
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembled chemiluminescent nanoprobes based on α,α-dicyanoolefins: leveraging a dioxetane “funnel” for high-contrast inflammation imaging
Li Liu†
ac,
Mu Kai†b,
Wenqing Lic,
Huijia Liuc,
Ying Wangc,
Jing Yang*c,
Peng Wang *c and
Wancun Zhang*a
*c and
Wancun Zhang*a
aHealth Commission of Henan Province Key Laboratory for Precision Diagnosis and Treatment of Pediatric Tumor, Children's Hospital Affiliated to Zhengzhou University, Zhengzhou, 450018, China. E-mail: zhangwancun@126.com
bHenan Key Laboratory of Rare Diseases, Endocrinology and Metabolism Center, The First Affiliated Hospital, College of Clinical Medicine of Henan University of Science and Technology, Luoyang, 471003, China
cDepartment of Biomedical Engineering, School of Engineering, China Pharmaceutical University, Nanjing 211198, China. E-mail: wangpeng@cpu.edu.cn; yangjing@cpu.edu.cn
First published on 28th May 2026
Abstract
Although peroxynitrite (ONOO−) chemiluminescence (CL) imaging offers near-zero background sensitivity for in vivo sensing, developing CL probes remains a formidable challenge due to the limited repertoire of chemiluminescent scaffolds and boronate-related cross-reactivity. Herein, we report the rational design of self-assembled chemiluminescent nanoprobes utilizing an α,α-dicyanoolefin scaffold. The optimal candidate, YMTPT-NPs, spontaneously forms stable nanoparticles in water and achieves orthogonal ONOO− specificity via its C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of α,α-dicyanoolefin, eliminating H2O2 interference. Experimental and density functional theory (DFT) calculations elucidate that ONOO− attack forms a dioxetane intermediate, triggering sequential O–O cleavage to access an S1–S0 near-degenerate “funnel” and subsequent excited-state C–C cleavage. This mechanism efficiently channels chemical energy to drive robust intermolecular CRET within the confined nano-architecture. In a murine acute arthritis model, YMTPT-NPs exhibited an extended functional imaging window (>15 min) and a superior signal-to-background ratio (SBR = 31.38). This study establishes a robust supramolecular paradigm for constructing CL probes that combine practical signal duration, superior specificity, and red-shifted emission for precision in vivo imaging of inflammation.
C bond of α,α-dicyanoolefin, eliminating H2O2 interference. Experimental and density functional theory (DFT) calculations elucidate that ONOO− attack forms a dioxetane intermediate, triggering sequential O–O cleavage to access an S1–S0 near-degenerate “funnel” and subsequent excited-state C–C cleavage. This mechanism efficiently channels chemical energy to drive robust intermolecular CRET within the confined nano-architecture. In a murine acute arthritis model, YMTPT-NPs exhibited an extended functional imaging window (>15 min) and a superior signal-to-background ratio (SBR = 31.38). This study establishes a robust supramolecular paradigm for constructing CL probes that combine practical signal duration, superior specificity, and red-shifted emission for precision in vivo imaging of inflammation.
Introduction
Peroxynitrite (ONOO−), a potent oxidant within the reactive oxygen and nitrogen species (RONS) family, has been identified as a pivotal mediator in the pathogenesis of diverse disorders,1,2 including arthritis,3,4 inflammatory bowel disease,1 neurodegenerative diseases,5 and cancer.6 As the generation of ONOO− is intimately associated with intracellular oxidative stress,7 elucidating its spatiotemporal dynamics provides critical biological and clinical insights into disease progression and facilitates early diagnosis.8 Consequently, there is a compelling need for diagnostic methods capable of detecting in vivo ONOO− in real time with high sensitivity9,10 and selectivity, thereby enabling precise in vivo imaging and mechanistic investigation.1,11While conventional fluorescence imaging is fundamentally limited by excitation-induced autofluorescence and shallow penetration, chemiluminescence (CL) circumvents these issues by operating without external light sources.12,13 Consequently, CL offers zero-background interference and superior contrast, making it an ideal modality for high-fidelity in vivo imaging.14–16
Despite their promising potential, advancing the broad preclinical application of current CL probes remains a formidable challenge.17,18 Classical CL scaffolds—such as luminol,19 Schaap's dioxetanes,20,21 and peroxyoxalates22—suffer from an inherent susceptibility to ambient auto-oxidation and hydrolytic degradation.23,24 Furthermore, their emission profiles frequently exhibit transient “flash-type” kinetics,25 which severely restricts their practical imaging window.26,27 While structural engineering strategies, such as those pioneered by Pu and co-workers, have notably extended emission half-lives, achieving a robust balance between a practical signal duration and exceptional recognition specificity remains a significant challenge.28–31 Beyond these kinetic issues, the paucity of biocompatible scaffolds restricts the effective modulation of energy levels, thereby hindering the development of deep-tissue-penetrating red-shifted probes.32,33 Although recent work by Li et al. utilizing electron-deficient acceptors has achieved longer-wavelength emission, the repertoire of adaptable red-shifted scaffolds remains limited.34,35 A final, critical limitation arises from the homogeneity of recognition moieties: the pervasive reliance on boronate esters for RONS detection often leads to persistent cross-reactivity with H2O2,36–38 compromising the selective quantification of ONOO− in complex inflammatory microenvironments.39
To address these critical limitations—specifically to circumvent the cross-reactivity of boronate esters and access longer-penetrating wavelengths—we rationally designed a library of chemiluminescent scaffolds with α,α-dicyanoolefin (SMTPT, YMTPT, QMTPT, and TMTPT). This design strategy strategically employs the carbon–carbon double bond (CC) of α,α-dicyanoolefin as a recognition site to ensure orthogonal specificity for ONOO− over H2O2. Notably, the lead candidate, YMTPT, spontaneously self-assembles into a nanoprobe in aqueous environments. Corroborated by experimental and theoretical evidence, we elucidated a distinct ONOO−-triggered chemiexcitation mechanism via a dioxetane intermediate, wherein O–O bond cleavage accesses an S1–S0 near-degenerate “funnel” to promote efficient chemiexcitation. Crucially, the confined nano-architecture establishes an ideal microenvironment for highly efficient intermolecular chemiluminescence resonance energy transfer (CRET), yielding robust, excitation-free red-shifted emission. Leveraging this robust mechanism, YMTPT-NPs demonstrated exceptional performance in an acute arthritis model, achieving a high SBR (31.38) and an extended practical imaging window (>15 min), thereby effectively mitigating the kinetic instability and poor selectivity characteristic of conventional probes (Scheme 1). As explicitly benchmarked against recent CL probes (Table S1), YMTPT-NPs distinctly stand out by combining absolute orthogonal selectivity, spontaneous self-assembly, and practical signal duration. This work establishes a versatile molecular paradigm for the development of high-fidelity preclinical imaging tools for inflammation.
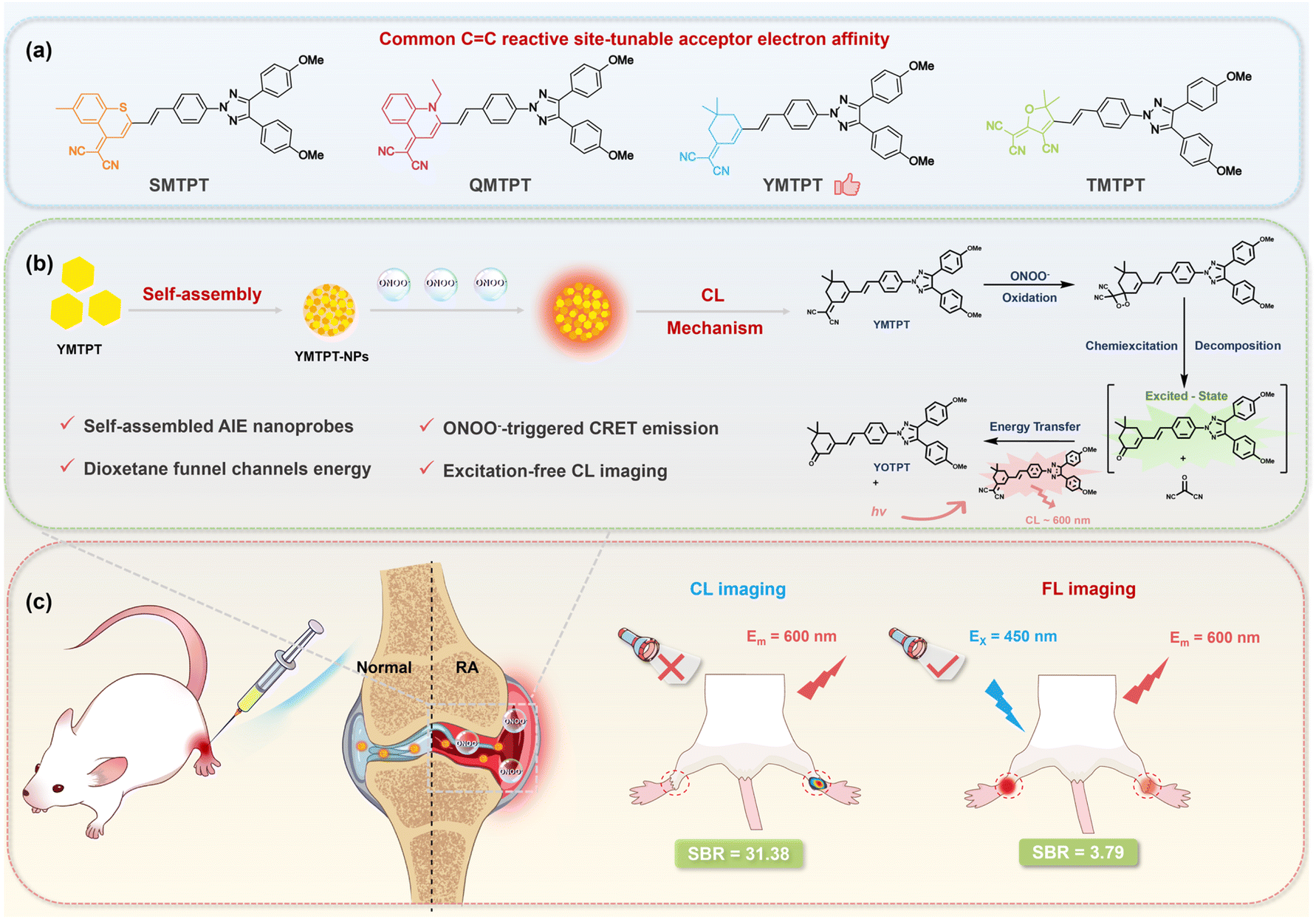 | ||
| Scheme 1 Design strategy, self-assembly, chemiluminescence mechanism, and in vivo application of the ONOO−-responsive nanoprobes. (a) Design and synthesis of a chemiluminescent molecule library with α,α-dicyanoolefins; YMTPT was identified as optimal via screening. (b) Schematic illustration of the spontaneous self-assembly of YMTPT into nanoparticles in aqueous media and the proposed ONOO−-triggered luminescence mechanism: reaction intermediate enables efficient chemiexcitation through an S1–S0 energy funnel, followed by bright emission via intermolecular CRET. (c) Successful high-contrast in vivo imaging (SBR = 31.38) of arthritis in mice using YMTPT-NPs. | ||
Results and discussion
Design and synthesis
The four chemiluminescent compounds—SMTPT, YMTPT, QMTPT, and TMTPT—share a common triazole backbone but feature distinct α,α-dicyanoolefin acceptors to modulate their photophysical properties (Fig. 1a) finely. The synthetic assembly was streamlined into a concise three-step protocol. Intermediate compounds 3–7 were not synthesized de novo herein, but were either obtained commercially or prepared following our previously reported protocols. Initially, the aldehyde-bearing precursor scaffolds were constructed via sequential Chan–Lam and Suzuki–Miyaura cross-couplings. Subsequently, the target molecules were afforded through Knoevenagel condensation with diverse active methylene-based acceptors, with isolated yields exceeding 65% for each step. Detailed synthetic procedures and structural characterization data (1H/13C NMR and HRMS) are provided in the SI (Fig. S1–S16). | ||
| Fig. 1 Photophysical properties of the synthesized chemiluminescent nanoprobes. (a) Chemical structures of the synthesized precursor molecules (SMTPT, QMTPT, YMTPT, and TMTPT). (b) Morphological and structural characterizations of the self-assembled nanoprobes in pure water, detected by scanning electron microscopy (SEM, scale bar: 200 nm) and dynamic light scattering (DLS). (c) Normalized UV-vis absorption spectra of the four nanoprobes (10 µM) in water. (d) Fluorescence (FL) spectra of the four nanoprobes (10 µM) in water. (e) Fold enhancement of CL intensity for each nanoprobe upon addition of ONOO−. (f) CL spectra of the four nanoprobes (10 µM). (g) Selectivity of YMTPT-NPs toward ONOO− (100 µM) and other ROS (1 mM). (h) CL intensity of YMTPT-NPs (10 µM) as a function of ONOO− concentration. | ||
Photophysical properties
To evaluate whether the highly conjugated and hydrophobic skeletons of the synthesized probes can induce the formation of nanoaggregates, we used SEM and DLS assays to analyze their self-assembly behaviors in aqueous media. As shown in Fig. 1b, employing YMTPT as a representative model, the molecules spontaneously assembled into stable nanoparticles with a well-defined spherical morphology in pure water. Furthermore, the average hydrodynamic diameter for the resulting YMTPT nanoprobes (YMTPT-NPs) was measured to be 89.42 nm. Notably, all four synthesized probes (SMTPT, QMTPT, YMTPT, and TMTPT) demonstrated this robust self-assembly capability. The spontaneous nanoprecipitation was likely due to the increased structural rigidity of the triazole backbones and the inherent hydrophobicity of their respective α,α-dicyanoolefin motifs.Detailed photophysical characterizations were subsequently performed to evaluate the optical properties of both the monomeric precursors and the self-assembled nanoprobes. UV-vis absorption spectra revealed maxima at approximately 450 nm for all nanoprobes, while FL emission extended from 600 nm into the near-infrared region (up to 700 nm), attributed to varying degrees of intramolecular charge transfer (Fig. 1c and d). Guided by these spectral data, optimal excitation and emission parameters as well as exposure times were selected for subsequent imaging. Both FL and CL signals were acquired using an IVIS Spectrum imaging system. Upon the addition of ONOO−, all compounds exhibited a marked increase in CL intensity and a concurrent decrease in FL intensity (Fig. S17). Quantitative analysis indicated that YMTPT-NPs exhibited the most pronounced response, with an approximately 50-fold increase in CL intensity (Fig. 1e). In contrast, QMTPT displayed the poorest performance, primarily because its inherently poor solubility led to severe precipitation in the aqueous system, thereby restricting its effective contact with ONOO−. Notably, YMTPT-NPs displayed a sharp CL peak immediately following ONOO− addition. This kinetic profile suggests an efficient, transient emission mechanism ideal for rapid, high-sensitivity imaging (Fig. S18). The CL emission maxima for all probes were centered mainly in the 550–600 nm range (Fig. 1f and S19), closely mirroring their respective fluorescence spectra. This spectral alignment is consistent with a potential mechanism wherein the specific dioxetane decomposition transfers chemical energy to populate the excited state; subsequently, intermolecular CRET within the confined nano-architecture likely contributes to the observed strong radiative relaxation.
Given that YMTPT-NPs exhibited the most robust CL response and superior photophysical stability, their selectivity was evaluated against a panel of reactive oxygen species (ClO−, 1O2, ˙OH, and H2O2). As depicted in Fig. 1g, YMTPT-NPs demonstrated remarkable selectivity toward ONOO−, yielding a significant CL signal compared to the negligible responses observed for other species. In particular, the inertness towards H2O2 validates our design strategy of utilizing the carbon–carbon double bond of the α,α-dicyanoolefin motif to circumvent the cross-reactivity inherent to traditional boronate-based probes. To assess sensitivity, 10 µM YMTPT-NPs were incubated with varying concentrations of ONOO− (0–150 µM) at 37 °C. The CL intensity correlated positively with ONOO− concentration, displaying a linear relationship across the 0–150 µM range (Fig. 1h). The limit of detection (LOD) was determined to be 163 nM (based on the 3σ/k method). Furthermore, co-incubation with various potential biological interferents—including amino acids (glycine, leucine, glutamic acid, cysteine), ions (Na+, Mg2+, Fe2+, F−, Br−, I−, HS−, CH3COO−, NO2−), and biomolecules/enzymes (galactosidase, catalase, horseradish peroxidase, sulfatase)—elicited negligible CL enhancement (Fig. S20), thereby confirming the superior specificity of YMTPT-NPs for ONOO−.
Chemiluminescence mechanism
Given that the self-assembled YMTPT-NPs exhibited the most robust CL signal, this formulation was selected for a detailed mechanistic investigation. To elucidate the photophysical impact of its self-assembly, the aggregation-induced emission (AIE) characteristics were systematically evaluated. As illustrated in Fig. 2a and b, the stepwise addition of water to a solution of YMTPT in DMF resulted in an initial decrease in FL intensity, followed by a sharp increase once the water fraction exceeded 50%. This remarkable fluorescence turn-on perfectly echoes the spontaneous nanoprecipitation process observed earlier, physically confirming that the formation of confined nano-aggregates effectively restricts intramolecular motions to suppress non-radiative decay. Notably, a comparable aggregation-triggered fluorescence enhancement was also observed for the other synthesized analogues (SMTPT, QMTPT, and TMTPT) (Fig. S21 and S22). This inherent AIE property is highly advantageous, providing a solid physical foundation for robust emission in aqueous biological environments.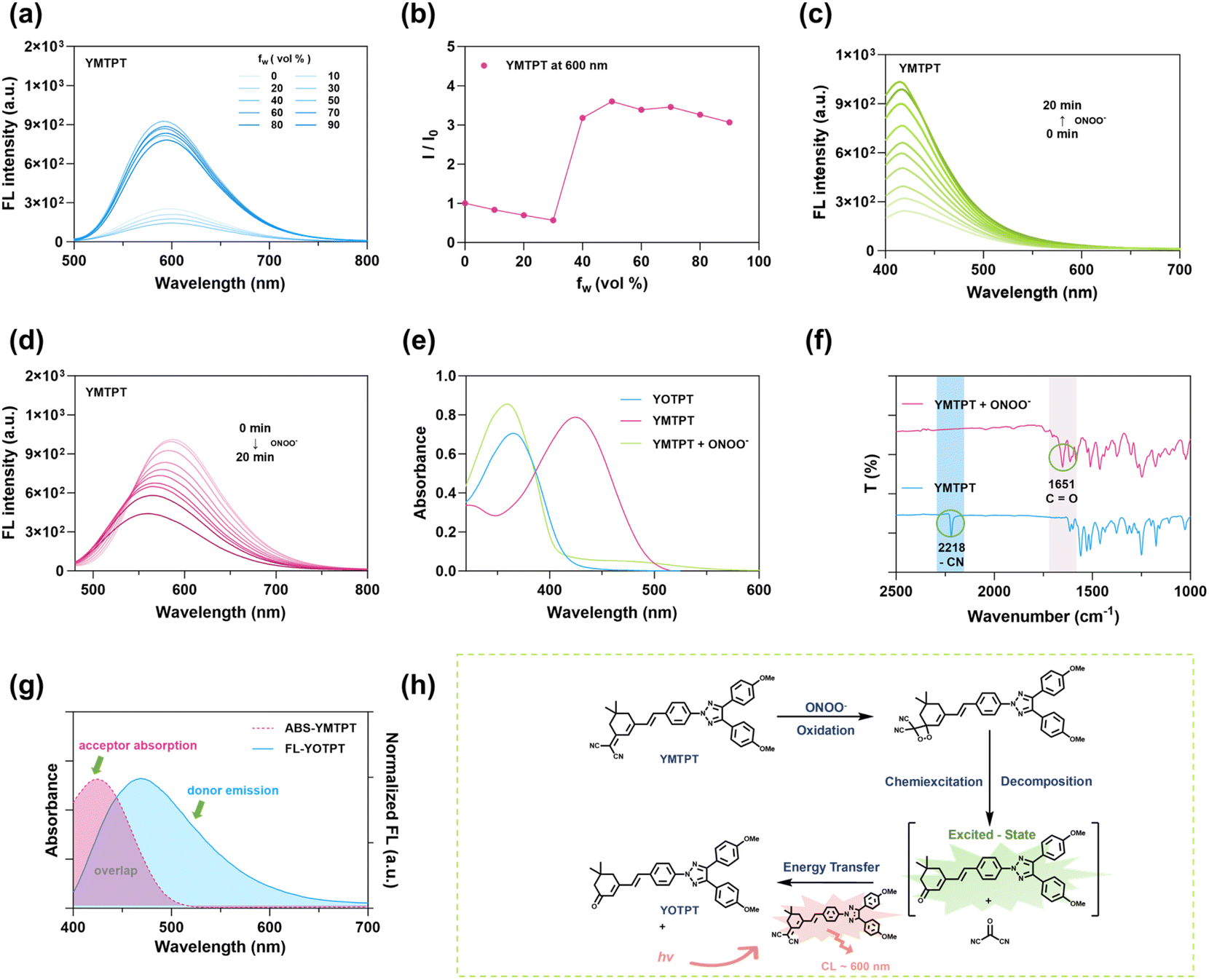 | ||
| Fig. 2 Photophysical characterization and mechanistic investigation of YMTPT. (a) FL spectra of YMTPT (10 µM) in DMF/water mixtures with varying water fractions (fw). Excitation wavelength (λex) = 450 nm. (b) Plot of the relative FL intensity (I/I0) of YMTPT versus water fraction (fw). (c) FL spectra of YMTPT in DMF following the addition of ONOO− (100 µM) (λex = 360 nm). (d) FL spectra of YMTPT in water upon reaction with ONOO− (100 µM) (λex = 450 nm). (e) Absorption spectra of YMTPT, the reaction mixture of YMTPT with ONOO− (100 µM), and the reference compound YOTPT in water. (f) FTIR spectra of YMTPT before and after the addition of ONOO−. (g) Spectral overlap between the absorption spectrum of the acceptor (YMTPT) and the emission spectrum of the donor (YOTPT) in water. (h) Schematic illustration of the proposed mechanism for ONOO−-activated chemiluminescence in YMTPT. | ||
To elucidate the CL mechanism, photophysical and spectroscopic variations were monitored following incubation with ONOO−. The absorption maximum of YMTPT at 450 nm underwent a hypsochromic shift to 360 nm upon ONOO− addition in DMF, indicating a chemical transformation of the molecular backbone and the generation of a new species. Concurrently, the fluorescence band at approximately 600 nm diminished with increasing incubation time. Upon excitation at 360 nm, a new emission peak centered at 420 nm emerged, accompanied by a visual transition from bright yellow to colorless. These spectral and visual alterations corroborate the formation of a distinct product (Fig. 2c–e and S23, S24). The reaction product was further characterized via Fourier Transform Infrared Spectroscopy (FTIR) and Mass Spectrometry (MS). The disappearance of the –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N band at 2218 cm−1 and the emergence of a carbonyl band at 1651 cm−1 following the reaction were observed, consistent with the oxidative cleavage of the terminal malononitrile-activated carbon–carbon double bond (Fig. 2f). MS corroborated this assignment: the YMTPT molecular ion at m/z 553.24 disappeared, and a new peak at m/z 528.24—matching the sodium adduct of the oxidized product ([M + Na]+ calc. m/z = 528.238)—was detected (Fig. S25).
N band at 2218 cm−1 and the emergence of a carbonyl band at 1651 cm−1 following the reaction were observed, consistent with the oxidative cleavage of the terminal malononitrile-activated carbon–carbon double bond (Fig. 2f). MS corroborated this assignment: the YMTPT molecular ion at m/z 553.24 disappeared, and a new peak at m/z 528.24—matching the sodium adduct of the oxidized product ([M + Na]+ calc. m/z = 528.238)—was detected (Fig. S25).
To validate this structural assignment, the putative oxidation product, YOTPT, was synthesized independently. Its UV-vis absorption profile was found to closely mirror that of the reaction product obtained from YMTPT and ONOO−. Significantly, the emission of YOTPT centered near 420 nm overlaps extensively with the absorption band of YMTPT in a DMF/water mixture (Fig. 2e and g). Collectively, these spectral overlaps, considered alongside the previously recorded CL spectrum of YMTPT following ONOO− treatment, point to a CRET process wherein the excited oxidation product (YOTPT) serves as the energy donor and the unreacted YMTPT serves as the acceptor. Since this system affords intense, red-shifted emission without the need for external excitation, it is particularly advantageous for high-contrast in vivo imaging applications with negligible background interference.
Crucially, to provide direct experimental evidence for the proposed chemiexcitation pathway, MS was employed to trace the reaction species. We successfully captured the key intermediate, with mass peaks clearly detected at m/z 586.24 and m/z 608.24, which perfectly match the [M + H]+ and [M + Na]+ adducts of the predicted dioxetane intermediate (calc. m/z = 585.238), respectively (Fig. S26). The direct capture of this intermediate provides compelling experimental validation for the initial ONOO−-triggered cyclization process.
Based on these empirical data, a mechanism is proposed wherein ONOO− attacks the carbon–carbon double bond of α,α-dicyanoolefin in YMTPT, initiating nucleophilic addition and subsequent cyclization to yield a dioxetane intermediate. Subsequent cleavage of the O–O bond and decomposition of the dioxetane moiety generate a high-energy excited species (YOTPT*). This excitation energy is subsequently transferred from the excited donor (YOTPT*) to the acceptor (YMTPT) via a CRET process, resulting in the observed CL emission (Fig. 2h). Crucially, the inherent AIE property of YMTPT facilitates the formation of nano-aggregates in aqueous environments, which spatially confines the excited donor and ground-state acceptor in proximity. This confinement ensures highly efficient intermolecular energy transfer that is not strictly diffusion-controlled, thereby maintaining robust luminescence even as the local concentration of the substrate changes. This proposed mechanism elucidates the kinetic profile characterized by a sharp initial CL peak followed by decay, and rationalizes the necessity of the self-assembled nano-architecture for high-contrast in vivo imaging.
Theoretical calculations
To theoretically validate the proposed oxidation pathway, the electronic states and energies of representative species—YMTPT, the dioxetane transition state (TS), and the oxidation product YOTPT—were computed at the B3LYP/6-31G(d,p) level, incorporating thermal corrections and zero-point energies.Fig. 3a illustrates the frontier molecular orbital (HOMO/LUMO) distributions and calculated optical gaps: reactant (YMTPT, ΔE = 2.66 eV, calculated λ ca. 466 nm), transition state (TS, ΔE = 2.97 eV), and product (YOTPT, ΔE = 3.23 eV, calculated λ ca. 384 nm). The experimentally determined UV-vis maxima for the reactant and product are approximately 450 nm and approximately 360 nm, respectively. Consequently, both theoretical predictions and experimental data consistently demonstrate a blue shift upon product formation. Two distinct features of the computed electronic-structure alterations warrant particular attention. First, the HOMO undergoes progressive stabilization along the reaction coordinate (−5.31 → −5.41 → −5.49 eV), reflecting enhanced thermodynamic stability. Second, the LUMO is progressively destabilized (−2.65 → −2.43 → −2.26 eV), resulting in an expansion of the HOMO–LUMO gap. Collectively, this simultaneous HOMO stabilization and LUMO destabilization suggests that oxidation and subsequent structural reorganization disrupt the original π-delocalization, thereby localizing the acceptor orbitals and inducing the observed blue shift in absorption.
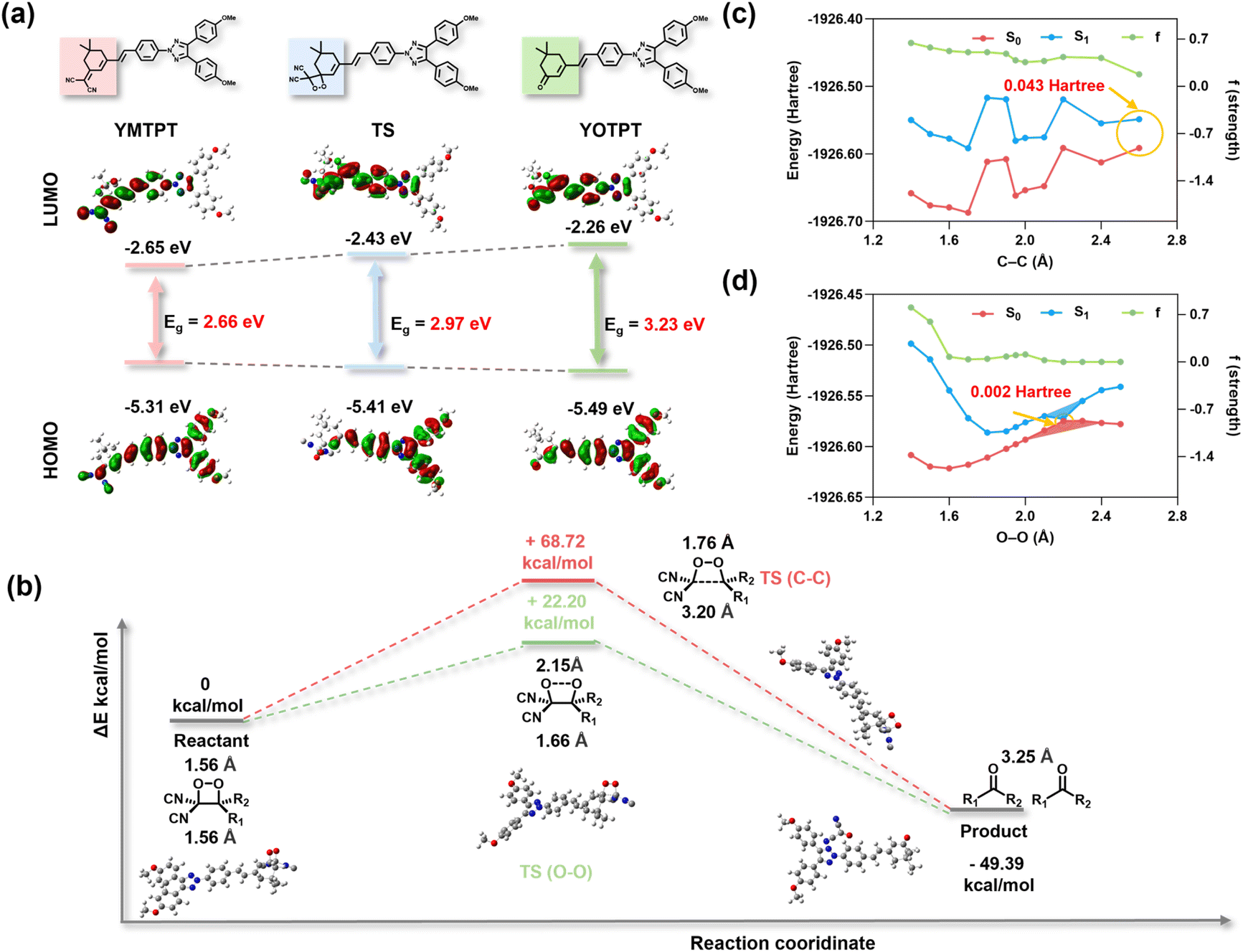 | ||
| Fig. 3 Theoretical investigation into the decomposition mechanism of YMTPT. (a) Chemical structures, frontier molecular orbital (HOMO/LUMO) distributions, and calculated energy levels for YMTPT (reactant), TS (the transition state), and YOTPT (product). Eg values represent the optical gaps derived from DFT calculations. (b) Calculated Gibbs free energy profiles for the oxidation of YMTPT by ONOO−. (c and d) Excited-state potential energy surface (PES) scans along the (c) C–C and (d) O–O bond cleavage coordinates. | ||
The potential coupling of the reaction to chemical excitation was subsequently evaluated by comparing two possible decomposition routes (O–O cleavage versus C–C cleavage) under a consistent theoretical framework. The computed Gibbs free-energy barriers and overall reaction free energy were determined as follows: ΔG(TSOO) = +22.20 kcal mol−1, ΔG(TSCC) = +68.72 kcal mol−1, and ΔG(product) = −49.39 kcal mol−1 (Fig. 3b), indicating a highly exergonic transformation. Consequently, the ground-state energy profile favors the initial O–O bond cleavage on kinetic grounds; furthermore, the chemical energy released in this step serves as the driving force for chemical excitation. In light of experimental evidence confirming the cleavage of the dioxetane C–C bond and the formation of YOTPT, the primary theoretical objectives were to rationalize the kinetic preference for O–O cleavage and to ascertain whether O–O rupture can produce a near-degenerate S1–S0 region (a “funnel”) capable of channeling chemical energy into an excited state.40,41 To elucidate these mechanisms, excited-state potential energy surface (PES) scans were performed at the same DFT level [B3LYP/6-31G(d,p), incorporating thermal and ZPE corrections] along the C–C and O–O dissociation coordinates. The S1 PES scan along the C–C stretching coordinate exhibits only a modest decrease in S1 energy with bond elongation, while the oscillator strength remains moderate (f ca. 0.3–0.6). Significantly, the S1–S0 gap does not approach near-degeneracy (remaining consistently larger than 0.04 hartree, ca. 1.1 eV), indicating that the C–C pathway does not facilitate efficient nonadiabatic population transfer to S1 (Fig. 3c). In sharp contrast, the S1 scan along the O–O coordinate unveils a steep descent of S1 with stretching: at elongated O–O distances (e.g., 2.20 Å), the S1–S0 energy gap drops to 0.002 hartree (ca. 0.05 eV), and the oscillator strength collapses from a large initial value to essentially zero (f ca. 10−4 at long O–O distances). These features unambiguously identify the presence of an S1–S0 near-degenerate region (the “funnel”) along the O–O coordinate (Fig. 3d).
Collectively, the experimental observations and theoretical calculations provide compelling support for the following plausible mechanistic pathway: first, the reaction is kinetically predisposed toward initial O–O bond cleavage. Second, O–O stretching induces the formation of an S1–S0 near-degenerate region (acting as an energy “funnel”), facilitating the nonadiabatic population of the S1 state. Subsequently, the intermediate undergoes adiabatic relaxation along the S1 potential energy surface. During this adiabatic evolution, the remaining dioxetane C–C bond cleaves, and the electronic structure continuously transitions to yield the fully localized excited state of the completely separated carbonyl product (YOTPT*). Finally, this excitation energy is transferred from the excited donor (YOTPT*) to the highly emissive acceptor (YMTPT) via an intermolecular energy transfer mechanism (CRET), resulting in the observed CL. Thus, both theory and experiment converge to suggest a sequential chemiexcitation mechanism: O–O cleavage establishes the crucial S1–S0 near-degeneracy (the “funnel”) for nonadiabatic excitation, followed by excited-state adiabatic C–C cleavage to generate the donor, driving the “funnel-energy transfer-acceptor emission” cascade.
Cytotoxicity and intracellular imaging
Motivated by its favorable photophysical properties in solution, the suitability of YMTPT-NPs for intracellular ONOO− sensing and their cytotoxicity were subsequently evaluated. Cytotoxicity was evaluated via a CCK-8 assay in 4T1 cells (Fig. S27). Cell viability exceeded 85% even at probe concentrations up to 40 µM, indicating low cytotoxicity and affirming the suitability of YMTPT-NPs for biological imaging applications.To evaluate the dynamic response of the nanoprobes in a physiological environment, time-dependent CL imaging was performed in LPS-treated cells. Upon the addition of YMTPT-NPs (20 µM), the intracellular CL signal rapidly intensified, reaching a peak within 2 min and maintaining a readily detectable signal over the 15 min observation window (Fig. S28).
Intracellular CL imaging experiments were conducted using four distinct treatment groups: PBS (control), LPS (to induce inflammation), LPS + UA (LPS treatment followed by incubation with uric acid, an ONOO− scavenger), and uric acid (UA) alone. Following incubation with YMTPT-NPs (20 µM), a negligible background CL signal was observed in the PBS group. In contrast, LPS treatment elicited a pronounced increase in intracellular CL intensity (ca. 3-fold higher than that of the PBS group), consistent with the upregulation of endogenous ONOO− production. Co-incubation with uric acid (LPS + UA) markedly attenuated the CL signal to near-baseline levels, whereas UA alone yielded signals comparable to those of the PBS control (Fig. S29). These results demonstrate the selective activation of YMTPT-NPs by inflammation-derived ONOO− within the cellular environment, thereby validating the utility of the excitation-free detection modality for achieving a high signal-to-noise ratio.
In vivo comparison of CL and FL imaging performance
To highlight the distinct advantages of YMTPT-NPs-mediated CL over conventional FL imaging in vivo, a comparative analysis of the two modalities was performed. All animal experiments were conducted in strict accordance with the Guidelines for the Care and Use of Laboratory Animals of China Pharmaceutical University, and were approved by the Institutional Animal Care and Use Committee. In contrast to conventional FL probes that rely on target-induced fluorescence enhancement (signal-on), YMTPT undergoes chemical decomposition upon reaction with ONOO−, thereby attenuating its intrinsic fluorescence (signal-off). Given that the decomposition product (YOTPT) exhibits hypsochromic emission and contributes negligibly to in vivo fluorescence enhancement, the subsequent analysis focused on reducing the FL signal following probe activation by ONOO−.To facilitate a direct comparison, YMTPT-NPs (20 µM) solutions containing either 0 µM (control) or 100 µM ONOO− were subcutaneously administered into the dorsal flanks of mice, after which corresponding CL and FL images were acquired (Fig. 4a and b). Quantitative analysis revealed a substantial CL response at the ONOO−-injected sites (SBR = 53.37), in sharp contrast to the negligible FL contrast observed (SBR = 1.95). Regarding signal modulation, CL intensity exhibited an approximately 10.65-fold enhancement upon ONOO− exposure, whereas FL intensity decreased to approximately 0.62-fold (Fig. 4c). These results demonstrate that YMTPT-NPs, when utilized in CL mode, afford markedly superior sensitivity and contrast relative to FL imaging, thereby facilitating the rapid and accurate localization of ONOO−-enriched regions. Collectively, these data underscore the potential of ONOO−-triggered CL probes for high-contrast, rapid in vivo imaging applications.
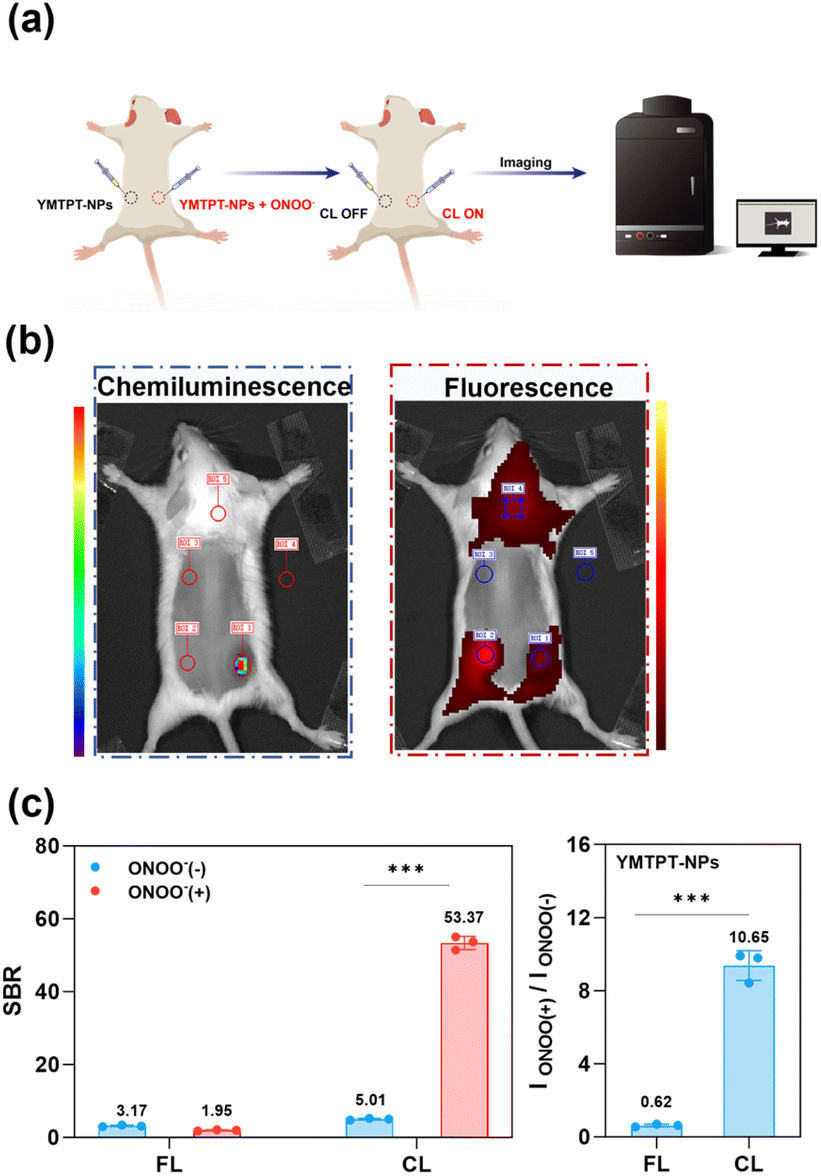 | ||
| Fig. 4 Comparative in vivo evaluation of CL and FL imaging modalities. (a) Schematic illustration of the subcutaneous imaging protocol. Physiological saline solutions of YMTPT-NPs (20 µM, formulated from a PBS stock) and YMTPT-NPs pre-treated with ONOO− were subcutaneously administered into the dorsal flanks of mice, followed by dual-modality image acquisition. (b) Representative CL and FL images obtained from the subcutaneous mouse model. (c) Quantitative analysis of fold enhancement and SBR for CL and FL modes corresponding to panel (b). Data are presented as mean ± SD (n = 3); ***p < 0.001. | ||
In vivo imaging of acute inflammation and biosafety evaluation
Acute inflammation was induced by injecting λ-carrageenan into the right hind limb of mice, while the contralateral limb received a saline injection as a negative control. Pronounced erythema and edema were observed at 6 h post-injection (Fig. S30). Subsequently, YMTPT-NPs (20 µM) were administered locally to both hind limbs, and images were acquired at multiple time points to monitor the temporal evolution of the signals (Fig. 5a).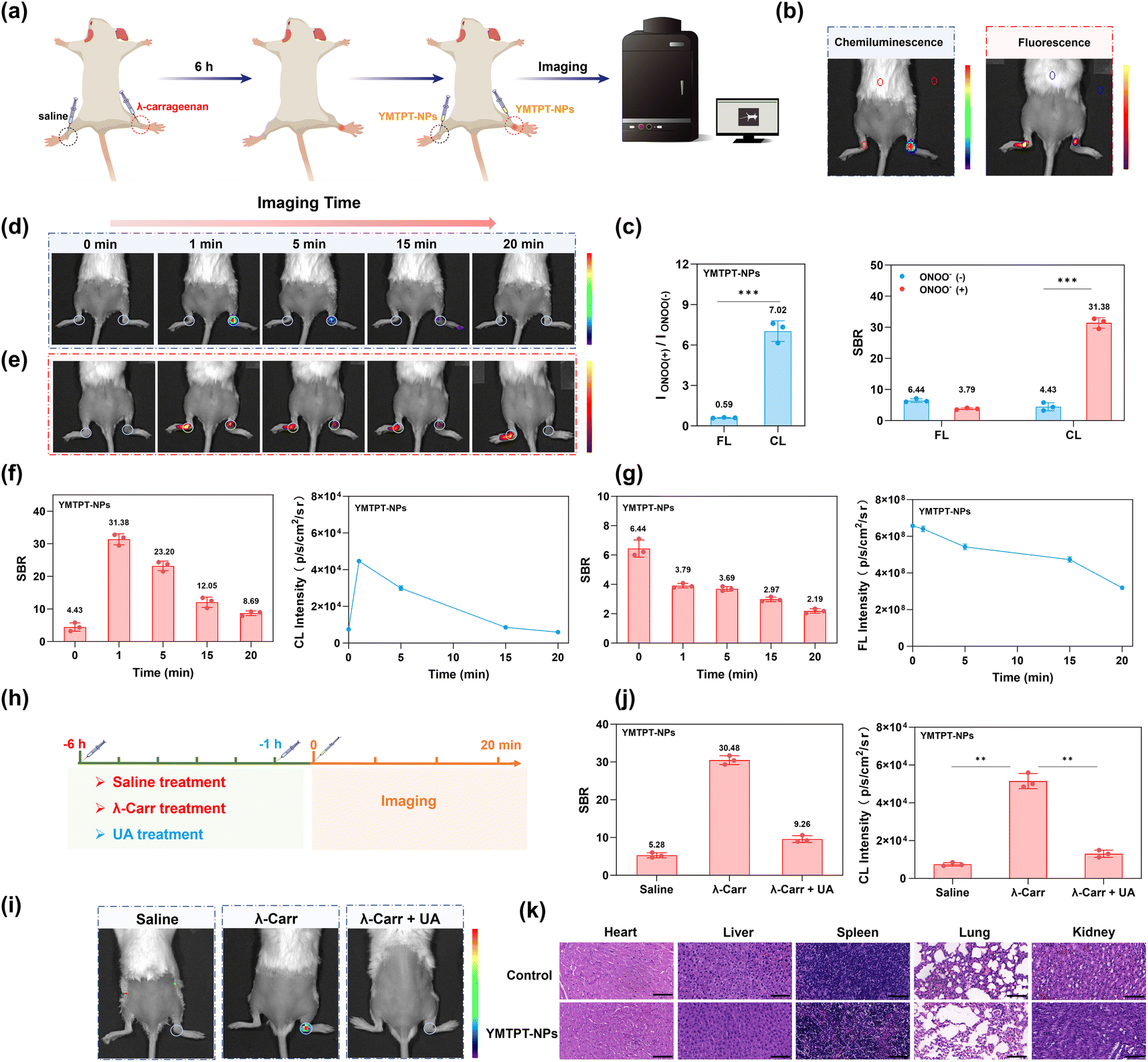 | ||
| Fig. 5 In vivo imaging of acute arthritis induced by λ-carrageenan. (a) Schematic illustration of the protocol for inducing acute arthritis via λ-carrageenan. (b) Representative FL and CL imaging of the mouse arthritis model. (c) Quantitative analysis of SBR and fold enhancement for CL and FL modes corresponding to panel (b). (d) Time-dependent CL images acquired at 0, 1, 5, 15, and 20 min following local administration of YMTPT-NPs (in physiological saline). (e) FL images acquired at the corresponding time points. (f) Corresponding quantification of SBR and CL intensity values. (g) Corresponding quantification of SBR and FL intensity values. (h) Schematic of the control-intervention study design. (i) Representative CL images of the three treatment groups: control, model, and intervention (UA scavenger). (j) Quantification of SBR and CL intensity for each group in (i). (k) Histological evaluation of potential toxicity. H&E staining images of major organs collected from mice in the control and YMTPT-NPs-treated groups. Data are presented as mean ± SD (n = 3); **p < 0.01, ***p < 0.001. | ||
Quantitative analysis revealed a marked increase in CL intensity at the inflamed site. Specifically, CL intensity was enhanced by approximately 7.02-fold relative to the contralateral control, yielding an SBR of 31.38 (Fig. 5b and c). In contrast, FL signals exhibited negligible variation over the same interval, presenting an initial FL SBR of only 3.79 (Fig. 5d–g). Temporal profiling indicated that the CL signal provided a practical functional imaging window of approximately 20 min post-injection before its characteristic decay, whereas FL variations were minimal. Collectively, these data demonstrate that YMTPT-NPs facilitate rapid, high-contrast lesion localization, underscoring the superior sensitivity of excitation-free CL imaging for preclinical in vivo applications. To verify that the observed CL activation was driven by inflammation-associated ONOO−, a control-intervention study involving specific scavenging was conducted. Mice were divided into three groups: a saline-treated control group, a λ-carrageenan-induced arthritis model group, and an intervention group administered uric acid (a specific ONOO− scavenger) locally 5 h post-carrageenan injection. At 6 h post-induction, YMTPT-NPs were administered, and CL imaging was subsequently performed (Fig. 5h). The model group exhibited substantially elevated CL intensity at the lesion site compared to both the control and intervention groups (Fig. 5i and j); notably, the SBR of the model group was approximately 3.3-fold higher than that of the intervention group, confirming the effective suppression of probe activation via ONOO− scavenging. These findings validate the capability of the probe to sensitively and selectively detect ONOO− accumulation within this acute arthritis model.
The biosafety profile of YMTPT-NPs was assessed via hematoxylin and eosin (H&E) staining of major organs (heart, liver, spleen, lungs, and kidneys). No significant pathological abnormalities were observed in tissues from YMTPT-NPs-treated animals relative to the control group (Fig. 5k), indicating acceptable short-term biocompatibility under the experimental conditions. Regarding blood compatibility, no observable hemolytic effect was detected even at YMTPT-NPs concentrations up to 60 µM (Fig. S31). This negligible hemolytic activity demonstrates the excellent hemocompatibility of the nanoprobes. While YMTPT-NPs combine red-shifted emission capabilities with structural tunability, we acknowledge that their kinetic profile remains essentially flash-type, presenting clear potential for further optimization regarding absolute signal output and emission lifetime.
Conclusions
In summary, we report the development of self-assembled chemiluminescent nanoprobes (YMTPT-NPs) to overcome the stability and selectivity limitations of current ONOO− sensing. By employing an α,α-dicyanoolefin motif as a specific recognition site, YMTPT-NPs fundamentally circumvent H2O2 cross-reactivity, ensuring high-fidelity detection. Both experimental and DFT investigations highlight a sequential chemiexcitation mechanism: ONOO−-triggered oxidation accesses a crucial S1–S0 energy “funnel” to channel chemical energy into the excited state, subsequently driving highly efficient intermolecular CRET within the confined nano-architecture. In vivo validations confirmed that YMTPT-NPs exhibit a favorable biosafety profile and deliver high-contrast imaging outcomes (SBR = 31.38) within a functional 15 min window post-administration, thereby facilitating the precise visualization of acute lesions. This study expands the repertoire of chemiluminescent scaffolds and establishes that integrating robust photophysical mechanisms with self-assembled nanoprobes is a highly effective paradigm for precision in vivo imaging.Author contributions
L. Liu synthesized the compounds and characterized the photophysical properties. M. Kai, W. Li, and H. Liu provided suggestions on experiments and test support. L. Liu, M. Kai, J. Yang, P. Wang, and W. Zhang wrote the manuscript. J. Yang, P. Wang, and W. Zhang supervised and directed this study.Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting this study are available in the supplementary information (SI). Additional data are available from the corresponding author upon reasonable request. Supplementary information is available. See DOI: https://doi.org/10.1039/d6sc03467e.Acknowledgements
This work was financially supported by the Fundamental Research Funds for the Central Universities (2632026XCZX01 and 2632026ZD06) and the Basic Research Program of Jiangsu (BK20252073).Notes and references
- M. P. Murphy, H. Bayir and V. Belousov, et al., Nat. Metab., 2022, 4, 651–662 CrossRef PubMed.
- Q. Li, S. Xiao and X. Ge, et al., Angew. Chem., Int. Ed., 2024, 63, e202408792 CrossRef CAS PubMed.
- Y. Deng, X. Shi and X. Hu, et al., Anal. Chem., 2023, 95, 6496–6500 CrossRef CAS PubMed.
- H. Liu, L. Liu and W. Li, et al., Biosens. Bioelectron., 2026, 292, 118079 CrossRef CAS PubMed.
- P. Wang, L. Yu and J. Gong, et al., Angew. Chem., 2022, 134, e202206894 CrossRef.
- E. N. Tcyganov, E. Sanseviero and D. Marvel, et al., Cancer Cell, 2022, 40, 1173–1189 CrossRef CAS PubMed.
- L. Yang, D. Wang and H. Jia, et al., Adv. Mater., 2023, 35, 2301455 CrossRef CAS PubMed.
- L. Wu, J. Liu and X. Tian, et al., J. Am. Chem. Soc., 2021, 144, 174–183 CrossRef PubMed.
- J. H. Wilde, Y.-Y. Sun and S. R. Simpson, et al., Nat. Biomed. Eng., 2025, 1–14 Search PubMed.
- Y. Tang, Y. Li and C. He, et al., Nat. Commun., 2025, 16, 278 CrossRef PubMed.
- X. Wang, Q. Ding and R. R. Groleau, et al., J. Nucl. Med., 2020, 61, 1419–1427 CrossRef PubMed.
- S. Yoon, S. Y. Cheon and S. Park, et al., Biomater. Res., 2022, 26, 57 CrossRef PubMed.
- C. Li, J. Du and G. Jiang, et al., Nat. Commun., 2024, 15, 5832 CrossRef CAS PubMed.
- Z. Chen, Q. Li and Y. Wu, et al., Nat. Commun., 2025, 16, 238 CrossRef PubMed.
- S. Huang, S. Bai and T. Luo, et al., Adv. Funct. Mater., 2024, 34, 2409292 CrossRef CAS.
- M. Yang, J. Huang and J. Fan, et al., Chem. Soc. Rev., 2020, 49, 6800–6815 RSC.
- J. H. Li, S. S. Bian and T. Liu, et al., Biosens. Bioelectron., 2025, 270, 116978 CrossRef CAS PubMed.
- X. Li, C. Yin and S. S. Liew, et al., Adv. Funct. Mater., 2021, 31, 2106154 CrossRef CAS.
- S. Ye, N. Hananya and O. Green, et al., Angew. Chem., Int. Ed., 2020, 59, 14326–14330 CrossRef CAS PubMed.
- N. Hananya and D. Shabat, ACS Cent. Sci., 2019, 5, 949–959 CrossRef CAS PubMed.
- Y. Wang, M. Fu and M. Sun, et al., Chem. Mater., 2024, 36, 6269–6275 CrossRef CAS.
- M. David, S. Gutkin and R. V. Nithun, et al., Angew. Chem., Int. Ed., 2025, 64, e202417924 CrossRef CAS PubMed.
- L. S. Xu, E. Liang and L. Y. Tan, et al., Anal. Chem., 2025, 97, 24468–24476 CrossRef CAS PubMed.
- Y. T. Liu, W. Shen and Q. Li, et al., Nat. Commun., 2017, 8, 1003 CrossRef PubMed.
- Z. X. Chen, Q. Li and Y. Wu, et al., Nat. Commun., 2025, 16, 238 CrossRef PubMed.
- Y. Zhang, Y.-T. He and X. He, et al., CCS Chem., 2025, 7, 3460–3472 CrossRef CAS.
- Y. Y. Jiang, J. G. Huang and X. Zhen, et al., Nat. Commun., 2019, 10, 2064 CrossRef PubMed.
- X. Z. Wang and K. Y. Pu, Chem. Soc. Rev., 2023, 52, 4549–4566 RSC.
- J. S. Huang, L. C. Su and C. Xu, et al., Nat. Mater., 2023, 22, 1421–1429 CrossRef PubMed.
- L. Z. Wei, Y. M. Wu and Y. H. Zhou, et al., J. Med. Chem., 2025, 68, 20506–20518 CrossRef CAS PubMed.
- L. Li, X. Y. Zhang and Y. X. Ren, et al., J. Am. Chem. Soc., 2024, 146, 5927–5939 CrossRef CAS PubMed.
- J. Li, X. Y. Zhao and Y. T. Zhang, et al., Chem. Sci., 2025, 16, 3228–3237 RSC.
- S. Y. Zhang, H. Yuan and S. C. Sun, et al., Adv. Sci., 2023, 10, 2207651 CrossRef CAS PubMed.
- Y. L. Cao, J. Q. Gu and Z. J. Chen, et al., Adv. Mater., 2025, 37, 2408941 CrossRef CAS PubMed.
- C.-C. Wu, S.-H. Lee and T.-J. Huang, et al., Anal. Chem., 2025, 97, 14912–14920 CrossRef CAS PubMed.
- B. Wang, Y. Kong and X. Tian, et al., J. Hazard. Mater., 2024, 469, 134094 CrossRef CAS PubMed.
- X. Chai, B. Li and C. Chen, et al., Anal. Chem., 2023, 95, 5747–5753 CrossRef CAS PubMed.
- Z. K. Wang, T. T. Geng and X. Y. Yue, et al., Anal. Chem., 2025, 97, 2367–2374 CrossRef CAS PubMed.
- Y. Chen, W. Li and S. Li, et al., ACS Sens., 2025, 10, 398–406 CrossRef CAS PubMed.
- L. Yue and Y.-J. Liu, Chem. Phys. Lett., 2022, 13, 10671–10687 CrossRef CAS PubMed.
- H. Isobe, Y. Takano and M. Okumura, et al., J. Am. Chem. Soc., 2005, 127, 8667–8679 CrossRef CAS PubMed.
Footnote |
| † L. Liu and M. Kai contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |