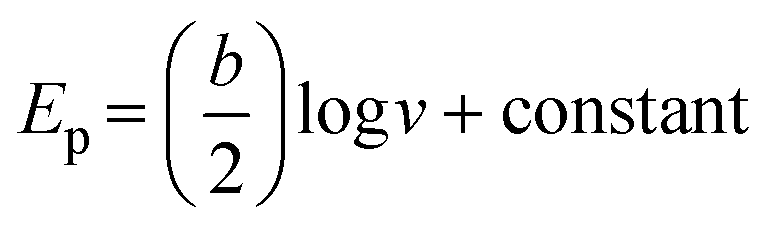DOI:
10.1039/D5SD00134J
(Paper)
Sens. Diagn., 2026,
5, 369-380
Electrochemical sensor for the detection of sunset yellow in food using a polyaniline/carbon nanopowder nanocomposite modified electrode
Received
18th July 2025
, Accepted 21st November 2025
First published on 19th December 2025
Abstract
Food items frequently face the risk of adulteration, which can lead to serious health consequences. An example of food fraud is the addition of synthetic azo dye sunset yellow (SY), for colouring food (SY-FCF). This study reports the design of an electrochemical sensor for the direct detection of SY at a glassy carbon electrode modified with a polyaniline (PANI) and carbon nanopowder (CNP) nanocomposite (GCE–PANI/CNP). The individual components of the sensor and the nanocomposite were characterised using FT-IR, UV-visible spectroscopy, XRD, SEM, and TEM. The developed electrochemical sensor (GCE/PANI/CNP) demonstrated optimal detection for SY using cyclic voltammetry and electrochemical impedance spectroscopy at approximately 0.8 V, compared to individual modified electrodes. The detection limit (LOD) was calculated to be 1.81 nM, with a broad linear detection range spanning from 1 to 194 nM, using square wave voltammetry. From the results obtained, it is concluded that the designed sensor can be a great tool for detecting sunset yellow in food samples.
1. Introduction
Consumers often base their food selection on a product's colour, which gives them a first impression of its flavour, texture, and freshness.1 Consequently, it has become the justification for the addition of additives to food items.1,2 Food adulteration, also known as food fraud, is in two primary forms: intentional acts carried out by dishonest producers, retailers, and processors seeking to make an economic profit, increase the weight and volume, improve the appearance, colour and flavor,3 and extend the food's shelf life, and incidental occurrences that may occur during production, handling, storage, and other processes. Both intentional and unintentional adulteration may reduce the safety and the quality of food products, posing health risks to consumers.3,4 Severe health effects such as chronic liver diseases, cancer, peptic ulcer diseases, kidney failure, electrolyte imbalance, bone marrow abnormalities, blood disorders, heart diseases, and skin problems can result from the use of food adulterants.5,6 An example of food fraud is the addition of synthetic azo-dye sunset yellow for colouring food (SY-FC) with E number E110. Sunset yellow consists of two sulfonate groups located at either end of the molecule,7 with two central aryl groups linked by an azo bridge (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N).7 The World Health Organization (WHO)8 and the Food and Agriculture Organization (FAO)8 have determined the acceptable daily intake (ADI)8 for sunset yellow to be 4 mg per kilogram of body weight per day.9 The maximum permitted levels of SY in non-alcoholic/alcoholic drinks are 50/200 mg L−1.10 When SY is consumed beyond the allowable daily intake, it can cause asthma, migraines, eczema, and other illnesses.11,12
N).7 The World Health Organization (WHO)8 and the Food and Agriculture Organization (FAO)8 have determined the acceptable daily intake (ADI)8 for sunset yellow to be 4 mg per kilogram of body weight per day.9 The maximum permitted levels of SY in non-alcoholic/alcoholic drinks are 50/200 mg L−1.10 When SY is consumed beyond the allowable daily intake, it can cause asthma, migraines, eczema, and other illnesses.11,12
Recently, numerous analytical methods have been developed in response to concerns about the sensitivity of reactions involving SY. These methods include thin-layer chromatography (TLC),13 spectrophotometry,1 high-performance liquid chromatography (HPLC),14,15 mass spectrometry,1 capillary electrophoresis, and electrochemical sensing.1,10,16,17 Among these, HPLC is commonly employed due to its speed, selectivity, and reliability in producing accurate results. The detection of synthetic dyes using HPLC is often combined with other techniques, such as diode array detection,1,18 UV-vis detection,19 and mass spectrometric detection.20 Although these techniques demonstrate commendable detection limits, none of these methodologies can lay claim to the combined advantages of cost efficiency, portability, rapidity, and facile manipulation inherent to electrochemical techniques.21
To enhance the performance of electrochemical techniques, nanomaterials are employed in designing electrochemical sensors.
Recently, carbon-based nanomaterials, including carbon nanopowder (CNP), have captivated significant attention due to their outstanding mechanical properties, high surface area, non-toxicity, and biocompatibility, opening numerous possibilities in the field of electrochemical devices, particularly electrochemical sensors and biosensors for detecting and quantifying various analytes. The unique electronic behaviour of these nanoparticles, which is dependent on their conducting–semiconducting structure, makes them highly suitable for these applications. Furthermore, their synthesis can be achieved through simple and low-cost procedures that are environmentally friendly.22–24 PANI generates the most attraction among other conducting polymers due to the simplicity of synthesis, low cost, higher theoretical conductivity, wider range of the working potential window, and better stability.25 Consequently, PANI has been used in the field of electrochemical sensors.25 Carbon based nanomaterials can be applied individually or combined with other materials as a composite. The integration of carbon materials and conducting polymers such as PANI in the development of electrochemical sensors is highly favoured due to its exceptional performance, leveraging the unique benefits of each constituent.25 For example, S. Jackson et al. (2025), developed a sensor using a GCE modified with PANI and carbon dots for alcohol detection with a low detection limit of 0.325 μM.26 Also, the literature reports the use of a carbon nanotube (CNT) and PANI composite for electrochemical sensing of L-ascorbic acid and ammonia gas.2,27 The literature search shows no report on the application of a CNP/PANI composite based electrochemical sensor for sunset yellow detection.
In this work, a carbon nanopowder (also known as carbon black) and polyaniline composite (PANI/CNP) is being explored in the development of an electrochemical sensor for the detection of SY.
2. Procedures and materials
2.1 Apparatus and chemicals
All reagents used in this study were of analytical grade. Sunset yellow (E110) with a dye content of 97%, N,N-dimethylformamide (99.8%), hydrogen chloride (HCl; 32%), aniline (C6H5NH2), ammonium persulfate ((NH4)2S2O8, AP), and potassium hexacyanoferrate(III) (K3Fe(CN)6) were procured from Merck Chemicals, South Africa. Sodium phosphate dibasic (NaH2PO4) and monobasic (Na2HPO4) were procured from Merck, South Africa. A glassy carbon electrode (GCE) with a 3 mm diameter, a reference electrode containing Ag/AgCl saturated with KCl, and a platinum disk counter electrode were supplied by CH Instrument Inc. Carbon nanopowder (CNP) was commercially obtained from Aldrich Chemicals.
2.2 Synthesis of PANI
Polyaniline was prepared following an established method in the literature.28,29 Approximately 0.312 g of aniline mass was added into a 1 M aqueous hydrochloric acid solution.28 About 0.19 g of ammonium persulfate (APS) was dissolved in 10 mL of the same 1 M hydrochloric acid solution.28,29 The two solutions were then combined together to achieve a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 APS to aniline ratio28 with a resulting PANI precipitate. The precipitate was collected through filtration, thoroughly washed with DI water, and subsequently dried in an oven at 65 °C for 24 h (Scheme 1).28,29
:4 APS to aniline ratio28 with a resulting PANI precipitate. The precipitate was collected through filtration, thoroughly washed with DI water, and subsequently dried in an oven at 65 °C for 24 h (Scheme 1).28,29
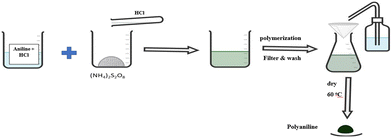 |
| | Scheme 1 Depiction of the synthesis method of PANI. | |
2.3 Preparation of the PANI/CNP nanocomposite
About 75 mg of the prepared PANI and 25 mg of the CNP were transferred into glass containers containing 3 mL DMF. The mixture was subjected to ultrasonication in a water bath at 25 °C for 48 h. The resulting paste was then used to modify the GCE using the drop-casting method.
2.4 Electrode pre-treatment
The bare GCE was cleaned by rubbing it in a spiral motion on a cleaning pad containing aqueous aluminium oxide, followed by an immediate rinse with distilled water. To remove any remaining organic impurities, the electrode was then immersed in ethanol and subsequently washed with distilled water under ultrasonication. After cleaning, the electrode was air-dried at room temperature.
2.5 Modification of the glassy carbon electrode
For electrode modification, the drop casting technique was employed. Specifically, approximately 10 μL of the nanomaterial and composite paste was deposited onto the surface of the clean bare GCE. The modified electrodes were subsequently heated in an oven at 50 °C for a duration of 3 min. The resulting modified electrodes were designated as GCE/PANI, GCE/CNP, and GCE/PANI/CNP.
2.6 Characterization of nanomaterials
Various spectroscopy techniques were employed for the characterization of electrode materials (PANI, CNP, and PANI/CNP).28 The functional groups present in the materials were identified using Fourier transform infrared (FT-IR) spectroscopy,30 their absorbance and wavelength properties were analysed using UV-visible (UV-vis) spectroscopy,30 the crystallinity of the nanomaterials was assessed on an X-ray diffractometer (XRD)30 and energy-dispersive X-ray (EDS) spectroscopy provided information on their elemental composition. Moreover, microscopic characterization was carried out using transmission electron microscopy (TEM) and scanning electron microscopy (SEM),31 with an emphasis on analysing their morphological and structural properties. Electrochemical analyses were performed on an Autolab potentiostat/galvanostat PGSAT-20 controlled by the software Nova 2.1.6, within a cell configured with a three-electrode21 system, including the reference, working (PANI/CNP-modified GCE), and counter electrodes. The electrochemical characterization and analysis of the analyte were performed in a phosphate-buffered saline solution (10 mM PBS, pH 7), prepared using Na2HPO4 and NaH2PO4.
2.7 Sample preparation
In this study, Skopas snacks purchased from a local supermarket were used as a real sample. 10 g of the crushed Skopas snack was transferred to a beaker containing 200 mL of PBS at pH 7, then stirred for 15 min at room temperature. The mixture was subsequently filtered through Whatman filter paper, transferred to a vial, and stored in a refrigerator until analysis.
3. Results
3.1 FT-IR analysis
The spectrum of the nanoparticles, shown in Fig. 1, reveals a characteristic peak at 1610 cm−1 corresponding to CC double bonds. The peak at 1783 cm−1 is attributed to the carboxylic group (CO) stretching vibration of COOH attached to CNP groups. Additionally, the fingerprint region shows bands associated with an aromatic structure for CNP.32,33 The PANI spectrum displays key peaks at 800 cm−1, 1100 cm−1, 1292 cm−1, 1469 cm−1, and 1540 cm−1. The peaks at 1469 cm−1 and 1540 cm−1 are linked to the stretching vibrations of CC bonds in the benzenoid and quinoid rings, respectively. The absorption bands observed at 1292 cm−1 and 1100 cm−1 are attributed to the stretching vibrations of C–N bonds in the secondary aromatic amine.28,34 The 800 cm−1 vibrational band corresponds to the C–H out-of-plane bending mode in substituted aromatic compounds.28,34–37 The PANI/CNP composite also shows the same characteristic peaks noticed in PANI, although with observable reduction in peak intensity. The decrease in peak intensity observed in the nanocomposite spectrum can indicate effective CNP integration into the polyaniline matrix.
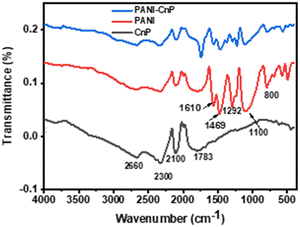 |
| | Fig. 1 FTIR spectra of CNP, PANI and the PANI/CNP nanocomposite. | |
3.2 Ultraviolet-visible spectroscopy
The optical properties of PANI, CNP, and PANI/CNP dissolved in DMF were evaluated across a wavelength range of 200 to 900 nm, as illustrated in Fig. 2. CNP exhibits a prominent absorption band with the highest vibrational intensity at 288 nm, attributed to the π–π* transition within the carbon material's structure.38 The PANI spectrum displayed peaks at 620 nm and 325 nm, corresponding to the π–π* transition of benzene ring charge and the electron transfer between quinoid and benzenoid rings, respectively.28,39 The shift observed in the peaks of the absorbance in the UV-vis spectrum of the nanomaterials and the PANI/CNP composite indicates the successful production of PANI/CNP nanocomposites and the existence of potential chemical interactions between the PANI and CNP nanoparticles. The interactions of the nanomaterials (CNP and PANI) can be seen in the nanocomposite with the appearance of a new peak which is absent in PANI. Additionally, the blue shift from 325 nm to 380 nm and the red shift from 620 nm to 530 nm, with reduction observed in the nanocomposite spectrum, suggest the interaction of the quinoid with the CNP.
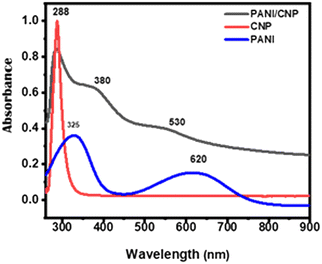 |
| | Fig. 2 UV-vis spectra of CNP, PANI and PANI/CNP. | |
3.3 XRD analysis
Fig. 3(a) presents the X-ray diffraction (XRD) pattern of polyaniline, showing four distinct peaks at 10.7°, 17.5°, 20.6°, and 25.5°, corresponding to the (001), (011), (020), and (200) planes, respectively. These sharp peaks indicate that the polymer is semi-crystalline, which is attributed to the presence of benzenoid and quinonoid groups in the polyaniline structure.40,41 Additionally, a characteristic peak at 26.1° is observed in the CNP and the nanocomposite (Fig. 3c), corresponding to the (002) plane, which is typical of carbon materials. Low-intensity peaks, characteristic of well-graphitized multi-walled carbon materials, appear at 43.6° (100) and 54.0° (004), consistent with the values reported in the literature.42,43
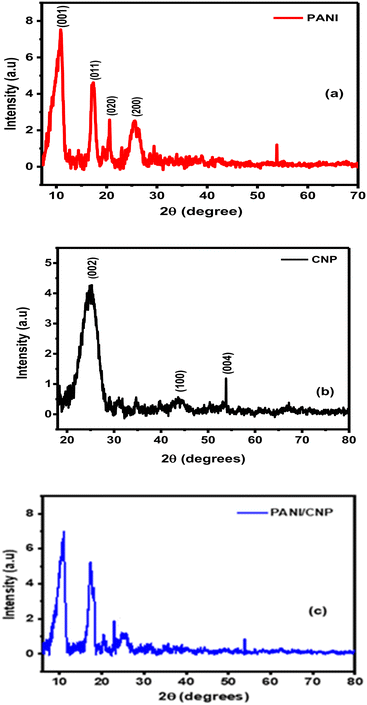 |
| | Fig. 3 XRD diffractograms of (a) PANI, (b) CNP and (c) PANI/CNP. | |
3.4 Scanning electron microscopy analysis
To evaluate the shape and morphology of the nanoparticles, SEM analysis was performed. Fig. 4(a–c) presents the SEM micrographs of PANI, CNP and PANI/CNP, respectively.44 In Fig. 4(b), the CNP micrograph reveals the agglomeration of spherical shaped particles with some voids. The same trend is noticed in the image of PANI, although less agglomerated spherical particles than CNP (Fig. 4(a)). The nanocomposite (PANI/CNP) morphology shows densely agglomerated spherical shaped particles with a noticeable reduction in the size of the particles and lesser voids in comparison to the CNP and PANI micrographs. The dense aggregation and decrease in particle size with minute voids suggest a strong interaction between PANI and CNP, and confirms that the PANI/CNP composite was successfully synthesized.
 |
| | Fig. 4 SEM micrographs of (a) PANI, (b) CNP and (c) PANI/CNP. | |
3.5 Energy dispersive X-ray spectroscopy
The EDS spectra for PANI, CNP, and PANI/CNP are presented in Fig. 5(a–c). The detection of nitrogen (N = 10.28%) and carbon (C = 67.4%) peaks verifies the successful synthesis of PANI molecules and the formation of the PANI/CNP nanocomposite. The presence of copper (Cu) and chlorine (Cl) in CNP and PANI, respectively, could probably have resulted from the instrument sample holder. Meanwhile, the existence of sulfur (S) in PANI and PANI/CNP could be residual sulfur from ammonium persulfate used in the PANI synthesis. The observable differences in weight (%) of C, N, and O in the nanomaterials indicate possible interactions between CNP and PANI.
 |
| | Fig. 5 EDS spectra showing the elemental composition of (a) CNP, (b) PANI and (c) PANI/CNP. | |
3.6 Transmission electron microscopy
TEM is an important tool in scientific study, particularly for determining the magnitude of produced nanomaterials.28Fig. 6a–c shows the TEM images of PANI, CNP and PANI/CNP accordingly, at a magnification of 200 nm. The TEM micrograph of PANI (Fig. 6a) shows a unique short-rod shape with spherical dark spots of PANI, agglomerated in certain points, likely as a result of π–π interactions between the PANI molecules.29 Such observations are made on the SEM micrograph of PANI as well.40 The TEM picture of CNP (Fig. 6b) shows that the carbon nanopowder has a pseudo-spherical structure with smaller dark spots. The darker spots in the micrograph indicate CNP aggregation, which results in two different aggregate types. The nanocomposite TEM image reveals the attachment of CNP on PANI (Fig. 6(c)), suggesting successful synthesis of PANI/CNP.
 |
| | Fig. 6 TEM images of (a) PANI, (b) CNP, and (c) PANI/CNP. | |
3.7 Electrochemical characterization
Electrochemical characteristics of the modified electrodes were assessed using cyclic voltammetry (CV) in a 10 mM redox probe ([Fe(CN)6]3−/4−) prepared in 0.1 M phosphate-buffered saline at pH 7 and a temperature of 25 °C (ref. 21) at 25 mV s−1. The oxidation current responses for the bare GCE, GCE/CNP, GCE/PANI, and GCE/PANI/CNP are 3.888, 53.464, 29.242, and 47.691 μA, respectively, as illustrated in Fig. 7. From the results, PANI/CNP demonstrated optimal current response, suggesting excellent electrical conductivity and surface properties, facilitating efficient transfer of electrons between the modified electrode and the redox probe. The bare GCE exhibited the lowest current response, in contrast to GCE/CNP and GCE/PANI, demonstrating that these materials significantly improve electron transfer kinetics compared to the unmodified electrode.| | | Ip = 2.69 × 105n3/2AD1/2Cv1/2 | (1) |
 |
| | Fig. 7 (a) Cyclic voltammograms of electrodes and (b) their current response in 10 mM [Fe(CN)6]3−/4− at a scan rate of 25 mV s−1. | |
According to the Randles–Sevcik equation21 (1), Ip, ν, n, A, C and D correspond to the peak current (A), scan rate (V s−1), number of electrons transferred, electrode surface area (cm2), concentration (mol cm−3), and diffusion coefficient (cm2 s−1), respectively.21 The surface areas (A) of the bare GCE, GCE/CNP, GCE/PANI, and GCE/PANI/CNP were calculated to be 0.005, 0.072, 0.039, and 0.064 cm2, respectively. The modified electrode exhibited the highest current response and the largest surface area, suggesting that the modification significantly improved the electrode's surface area and potentially its conductivity. Table 1 summarises the parameters obtained from CV measurements for all four electrodes (GCE, GCE/CNP, GCE/PANI, and GCE/PANI/CNP). The anodic to cathodic peak current ratio for all the electrodes is approximately 1, suggesting a reversible electrochemical process. This enhanced electronic conductivity promotes more effective electron transfer between the modified electrode and the redox probe.
Table 1 The comparative parameters from the cyclic voltammograms of the unmodified and modified electrodes obtained in the 10 mM [Fe(CN)6]3−/4− redox couple in PBS (pH 7) at a scan rate of 25 mV s−1
| Electrodes |
I
pa (μA) |
E
pa (V) |
I
pc (μA) |
E
pc (V) |
ΔEp (V) |
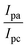
|
A (cm2) |
| GCE |
30.888 |
0.0817 |
−3.0730 |
0.2083 |
0.29 |
1.27 |
0.005 |
| GCE–CNP |
53.464 |
0.1076 |
−43.4239 |
0.0857 |
0.19 |
1.23 |
0.072 |
| GCE–PANI |
29.242 |
0.2192 |
−26.6723 |
0.1266 |
0.35 |
1.10 |
0.039 |
| GCE–PANI/CNP |
47.691 |
0.2303 |
−46.3101 |
0.1043 |
0.33 |
1.03 |
0.064 |
Electrochemical impedance spectroscopy (EIS) was employed to further understand the charge resistivity of the electrodes in a 10 mM [Fe(CN)6]3−/4− solution. The Nyquist plots for GCE, GCE/CNP, GCE/PANI, and GCE/PANI/CNP recorded in a 10 mM [Fe(CN)6]3−/4− solution are presented in Fig. 8(a). Additionally, Fig. 8(b) illustrates the analogous circuits used to fit the EIS data for these electrodes (GCE/CNP, GCE/PANI, and GCE–PANI/CNP). The numerical values of the fitted parameters, including solution resistance (Rs), charge transfer resistance (Rct), the exponent of the constant phase element (N), and the Warburg impedance (W), are listed in Table 2. The errors associated with these parameters, as calculated from the EIS data fitting, are provided in parentheses in Table 2.
 |
| | Fig. 8 EIS spectra (Nyquist plot) of (a) bare GCE and modified GCE/CNP, GCE/PANI and GCE/PANI/CNP in 10 mM [Fe(CN)6]3−/4− (pH 7); (b) equivalent electrochemical circuit of the EIS data in (a). | |
Table 2 EIS parameters of the bare GCE and modified electrodes with errors in parentheses
| Electrode |
R
s (kΩ) |
R
ct (kΩ) |
CPE (μF) |
W (F)/*C (μF) |
N
|
X
2
|
| GCE |
0.19 (3.68) |
16.44 (5.77) |
19.4 (3.27) |
7491 (10.0) |
0.86 (0.63) |
0.0 |
| GCE–CNP |
5.4 × 10−14 (2.38) |
6.10 (8.30) |
7.35 (4.50) |
1434 (9.36) |
0.94 (0.62) |
0.0007 |
| GCE–PANI |
0.18 (0.34) |
3.2 (0.56) |
8.41 (2.51) |
752.5 (7.46) |
0.95 (0.35) |
0.0 |
| GCE–PANI/CNP |
0.16 (0.12) |
1.36 (0.24) |
21.6 (3.13) |
797.4 (9.9) |
0.87 (0.54) |
1.77 × 10−0.5 |
3.8 Electrocatalytic response of PANI/CNP to SY oxidation
The electrochemical behaviour of SY on the bare GCE and the modified electrodes was assessed through CV at a sweep rate of 25 mV s−1 in 0.1 mM SY solution prepared in 10 mM PBS (pH 7). The voltammetric responses of the electrodes as shown in Fig. 9(a) demonstrate a reversible electrochemical process with well-defined redox peaks. All cyclic voltammograms exhibited two key peaks: an anodic peak corresponding to SY oxidation at a potential range of 0.29–0.34 V and a cathodic peak occurring between 0.09 and 0.125 V. The oxidation peak currents (Ipa) with the associated potentials in parentheses for SY at the bare GCE, GCE/CNP, GCE/PANI, and GCE/PANI/CNP were recorded as 26.995 μA (0.347 V), 235 μA (0.292 V), 192.529 μA (0.294 V), and 294 μA (0.294 V), respectively. Fig. 9(b) presents a bar graph of the current responses for the bare and modified electrodes. The oxidation peak current at the GCE/PANI/CNP electrode was approximately ten times higher than that of the unmodified GCE, suggesting that the combination of CNP and PANI provided a synergistic effect that significantly enhanced the electrocatalytic activity of the composite for SY oxidation. Overall, the electrodes' current responses followed the trend: GCE/PANI/CNP > GCE/CNP > GCE/PANI > GCE, as shown in Fig. 9(b).
 |
| | Fig. 9 (a) CVs and (b) bar graph of the oxidation peak current response for 0.1 mM SY at the unmodified and modified electrodes recorded at a 25 mV s−1 scan rate. | |
3.9 Influence of pH on SY oxidation
The influence of pH on the SY oxidation peak current and potential was evaluated in 0.1 mM PBS with varying pH ranges of 3–9, employing CV at a 25 mV s−1 scan rate. Fig. 10 shows the maximum oxidation current response for SY at pH 7, suggesting that pH 7 is the best for determining the sensitivity of SY. Consequently, pH 7 was used in this study. Also, as the pH increase, the oxidation peak potential shifted progressively towards the negative potentials, indicating the participation of protons in the oxidation process of SY.2,45,46
 |
| | Fig. 10 Cyclic voltammograms of 0.1 mM SY at different pH values. | |
3.10 Influence of the scan rate on the oxidation of SY at GCE–PANI/CNP
The influence of the scan rate on the oxidation of SY was examined at different scan rates from 5–850 mV s−1 in PBS at pH 7. Fig. 11(a) shows a progressive increase in the oxidation peak current with rising scan rates. Additionally, the voltammograms demonstrated a rightward shift in potential with each increment in the scan rate. The linear correlations between the square root of the scan rate (ν1/2) and peak currents (Ipa and Ipc) were described by the equations:
| Ipa (μA) = 10.876x − 50.787ν1/2(v)1/2 (R2 = 0.9905) and |
| Ipc (μA) = −1.7654x − 5.13ν1/2(v)1/2 (R2 = 0.9373). |
 |
| | Fig. 11 (a) Cyclic voltammetric response of the PANI/CNP electrode in 0.1 mM SY in PBS, pH 7, at varying scan rates of 25 to 850 mV s−1, (b) plots of I (μA) vs. ν1/2, and (c) plots of E (V) versus logv. | |
These results indicate the occurrence of both diffusion-controlled and surface-confined processes at the electrode.21 The number of electrons transferred (n) and the electron transfer coefficient (α) determined from the relationship between the anodic peak potential (Epa) and the natural logv, in accordance with Laviron's eqn (2) and (3),47,48 were found to be 2 and 0.63, respectively. The corresponding reaction mechanism of SY at the modified PANI/CNP electrode is presented in Scheme 2.
| | 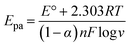 | (2) |
| | 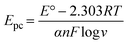 | (3) |
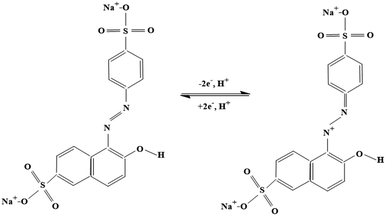 |
| | Scheme 2 Proposed mechanism of SY. | |
The correlation between the logv and peak potentials is essential for elucidating the kinetics and mechanisms underlying electrochemical processes on electrodes. Linear plots of logv against the peak potentials, such as those described in Fig. 11, typically indicate that the electrochemical process is controlled by a kinetic mechanism. The Tafel value for the PANI/CNP nanocomposite modified electrode was determined based on the slope of the plot Ep against logv (the linear equations presented in eqn (4) and (5) from Fig. 11(c)) using eqn (6)28 was found to be 0.052.V dec−1 and 0.0313 V dec−1 for Epa and Epc, respectively. The Tafel values (b) are less than the theoretical value (0.118 V dec−1) for a one-electron process, suggesting non-occurrence of adsorption on the modified electrode's surface.28
| | | Epa = 0.1047x + 0.023 (R2 = 0.987) | (4) |
| | | Epc = 0.0625x − 0.2921 (R2 = 0.9857) | (5) |
| |  | (6) |
3.11 Electroanalysis of SY at the PANI/CNP modified GC electrode
The influence of varying concentrations on the current response of SY was determined using SWV under 0.01 V potential step, 0.001 V amplitude, a deposition time of 10 seconds, and a frequency of 25 Hz. The oxidation peak current (Ipa) increased with increasing concentration of SY over a dynamic linear range of 1–194 nM as shown in Fig. 12(a), showing the dependence of the peak current of SY with the increase in the SY concentration. The linear relationship for SY concentrations and Ipa is presented in Fig. 12(b) in PBS with a pH of 7 with the linear regression equation of I (μA) = 9.1159 × 108 + 30.025 [SY] and a 0.9909 correlation coefficient. The limits of detection and quantification (LOD and LOQ) were computed to be 1.81 nM and 5.49 nM employing eqn (7) and (8), respectively, where SD represents the standard deviation of the peak current, over a slope of the calibrated plot. This LOD is favorably comparable with some SY sensors reported in studies (see Table 4).| | 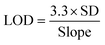 | (7) |
| | 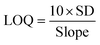 | (8) |
 |
| | Fig. 12 (a) Square wave voltammograms at different SY concentrations (1–194 nM) in PBS at pH 7. (b) The corresponding calibration plot depicting the relationship between of Ip (μA) and SY concentrations. | |
3.12 Real sample analysis
To check the practical rationality of the designed electrode, SY was determined in a real sample (Skopas snacks) following a well-established procedure (standard addition) using SWV. The SWV results of unspiked and spiked samples were obtained under optimal conditions: a deposition time of 10 seconds, a potential step of 0.01 V, a frequency of 25 Hz, and an amplitude of 0.001 V.54Table 3 shows the result obtained for real SY sample analysis with recovery values between 95 and 99% with an average RSD (%) value of 2.139 (n = 3). The good recoveries confirm that the detection of SY at the modified GCE–PANI/CNP electrode is reliable for the direct determination of SY in food samples.
Table 3 Peak summary for the comparative voltammograms of the different electrodes in 0.1 mM SY in PBS at pH 7 and 25 mV s−1 scan rate
| Electrodes |
I
pa (μA) |
E
pa (V) |
I
pc (μA) |
E
pc (V) |
ΔEp (V) |

|
A (cm2) |
| GCE |
26.995 |
0.347 |
−29.881 |
0.094 |
0.253 |
0.903 |
0.036 |
| GCE–CNP |
235.823 |
0.292 |
−302.094 |
0.125 |
0.167 |
0.780 |
0.315 |
| GCE–PANI |
192.530 |
0.294 |
−98.642 |
0.117 |
0.177 |
1.952 |
0.257 |
| GCE–PANI/CNP |
294.397 |
0.294 |
−148.727 |
0.117 |
0.177 |
1.979 |
0.394 |
Table 4 Comparison with other chemically modified electrodes towards the detection of sunset yellow
| Modified electrode |
Technique |
Linear range (μM) |
LOD (μM) |
Ref. |
|
Graphene oxide.
Gold.
Reduced graphene oxide.
Graphene.
Titanium dioxide.
Graphitic carbon nitride.
Zinc oxide.
Gold nanoparticles.
|
| GOa/MWCNTs/GCE |
LSV |
0.09–30 |
0.025 |
49
|
| MWNT film-modified GCE |
DPV |
0.0553–11.05 |
0.0244 |
50
|
| Aub/RGOc/GCE |
DPV |
0.002–109.14 |
0.002 |
51
|
| Chitosan/graphene/GCE |
CV |
0.2–100 |
0.0666 |
52
|
| GNd/TiO2e-CPE |
SWV |
0.02–2.05 |
0.006 |
53
|
| rGO-g-CNf/ZnOg-AuNPsh/GCE |
SWV |
0.005–0.085 |
0.0013 |
54
|
| GCE/CNP/PANI |
SWV |
0.001–0.194 |
0.00181 |
This work |
3.13 Interference studies
Since SY is an anionic dye commonly found in food alongside other anionic and cationic dyes, the selectivity of GCE–PANI/CNP sensor electrodes was examined in the presence of methylene blue (MB), a cationic dye. This assessment was conducted using square wave voltammetry (SWV). Fig. 13(a) shows the square wave voltammogram of 0.01 mM of MB used against 0.1 mM SY with the MB peak appearing around −0.2 V and that of SY at around 0.7 V.55,56Fig. 13(b) shows that the electrode exhibited strong selectivity with a constant methylene blue concentration of 0.01 mM. The electrode was able to detect SY as the concentrations increased from 14.7 μM to 32 μM in the presence of MB (Table 5).
 |
| | Fig. 13 (a) Square wave voltammogram displaying the characteristic peaks of methylene blue (MB) and SSY. (b) SWV voltammograms recorded while maintaining a constant MB concentration of 0.1 mM, with a gradual increase in SY concentration from 14.7 to 32 μM on the GCE–PANI/CNP-modified electrode. | |
Table 5 Recovery studies of sunset yellow (SY) at GCE–PANI/CNP
| Sample (Skopas snacks) |
Amount added (μM) |
Amount found (μM) |
% Recovery |
RSD (% n = 3) |
| 1 |
9.09 |
8.52 |
95 |
2.139 |
| 2 |
9.09 |
8.80 |
98 |
2.139 |
| 3 |
9.09 |
8.87 |
99 |
2.139 |
3.14 Stability studies
The stability and reproducibility of the GCE–PANI/CNP-modified electrode were evaluated using cyclic voltammetry over twenty-five consecutive scans at a 25 mV s−1 sweep rate in a 10 mM K3[Fe(CN)6] solution prepared in 10 mM PBS (pH 7.0). A 29.8% increase in the oxidation peak current was observed with no observable shift of peak potential (Fig. 14). The increase in oxidation current could be potentially due to an increase in the electroactive surface area, allowing a greater interaction with the probe solution over time.
 |
| | Fig. 14 The stability of the GCE–PANI/CNP modified electrode in 0.1 mM SY prepared in 10 mM PBS, pH 7, at a 25 mV s−1 scan rate. | |
4. Conclusions
The addition of adulterants, such as sunset yellow, in food has serious health effects on consumers; hence, there is a need to develop a sensor to detect the concentrations of such adulterants in food. PANI functionalized with CNP to form nanocomposites was used in detecting sunset yellow (SY) in a food sample. To confirm the correct synthesis of this material, various characterization techniques, including morphological, spectroscopic, microscopic, and structural analyses, were employed. Electrochemical experiments were performed using SWV, CV and EIS, demonstrating that the GCE–PANI/CNP nanocomposite-modified electrodes exhibit enhanced electron transport and a superior response to SY detection compared to other tested electrodes. The SY detection process at the GCE–PANI/CNP nanocomposite-modified electrode was found to be diffusion-controlled. The sensor provides a low LOD of 1.8 nM and a broad linear range from 1 to 194 nM, indicating its improved sensitivity over previously fabricated sensors.
Additionally, when tested against MB, the sensor could reliably distinguish SY from MB, showing good selectivity. Further validation of the sensor was conducted by testing it on real food samples. The sensor achieved high recovery rates when used to detect SY in Skopas snacks, highlighting its potential for application in the food industry. Despite moderate stability observed in comparing the first and last of the 25 scans in the PANI/CNP nanocomposite voltammogram, other favourable performance metrics suggest that this sensor is a strong candidate for commercialisation. It shows promise for point-of-care SSY detection in food products and other real samples at nanomolar levels, supporting consumer safety and regulatory compliance.
Author contributions
OEF: conceptualization, supervision, reviewing, and editing. JL: synthesis and electrochemical investigations, data curation, interpretation of data, and manuscript writing. All authors OEF, JL and GEU reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
Data used in this study will be made available upon request from the corresponding author.
Acknowledgements
The authors acknowledge the National Foundation Research (NRF), the North-West University and MaSim.
References
- K. Rovina, L. A. Acung, S. Siddiquee, J. H. Akanda and S. M. Shaarani, Food Anal. Methods, 2017, 10, 773–787 CrossRef.
- F. S. Lisboa, E. G. C. Neiva, M. F. Bergamini, L. H. Marcolino Junior and A. J. G. Zarbin, J. Braz. Chem. Soc., 2020, 31, 1093–1100 Search PubMed.
- G. Kaur, R. Bhari and K. Kumar, Crit. Rev. Biotechnol., 2024, 44, 547–561 CrossRef.
- M. Momtaz, S. Y. Bubli and M. S. Khan, Foods, 2023, 12, 199 CrossRef CAS.
- D. Yeasmin, M. Baker, A. H. M. Kamal, M. S. Islam, M. Rahman, P. J. Winch and L. Unicomb, BMC Public Health, 2022, 23, 206 CrossRef.
- N. Raturi, J. Aman and C. Sharma, Octa J. Biosci., 2022, 10, 37–50 CAS.
- N. Kizil, E. Basaran, D. Erbilgin, M. L. Yola, F. Uzcan and M. Soylak, Microchem. J., 2022, 181, 107734 CrossRef CAS.
- K. Rovina, S. Siddiquee and S. M. Shaarani, Trends Food Sci. Technol., 2017, 65, 68–79 CrossRef CAS.
- L. C. A. Feitosa, P. D. S. Rodrigues, A. S. Da Silva, A. D. O. Rios and F. Cladera-Olivera, Food Addit. Contam.,: Part A, 2017, 34, 687–694 CAS.
- K. Rovina, P. P. Prabakaran, S. Siddiquee and S. Md. Shaarani, TrAC, Trends Anal. Chem., 2016, 85, 47–56 CrossRef CAS.
- M. N. N. Begam, K. Muthukumaran, P. Thamarai and J. P. Joshua, Global NEST J., 2022, 24, 276–285 Search PubMed.
- Y. Zheng, S. Mao, J. Zhu, L. Fu, N. Zare and F. Karimi, Food Chem. Toxicol., 2022, 164, 113019 Search PubMed.
- F. I. de Andrade, M. I. F. Guedes, Í. G. P. Vieira, F. N. P. Mendes, P. A. S. Rodrigues, C. S. C. Maia, M. M. M. Ávila and L. de Matos Ribeiro, Food Chem., 2014, 157, 193–198 CrossRef.
- S. Bonan, G. Fedrizzi, S. Menotta and C. J. D. Elisabetta, Dyes Pigm., 2013, 99, 36–40 CrossRef CAS.
- M. A. Prado, L. F. Vilas Boas, M. R. Bronze and H. T. Godoy, J. Chromatogr. A, 2006, 1136, 231–236 Search PubMed.
- K. Yamjala, M. S. Nainar and N. R. Ramisetti, Food Chem., 2016, 192, 813–824 CrossRef CAS PubMed.
- K. Hroboňová, J. Lehotay, J. Čižmárik and J. Sádecká, J. Liq. Chromatogr. Relat. Technol., 2013, 36, 486–503 CrossRef.
- P. Qi, Z.-h. Lin, G.-y. Chen, J. Xiao, Z.-a. Liang, L.-n. Luo, J. Zhou and X.-w. Zhang, Food Chem. Mass Spectrom., 2015, 181, 101–110 CAS.
- Y. P. Zhang, Y. J. Zhang, W. J. Gong, A. I. Gopalan and K.-P. Lee, J. Chromatogr. A, 2005, 1098, 183–187 CrossRef CAS PubMed.
- F. Gosetti, P. Frascarolo, E. Mazzucco, V. Gianotti, M. Bottaro and M. C. Gennaro, J. Chromatogr. A, 2008, 1202, 58–63 Search PubMed.
- S. E. Elugoke, O. E. Fayemi, A. S. Adekunle, B. B. Mamba, T. T. Nkambule and E. E. J. F. Ebenso, FatChem, 2022, 33, 100372 CrossRef CAS.
- M. Antunes and J. I. Velasco, Prog. Polym. Sci., 2014, 39, 486–509 CrossRef CAS.
- E. Asadian, M. Ghalkhani and S. Shahrokhian, Sens. Actuators, B, 2019, 293, 183–209 CrossRef CAS.
- N. L. Teradal, R. D. Tandel and J. Seetharamappa, Mater. Sci. Energy Technol., 2019, 2, 337–344 Search PubMed.
- H. Wang, J. Lin and Z. X. Shena, J. Sci.: Adv. Mater. Devices, 2016, 1, 225–255 Search PubMed.
- S. Jackson, M. Taylor, R. Kumar, A. K. Shringi, T. Leung and U. Riaz, Nanomaterials, 2025, 15, 593 CrossRef CAS.
- G. Wu, H. Du, Y. L. Cha, D. Lee, W. Kim, F. Feyzbar-Khalkhali-Nejad, T. S. Oh, X. Zhang and D. J. Kim, Sens. Actuators, B, 2023, 375, 132858 CrossRef CAS.
- O. E. Fayemi, J. Makgopa and E. Elugoke, Mater. Res. Express, 2024, 10, 125603 CrossRef.
- O. E. Fayemi, A. S. Adekunle, B. E. K. Swamy and E. E. Ebenso, J. Electroanal. Chem., 2018, 818, 236–249 CrossRef CAS.
-
J. T. Mhlongo, Master's thesis, University of the Witwatersrand, 2023 Search PubMed.
- S. E. Elugoke, O. E. Fayemi, A. S. Adekunle, P.-S. Ganesh, S. Y. Kim and E. E. Ebenso, J. Electroanal. Chem., 2023, 929, 117120 CrossRef CAS.
- T. L. Ba, M. Bohus, I. E. Lukács, S. Wongwises, G. Gróf, K. Hernadi and I. M. Szilágyi, Nanomaterials, 2021, 11, 608 CrossRef PubMed.
- V. Ţucureanu, A. Matei and A. M. Avram, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef.
- B. K. Sharma, A. K. Gupta, N. Khare, S. K. Dhawan and H. C. Gupta, Synth. Met., 2009, 159, 391–395 CrossRef CAS.
- M. Ibrahim and E. Koglin, Acta Chim. Slov., 2005, 52, 159–163 Search PubMed.
- X. Gu, Z. Chen, Y. Li, J. Wu, X. Wang, H. Huang, Y. Liu, B. Dong, M. Shao and Z. Kang, ACS Appl. Mater. Interfaces, 2021, 13, 24814–24823 CrossRef CAS.
- T. S. Kuznetsova, A. E. Burakov, O. A. Ananyeva, I. V. Burakova, A. E. Memetova, V. O. Yarkin and A. G. Tkachev, Colloid J., 2024, 86, 408–417 CrossRef CAS.
- F. Mocci, L. de Villiers Engelbrecht, C. Olla, A. Cappai, M. F. Casula, C. Melis, L. Stagi, A. Laaksonen and C. M. Carbonaro, Chem. Rev., 2022, 16, 13709–13799 CrossRef PubMed.
- A. Rahy and D. J. Yang, Mater. Lett., 2008, 62, 4311–4314 CrossRef CAS.
- S. Yang, S. Zhu and R. J. C. Hong, Coatings, 2020, 10, 1215 CrossRef CAS.
- S. B. Kondawar, M. D. Deshpande and S. P. Agrawal, Int. J. Compos. Mater., 2012, 2, 32–36 Search PubMed.
- F. A. Adesanya and O. E. Fayemi, Int. J. Electrochem. Sci., 2023, 18, 100382 Search PubMed.
- R. Atchudan, A. Pandurangan and J. Joo, J. Nanosci. Nanotechnol., 2015, 15, 4255–4267 CrossRef CAS PubMed.
- S. Zhu, A. Xie, B. Wei, X. Tao, J. Zhang, W. Peng, C. Liu, L. Gu, C. Xu and S. Luo, New J. Chem., 2020, 44, 9288–9297 Search PubMed.
- S. M. Ghoreishi, M. Behpour and M. Golestaneh, Food Chem., 2012, 132, 637–641 Search PubMed.
- E. C. Hagan, W. H. Hansen, O. G. Fitzhugh, P. M. Jenner, W. I. Jones, J. M. Taylor, E. L. Long, A. A. Nelson and J. B. Brouwer, Food Cosmet. Toxicol., 1967, 5, 141–157 Search PubMed.
- S. J. Mokole and O. E. Fayemi, Mater. Res. Express, 2024, 11, 105008 CrossRef CAS.
- M. N. Ranku, G. E. Uwaya and O. E. Fayemi, Molecules, 2021, 26, 5357 CrossRef CAS PubMed.
- X. Qiu, L. Lu, J. Leng, Y. Yu, W. Wang, M. Jiang and L. Bai, Food Chem., 2016, 190, 889–895 Search PubMed.
- W. Zhang, T. Liu, X. Zheng, W. Huang and C. Wan, Colloids Surf., B, 2009, 74, 28–31 CrossRef CAS.
- J. Wang, B. Yang, H. Wang, P. Yang and Y. Du, Anal. Chim. Acta, 2015, 893, 41–48 Search PubMed.
- L. Magerusan, F. Pogacean, M. Coros, C. Socaci, S. Pruneanu, C. Leostean and I. O. Pana, Electrochim. Acta, 2018, 283, 578–589 CrossRef CAS.
- T. Gan, J. Sun, W. Meng, L. Song and Y. Zhang, Food Chem., 2013, 141, 3731–3737 CrossRef CAS.
- A. T. E. Vilian, S. M. Kang, S. Y. Oh, C. W. Oh, R. Umapathi, Y. S. Huh and Y. K. Han, Food Chem., 2020, 323, 126848 Search PubMed.
- M. Wang, Q. Sun, Y. Gao, X. Yang and J. Zhao, Anal. Methods, 2014, 6, 8760–8766 RSC.
- M. Hayat, A. Shah, J. Nisar, I. Shah, A. Haleem and M. N. Ashiq, Catalysts, 2022, 12, 306 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ab,
Gloria E.
Uwaya
c and
Omolola E.
Fayemi
ab,
Gloria E.
Uwaya
c and
Omolola E.
Fayemi