excited state also enhances solute–solvent interactions, stabilizing the CT state in polar environments. This further lowers the excited-state energy, leading to red-shifted emission and a larger Stokes shift, while decreasing fluorescence quantum yield in polar solvents, as evidenced in Table S1 for compound 8 relative to the phenyl derivative. Moreover, DFT calculations further support these observations: in compound 7, both the HOMO and LUMO are localized predominantly on the BODIPY core, consistent with LE-type transitions and a small Stokes shift, whereas in compound 8, the HOMO is localized on the donor thienyl group and the LUMO on the BODIPY acceptor core. This spatial separation generates a polarized ICT state with a significant dipole moment, promoting charge separation and excited-state relaxation, consistent with the experimentally observed polarity-sensitivity and large Stokes shift.21 Additionally, the HOMO energy of the thienyl-substituted derivative is higher than that of the phenyl analogue, reflecting stronger electron-donating character, which contributes to a reduced HOMO–LUMO gap (2.61 eV for compound 8vs. 2.80 eV for compound 7) due to enhanced ICT and improved coplanarity with extended conjugation.32a In contrast, compound 7 may also undergo geometric relaxation but the extent of this relaxation may not significantly impact the energy levels, resulting in a comparatively smaller Stokes shift. For the control compound, the rigid molecular framework ensures minimal structural changes between the ground and excited states, leading to limited geometry relaxation and a correspondingly small Stokes shift.33 Further, the unsubstituted BODIPY exhibits green colour emission, while compounds 7 and 8 exhibit orangish red and purple colour emission, respectively, as shown in Fig. S17. This clearly demonstrates that extension of conjugation through substitution at the 2,6-positions of the BODIPY core leads to a pronounced bathochromic shift in the emission profile, effectively tuning the fluorescence from green to deep red. The red-shifted absorption and emission of the target fluorophores particularly compound 8, which exhibits a large Stokes shift, make them highly promising for bioimaging applications, as such features minimize self-absorption and suppress the inner-filter effect commonly observed in fluorophores with small Stokes shifts.
 |
| | Chart 1 Chemical structures of the control and compounds 7 and 8. | |
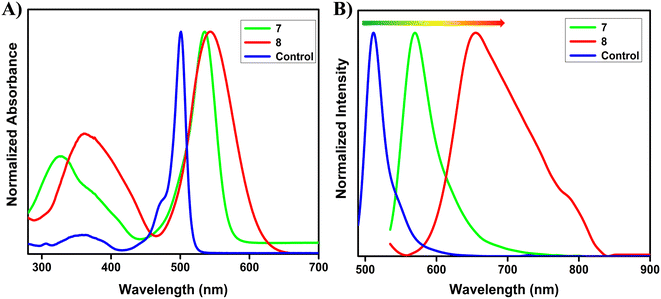 |
| | Fig. 1 A) UV-vis absorption (10 μM) and B) emission spectra of compounds 7 and 8 (1.6 μM, λex = 530 nm), and the control (1.6 μM, λex = 485 nm) in DCM. | |
Table 1 Optical properties of the control and compounds 7 and 8
| S no |
Compounds |
Absorbance λabs (nm)/ε (× 104) |
Emission λem (nm) |
Φ
F
|
Stokes shift (cm−1) |
|
Quantum yields were determined using rhodamine B in ethanol as a standard (ΦF = 0.49 in ethanol) and using the following formula ΦF = ΦF × I/IR × AR/A × η2/ηR2 where ΦF = quantum yield, I = integral area of the emission peak, A = absorbance at λex, η = refractive index of the solvent.
|
| 1 |
Control
|
350 (0.4), 501 (5.4) |
512 |
0.43 |
429 |
| 2 |
7
|
329 (2.0), 535 (4.9) |
570 |
0.19 |
1148 |
| 3 |
8
|
361 (1.7), 543 (3.1) |
655 |
0.04 |
3149 |
2.3 Solvent-dependent studies
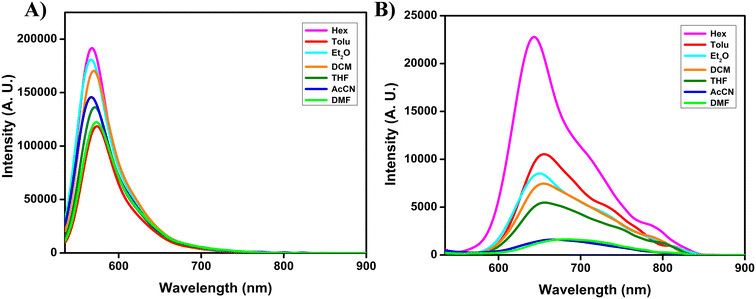
To gain deeper insight into the D–A characteristics and the ICT behavior of the synthesized fluorophores, we examined their solvatochromic responses. The results are summarized in Table S1. For compound 7, the absorption and emission spectra were mostly insensitive to solvent polarity, showing a variation of less than 5 nm when moving from non-polar hexane to polar DMF (Fig. 2, S18 and S19). This observation indicates that both the ground and excited states of compound 7 are relatively unaffected by the surrounding medium, suggesting minimal ICT contribution in its photophysical behavior. In contrast, compound 8 exhibited a clearly different response. While its absorption profile remained nearly unchanged across solvents of varying polarity (Fig. S18), its emission spectra displayed a substantial red shift of ∼39 nm, accompanied by a significant reduction in fluorescence intensity, broader band shapes, and diminished vibronic features as solvent polarity increased (Fig. 2 and S19). These findings strongly suggest stabilization of charge-transfer excited states in polar solvents, consistent with a pronounced ICT character for compound 8. To further explore their photophysical behavior, we investigated the aggregation properties of compounds 7 and 8 in THF/water mixed solvent systems (Fig. S20). For compound 7, fluorescence intensity remained largely unchanged up to a water fraction (fw) of 50%, beyond which it dramatically decreased. This quenching can be attributed to aggregation-caused quenching (ACQ) effects, likely arising from intermolecular π–π stacking interactions and enhanced non-radiative decay pathways. Similarly, compound 8 also exhibited a sharp reduction in emission intensity with increasing fw values, again consistent with ACQ behavior.
 |
| | Fig. 2 Emission spectra of compounds 7 (A) and 8 (B) in solvents of different polarity (1.6 μM, λex = 530 nm). | |
2.4 Competitive and stability studies
Next, we evaluated the stability and performance of our target fluorophores in the presence of biologically relevant factors, including physiologically important metal ions, amino acids, reactive oxygen species, varying pH conditions, and common anions. As shown in Fig. 3 and S21, both compounds exhibited negligible variations in fluorescence intensity under these conditions. Such excellent tolerance towards external perturbations underscores their strong anti-interference nature, a critical prerequisite for reliable use in complex biological systems and bioimaging applications. Since different cellular organelles maintain distinct pH environments, we further examined the effect of pH fluctuations on the emission characteristics of our probes. As depicted in Fig. 3 and S22, the fluorescence of both compounds remained essentially unaltered across a wide pH range (4–10), demonstrating their excellent pH stability. This property is particularly advantageous, as it enables consistent fluorescence signals irrespective of intracellular localization or pH microenvironments. Given that conventional BODIPY dyes are often susceptible to nucleophilic attack, leading to core instability, we also assessed the chemical robustness of our probes towards strong anionic species. Specifically, compounds 7 and 8 were dissolved in DMSO with 10 equivalents of TFA for 24 h. Remarkably, their emission spectra (Fig. S23) remained unchanged, confirming that the BODIPY scaffold preserved its structural integrity even under acidic conditions, without undergoing BF2 dissociation or pyrrolic core degradation.36 Collectively, these results highlight the superior chemical and photophysical stability of compounds 7 and 8. Their resistance to environmental factors such as solvent polarity, pH fluctuations, nucleophiles, and biologically relevant species establishes them as robust fluorophores with significant potential for bioimaging applications, where maintaining signal integrity and environmental resilience is crucial.
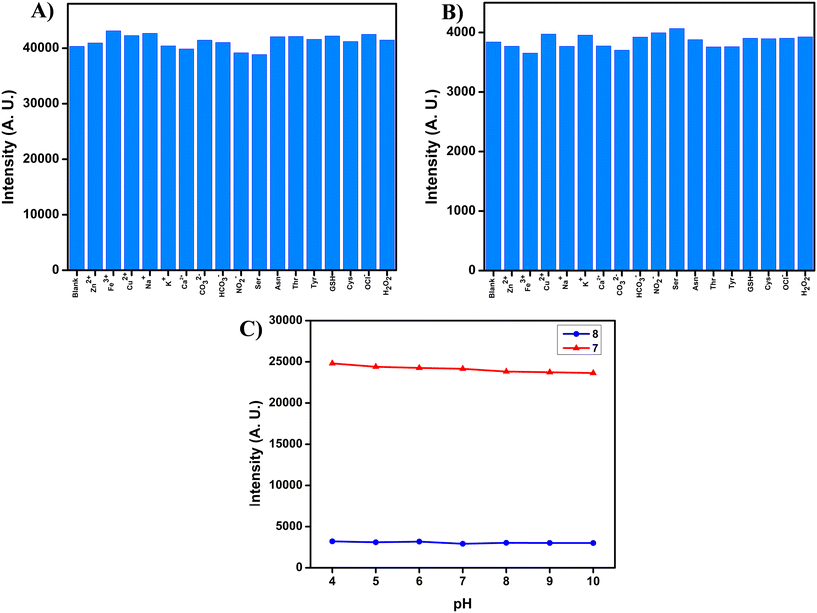 |
| | Fig. 3 Emission spectra of compounds (A) 7 and (B) 8, in a mixture of THF/PBS (pH 7.4, 1 mM) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio) with various competitive species (100 μM) (blank, Zn(OAc)2, FeCl3, Cu(OAc)2, NaCl, KCl, CaCl2, K2CO3, NaHCO3, NaNO2, Ser, Asn, Thr, Tyr, GSH, Cys, OCl−, and H2O2). (C) Emission studies of compounds 7 and 8 at different pH values in DMSO solvent (1.6 μM, λex = 530 nm). :4 ratio) with various competitive species (100 μM) (blank, Zn(OAc)2, FeCl3, Cu(OAc)2, NaCl, KCl, CaCl2, K2CO3, NaHCO3, NaNO2, Ser, Asn, Thr, Tyr, GSH, Cys, OCl−, and H2O2). (C) Emission studies of compounds 7 and 8 at different pH values in DMSO solvent (1.6 μM, λex = 530 nm). | |
2.5 Theoretical calculations
Furthermore, to correlate the photophysical properties with the electronic and structural factors, we have carried out density functional theory (DFT) calculations using the Gaussian 09W suite without any symmetry constraints at the B3LYP/6-31G(d) level of theory in the gas phase. In order to understand the extent of electronic coupling or communications between the donor and acceptor chromophores, we have calculated the dihedral angle between the BODIPY core and the aryl/thienyl moiety. As shown in Fig. S24, the dihedral angle between the BODIPY and the phenyl ring is around ∼51° while in the case of BODIPY and the thienyl moiety it is around ∼44°. The data suggest that compound 8 with a more electron rich thienyl group acts as a better conjugation linker as compared to phenyl because of the smaller torsional angle which results in better coplanarity between the two chromophores and large Stokes shift.33 This is further supported by the analysis of electronic distributions in the frontier molecular orbitals (FMOs), as illustrated in Fig. 4 and S25. In the ground state, both the HOMO and LUMO of compound 7 are predominantly localized on the BODIPY core, consistent with LE-type transitions and a small Stokes shift. In contrast, for compound 8, the HOMO is concentrated across the entire molecule with significant contributions from the thienyl donor groups, while the LUMO remains confined to the BODIPY acceptor core. This spatial separation leads to a polarized ICT state with a pronounced dipole moment, facilitating charge separation and excited-state relaxation, in line with the experimentally observed polarity sensitivity and large Stokes shift.21 Additionally, the HOMO energy level of the thienyl-substituted derivative is higher than that of the phenyl analogue, reflecting the stronger electron-donating nature of the thienyl units at the 2,6-positions. This results in a reduced HOMO–LUMO energy gap (Eg) for compound 8 (2.61 eV) compared to compound 7 (2.80 eV), which arises from enhanced ICT and improved coplanarity with extended conjugation.32a These theoretical findings are in excellent agreement with experimental data, highlighting the critical role of π-spacer modification in tuning the photophysical properties of the designed probes.
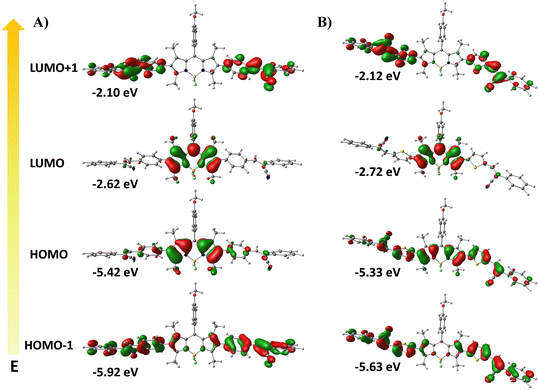 |
| | Fig. 4 Frontier molecular orbitals (MOs) for compounds (A) 7 and (B) 8 in the ground state at the B3LYP/6-31G(d) level with Gaussian 09. | |
2.6 Co-localization imaging
To assess the biological applicability of our target molecules, we first evaluated their cytotoxicity using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. HeLa cells (5 × 103 cells per well) were seeded in 96-well plates. After 24 h, the cells were treated with varying concentrations of the target probes and incubated for 16 h under controlled conditions. After incubation, the cells were washed three times with PBS, treated with MTT solution (0.5 mg mL−1) for 2 h, and the resulting absorbance was recorded at 590 nm. As shown in Fig. S26, the cell viability remained above 50% up to 5 μM for both target probes, indicating low cytotoxicity at imaging concentrations. Although a gradual decline in viability was observed at higher concentrations, it is noteworthy that effective cellular imaging was achieved at 500 nM, well within the biocompatible range. We then explored the intracellular distribution of the probes via confocal laser scanning microscopy (CLSM). After 30 min of incubation in HeLa cells, both probes displayed distinct red-fluorescent spots dispersed throughout the cytoplasm, similar to LD targeting. To verify this, we performed co-localization experiments with commercial LD-specific tracker BODIPY 493/503 (LDs), as shown in Fig. 5 and S27. The red fluorescence signals from the probes exhibited excellent overlap with the green fluorescence of BODIPY 493/503, with Pearson correlation coefficients (PCC) of approximately 0.91 for compound 7 and 0.86 for compound 8, confirming their LD-targeting behavior. To further evaluate the organelle specificity, additional co-localization experiments were conducted using commercial Lysotracker-Green (lysosomes) and DAPI (nucleus). No overlap with DAPI was observed, indicating that both compounds were excluded from the nucleus. In contrast, partial overlap with Lysotracker-Green was detected, with PCC values of 0.70 and 0.67 for compounds 7 and 8, respectively. These observations suggest that the probes possess dual-organelle targeting characteristics toward LDs and lysosomes. These findings demonstrate that the π-spacer unit not only influences photophysical properties but also governs organelle-targeting behavior. Furthermore, we have conducted a detailed evaluation of the photostability of the target probes under continuous laser irradiation for up to 100 minutes, until cell rupture occurred as illustrated in Fig. S28. HeLa cells stained with the target probes (500 nM) and the reference dye BODIPY 493/503 (200 nM) were continuously excited at 568 nm, and the fluorescence intensity was recorded at 5-minute intervals. Both probes exhibited high photostability, with minimal changes in fluorescence intensity for more than 60 minutes. A subsequent decrease in fluorescence intensity observed after this period was attributed to cell bursting rather than photodegradation. Collectively, these results highlight that rational π-spacer engineering enables simultaneous fine-tuning of photophysical characteristics and bioimaging performance, providing a strategic foundation for the development of next-generation organelle-specific fluorescent probes.
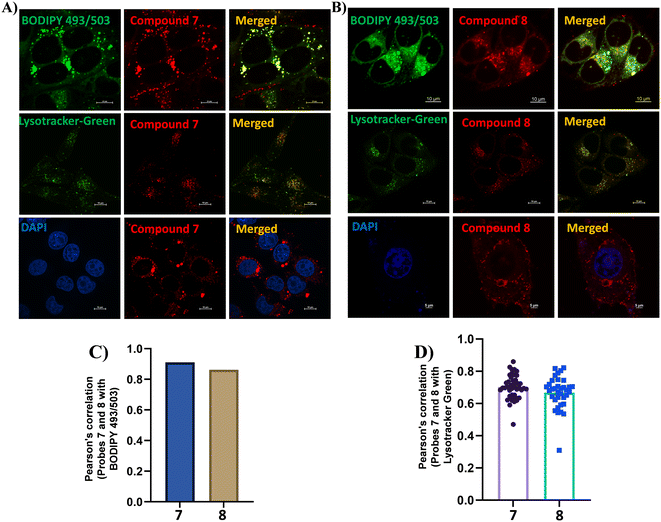 |
| | Fig. 5 Confocal fluorescence microscopy images showing intracellular co-localization in HeLa cells. Cells were stained with 200 nM commercially available trackers such as LD-targeting BODIPY 493/503 (Ex: 488 nm, Em: 503–551 nm), lysosome-targeting Lysotracker-Green (Ex: 488 nm, Em: 503–551 nm), and nucleus-staining DAPI (Ex: 405 nm, Em: 420–488 nm) (left), 500 nM compounds 7 (A) and 8 (B) (Ex: 561 nm, Em: 585–628 nm) (middle), and the merged images (right). (C) Pearson correlation coefficient quantifying the extent of co-localization between compounds 7 and 8 with BODIPY 493/503. (D) Pearson correlation coefficient quantifying the extent of co-localization between compounds 7 and 8 with Lysotracker-Green. Scale bar, 10 μm and 3% laser power. | |
2.7 Tracking changes of LDs and lysosomes during cellular dysregulation
Next, we investigated the dynamic changes of LDs and lysosomes in living HeLa cells using our target probes under oleic acid (OA) and chloroquine (CQ) treatments. CQ, a lysosomotropic weak base, is known to elevate the lysosomal pH from approximately 4.5 to 6.5. By accumulating within lysosomes, CQ neutralizes their acidic environment and disrupts the autophagy-lysosomal pathway, leading to a marked reduction in lysosomal numbers.37 In this experiment, HeLa cells were pretreated with CQ (50 μM) for 30 minutes, followed by incubation with the target probes for an additional 30 minutes. As shown in Fig. 6, CQ treatment led to a pronounced reduction in fluorescence intensity, which can be attributed to the decreased number of lysosomes in comparison with the untreated control group. Further, the dynamic behavior of LDs was evaluated under OA treatment. As a nutrient-rich fatty acid, OA is known to stimulate lipid droplet biogenesis and promote lipid storage.38 HeLa cells were pretreated with OA (100 μM) for 30 minutes and then incubated with the probes (500 nM) for another 30 minutes. As illustrated in Fig. 7, OA-treated cells exhibited intense red fluorescence signals, indicating a pronounced increase in LD formation compared to the control group (cells incubated with probes alone).25 Collectively, these findings demonstrate that the developed probes can effectively visualize both the CQ-induced reduction of lysosomes and the OA-induced accumulation of LDs. This dual functionality highlights their potential as versatile fluorescent tools for real-time monitoring of dynamic organelle changes associated with cellular homeostasis.
 |
| | Fig. 6 Fluorescence images of HeLa cells co-stained with probes 7 (A) and 8 (B) (500 nM). (a) Control group treated with the probe alone for 30 min. (b) Cells were treated with CQ (50 μM) for 30 min and then followed by the probe for 30 min. Bar graph depicting the mean fluorescence intensity of probe 7 (C) and probe 8 (D) in the control versus CQ-treated HeLa cells, with error bars representing mean ± SEM, statistical significance annotations (p < 0.0001), and a note about sample sizes (n = 40 untreated, n = 45 treated) and the image scale bar (10 μm). Statistical significance was determined by unpaired two-tailed t-test. | |
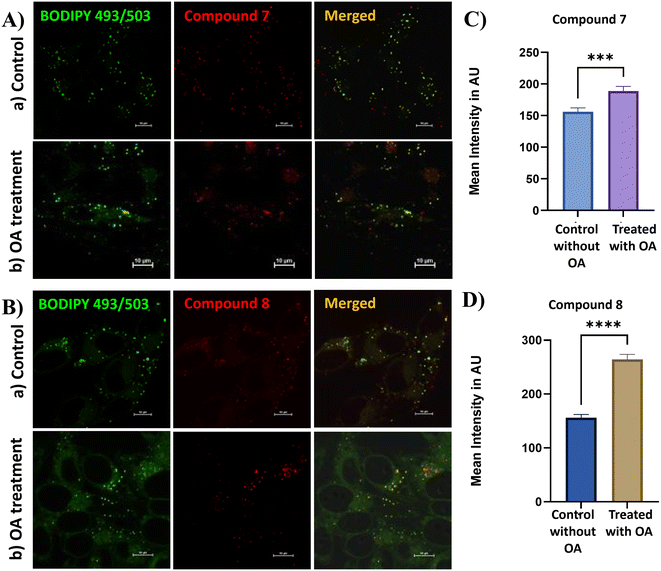 |
| | Fig. 7 Fluorescence images of HeLa cells co-stained with probes 7 (A) and 8 (B) (500 nM). (a) Control group treated with the probe alone for 30 min. (b) Cells were treated with OA (100 μM) for 30 min and then followed by the probe for 30 min. Bar graph depicting the mean fluorescence intensity of probe 7 (C) and probe 8 (D) in the control versus OA-treated HeLa cells, with error bars representing mean ± SEM, statistical significance annotations (p < 0.0001), and a note about sample sizes (n = 40 untreated, n = 45 treated) and the image scale bar (10 μm). Statistical significance was determined by unpaired two-tailed t-test. | |
Conclusions
In summary, we have successfully developed two 2,6-di-substituted BODIPY-cyanostilbene fluorophores through strategic modulation of π-spacer linkages with phenyl and thiophene units, in order to study how spacer selection influences photophysical behavior and biological applicability. The thiophene-based derivative (compound 8) displayed a remarkably large Stokes shift (∼112 nm) and far-red/NIR emission (600–800 nm). These features can be attributed to the enhanced conjugation and intrinsic molecular flexibility of the thienyl spacer, which facilitate substantial geometric relaxation in the excited state and ICT characteristics, ideal for high-contrast bioimaging. In comparison, the phenyl analogue (compound 7) exhibited emission at 570 nm with a smaller Stokes shift of 35 nm, consistent with its more rigid π-conjugated framework and reduced capacity for structural relaxation. Furthermore, both target probes demonstrated dual localization toward LDs and lysosomes, enabling tracking of the dynamic changes of these organelles upon CQ and OA treatment. This dual-targeting property, coupled with its far-red/NIR emission, highlights compound 8 as a promising candidate for multiplexed imaging and monitoring of subcellular processes. DFT calculations matches well with the experimental observations, revealing distinct conformational geometries and electronic distributions that emphasize the influence of structural rigidity on fluorescence behaviour. Importantly, both fluorophores exhibited high photostability, negligible cytotoxicity, robust chemical and pH stability, and strong resistance to interference from competing biological species. These features ensure reliable fluorescence signals in complex cellular environments a prerequisite for practical bioimaging applications. Overall, this work demonstrates that π-spacer engineering is a powerful strategy to fine-tune the optoelectronic properties of BODIPY-based fluorophores. This study not only enriches the design principles of functional BODIPY fluorophores but also paves the way for next-generation molecular probes with tailored photophysical and biological performance.
Conflicts of interest
The authors declare no conflict of interest.
Data availability
The data that support the findings of this study are available in the supplementary information (SI) of this article.
Supplementary information: these include 1H NMR, 13C NMR, HRMS characterization data for all the compounds, photophysical data, bioimaging results (cytotoxicity, co-localization, photostability), and DFT data. See DOI: https://doi.org/10.1039/d5sd00155b.
Acknowledgements
C. A. S. P. gratefully acknowledges the Science and Engineering Research Board (SERB) for the EEQ grant (EEQ/2021/000180). C. K. sincerely thanks the Department of Science and Technology, New Delhi, for awarding the DST-Inspire Fellowship (IF210349). C. K. and C. A. S. P. also thank NIT Calicut for the NMR (CMC), HRMS (DST-FIST) and HPC (CCMS) facility. K. M. acknowledges the funding support from the Science and Engineering Research Board (SERB), Government of India, for the Core Research Grant (SERB/CRG/2022/006517) and SNIoE core funding. S. B. sincerely thanks the SNIoE PhD fellowship. S. B. and K. M. also acknowledge the SNU DST-FIST grant [SR/FST/LS-1/2017/59(c)] for the confocal microscopy facility. We also thank Rajan Singh for his help at the confocal microscopy facility at SNIoE.
References
- H. Zhu, J. Fan, J. Du and X. Peng, Acc. Chem. Res., 2016, 49, 2115–2126 CrossRef CAS PubMed.
- D. Seah, Z. Cheng and M. Vendrell, ACS Nano, 2023, 17, 19478–19490 CrossRef CAS PubMed.
- T. Kowada, H. Maeda and K. Kikuchi, Chem. Soc. Rev., 2015, 44, 4953–4972 RSC.
- Y. Ye, J. Sun, F. Tang, R. Xie, H. Wang, A. Ding, S. Pan and L. Li, Adv. Sens. Res., 2023, 2, 2300057 CrossRef CAS.
- S. Wang, L. Gai, Y. Chen, X. Ji, H. Lu and Z. Guo, Chem. Soc. Rev., 2024, 53, 3976–4019 RSC.
-
(a) J. Karolin, L. B.-A. Johansson, L. Strandberg and T. Ny, J. Am. Chem. Soc., 1994, 116, 7801–7806 CrossRef CAS;
(b) R. Ziessel, G. Ulrich and A. Harriman, New J. Chem., 2007, 31, 496–501 RSC;
(c) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed;
(d) N. Boens, V. Leen and W. Dehaen, Chem. Soc. Rev., 2012, 41, 1130–1172 RSC.
-
(a) A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891–4932 CrossRef CAS PubMed;
(b) G. Ulrich, R. Ziessel and A. Harriman, Angew. Chem., Int. Ed., 2008, 47, 1184–1201 CrossRef CAS PubMed.
-
(a) P. Kaur and K. Singh, J. Mater. Chem. C, 2019, 7, 11361–11405 RSC;
(b) M. Poddar and R. Misra, Coord. Chem. Rev., 2020, 421, 213462–213683 CrossRef CAS;
(c) B. M. Squeo, L. Ganzer, T. Virgili and M. Pasini, Molecules, 2021, 26, 153–182 CrossRef CAS PubMed;
(d) F.-Z. Li, Z. Wu, C. Lin, Q. Wang and G.-C. Kuang, Results Chem., 2022, 4, 100384–100398 CrossRef CAS.
- H. Klfout, A. Stewart, M. Elkhalifa and H. He, ACS Appl. Mater. Interfaces, 2017, 9, 39873–39889 CrossRef CAS PubMed.
- I. S. Yadav and R. Misra, J. Mater. Chem. C, 2023, 11, 8688–8723 RSC.
- J. Bañuelos, F. L. Arbeloa, V. Martinez, M. Liras, A. Costela, I. G. Moreno and I. L. Arbeloa, Phys. Chem. Chem. Phys., 2011, 13, 3437–3445 RSC.
- A. Prakash, J. C. Janardhanan, V. K. Praveen, P. Radhakrishnan and A. Mujeeb, J. Lumin., 2022, 252, 119343 CrossRef CAS.
- L. Bonardi, H. Kanaan, F. Camerel, P. Jolinat, P. Retailleau and R. Ziessel, Adv. Funct. Mater., 2008, 18, 401–413 CrossRef CAS.
- X. Qi, E. J. Jun, L. Xu, S.-J. Kim, J. S. J. Hong, Y. J. Yoon and J. Yoon, J. Org. Chem., 2006, 71, 2881–2884 CrossRef CAS PubMed.
- A. V. Raveendran, P. A. Sankeerthana, A. Jayaraj and P. C. A. Swamy, Results Chem., 2022, 4, 100297–100351 CrossRef CAS.
- S. Chakraborty, J. Fluoresc., 2025, 35, 1875–1894 CrossRef CAS PubMed.
-
(a) P. Liu, X. Mu, X.-D. Zhang and D. Ming, Bioconjugate Chem., 2019, 31, 260–275 CrossRef PubMed;
(b) C. Kalarikkal and P. C. A. Swamy, J. Mater. Chem. B, 2025, 13, 12831–12868 RSC.
- C. S. Abeywickrama, Chem. Commun., 2022, 58, 9855–9869 RSC.
- S. Wu, Z. Ban, H. Tang, N. Ma, X. Ran, Q. Zhou, Y. Zhang, Z. Wang and X. Yu, Chem. Commun., 2025, 61, 10574–10577 RSC.
- W. Zheng, Z. Zhou, T. Zhu, Z. Zou, Q. Shan, Q. Huang, G. Wang and Y. Wang, Microchem. J., 2025, 210, 112968 CrossRef CAS.
- H. Chen, N. An, Y. Wang, G. Wang, S. Mukherjee, H. Bian, J. Ma, J. Liu and Y. Fang, J. Phys. Chem. B, 2023, 127, 2044–2051 CrossRef CAS PubMed.
- Y. Wu, C. Ge, Y. Zhang, Y. Wang and D. Zhang, Front. Chem., 2023, 11, 2296–2646 Search PubMed.
- C. S. Abeywickrama, K. A. Bertman and Y. Pang, Chem. Commun., 2019, 55, 7041–7044 RSC.
- C. Kalarikkal, Anjali, S. Bhattacharjee, K. Mapa and P. C. A. Swamy, J. Mater. Chem. B, 2025, 13, 1474–1486 RSC.
- C. Kalarikkal, Anjali, K. Mapa and P. C. A. Swamy, New J. Chem., 2025, 49, 16914–16924 RSC.
-
(a) J. A. Olzmann and P. Carvalho, Nat. Rev. Mol. Cell Biol., 2019, 20, 137–155 CrossRef CAS PubMed;
(b) A. Zadoorian, X. Du and H. Yang, Nat. Rev. Endocrinol., 2023, 19, 443–459 CrossRef CAS PubMed;
(c) F. Geltinger, L. Schartel, M. Wiederstein, J. Tevini, E. Aigner, T. K. Felder and M. Rinnerthaler, Molecules, 2020, 25, 5053–5083 CrossRef CAS PubMed;
(d) E. Jarc and T. Petan, Yale J. Biol. Med., 2019, 92, 435–452 CAS;
(e) M. Gao, X. Huang, B.-L. Song and H. Yang, Prog. Lipid Res., 2019, 75, 100989–100999 CrossRef CAS PubMed;
(f) M. F. Renne and H. Hariri, Front. Cell Dev. Biol., 2021, 9, 726261–726271 CrossRef PubMed;
(g) Y. Jin, Y. Tan, J. Wu and Z. Ren, Cell Death Discovery, 2023, 9, 254–262 CrossRef CAS PubMed;
(h) J. Z. Hsia, D. Liu, L. Haynes, R. Cruz-Cosme and Q. Tang, Microorganisms, 2024, 12, 647–670 CrossRef CAS PubMed.
-
(a) C. De Duve, B. C. Pressman, R. Gianetto, R. Wattiaux and F. Appelmans, Biochem. J., 1955, 60, 604–617 CrossRef CAS PubMed;
(b) F. Wang, R. Gómez-Sintes and P. Boya, Traffic, 2018, 19, 918–931 CrossRef CAS PubMed;
(c) S. R. Bonam, F. Wang and S. Muller, Nat. Rev. Drug Discovery, 2019, 18, 923–948 CrossRef CAS PubMed.
-
(a) F. Meng, J. Niu, H. Zhang, R. Yang, Q. Lu, Y. Yu, Z. Liu, G. Niu and X. Yu, Sens. Actuators, B, 2020, 329, 129148 CrossRef;
(b) M. Liu, Y. Bu, D. Wang, L. Tang, D. Hu, L. Li and X. Gan, Analyst, 2024, 149, 4953–4959 RSC;
(c) X. Zheng, W. Zhu, F. Ni, H. Ai, S. Gong, X. Zhou, J. L. Sessler and C. Yang, Chem. Sci., 2019, 10, 2342–2348 RSC;
(d) J. Zhuang, Y. Yu, R. Su, Q. Ma, N. Li and N. Zhao, Dyes Pigm., 2022, 208, 110809 CrossRef CAS.
- J. Hwang, K. Yang, I. S. Koo, D. D. Sung and I. Lee, Bull. Korean Chem. Soc., 2006, 27, 733–738 CrossRef CAS.
- M. Wang, Y. Zhang, T. Wang, C. Wang, D. Xue and J. Xiao, Org. Lett., 2016, 18, 1976–1979 CrossRef CAS PubMed.
- X. Li, X. Sun, H. Chen, X. Chen, Y. Li, D. Li, Z. Zhang, H. Chen and Y. Gao, Eur. J. Med. Chem., 2024, 264, 116035–116047 CrossRef CAS PubMed.
-
(a) S. Xuan, N. Zhao, X. Ke, Z. Zhou, F. R. Fronczek, K. M. Kadish, K. M. Smith and M. G. H. Vicente, J. Org. Chem., 2017, 82, 2545–2557 CrossRef CAS PubMed;
(b) N. Zhao, S. Xuan, B. Byrd, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Org. Biomol. Chem., 2016, 14, 6184–6188 RSC;
(c) N. Zhao, M. G. H. Vicente, F. R. Fronczek and K. M. Smith, Chem. – Eur. J., 2015, 21, 6181–6192 CrossRef CAS PubMed;
(d) N. Zhao, S. Xuan, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, J. Org. Chem., 2015, 80, 8377–8383 CrossRef CAS PubMed;
(e) H. Wang, F. R. Fronczek, M. G. H. Vicente and K. M. Smith, J. Org. Chem., 2014, 79, 10342–10352 CrossRef CAS PubMed.
-
(a) Y. Chen, J. Zhao, L. Xie, H. Guo and Q. Li, RSC Adv., 2012, 2, 3942–3953 RSC;
(b) Y. Chen, J. Zhao, H. Guo and L. Xie, J. Org. Chem., 2012, 77, 2192–2206 CrossRef CAS PubMed.
- J. Liao, Y. Xu, H. Zhao, Y. Wang, W. Zhang, F. Peng, S. Xie and X. Yang, RSC Adv., 2015, 5, 86453–86462 RSC.
- M. Essid and E. U. Mughal, RSC Adv., 2025, 15, 37609–37644 RSC.
-
(a) M. Wang, M. G. H. Vicente, D. Mason and P. Bobadova-Parvanova, ACS Omega, 2018, 3, 5502–5510 CrossRef CAS PubMed;
(b) T. Stemler, C. Hoffmann, I. M. Hierlmeier, S. Maus, E. Krause, S. Ezziddin, G. Jung and M. D. Bartholomä, ChemMedChem, 2021, 16, 2535–2545 CrossRef CAS PubMed.
- L. E. Gallagher, O. A. Radhi, M. O. Abdullah, A. G. McCluskey, M. Boyd and E. Y. W. Chan, Cell Death Discovery, 2017, 8, e3014 CrossRef CAS PubMed.
-
(a) H. Zheng, G. Li, L. Zhang, M. Fan and W. Lin, New J. Chem., 2024, 48, 16710–16717 RSC;
(b) D. Wei, Y. Dai, J. Cao and N. Fu, Anal. Chim. Acta, 2024, 1299, 342422 CrossRef CAS PubMed;
(c) X. Wu, X. Wang, Y. Li, F. Kong, K. Xu, L. Li and B. Tang, Anal. Chem., 2022, 94, 4881–4888 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a
*a