
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA unified electrocatalytic platform for the chemical–H2–electricity triad from biomass via interfacial electronic engineering
Wei Wanga,
Xiaoyang Hea,
Shuai Donga,
Jianying Wanga,
Deli Wu b and
Zuofeng Chen*a
b and
Zuofeng Chen*a
aSchool of Chemical Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, China. E-mail: zfchen@tongji.edu.cn
bState Key Laboratory of Pollution Control and Resources Reuse, College of Environmental Science and Engineering, Tongji University, 1239 Siping Road, Shanghai 200092, China
First published on 4th June 2026
Abstract
The selective electrooxidation of biomass-derived 1,3-propanediol (1,3-PDO) to high-value 3-hydroxypropionic acid (3-HP) offers a sustainable route for chemical synthesis but is severely hindered by competitive C–C bond cleavage and sluggish reaction kinetics. Here, we propose an electronic structure tuning strategy through interfacial engineering, constructing a Pd–Ni(OH)2 heterostructure to enhance the catalytic performance of Pd nanoparticles. The resulting catalyst exhibits exceptional activity toward the 1,3-PDO oxidation reaction (POR) in alkaline media, requiring a low potential of only 0.645 V (vs. RHE) to achieve a current density of 100 mA cm−2 while retaining a high faradaic efficiency of ∼94.9%. Comprehensive characterization and density functional theory (DFT) calculations reveal that strong interfacial interaction induces a downshift in the d-band center of Pd. This electronic restructuring optimizes the adsorption energetics of key intermediates, facilitating rapid desorption of 3-HP from active Pd sites and thereby suppressing overoxidation and carbon backbone degradation. A membrane electrode assembly (MEA) electrolyzer towards the POR demonstrates robust stability, retaining faradaic efficiencies above 90% during continuous operation. Furthermore, we demonstrate an integrated synthesis–energy device by coupling the POR workflow with the hydrogen evolution reaction (HER) and oxygen reduction reaction (ORR). This hybrid system, powered by the multifunctional Pd–Ni(OH)2 catalyst, enables flexible temporal deployment via a mode-switching scheme between daytime (HER‖POR) and nighttime (ORR‖POR). Collectively, this work elucidates an effective strategy for selective biomass valorization through electronic structure tailoring, offering an appealing multidimensional solution for sustainable electrochemical synthesis.
1 Introduction
The exponential growth of synthetic plastic materials over the past century has been a critical driver of industrial advancement and modern conveniences. However, this progress has also resulted in a global plastic waste crisis. Research indicates that the global annual production of plastics exceeds 430 million tons, with approximately two-thirds (over 280 million tons) ending up as waste.1–6 Plastics, commonly found in beverage bottles, textiles, and packaging, are highly durable and resistant to natural degradation, leading to their accumulation in terrestrial and marine ecosystems. This plastic waste not only pollutes oceans, severely impacting wildlife and climate, but also enters the human food chain, posing significant health risks.7,8The mitigation of plastic waste pollution currently follows two primary pathways: the 3R strategy (reduce, reuse, and recycle) and the development of biodegradable plastics, with the latter representing one of the most dynamic research frontiers in polymer science in recent decades.9 Although biodegradable plastics such as polylactic acid (PLA) and polybutylene succinate (PBS) have achieved commercial viability, their widespread application remains significantly constrained by several inherent drawbacks, including high production costs and slow degradation kinetics upon disposal.10–12 Consequently, the development of superior alternatives to existing biodegradable plastics has become an imperative research priority. In this context, poly(3-hydroxypropionate) (P3HP) has emerged as an ideal biodegradable bioplastic, demonstrating enhanced biodegradability, superior biocompatibility, and improved overall performance characteristics.13,14
3-Hydroxypropionic acid (3-HP), the precursor for P3HP synthesis, has emerged as a highly promising platform chemical with extensive industrial applications. Recognized by the U.S. Department of Energy as a top-value platform chemical, 3-HP demonstrates remarkable versatility as both a chemical intermediate and a functional end product.15,16 Its commercial significance stems from its dual utility as a renewable precursor for valuable derivatives including acrylic acid, malonic acid, acrylonitrile, and acrylamide, and as a key ingredient in coatings, adhesives, and antimicrobial formulations for diverse industries.17 Despite its substantial market potential projected, current production methods face considerable technical and economic challenges that limit commercial viability. Conventional chemical synthesis routes through acrylic acid hydration, β-hydroxypropionitrile hydrolysis, or various oxidation processes suffer from inherent limitations including high feedstock costs, low yields, complex purification requirements, and reliance on hazardous reagents. Microbial production platforms that utilize renewable feedstocks such as glycerol or glucose face fundamental challenges, as substantial byproduct formation and suboptimal yields continue to hinder the commercial viability of the biological production of 3-HP.18–21 Therefore, there is an urgent need to develop an efficient strategy for 3-HP production.
Although renewable energy-powered water electrolysis shows promise for green hydrogen production, its efficiency remains fundamentally limited by the sluggish kinetics of the anodic oxygen evolution reaction (OER).22–24 This fundamental challenge has driven the development of electrochemical hydrogen production coupled with alcohol oxidation (EHCO) as an innovative alternative strategy. The EHCO process offers dual advantages by replacing the kinetically sluggish OER with more favorable alcohol oxidation reactions: (1) significantly reduced overpotential for the oxygen evolution reaction and (2) simultaneous electrochemical upgrading of alcohols to value-added products at the anode.10,25–28 While the electrorefining of ethylene glycol to glycolic acid (C2) has been extensively documented,29–31 the electrocatalytic synthesis of 3-HP (C3) via the selective 1,3-PDO oxidation reaction (POR), as an alternative to the OER coupled with hydrogen production, remains unexplored to date.
In this work, we present an electronic structure tuning strategy to enhance the performance of the POR over Pd nanoparticles, as shown in Scheme 1. By introducing oxyphilic Ni(OH)2 as a support and employing a noble metal–transition metal combination strategy, we effectively reduce the Pd loading while modulating its electronic structure, electron transfer kinetics, and intermediate adsorption behavior, thereby enhancing the catalytic activity of the Pd-based electrocatalyst. Notably, the Ni(OH)2 matrix promotes the generation of abundant ˙OH on the Pd surface, which simultaneously boosts POR activity while suppressing product overoxidation. The Pd–Ni(OH)2/NF catalyst requires only 0.645 V (vs. RHE) to achieve 100 mA cm−2 for the POR in alkaline medium, with a high faradaic efficiency of ∼94.9% for 3-HP production. Comprehensive characterization and density functional theory (DFT) calculations confirm that interfacial interaction induces a downshift in the d-band center of Pd and enhances the oxophilicity of Ni species. This synergistic electronic restructuring facilitates rapid 3-HP desorption from active Pd sites and subsequent stabilization on adjacent Ni sites, effectively suppressing both over-oxidation and C–C bond cleavage. In a membrane electrode assembly (MEA) electrolyzer integrating the POR with the HER, the system demonstrates excellent stability, retaining faradaic efficiencies above 90% over 100 h of continuous operation. Moreover, the POR process can be further extended into an integrated synthesis–energy device by coupling with the ORR in a direct alcohol fuel cell configuration. Consequently, this work establishes a promising strategy for the mild, efficient, and selective electrosynthesis of 3-HP through mode-switching operation between daytime (HER-coupled) and nighttime (ORR-coupled) processes.
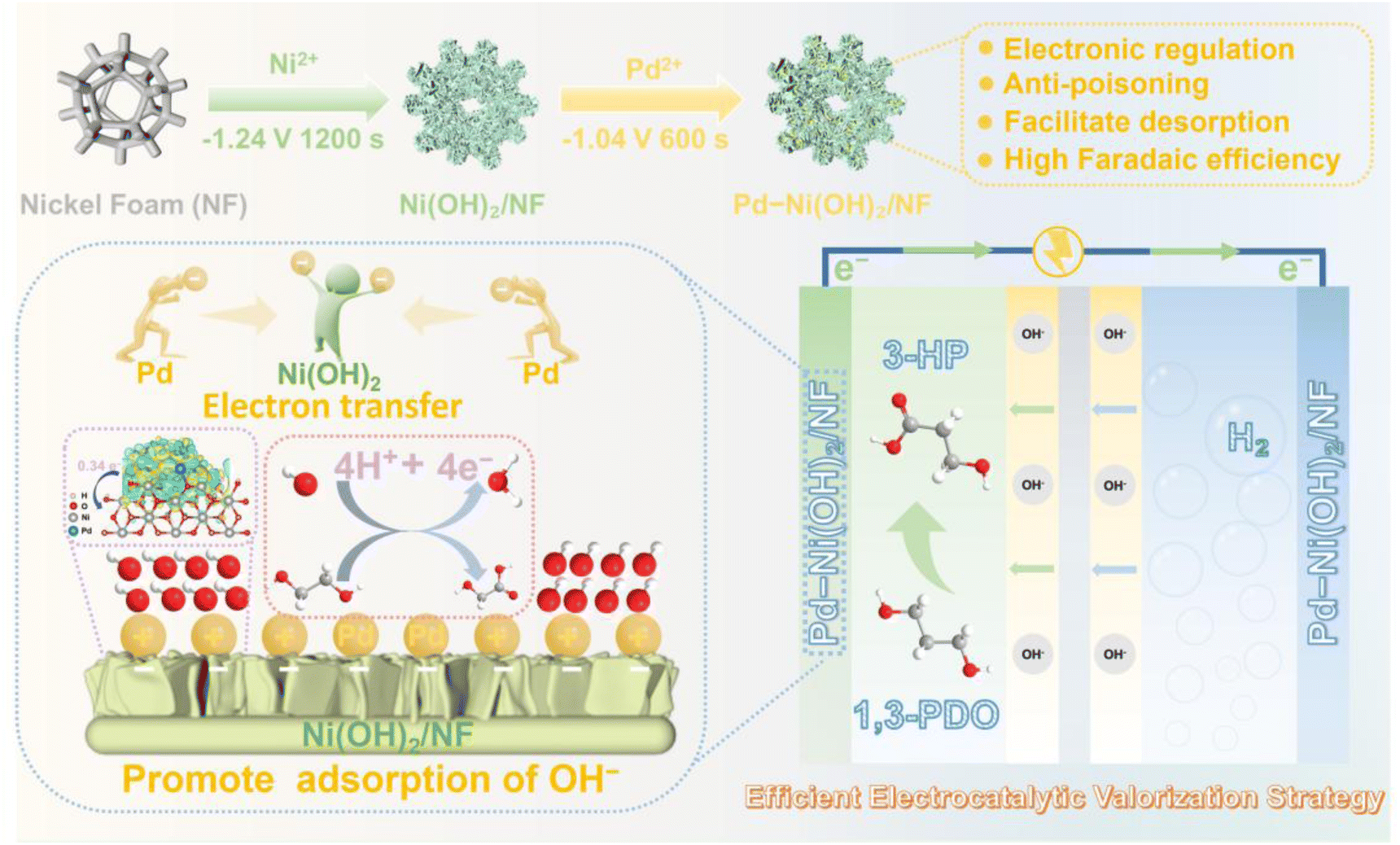 | ||
| Scheme 1 Schematic illustration of the preparation of Pd–Ni(OH)2/NF and the synergistic catalyst design, wherein interfacial engineering tunes the d-band center for highly selective conversion of 1,3-propanediol to 3-HP. | ||
2 Results and discussion
2.1 Synthesis and structural characterization of Pd–Ni(OH)2/NF
The Pd–Ni(OH)2/NF catalyst was prepared on a three-dimensional (3D) NF substrate via a two-step process, as detailed in the Experimental section. While optical images in Fig. S1 reveal clear visual distinctions among NF, Ni(OH)2/NF, and Pd–Ni(OH)2/NF, the morphology and microstructure of the prepared electrodes were examined using scanning electron microscopy (SEM). Fig. S2 shows in situ growth of Ni(OH)2 nanosheets on the three-dimensional NF scaffold, and Fig. 1A and B confirm the uniform deposition of Pd nanoparticles on these nanosheets. BET measurements reveal specific surface areas of 0.9162 m2 g−1 for Pd/NF and 4.0751 m2 g−1 for Pd–Ni(OH)2/NF, suggesting that the introduction of Ni(OH)2 nanosheets substantially enhances the catalyst surface area, offering abundant active sites and favorable mass transport.32 Transmission electron microscopy (TEM, Fig. 1C) verifies the irregular Pd nanoparticles anchored on Ni(OH)2. High-resolution TEM (HRTEM, Fig. 1D) displays lattice spacings of 0.223 nm, 0.195 nm, and 0.232 nm, corresponding to Pd (111), Pd (200), and Ni(OH)2 (002), respectively.33 TEM energy-dispersive X-ray spectroscopy (TEM-EDS) mapping (Fig. 1E and S3) illustrates a homogeneous distribution of Ni, Pd, and O, while inductively coupled plasma-optical emission spectrometry (ICP-OES) gives a Pd loading of 8.04 wt%. The selected area electron diffraction (SAED) pattern (inset) confirms the polycrystalline nature of deposited Pd. The XRD pattern of Ni(OH)2/NF (Fig. 1F) shows only peaks from the Ni substrate, consistent with low-crystallinity surface hydroxides. For Pd–Ni(OH)2/NF, additional peaks at 40.3°, 46.7°, and 68.1° are indexed to Pd (111), Pd (200), and Pd (220), respectively, alongside the Ni substrate signals. | ||
| Fig. 1 (A and B) SEM images, (C and D) TEM images, and (E) TEM-EDS mapping of Pd–Ni(OH)2/NF; the inset in (D) shows the SEAD pattern. (F) XRD patterns, (G) Pd 3d and (H) Ni 2p XPS spectra, and (I) XPS valence band spectra of Pd–Ni(OH)2/NF with reference samples. | ||
The chemical composition and valence states of the Pd–Ni(OH)2/NF electrode were analyzed by X-ray photoelectron spectroscopy (XPS). The survey spectrum (Fig. S4) confirms the presence of Pd, Ni, and O, consistent with the EDS results. In the Pd 3d region (Fig. 1G), peaks at 335.6 eV (3d5/2) and 340.8 eV (3d3/2) are assigned to metallic Pd, while those at 336.6 eV and 342.1 eV correspond to surface PdO. Notably, the Pd 3d peaks at Pd–Ni(OH)2/NF shift positively by 0.6 eV relative to Pd/NF (a control sample by direct electrodeposition of Pd nanoparticles on pristine NF), indicating strong electronic interaction between Pd and Ni(OH)2.34,35 The positive shift in the Pd 3d binding energy corresponds to a downshift of the Pd d-band center and consequently weakens the adsorption of key intermediates on Pd active sites.36–38 The Ni 2p spectrum (Fig. 1H) shows characteristic peaks of Ni2+ (854.5 eV and 872.3 eV) and Ni3+ (855.6 eV and 873.6 eV), along with satellite features at 860.9 eV and 879.6 eV.39–41 Compared with Ni(OH)2/NF, the Ni 2p peaks in Pd–Ni(OH)2/NF show an obvious negative shift, further confirming strong electronic coupling in the heterostructure. The O 1s spectrum (Fig. S5) displays contributions from lattice O2− (530.7 eV), OH− (532.1 eV), and adsorbed H2O (533.5 eV).
To investigate how interfacial electronic interaction affects adsorbate binding, surface valence-band photoemission spectroscopy and DFT calculations were performed on Pd/NF and Pd–Ni(OH)2/NF. Valence-band spectra (Fig. 1I) show a clear downshift of the Pd d-band center from −3.02 eV (Pd/NF) to −3.53 eV (Pd–Ni(OH)2/NF). DFT calculations corroborate this trend, yielding d-band centers of −0.898 eV and −1.065 eV for Pd/NF and Pd–Ni(OH)2/NF, respectively. The electron transfer from Pd to Ni(OH)2 not only renders the Pd surface positively charged, which favors the adsorption of small alcohol molecules and boosts catalytic activity, but also lowers the d-band center of Pd, as confirmed by the consistent downshift trend at the Pd–Ni(OH)2/NF interface. In line with the d-band model, this downshift weakens the adsorption of key carbonyl intermediates (e.g., 3-HP) and promotes their desorption, thereby explaining the enhanced long-term stability and high product selectivity of Pd–Ni(OH)2/NF in subsequent POR studies.10
2.2 Electrocatalytic performance for 1,3-PDO oxidation
The electrocatalytic performance of Pd–Ni(OH)2/NF for the 1,3-PDO oxidation reaction (POR) was systematically evaluated in alkaline medium using a three-electrode system. LSV curves in 1 M KOH with and without 0.1 M 1,3-PDO are shown in Fig. 2A. The Pd–Ni(OH)2/NF electrode requires only 0.645 V (vs. RHE) to reach 100 mA cm−2 in the presence of 1,3-PDO, whereas without the substrate a much higher potential of 1.66 V is needed, reflecting a ∼1 V lowering due to the POR. As summarized in Fig. 2B, Pd–Ni(OH)2/NF requires lower potentials to reach the target current densities for the POR than for the OER. The Tafel slope of Pd–Ni(OH)2/NF in POR medium is 194.8 mV dec−1 (Fig. 2C), confirming favorable reaction kinetics compared to the oxygen evolution reaction (OER). Control experiments show that Ni(OH)2/NF exhibits no activity below the onset potential of the Ni2+/Ni3+ redox process (Fig. S6), whereas Pd–Ni(OH)2/NF and Pd/NF generate significant POR currents from 0.38 V onward, confirming Pd as the primary active site (Fig. S7). Notably, at 1.0 V, the current density on Pd–Ni(OH)2/NF is 1.6 times that on Pd/NF. EIS measurements further reveal a significantly lower charge-transfer resistance (Rct) for Pd–Ni(OH)2/NF than for other electrodes (Fig. S8 and S9). These results demonstrate that although Ni(OH)2 alone has almost no oxidation ability toward 1,3-propanediol at low potentials, its incorporation as a support greatly enhances the catalytic activity of Pd-based electrocatalysts through electronic coupling, highlighting the crucial role of the Ni(OH)2 support in boosting the POR activity of Pd. | ||
| Fig. 2 (A) LSV curves and (C) Tafel plots of Pd–Ni(OH)2/NF in 1 M KOH solution with and without 0.1 M 1,3-PDO. (B) Comparisons of the potentials needed to achieve designated current densities for Pd–Ni(OH)2/NF in 1.0 M KOH with and without 0.1 M 1,3-PDO. (D) 1H NMR and (E) 13C NMR spectra of the electrolyte before and after electrolysis. (F) 1H NMR spectra and (G) HPLC curves of the electrolyte after electrolysis with different amounts of charge passed. (H) FEs of the POR at different potentials. (I) Calculated FEs for the 3-HP product in five successive electrolysis cycles. | ||
To identify the catalytic products of 1,3-PDO oxidation on Pd–Ni(OH)2/NF, the post-electrolysis electrolyte was analyzed by 1H and 13C nuclear magnetic resonance (NMR) as well as high-performance liquid chromatography (HPLC). 1H NMR (Fig. 2D) of the electrolyte after electrolysis at 0.8 V (vs. RHE) shows new signals at δ = 3.12, 2.43, and 3.80 ppm, corresponding to the target product 3-HP and a small amount of the malonic acid byproduct. 13C NMR (Fig. 2E) further confirms 3-HP with characteristic peaks at 40.4, 59.1, and 180.8 ppm. Together, these spectra indicate that oxidation proceeds selectively at one hydroxyl group while leaving the C–C bond intact. Time-dependent 1H NMR (Fig. 2F) reveals a gradual decrease in 1,3-PDO signals and a concurrent increase in 3-HP peaks with increasing charge passed, consistent with progressive conversion. Quantitative HPLC analysis (Fig. 2G) corroborates the product distribution. The faradaic efficiency (FE, Fig. 2H) for 3-HP, determined via HPLC calibration (Fig. S10), remains high (∼90%) over the studied potential range and reaches a maximum of 94.9% at 0.8 V (vs. RHE), which was therefore selected as the optimal potential for this transformation.
To assess the operational stability of the Pd–Ni(OH)2/NF electrode for 1,3-PDO oxidation, chronopotentiometry (CP) measurements were performed (Fig. 2I). The catalyst demonstrated excellent durability, retaining a consistently high faradaic efficiency (∼90%) for 3-HP production over five consecutive batch cycles without noticeable activity decay. Although the Pd–Ni(OH)2/NF electrode demonstrates improved stability, gradual current decay during prolonged electrolysis remains a challenge. In situ Raman spectroscopy measurements on Pd–Ni(OH)2/NF in 1 M KOH containing 0.1 M 1,3-PDO revealed characteristic peaks corresponding to Pd–O and Pd–OH emerging at 0.9 V with increasing potential, confirming electrode deactivation due to the formation of PdOx (Fig. S11). To address this issue, we introduce an intermittent potential (IP) strategy that alternates between anodic polarization for 1,3-PDO oxidation and open-circuit periods to facilitate regeneration of Pd sites. Under IP operation, Pd–Ni(OH)2/NF maintains 80 mA cm−2 over 45 h without electrolyte renewal (Fig. S12), representing a marked improvement over continuous-potential operation. Post-electrolysis SEM/TEM (Fig. S13) confirms the preserved nanostructure, while XPS and XRD (Fig. S14 and S15) show retained phase composition and chemical states, collectively verifying the electrode's structural and compositional robustness.
2.3 Mechanistic insights
To elucidate the reaction mechanism, we performed potential-dependent in situ FTIR spectroscopy on the Pd–Ni(OH)2/NF electrode during 1,3-PDO oxidation in 1 M KOH solution (Fig. 3A and B). A band at 1640 cm−1, assigned to the bending mode of adsorbed H2O (δ(HOH)), confirms water adsorption on the catalyst surface.42 As the potential increases, the bands at 1473 cm−1, characteristic of ν(OCO) vibrations in adsorbed 3-HP, grow progressively. Simultaneously, the inverse absorption peak at 1445 cm−1, corresponding to the –OH group of 1,3-PDO, intensifies with potential, reflecting the continuous consumption of the reactant.43,44 | ||
| Fig. 3 (A and B) In situ FTIR spectra of Pd–Ni(OH)2/NF at various applied potentials for the POR. (C) OCPs of Pd/NF and Pd–Ni(OH)2/NF in 1 M KOH solution before and after adding 1 M 1,3-PDO. (D) OCPs of Pd/NF and Pd–Ni(OH)2/NF in 1 M KOH solution before and after adding 1 M 3-HP. (E) CV curves of Pd–Ni(OH)2/NF and Pd/NF in 1 M KOH. (F) Fluorescence spectra of ˙OH detected in the electrolyte using 0.4 mM terephthalic acid (TPA) as a probe molecule for Pd–Ni(OH)2/NF and Pd/NF electrodes. EPR spectra from the electrolyte using DMPO as a trapping agent: (G) OER and (H) POR. | ||
The selectivity and pathway of 1,3-PDO electrooxidation in alkaline medium are governed by the distinct adsorption of the reactant and product, as revealed by open-circuit potential (OCP) measurements. The OCP of Pd–Ni(OH)2/NF (Δ = 0.61 V) is significantly higher than that of Pd/NF (Δ = 0.56 V) after adding 1,3-PDO in 1 M KOH solution (Fig. 3C), indicating stronger adsorption of 1,3-PDO on Pd–Ni(OH)2/NF. Conversely, the addition of 3-HP causes a less negative OCP shift on Pd–Ni(OH)2/NF (−0.02 V) than on Pd/NF (−0.08 V), reflecting weaker adsorption of 3-HP on the Pd–Ni(OH)2/NF surface, a feature that promotes its desorption and suppresses over-oxidation (Fig. 3D). Furthermore, the smaller OCP change (Δ = 0.02 V) by adding 3-HP, compared to the pronounced shift (Δ = 0.61 V) by adding 1,3-PDO, confirms that 3-HP is only weakly adsorbed in the inner Helmholtz plane, effectively hindering its further oxidation to malonate.34
Adsorption of OH− species is crucial for electrocatalytic 1,3-PDO oxidation in alkaline media. As shown in Fig. 3E, Pd–Ni(OH)2/NF exhibits markedly enhanced OH− adsorption/desorption compared with Pd/NF. This enhancement originates from oxyphilic Ni sites in Ni(OH)2, which promote the oxidation of OH− to adsorbed ˙OH radicals on Pd at relatively low potentials. To quantify the production of ˙OH radicals, we utilized a terephthalic acid (TPA) fluorescence probe (Fig. 3F). The non-fluorescent probe reacts with ˙OH radicals to yield highly fluorescent 2-hydroxyterephthalic acid, whose emission intensity scales with ˙OH radical concentration (Fig. S16). The fluorescence signal increases for Pd–Ni(OH)2/NF, indicating higher ˙OH production than on Pd/NF. When 1,3-PDO is added, the fluorescence intensity drops substantially, confirming that ˙OH radicals act as the active species for 1,3-PDO oxidation. Together, these results demonstrate that oxyphilic Ni promotes abundant ˙OH formation on Pd, thereby driving the POR.
Quasi in situ electron paramagnetic resonance (EPR) measurements with 5,5-dimethyl-1-pyrroline N-oxide (DMPO) as a trapping agent were performed to detect radical intermediates during 1,3-PDO oxidation. As shown in Fig. 3G, Pd–Ni(OH)2/NF produces a much stronger ˙OH signal than Pd/NF in the absence of substrate, confirming that Ni(OH)2 promotes OH− oxidation to ˙OH. Upon addition of 1,3-PDO, the DMPO–˙OH signal sharply decreases while new signals assigned to alkoxy (DMPO–˙OR) and hydroperoxyl (DMPO–˙OOH) radicals appear (Fig. 3H), indicating that ˙OH attacks the O–H bond of 1,3-PDO to generate ˙OR intermediates. The ˙OOH signal, absent in pure KOH (likely due to its rapid conversion to DMPO–˙OH), emerges presumably from further oxidation of ˙OH; however, ˙OOH is unstable and tends to decompose spontaneously, thus playing no direct role in substrate oxidation.35 In contrast, ˙OH serves as the primary oxidizing species: it not only initiates alcohol dehydrogenation to form ˙OR but also helps oxidize adsorbed carbonaceous species and remove poisoning *CO from Pd sites, thereby regenerating the catalyst surface for sustained 1,3-PDO oxidation. Therefore, while all three radicals are detectable, ˙OH is the key reactive species responsible for substrate conversion, ˙OR is a reaction intermediate formed from alcohol O–H cleavage, and ˙OOH is a minor byproduct that does not contribute meaningfully to the reaction pathway.45,46
To elucidate the promoting role of Ni(OH)2 in 1,3-PDO oxidation, DFT calculations were performed on the Pd and Pd–Ni(OH)2 surfaces (Fig. S17). Structural optimization reveals an interfacial charge transfer of 0.34e− from Pd to Ni(OH)2 (Fig. 4A), attributed to strong Pd–O–Ni interactions that lower the electron density of Pd sites. Projected density of states (PDOS) analysis shows a downshift of the Pd d-band center from −0.898 eV (Pd) to −1.065 eV (Pd–Ni(OH)2) (Fig. 4B), which weakens product binding and facilitates desorption.47 Adsorption energies (Eads) of key intermediates further illustrate this modulation (Fig. 4C, S18 and S19): *1,3-PDO and *OH bind more strongly on Pd–Ni(OH)2 (Eads = −0.174 eV and −0.652 eV, respectively) than on Pd, promoting reactant activation. In contrast, *CO (−0.909 eV) and *3-HP (−0.138 eV) exhibit weaker adsorption, enhancing CO tolerance and favoring 3-HP release. Crystal orbital Hamilton population (COHP) analysis confirms further the trend: the Pd–O bond strengthens for *1,3-PDO (ICOHP from −0.278 to −0.538) but weakens for *3-HP (ICOHP from −0.639 to −0.342), providing direct electronic evidence for optimized adsorption–desorption energetics (Fig. 4D). Moreover, the C–C bond in adsorbed *HOOC–CH2CH2OH is more stable on Pd–Ni(OH)2 than on Pd (Fig. S20), corroborating that Ni(OH)2 suppresses C–C cleavage and thus prevents over-oxidation, ensuring high selectivity.
 | ||
| Fig. 4 (A) Charge density differences of Pd–Ni(OH)2. (B) PDOS for the d-band of Pd and Pd–Ni(OH)2. (C) Calculated adsorption energies of 1,3-PDO, CO, OH, and 3-HP on Pd and Pd–Ni(OH)2 surfaces. (D) COHP analyses of 1,3-PDO and 3-HP adsorption on Pd and Pd–Ni(OH)2. (E) The optimized configurations of POR intermediates on Pd–Ni(OH)2. (F) Free-energy diagram of the POR on Pd and Pd–Ni(OH)2. | ||
Furthermore, the reaction pathway and Gibbs free-energy profiles (ΔG) for 1,3-PDO oxidation to 3-HP on Pd and Pd–Ni(OH)2 are shown in Fig. 4E, F and S21. The mechanism involves sequential dehydrogenation and hydroxylation steps. Although both catalysts oxidize OH− to *OH active species and share this as the rate-determining step (RDS), the Pd–Ni(OH)2 interface exhibits a substantially lower energy barrier for this process. This reduction originates from the oxophilic Ni(OH)2 sites, which promote the supply of active *OH species and thus lower the overpotential for intermediate conversion. Moreover, product desorption (HOOC–CH2CH2OH) is significantly accelerated on Pd–Ni(OH)2 due to the downshifted Pd d-band center induced by the interfacial interaction, which weakens product binding. The resulting rapid release of 3-HP helps preserve the C–C backbone and suppress over-oxidation.
2.4 HER performance and a MEA electrolyzer
The HER performance of Pd–Ni(OH)2/NF and reference catalysts was evaluated in 1.0 M KOH. Pd–Ni(OH)2/NF exhibits an overpotential of only 168 mV at 100 mA cm−2 (Fig. 5A), comparable to Pt/C and superior to most reported noble-metal catalysts in alkaline media (Table S1). Tafel analysis (Fig. 5B) reveals a slope of 29.2 mV dec−1 for Pd–Ni(OH)2/NF, significantly lower than those of Pt/C (64.6 mV dec−1) and Ni(OH)2/NF (180.9 mV dec−1), indicating faster proton-coupled kinetics. EIS further confirms its favorable charge-transfer characteristics (Fig. S22). During a 26 h stability test at −0.3 V (vs. RHE), the catalyst retains 98.7% of its initial current density, with nearly identical LSV curves before and after operation (Fig. 5C), demonstrating both high activity and robust durability. | ||
| Fig. 5 (A) LSV curves and (B) Tafel plots of various electrodes for the HER. (C) j–t curve (inset) of Pd–Ni(OH)2/NF and LSV curves before and after the stability test. (D) Schematic illustration of the MEA flow reactor of the HER‖POR system, using 1 M KOH solution containing 0.1 M 1,3-PDO as the anode electrolyte and 1 M KOH solution as the cathode electrolyte, respectively. (E) LSV curves of HER‖POR in 1 M KOH solution with and without 0.1 M 1,3-PDO, using Pd–Ni(OH)2/NF as both the anode and cathode. (F) Stability test of the HER‖POR system in a MEA setup using an intermittent potential strategy. | ||
To assess practical feasibility in a two-electrode setup, a membrane electrode assembly (MEA) electrolyzer was constructed with Pd–Ni(OH)2/NF as both the anode and cathode (active area: 1 cm2; Fig. 5D and S23). The anode was fed with 1 M KOH solution containing 0.1 M 1,3-PDO, and the cathode with 1 M KOH; both electrolytes were continuously recirculated by a peristaltic pump. Linear sweep voltammetry (LSV) was first performed on the coupled HER‖POR and HER‖OER systems to evaluate their electrochemical performance. The LSV curves in Fig. 5E show that the HER‖POR system has a much lower onset potential than the conventional HER‖OER system. Specifically, it requires only 0.84 V to reach 100 mA cm−2, highlighting its superior energy efficiency. As further shown in Fig. S24, the integrated HER‖POR configuration allows simultaneous hydrogen production and valuable chemical synthesis at significantly lower energy input, offering a promising route toward sustainable electrochemical manufacturing.
To improve the long-term stability of the MEA electrolyzer, we applied an intermittent potential (IP) strategy that cycles between operation and open-circuit periods. Under oxidizing potential, PdOx forms on the catalyst surface, while during the open-circuit periods, PdOx is reduced back to metallic Pd. This periodic regeneration of Pd active sites greatly enhances both the long-term stability and catalytic activity of the Pd catalyst.48 After 48 h, the current density under continuous-potential (CP) operation decays to 23 mA cm−2, whereas IP operation sustains a much higher current (Fig. 5F). The system also delivers a stable faradaic efficiency of ∼90% for 1,3-PDO to 3-HP conversion over 100 h, with product identity confirmed by 1H/13C NMR (Fig. S25). These results demonstrate that the IP strategy markedly enhances operational durability while enabling efficient and selective oxidation of 1,3-PDO to value-added 3-HP.
2.5 DPFC for combined power and chemical generation
Fig. 6A schematically illustrates the configuration and operating principle of the direct 1,3-PDO fuel cell (DPFC) powered by the multifunctional Pd–Ni(OH)2/NF catalyst. In this integrated device, the anodic compartment is fed with 1,3-PDO in alkaline electrolyte, where the POR selectively converts 1,3-PDO to 3-HP, while the cathodic compartment hosts the ORR, completing the direct 1,3-PDO fuel cell circuit. This design enables the DPFC to simultaneously achieve selective electrosynthesis of high-value 3-HP from biomass-derived 1,3-PDO at the anode and electrical power generation from the spontaneous redox reaction. Notably, this configuration complements the daytime HER‖POR electrolysis mode by providing a nighttime energy-harvesting pathway, thereby enabling round-the-clock operation of the unified electrocatalytic platform for the chemical–H2–electricity triad. | ||
| Fig. 6 (A) Scheme of an integrated system that converts 1,3-PDO into high-value 3-HP, producing H2 simultaneously during the daytime (HER-coupled) and generating electrical energy simultaneously during the nighttime (ORR-coupled). (B) ORR LSV curves of Pd–Ni(OH)2/NF and other reference electrodes in 1.0 M KOH; (C) OCV of the direct 1,3-PDO fuel cell; (D) polarization curve and power density curve of the direct 1,3-PDO fuel cell; (E) 1H NMR spectrum after electrolyte discharge. | ||
The electrocatalytic ORR performance of Pd–Ni(OH)2/NF was initially assessed in a 1.0 M KOH solution saturated with O2. As depicted in Fig. 6B, in O2-saturated 1 M KOH, Pd–Ni(OH)2/NF shows an onset potential of 0.95 V and a half-wave potential of 0.71 V for the ORR, significantly outperforming Pd/NF (Fig. 6B). The assembled DPFC delivers a stable open-circuit voltage of 0.59 V (Fig. 6C) and a peak power density of 2.46 mW cm−2 (Fig. 6D). 1H NMR analysis of the anolyte after discharge confirms selective conversion of 1,3-PDO to 3-HP (Fig. 6E). Together, these results demonstrate the feasibility of simultaneous electricity generation and valuable chemical production in a single integrated device.
3 Conclusions
In summary, we developed an electronic-structure tuning strategy to enhance the selective electrooxidation of 1,3-PDO to 3-HP on Pd nanoparticles. By constructing a Pd–Ni(OH)2/NF heterointerface, the Pd d-band center is downshifted while Ni sites gain enhanced oxophilicity. This synergistic restructuring optimizes intermediate adsorption energetics and promotes 3-HP desorption, thereby suppressing C–C cleavage and over-oxidation. As a result, the catalyst exhibits outstanding activity for the POR, requiring only 0.645 V (vs. RHE) to deliver 100 mA cm−2 with a faradaic efficiency of ∼94.9% for 3-HP production. The practical viability of this approach is demonstrated in a MEA electrolyzer that integrates both the POR and HER. The system operates stably for over 100 h with faradaic efficiencies above 90%, and its durability is further enhanced by an intermittent-potential strategy that enables periodic regeneration of active sites. Moreover, the same Pd–Ni(OH)2/NF electrode serves as a robust bifunctional catalyst in a direct 1,3-PDO fuel cell, enabling simultaneous electricity generation and selective chemical production. By synergistically integrating the HER (daytime mode) and ORR (nighttime mode) with the same Pd–Ni(OH)2/NF platform, our system enables a flexible mode-switching operation that produces green hydrogen during the daytime and switches to electricity generation at night, maximizing renewable energy utilization. Collectively, this work establishes a general design principle of interfacial electronic-structure tailoring to develop multifunctional catalysts and offers a competitive alternative for sustainable electrochemical synthesis.4 Experimental section
4.1 Chemicals
1,3-Propanediol (1,3PDO), sulfuric acid (H2SO4, 98%), and potassium hydroxide (KOH, 99%) were purchased from Sinopharm Chemical Reagent Co., Ltd. Nickel nitrate hexahydrate (Ni(NO3)2·6H2O), orthoboric acid (H3BO3), malonic acid (HOOCCH2COOH, 98%) and sodium tetrachloropalladate (Na2PdCl4, 98%) were purchased from Shanghai Aladdin Biochemical Technology Co., Ltd. 3-Hydroxypropionic acid (sodium salt, 95%) was obtained from Beijing Mreda Technology Co., Ltd. All other chemical reagents were of analytical grade and used as received without further purification. All electrolyte solutions were prepared using Milli-Q ultrapure water (18 MΩ cm).4.2 Electrocatalyst synthesis
Pd–Ni(OH)2/NF was prepared on three-dimensional nickel foam (NF, 1 × 2 cm2) via a two-step electrodeposition process. The NF substrate was first ultrasonically cleaned in 0.1 M HCl, ethanol, and deionized water (10 min each) and dried under Ar at 25 °C. Ni(OH)2 nanosheets were then electrodeposited at −1.244 V (vs. SCE) for 1200 s in an aqueous 300 mM Ni(NO3)2 solution, using a Pt plate as the counter electrode and a SCE as the reference electrode. Subsequently, Pd nanoparticles were electrodeposited onto the Ni(OH)2/NF electrode at −1.0244 V (vs. SCE) for 600 s in a solution of 10 mM Na2PdCl4 dissolved in 10 mL of boric acid aqueous solution. The resulting composite was washed sequentially with deionized water and absolute ethanol and then vacuum-dried at 60 °C for 12 h. For comparison, control electrodes (Pd/NF and Ni(OH)2/NF) were synthesized under identical conditions using only the corresponding precursor.4.3 Material characterization
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy analysis (EDX) mapping images were obtained using a JSM 7900F (JEOL, Japan) equipped with an EDX system. Transmission electron microscopy (TEM) images, high-resolution TEM (HRTEM) images and selective electron diffraction (SAED) patterns were obtained using a JEM-2100F (JEOL, Japan). X-ray diffraction (XRD) analysis was conducted on a Bruker Foucs D8 diffractometer with Cu Kα radiation (40 kV, 40 mA), scanning from 10° to 80° at a rate of 10° per minute. X-ray photoelectron spectroscopy (XPS) was conducted on a Kratos Axis Ultra DLD X-ray photoelectron spectrometer using 60 W monochromated Mg Kα radiation as the excitation source. The C 1s peak (284.6 eV) was used for internal standard calibration, and XPS Peak 41 software was employed for peak deconvolution and fitting. The oxidation products in solution were analyzed using an Agilent 1260 HPLC system with a UV-Vis detector and a BioRad Aminex 87H column. 1H and 13C NMR spectra were recorded on a Bruker-DRX 600 MHz instrument. The photoluminescence (PL) spectra were measured at room temperature using an Edinburgh FLS1000 spectrofluorometer with an excitation wavelength of 320 nm.4.4 Electrochemical testing
All electrochemical tests were conducted using a CHI 760e electrochemical workstation (CH Instrument Co., Ltd., Shanghai, China). Electrochemical experiments were carried out in a standard three-electrode cell to evaluate the activities of electrocatalysts. A prepared electrode was used as the working electrode, while a graphite rod and Hg/HgO (1 M KOH) were used as counter and reference electrodes, respectively. The measured potentials were converted to the reversible hydrogen electrode (RHE) scale using the equation: ERHE = EHg/HgO + 0.098 + 0.059 pH. Linear scanning voltammetry (LSV) and cyclic voltammetry (CV) measurements were performed at a scan rate of 10 mV s−1 with 80% ohmic compensation. Electrochemical impedance spectroscopy (EIS) measurements were performed using an amplitude potential of 5 mV over a frequency range of 1 MHz to 0.01 Hz. Tafel slopes were derived from LSV plots obtained at a very slow scan rate of 0.1 mV s−1 by linear fitting of the points in the Tafel region. The membrane electrolysis tests were performed with Pd–Ni(OH)2/NF serving as both the anode and cathode. The FAA-3-50 membrane was employed to separate the electrolytes. Prior to use, the FAA-3-50 membrane was soaked in 1 M KOH solution for 12 h and then rinsed with deionized water.4.5 3-HP product analysis (NMR and HPLC)
To identify the oxidation products of 1,3-PDO and calculate the faradaic efficiency, a long-term bulk electrolysis process was carried out in a three-electrode system at a potential of 0.8 V (vs. RHE) in 1.0 M KOH solution with 0.1 M 1,3-PDO under vigorous stirring at room temperature. After electrolysis, the electrolyte solution was collected for qualitative analysis and quantitative analysis by NMR and HPLC. For 1H NMR and 13C NMR, 500 µL of the electrolyte was mixed with 100 µL of D2O to prepare the sample. For each HPLC measurement, 50 µL of electrolyte solution containing the product was diluted to 1 mL with diluted H2SO4 solution. Then 20 µL of the prepared sample was injected into a BioRad Aminex 87H column. The mobile phase consisted of a 5 mM H2SO4 solution delivered at a constant flow rate of 0.6 mL min−1. Product identification was achieved by comparing the retention time of the elution peak with those of single standard solutions. The concentrations of the product were determined using calibration curves generated from standard solutions of known concentrations.4.6 FE calculations
The FE of product formation was calculated using the following equation: FE = (n × z × F/Q) × 100%, where n is the number of moles of product obtained, z is the number of electrons required to form one molecule, Q is the charge passed (C), and F is the Faraday constant (96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 485 C mol−1).
485 C mol−1).
4.7 Theoretical calculations
Based on DFT, all electronic structure calculations were implemented using the Vienna Ab initio Simulation Package (VASP).49 The exchange–correlation potential was described by using the generalized gradient approximation of Perdew–Burke–Ernzerhof (GGA-PBE).50 The projector augmented wave (PAW) potential is employed to describe the interactions between ions and electrons.51 The plane-wave cutoff energy was set to 450 eV. The convergence of energy was set to 10−5 eV and that of forces for geometry optimizations was set to 0.03 eV Å−1. The Brillouin zone was sampled with a 3 × 3 × 1 Gamma-centered special k-point grid for geometry optimization. A vacuum layer of around 15 Å was added in the c direction perpendicular to the surface to eliminate the spurious interlayer interaction. The free energy (ΔG) calculations of each elementary step were based on the standard hydrogen electrode model and were determined using the following equation:| ΔG = ΔE + ΔEZPE − TΔS − neU |
Author contributions
Wei Wang: conceptualization, data curation, formal analysis, investigation, methodology, writing – original draft and writing – review and editing; Xiaoyang He and Shuai Dong: methodology, data curation, formal analysis, investigation; Jianying Wang and Deli Wu: conceptualization, resources, supervision; Zuofeng Chen: conceptualization, formal analysis, investigation, methodology, resources, funding acquisition, supervision, project administration, and writing – review and editing.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: theoretical models of Pd and Pd–Ni(OH)2; stability characterization studies; standard curves of product analysis; SI figures and tables. See DOI: https://doi.org/10.1039/d6sc03316d.Acknowledgements
This work was supported by the National Key R&D Program of China (2024YFA1211004) and the Natural Science Foundation of Shanghai Municipality (23ZR1464800).References
- J. R. Jambeck, R. Geyer, C. Wilcox, T. R. Siegler, M. Perryman, A. Andrady, R. Narayan and K. L. Law, Science, 2015, 347, 768–771 Search PubMed.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A. Rahimi and J. M. Garcia, Nat. Rev. Chem., 2017, 1, 0046 Search PubMed.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Roman-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole, Y. Zhang, N. A. Lynd, A. D. Ellington and H. S. Alper, Nature, 2022, 604, 662 CrossRef CAS PubMed.
- K. Zheng, Y. Wu, Z. Hu, S. Wang, X. Jiao, J. Zhu, Y. Sun and Y. Xie, Chem. Soc. Rev., 2023, 52, 8–29 RSC.
- R. Nistico, Polym. Test., 2020, 90, 106707 CrossRef CAS.
- J. Wang, X. Li, M. Wang, T. Zhang, X. Chai, J. Lu, T. Wang, Y. Zhao and D. Ma, ACS Catal., 2022, 12, 6722–6728 CrossRef CAS.
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2022, 7, 104–116 CrossRef CAS.
- Y. Yan, Q. Wang, J. Yang, Y. Fu, Q. Shi, Z. Li, J. Zhang, M. Shao and X. Duan, Small, 2025, 21, 2406782 Search PubMed.
- M. Barletta, C. Aversa, M. Ayyoob, A. Gisario, K. Hamad, M. Mehrpouya and H. Vahabi, Prog. Polym. Sci., 2022, 132, 101579 Search PubMed.
- B. Andreessen, N. Taylor and A. Steinbuechela, Appl. Environ. Microbiol., 2014, 80, 6574–6582 CrossRef PubMed.
- Z.-Y. Lu, Z. Chen, Z. Li, Z.-Y. Chi, X.-G. Li, S.-Y. Wei, C.-W. Liu and W.-D. Xiao, Chem. Eng. J., 2025, 507, 160382 CrossRef CAS.
- S. S. Bhagwat, Y. Li, Y. R. Cortes-Pena, E. C. Brace, T. A. Martin, H. Zhao and J. S. Guest, ACS Sustainable Chem. Eng., 2021, 9, 16659–16669 Search PubMed.
- J. Zhu, J. Xie, L. Wei, J. Lin, L. Zhao and D. Wei, Bioresour. Technol., 2018, 265, 328–333 CrossRef CAS PubMed.
- Y. Zhang, J. Yun, H. M. Zabed, Y. Dou, G. Zhang, M. Zhao, M. J. Taherzadeh, A. Ragauskas and X. Qi, Bioresour. Technol., 2023, 369, 128438 CrossRef CAS PubMed.
- M. Moussa, G. Burge, F. Chemarin, R. Bounader, C. Saulou-Berion, F. Allais, H.-E. Spinnler and V. Athes, J. Chem. Technol. Biotechnol., 2016, 91, 2276–2285 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Green Chem., 2011, 13, 1624–1632 RSC.
- J. W. Kim, Y.-S. Ko, T. U. Chae and S. Y. Lee, Biotechnol. Bioeng., 2020, 117, 2139–2152 Search PubMed.
- H. M. Zabed, S. Akter, P. F. Rupani, J. Akor, Y. Zhang, M. Zhao, C. Zhang, A. J. Ragauskas and X. Qi, Biotechnol. Adv., 2023, 62, 108075 CrossRef CAS PubMed.
- S. Zhou, Y. Zhang, Z. Wei and S. Park, Bioresour. Technol., 2023, 377, 128973 CrossRef CAS PubMed.
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 Search PubMed.
- J. Kibsgaard and I. Chorkendorff, Nat. Energy, 2019, 4, 430–433 CrossRef.
- T. Wang, L. Tao, X. Zhu, C. Chen, W. Chen, S. Du, Y. Zhou, B. Zhou, D. Wang, C. Xie, P. Long, W. Li, Y. Wang, R. Chen, Y. Zou, X.-Z. Fu, Y. Li, X. Duan and S. Wang, Nat. Catal., 2022, 5, 66–73 Search PubMed.
- J. Liu, X. Sun, K. Lin, Z. Liang, W. Xing, J. Chen and X. F. Lu, Nano Res., 2026, 19, 94908088 Search PubMed.
- G. Wang, J. Chen, F. Qiao, J. Wang and Z. Wen, Chem. Sci., 2025, 16, 12651–12678 RSC.
- B. Huang, J. Yan, Z. Li, L. Chen and J. Shi, Angew. Chem., Int. Ed., 2024, 63, e202409419 Search PubMed.
- J. Liu, Y. Du, D. Zheng, S. Wang, Y. Hou, J. Zhang and X. F. Lu, ACS Mater. Lett., 2023, 6, 466–481 Search PubMed.
- L. Sun, H. Lv, J. Xiao and B. Liu, Adv. Mater., 2024, 36, 2402767 Search PubMed.
- S. Zhang, K. Li, X. Zhang, Y. Ye, T. Shi, Y. Jiang, L. Zheng, Y. Lin and H. Zhang, Adv. Funct. Mater., 2025, 35, 2415046 Search PubMed.
- T. Ren, Z. Duan, H. Wang, H. Yu, K. Deng, Z. Wang, H. Wang, L. Wang and Y. Xu, ACS Catal., 2023, 13, 10394–10404 Search PubMed.
- W. Wang, X. He, Z. Tu, D. Xiong, S. Dong, T. Zhang, D. Wu, J. Wang and Z. Chen, ACS Catal., 2025, 15, 9574–9583 Search PubMed.
- X. Jin, X. Chen, C. Chen, X. Xiao, L. Chen, Z. Wang, B. Sun and D. Sun, J. Colloid Interface Sci., 2025, 688, 403–410 CrossRef CAS PubMed.
- K. Shi, D. Si, X. Teng, L. Chen and J. Shi, Nat. Commun., 2024, 15, 2899 Search PubMed.
- F. Liu, X. Gao, R. Shi, Z. Guo, E. C. M. Tse and Y. Chen, Angew. Chem., Int. Ed., 2023, 62, e202300094 Search PubMed.
- C. Chen, X. Zhu, X. Wen, Y. Zhou, L. Zhou, H. Li, L. Tao, Q. Li, S. Du, T. Liu, D. Yan, C. Xie, Y. Zou, Y. Wang, R. Chen, J. Huo, Y. Li, J. Cheng, H. Su, X. Zhao, W. Cheng, Q. Liu, H. Lin, J. Luo, J. Chen, M. Dong, K. Cheng, C. Li and S. Wang, Nat. Chem., 2020, 12, 717–724 Search PubMed.
- Q. Gao, H. S. Pillai, Y. Huang, S. Liu, Q. Mu, X. Han, Z. Yan, H. Zhou, Q. He, H. Xin and H. Zhu, Nat. Commun., 2022, 13, 2338 CrossRef CAS PubMed.
- Z. Tu, X. He, X. Liu, D. Xiong, S. Xue, D. Wu, J. Wang and Z. Chen, Chem. Mater., 2025, 37, 1195–1204 Search PubMed.
- R. Sainju, T. Li, M. R. Nielsen and Y. Zhu, Nano Lett., 2025, 25, 10521–10527 CrossRef CAS PubMed.
- J. Liu, X. Liu, J. Zhang, Z. Liang, W. Lin and X. F. Lu, ACS Catal., 2026, 16, 7726–7737 CrossRef CAS.
- X. Sun, J. Liu, Y. Du, M. Shen, K. Liu, Z. Liang, W. Cheng, J. Chen and X. F. Lu, Nano Lett., 2025, 25, 11680–11688 Search PubMed.
- J. Li, Y. Zhang, L. Zhang, Q. Zhou, J. Yang, Y. Fu, L. Yu, Y. Sun, Z. Li, Y. Qin, D. Yi, H. Zhang, D. Wang and Z. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202425006 CrossRef CAS PubMed.
- D. Si, M. Wang, X. Yang, C. Wang, K. Shi, B. Huang, L. Chen and J. Shi, Appl. Catal., B, 2023, 331, 122664 Search PubMed.
- X. Liu, X. He, D. Xiong, G. Wang, Z. Tu, D. Wu, J. Wang, J. Gu and Z. Chen, ACS Catal., 2024, 14, 5366–5376 CrossRef CAS.
- W. Huang, H. Wang, J. Zhou, J. Wang, P. N. Duchesne, D. Muir, P. Zhang, N. Han, F. Zhao, M. Zeng, J. Zhong, C. Jin, Y. Li, S.-T. Lee and H. Dai, Nat. Commun., 2015, 6, 10035 CrossRef CAS PubMed.
- W. Huang, X.-Y. Ma, H. Wang, R. Feng, J. Zhou, P. N. Duchesne, P. Zhang, F. Chen, N. Han, F. Zhao, J. Zhou, W.-B. Cai and Y. Li, Adv. Mater., 2017, 29, 1703057 Search PubMed.
- D. Du, P. Liu, Z. Teng, T. Chen, J. Zhu, B. Shao and J. Luo, ACS Catal., 2025, 15, 3038–3045 Search PubMed.
- Y. Yan, H. Zhou, S.-M. Xu, J. Yang, P. Hao, X. Cai, Y. Ren, M. Xu, X. Kong, M. Shao, Z. Li and H. Duan, J. Am. Chem. Soc., 2023, 145, 6144–6155 Search PubMed.
- G. Kresse and J. Furthmuller, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 Search PubMed.
- P. E. Blochl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |