
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAddressing interfacial chemical corrosion in lithium metal batteries: a ferroelectric-dipole-regulation route
Baolei Xu
ab,
Zeqiang Xiea,
Chunxiao Zhang b,
Liangjun Zhoub,
Libao Chenb and
Weifeng Wei
*b
b,
Liangjun Zhoub,
Libao Chenb and
Weifeng Wei
*b
aSchool of Frontier Crossover Studies, Hunan University of Technology and Business, Changsha 410205, China
bNational Research & Development Center of Powder Metallurgy, Powder Metallurgy Research Institute, Central South University, Changsha, Hunan 410083, China. E-mail: weifengwei@csu.edu.cn
First published on 14th May 2026
Abstract
Lithium (Li) metal batteries are hailed as one of the ultimate choices for next-generation high-energy-density energy storage systems. However, their commercialization has been persistently hindered by the bottlenecks of short cycle life and high safety risks. Research predominantly focuses on the electrochemical growth and suppression of Li dendrites, often overlooking the spontaneous chemical corrosion between the Li anode and electrolyte during battery assembly, resting, and storage, which can initiate catastrophic chain-reaction failures. This perspective article aims to systematically elucidate the critical role of interfacial chemical corrosion as the “initiating factor” in battery failure. We will delve into the intrinsic mechanisms by which chemical corrosion induces initial surface-tip-electric fields, triggers heterogeneous formation of a solid electrolyte interphase (SEI), and ultimately leads to Li dendrite flooding and battery failure. More importantly, we propose a novel paradigm for precise interphase environment regulation based on ferroelectric dipole (FD) engineering. This involves a detailed discussion on the active regulatory effects of FDs on interfacial ion distribution, solvation structure, and SEI composition, particularly the mechanisms by which they enrich and activate anions. We also innovatively introduce the concept of “pre-adsorbed anion-type FDs”, offering a fresh theoretical perspective and technical pathway for the targeted design and controllable fabrication of the SEI.
 Baolei Xu | Baolei Xu is an assistant professor at the School of Frontier Crossover Studies, Hunan University of Technology and Business. He obtained a master's degree in Materials Science and Engineering from Xiangtan University in 2017. He received his PhD in Materials Science and Engineering from Central South University under the supervision of Prof. Weifeng Wei in 2025. His research focuses on new energy storage materials and devices. |
 Weifeng Wei | Weifeng Wei a professor at Central South University, where he also serves as Vice Dean of the Powder Metallurgy Research Institute and Deputy Director of the National Research & Development Center of Powder Metallurgy. After receiving his PhD in Materials Engineering from the University of Alberta in 2009, he joined the Sadoway Group as a postdoctoral research associate at MIT (2009–2011). His research focuses on solid-state battery materials and device technologies. |
1. Introduction
The lithium (Li) metal anode has garnered significant attention due to its extremely high theoretical specific capacity (3860 mA h g−1) and the most negative electrochemical potential (−3.04 V vs. the standard hydrogen electrode), making it a key electrode material for constructing battery systems with energy densities as high as 500 Wh kg−1.1,2 However, for decades, the practical application of Li metal batteries (LMBs) has remained challenging, with the core obstacle stemming from the highly reactive and complex interfacial reactions between Li metal and organic liquid electrolytes.3,4 This interfacial issue not only limits battery cycle life but also poses serious safety hazards, becoming the “Achilles' heel” constraining their commercialization.5,6Currently, the vast majority of research focuses on dynamic issues during electrochemical cycling, such as dendrite growth caused by uneven Li-ion flux,7,8 significant volume changes during charge/discharge,9,10 and the accumulation of “dead” Li.11,12 These studies have undoubtedly enhanced our understanding of LMB failure mechanisms and spurred a series of improvement strategies, including three-dimensional current collector design,13,14 electrolyte engineering,15,16 and artificial interphase layer construction.17,18 However, a relatively neglected yet crucial problem, which sows the seeds of failure from the very “birth” of the battery, is the spontaneous chemical corrosion of Li metal at open-circuit potential in contact with the electrolyte.19–21 This corrosion is not driven by external circuit current but originates from the inherent, strong chemical reaction tendency between Li and electrolyte components (e.g., solvent molecules, trace water/oxygen impurities, and Li salt anions).22,23 The extremely high reactivity of metallic Li makes it akin to a piece of “alkali metal” placed in the electrolyte, spontaneously forming an initial, heterogeneous solid electrolyte interphase (SEI) on the electrode surface.24
The importance of this initial chemical corrosion process lies in the fact that it sets the “initial state” of the electrode/electrolyte interface. An undesirable initial interfacial state, like a cornerstone with inherent defects, makes it difficult to build a stable structure regardless of subsequent electrochemical process optimization. Specifically, the uneven morphology and SEI heterogeneity caused by chemical corrosion predetermine the distribution of Li-ion flux, induce uneven Li deposition, and trigger continuous side reactions, ultimately leading to rapid battery performance degradation.25,26 Therefore, a thorough understanding and effective regulation of this initial chemical corrosion process are of paramount significance for breaking the failure chain of LMBs.
This article highlights a core viewpoint: the initial chemical corrosion behavior is the “igniter” and “amplifier” for a series of subsequent electrochemical failures. To this end, we will first systematically dissect the behavior chain of chemical corrosion-induced battery failure (Chapter 2), from the formation of initial surface tip electric fields to the generation of SEI heterogeneity and the continuous side reactions triggered by an unstable SEI, constructing a comprehensive failure mechanism map. Subsequently, we propose a disruptive “active defense” strategy, utilizing ferroelectric dipoles (FDs) for the precise design and directional regulation of the Li anode/electrolyte interfacial phase environment (Chapter 3). We will focus on explaining how FDs, through their unique field effects, optimize interfacial ion distribution, regulate the solvation structure by binding, enriching, polarizing and activating anions, and thus drive the formation of an ideal SEI. We will further introduce a forward-looking concept—“pre-adsorbed anion-type FDs”—aiming to achieve targeted design and controllable fabrication of SEI components. This strategy seeks to intervene at the source of the failure chain initiation, providing new insights and prospects for constructing highly stable LMBs.
2. Chemical corrosion-induced failure in LMBs
It is important to note that Li metal degradation arises from multiple coupled factors, including electrochemical deposition instability, SEI mechanical fracture, non-uniform current distribution due to current collector defects, and intrinsic overpotential effects.4,27,28 Chemical corrosion acts as one critical initiating factor that sets the initial state of the Li/electrolyte interface. This initial state then couples with subsequent electrochemical processes to amplify or accelerate failure. In this section, we focus on how chemical corrosion contributes to this multi-factor degradation network, while acknowledging that other mechanisms operate in parallel.2.1. Corrosion-induced tip-electric-field and dendrite initiation
Ideally, the Li metal surface should be perfectly smooth to ensure uniform electric field and ion flux distribution. However, actual metallic Li foil or deposited Li layers always exhibit uneven topography at the microscale, including natural scratches, grain boundaries, dislocation outcrops, and defects introduced during preparation (Fig. 1a). These surface inhomogeneities set the stage for subsequent chemical corrosion.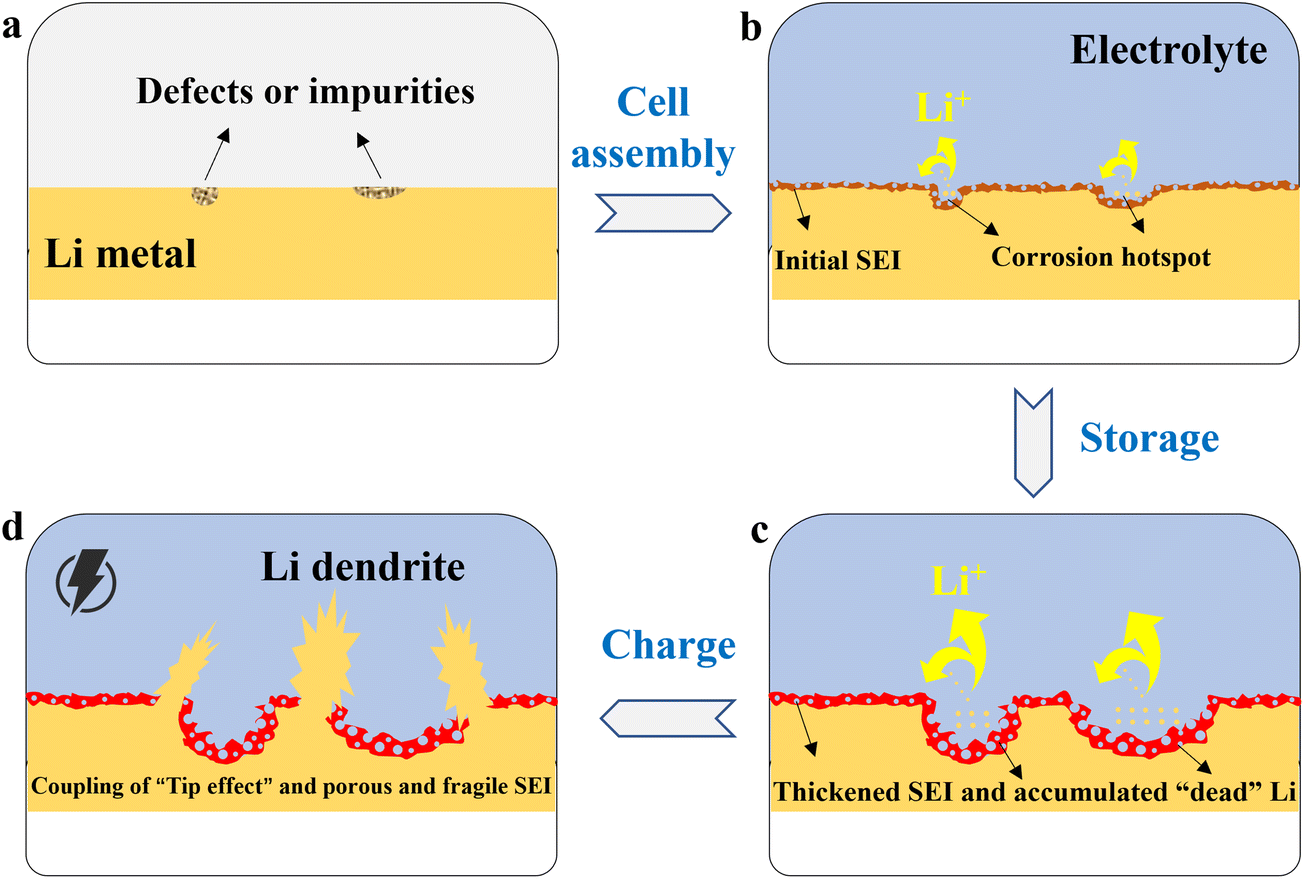 | ||
| Fig. 1 Schematic illustration of Li dendrite flooding induced by chemical corrosion in LMBs. (a) Initial defects and impurities on the Li anode surface. (b) Defects are preferred to be corrosion hotspots. (c) The corrosion pit expands to form “valleys” and “peaks”, accompanied by thick SEI growth and “dead” Li accumulation. (d) The Li dendrite flooding in service. | ||
When the non-uniform surface contacts the electrolyte, chemical corrosion first intensifies in these “hotspot” regions with high surface energy (Fig. 1b). The mechanism lies in the fact that atoms in these defect regions have incomplete coordination and higher reactivity29,30 and are more prone to react with reductive components in the electrolyte. From a thermodynamic perspective, the Gibbs free energy in these regions is higher, making corrosion reactions more likely.31 From a kinetic perspective, these regions provide more reaction sites, accelerating the corrosion process.32 This selective corrosion leads to two direct consequences:
(1) Geometrical morphology deterioration. Initial micron- or nano-scale depressions or scratches may deepen and widen upon corrosion, forming sharper “valleys” and “peaks” (Fig. 1c). This morphological evolution is not a smooth process but exhibits significant spatial heterogeneity.
(2) Surface composition inhomogeneity. Due to different corrosion rates, the thickness and composition of the initial passivation film (chemical SEI) formed vary significantly across regions. High-reactivity regions may form a thicker but porous SEI, while low-reactivity regions form a thinner but potentially denser SEI.
The morphological and chemical heterogeneity induced by chemical corrosion collectively induces the initial surface tip electric field. According to classical electrodynamics theory, on a conductor surface, charges preferentially accumulate in regions with small curvature radii.33 This charge redistribution leads to a significant enhancement of the electric field at the tips, forming the so-called “tip effect”.34,35 Consequently, during subsequent electrochemical cycling, Li ions (Li+) are strongly attracted to these tips or protrusions generated by corrosion, resulting in a much higher local ion flux than in flat or depressed regions.36,37 The excessively high local ion flux causes the Li deposition rate at these spots to far exceed other regions, rapidly forming “nuclei” for Li dendrites and further self-amplifying, eventually developing into dangerous dendritic structures (Fig. 1d). In short, chemical corrosion creates a “breeding ground” for electric field concentration, laying the groundwork for electrochemical dendrite growth. This process establishes a positive feedback loop: corrosion-induced tip formation → local electric field enhancement → preferential Li+ attraction and dendrite nucleation → further tip sharpening upon deposition → even stronger electric field enhancement. Such a chemo-electrochemical feedback loop accelerates dendrite proliferation and is a key reason why early-stage chemical corrosion can have catastrophic consequences. Therefore, the chemical corrosion-induced tip electric field and the subsequent dendrite growth should be understood as a coupled chemo-electrochemical process, rather than a purely chemical corrosion-driven chain.
2.2. Corrosion-induced SEI heterogeneity deteriorates interfacial transport
The SEI is hailed as the “lifeline” of LMBs.38 An ideal SEI should possess uniform thickness, excellent ionic conductivity and electronic insulation, and good mechanical toughness.39,40 However, the initial SEI formed spontaneously by chemical corrosion is inherently inhomogeneous, manifesting in multiple dimensions.24,41As described in Section 2.1, the regional differences in corrosion reaction activity directly led to different growth rates of the SEI during the initial stage of chemical corrosion. One might argue that once an electronically insulating, Li+-conducting SEI forms, further reaction should be suppressed, limiting thickness variation. However, the first-formed, organic-rich SEI (typical of spontaneous reactions in carbonate electrolytes) is often porous and exhibits weak electronic insulation.41,42 Consequently, electrolyte reduction can continue beneath or through this nascent layer, called “continuous corrosion”. As a result, high-activity regions may form a thicker but still porous SEI, while low-activity regions form a thinner but potentially denser SEI. Although the steady-state thickness difference may eventually be limited, the transient heterogeneity during the first hours/days of contact is sufficient to imprint uneven Li+ flux patterns during subsequent cycling. This thickness inhomogeneity has fatal effects on subsequent battery performance:
(1) Uneven ion transport impedance. According to Ohm's law, under the same electric field drive, the impedance that Li+ experiences when passing through the SEI of different thicknesses varies. The thin SEI regions exhibit fast ion transport and low overpotential, while the thick SEI regions show slow ion transport and higher overpotential. This leads to secondary inhomogeneity in interfacial ion flux, which superimposes with the tip electric field effect, further exacerbating the selectivity of Li deposition.
(2) Local current density imbalance. Regions with faster ion transport have higher effective current density, thereby accelerating Li deposition at those sites, forming positive feedback. This uneven distribution of current density intensifies with cycling.
More complexly, chemical corrosion also causes the chemical composition of the SEI to be spatially distributed unevenly. Typically, organic components (e.g., ROCO2Li) primarily formed by solvent molecule reduction have poor mechanical properties and low ionic conductivity,41,43 whereas inorganic components (e.g., Li2O, LiF, and Li3N) are denser and harder and have higher ionic conductivity.44,45 The randomness of chemical corrosion results in a disorderly distribution of organic and inorganic phases within the SEI. This chemical heterogeneity further distorts the Li+ concentration field at the interface because Li+ tends to migrate through regions with higher ionic conductivity (inorganic phases). Consequently, even on a macroscopically uniform electrode surface, microscopic “shortcuts” and “blockage points” for Li+ flux appear.
Furthermore, the mechanical properties of the SEI also vary significantly due to its compositional and thickness inhomogeneity. Under the volume changes during Li deposition/stripping, mechanically weaker regions are more prone to fracture, exposing fresh Li surfaces and triggering new rounds of corrosion reactions, leading to continuous SEI thickening and electrolyte consumption.12,46 Ultimately, an initial SEI with a “mosaic-like” pattern in both thickness and composition severely deteriorates the uniformity of interfacial ion flux and concentration field, making uniform deposition nearly impossible.41
2.3. Unstable SEI triggers continuous side reactions and failure
Before battery cycling, the chemical corrosion process is primarily thermodynamic-driven, with its reaction pathway determined by the reduction potentials of various components.22 In most conventional carbonate-based electrolytes, the reduction potential of solvent molecules (e.g., ethylene carbonate and dimethyl carbonate) at the interface is typically higher than that of Li salt anions (e.g., PF6−), generally determined by the lower unoccupied molecular orbital (LUMO) values of electrolyte components and the Li+ solvation environment.22,47–49 Therefore, the initial SEI is mainly composed of reduction products of solvent molecules, rich in organic components like polycarbonates and Li alkoxides.41 This SEI, dominated by chemical corrosion, forms without the regulation of an electrochemical field, making it disordered and random.This type of solvent-derived SEI has three inherent defects:
(1) Poor chemical and electrochemical stability. Organic components are not fully passivated at low potentials, and their side reactions with Li metal can still proceed spontaneously. During subsequent electrochemical cycling, the volume-changing Li metal repeatedly tears this mechanically poor SEI, exposing fresh Li surfaces. Newly exposed Li reacts with the electrolyte in new rounds of side reactions, continuously consuming active Li and electrolyte, leading to low coulombic efficiency and battery capacity fading. This is the so-called “continuous corrosion” process. The cumulative effect of this process results in continuous SEI thickening and rising interfacial impedance.
(2) Weak resistance to electron tunneling. An ideal SEI should effectively block electron migration from the electrode to the electrolyte.42 However, the electronic conductivity of an organic SEI is usually higher than that of an inorganic SEI.50 Weaker electronic insulation allows electrons to tunnel through the SEI, reaching its interface with the electrolyte, thereby reducing and decomposing electrolyte components, leading to continuous SEI thickening and pore generation. This electron leakage process is a significant cause of dynamic SEI growth and continuous electrolyte decomposition.
(3) Limited Li+ conduction. Due to the thickening of the SEI and the accumulation of side reaction products caused by continuous interface corrosion, an organic-rich SEI usually shows unsatisfactory Li+ conduction. This leads to greater concentration polarization and higher battery overpotentials, especially under high-rate charge/discharge conditions, further deteriorating battery performance.
In summary, the initial, unstable, heterogeneous SEI generated from chemical corrosion not only fails to effectively protect the Li anode but instead becomes a “dynamic wound”. It induces uneven Li deposition, and its own continuous fracturing and regeneration persistently consume the battery's precious internal resources. Ultimately, this vicious cycle leads to rampant Li dendrite growth, a sharp increase in interfacial impedance, and rapid battery failure. Therefore, intervening in the initial chemical corrosion process and constructing an ideal artificial/natural hybrid SEI is key to breaking this failure chain.
3. FD-based interfacial regulation strategies
Facing the severe challenges posed by chemical corrosion, traditional “passive” optimization strategies (e.g., electrolyte additives and three-dimensional hosts) often address symptoms rather than the root cause. We propose that utilizing the inherent spontaneous polarization characteristics of ferroelectric materials to construct an interface layer with active regulatory capability on the Li metal surface can fundamentally improve the interfacial environment from a physical field perspective. Ferroelectric materials, such as barium titanate (BTO), lead zirconate titanate (PZT), and ferroelectric polymer poly(vinylidene fluoride-trifluoroethylene) (P(VDF-TrFE)), are characterized by stable spontaneous polarization below the Curie temperatures, meaning that internal positive and negative charge centers separate, forming a macroscopic electric dipole moment.51,52 Moreover, this polarization state can be oriented by an external electric field and retained after its removal, creating a persistent built-in electric field.533.1. FDs homogenize ion distribution and electric field
When ferroelectric materials are introduced onto the Li metal surface (as a coating or composited in the separator/host), their surface dipoles result in fixed bound charges and a polarization electric field.54 This property enables active regulation of interfacial ion distribution:(1) Binding/enriching anions and electrostatic attraction of Li+. The positively charged surface of ferroelectric materials binds/enriches anions from the electrolyte (e.g., PF6− and TFSI−) in the interfacial region (Fig. 2). Due to electrostatic interactions, Li+ in the electrolyte is also attracted/enriched in the interfacial region, forming a pre-existing, high-concentration layer of Li+.55,56 This combined effect of “binding” and “enriching” anions and “attracting” Li+ co-induces a “local high concentration” region of Li salt near the ferroelectric interface. The salient feature of this region is the reconstructed distribution of Li+ and anions, with significantly elevated and uniformly distributed concentrations. This ion distribution state offers three key advantages: first, it prepares ample and uniformly distributed “raw material” (Li+) for subsequent Li deposition. Second, it suppresses the decrease in the Li+ transference number and the increase in concentration polarization caused by anion back-migration under an applied electric field. Third, it fundamentally mitigates local electric field distortion induced by uneven spatial distribution of charge carriers (e.g., enrichment at tips).
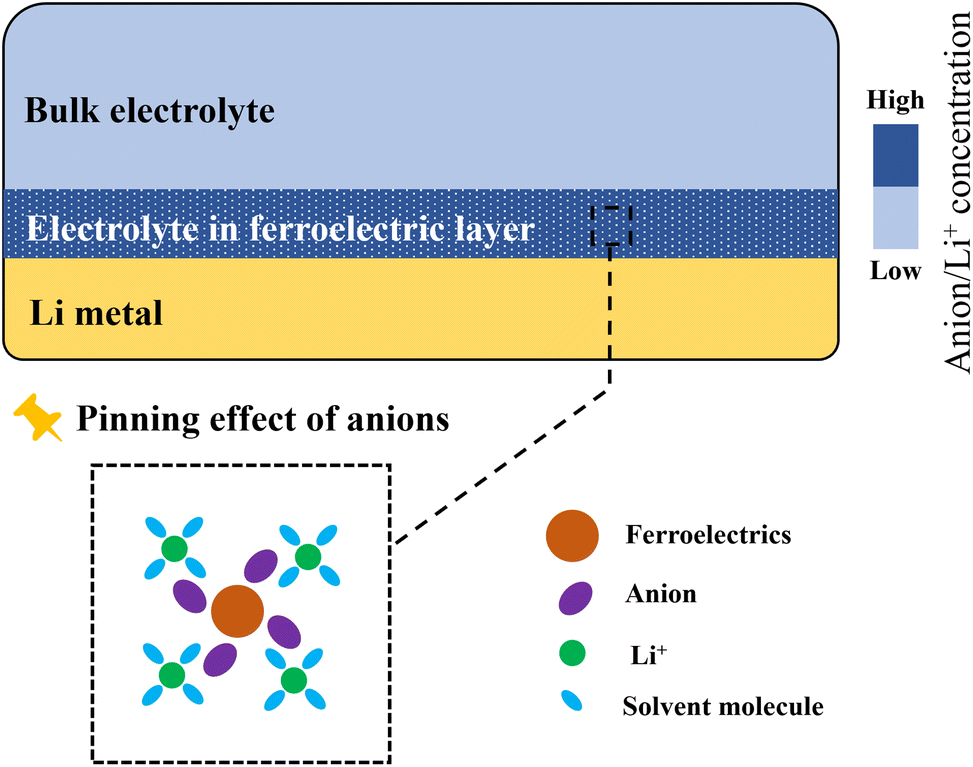 | ||
| Fig. 2 Schematic illustration of the pinning effect of the ferroelectric layer on anions and the resulting enrichment of Li+. | ||
It should be noted, however, that the long-term chemical and electrochemical stability of ferroelectric materials (especially oxide-based ones like BTO and PZT) in direct contact with Li metal remains to be systematically evaluated. Potential issues include the reduction of transition metals (e.g., Ti4+) or dissolution of cations (e.g., Ba2+ and Pb2+) into the electrolyte. These concerns are further discussed in Chapter 4.
(2) Shielding the tip electric field. When tips exist on the electrode surface, their inherent electric field concentration effect can be partially counteracted or weakened by the uniform, opposing electric field generated by the ferroelectric layer – a plausible mechanism rather than a directly proven fact (Fig. 3). The experimental evidence57 shows that BTO ceramic nanofiber films lead to more uniform Li deposition morphology, which is consistent with this tip-shielding hypothesis. However, direct visualization of the local electric field distribution near the ferroelectric interface remains a challenge for future in situ characterization. The homogenized anion distribution further aids this shielding effect.
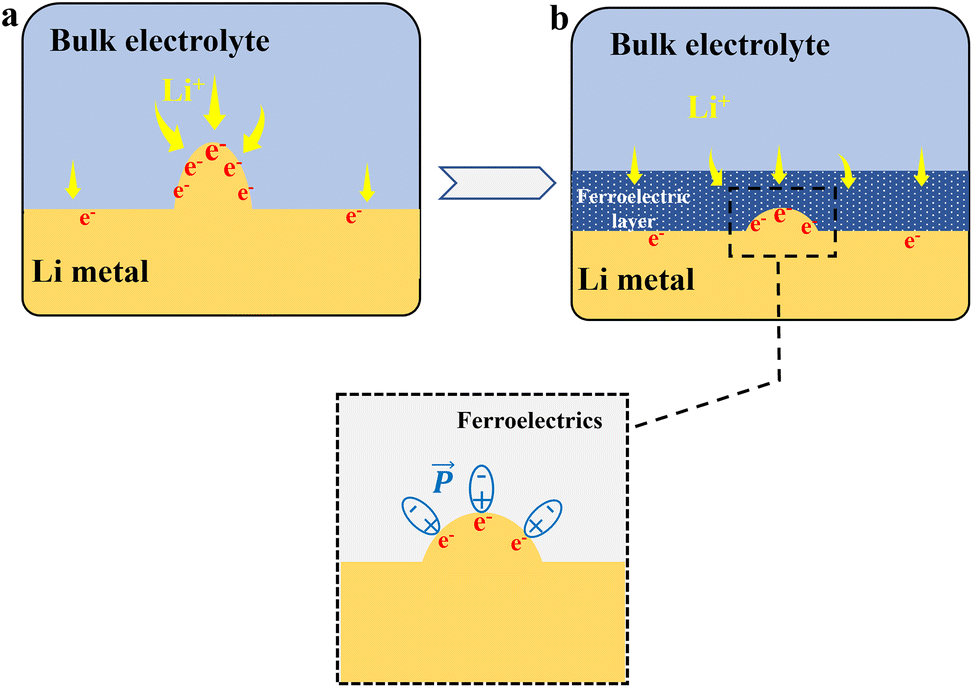 | ||
| Fig. 3 Schematic illustration of ferroelectrics shielding the tip electric field. (a) Tip electric field on the Li anode surface. (b) The strong tip electric field excites the polarization reversal of the ferroelectric surface, and the generated reverse polarization electric field shields and weakens the electric field at the tip. | ||
(3) Guiding uniform nucleation. At the onset of Li deposition, due to the uniform concentration distribution of Li+ and anions at the interface and the absence of strong local electric field attraction, the Li nucleation process occurs more randomly and uniformly across the entire electrode surface, avoiding preferential growth at certain points. Research indicates that uniform nucleation is the primary condition for obtaining dense Li deposition layers.58–60
3.2. FDs regulate the solvation structure
Li ions do not exist in the electrolyte as bare entities but coordinate with solvent molecules and anions, forming a solvation structure61 (Fig. 4a). The stability of this structure directly determines its reduction decomposition pathway at the electrode interface, thereby affecting SEI composition.62 The strong local electric field and high dielectric environment generated by FDs can exert a profound dielectric regulatory effect on the solvation structure at the interface (Fig. 4b).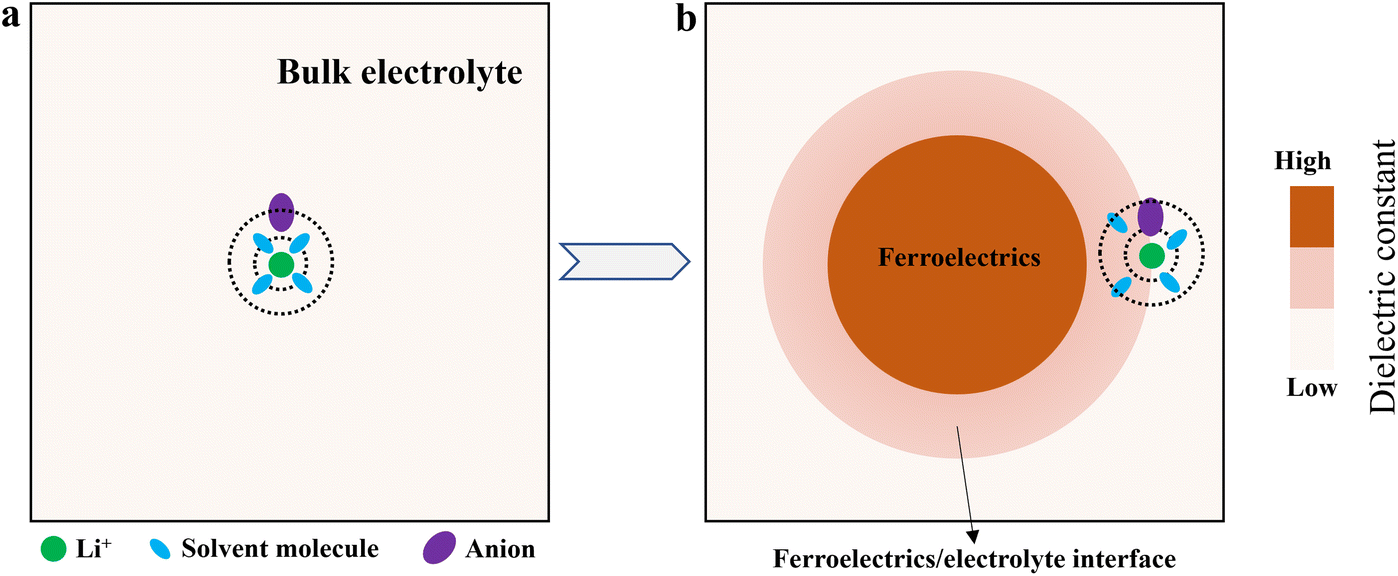 | ||
| Fig. 4 Schematic illustration of ferroelectrics regulating the solvation structure. (a) Solvation structure in bulk electrolyte. (b) The high dielectric environment generated by ferroelectrics loosens the solvation sheath, allowing anions to approach the inner-layer Li+ more closely. | ||
(1) Weakening Li+–solvent interactions. Ferroelectric materials typically possess extremely high dielectric constants (e.g., εr > 1000 for BTO).51,52 According to dielectric theory, a high dielectric environment weakens the coulombic force between point charges.63 Therefore, near the ferroelectric interface layer, the binding force between Li+ and polar solvent molecules is weakened, loosening the solvation sheath. This hypothesized “sheath loosening” effect, predicted from classical dielectric theory, would reduce the energy barrier for solvent molecules to detach from Li+. Direct spectroscopic evidence (e.g., Raman) is still needed to confirm this effect at ferroelectric interfaces, but it provides a plausible mechanism that is consistent with observed changes in SEI composition.
(2) Promoting anion participation in the inner solvation sheath. When Li+–solvent interactions are weakened, anions with lower reduction potentials (e.g., PF6− and TFSI−) more easily “approach” Li+ and even enter its primary solvation sheath. This is referred to as changing the anion coordination number. The ferroelectric interface layer effectively shifts the thermodynamic and kinetic equilibrium for various components competing to coordinate with Li+ at the interface in favor of anions.
(3) Prioritizing anion decomposition. In subsequent electrode reactions, due to the increased proportion of anions in the inner sheath, they will be preferentially reduced at the electrode surface.62,64 This process favors the generation of an inorganic-rich (e.g., LiF) SEI.65
In essence, FDs, through their high dielectric environment and surface electric field, reshape the interfacial solvation structure, shifting it from “solvent-dominated” to “anion-dominated”, laying a thermodynamic foundation for constructing a high-performance SEI.
3.3. FDs drive precise SEI construction
Based on the mechanisms in Sections 3.1 and 3.2, FDs enable “top-down” precise design of the SEI, overcoming the unstable SEI generated by chemical corrosion at the source. Crucially, FDs not only alter the solvation structure but also directly polarize and activate anions, promoting their preferential reduction and decomposition, thereby dominating SEI formation.55(1) Polarization and activation of anions by FDs. The strong local electric field on the surface of ferroelectric materials can exert a polarization effect on the electron cloud distribution of anions, causing a certain degree of relaxation and weakening of their chemical bonds, thereby lowering the energy barrier for their reduction decomposition (Fig. 5). Taking the PF6− anion as an example, the electric field at the ferroelectric interface may influence the polarity of P–F bonds, making F atoms more easily detach as F−, subsequently combining with Li+ to form LiF. This pre-activation effect places anions in a state more prone to reduction upon reaching the electrode surface. This effect goes beyond simple thermodynamic competition, introducing a kinetically promoting factor of electric field catalysis.
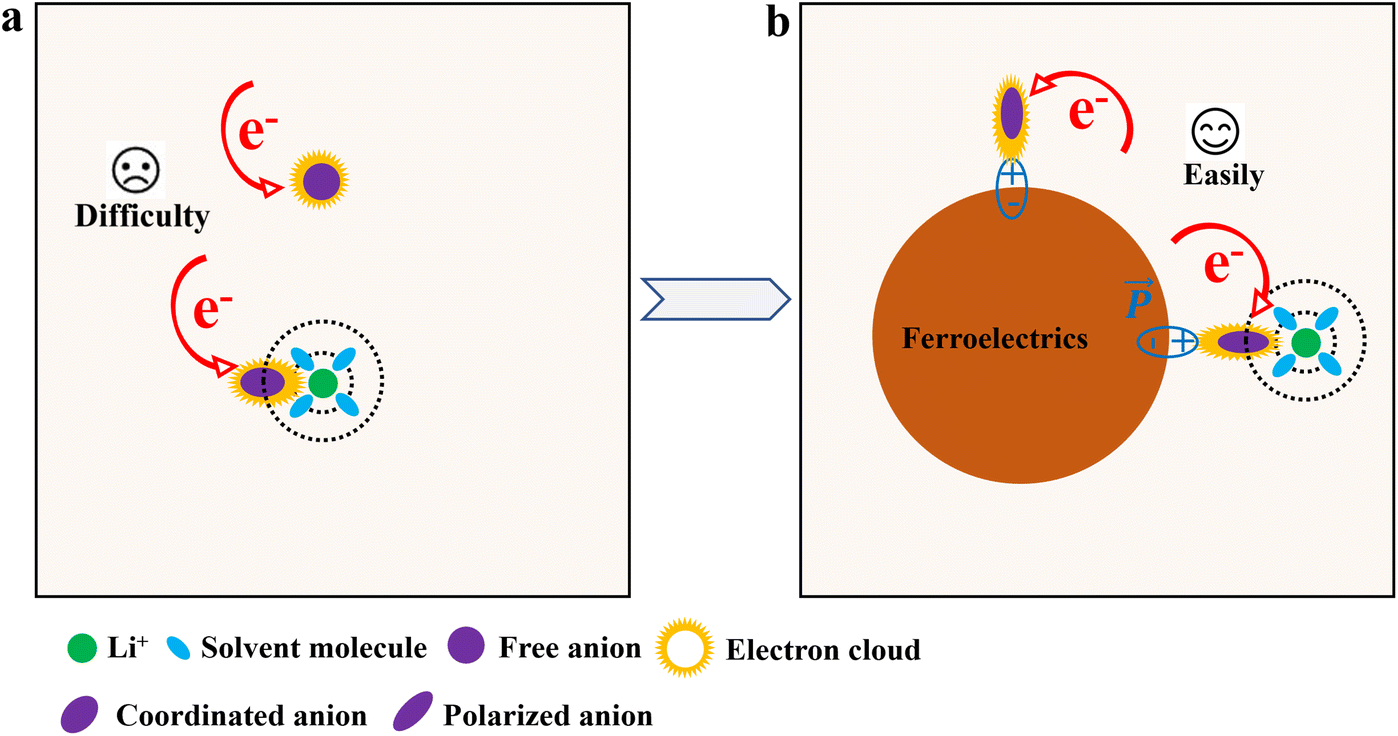 | ||
| Fig. 5 Schematic illustration of the polarization effect of ferroelectrics on anions in electrolyte. (a) Both free anions and coordinating anions in the electrolyte undergo reductive decomposition by capturing electrons, a process that is relatively difficult. (b) Due to the perturbation of electron clouds by FDs, anions polarized by ferroelectrics are more likely to capture electrons and undergo reductive decomposition. | ||
(2) Guiding the formation of an inorganic-rich SEI. As described above, the ferroelectric interface-induced preferential reduction and activated decomposition of anions (e.g., PF6−, TFSI−, and NO3−) directly lead to the generation of substantial inorganic components (e.g., LiF, Li2O, and Li3N). LiF, with its extremely high interfacial energy, excellent electronic insulation, and moderate Li+ conductivity, is considered one of the most ideal components in the SEI.65,66 Li2O, with good mechanical strength, excellent chemical stability, and outstanding electronic insulation, can effectively block electron tunneling and continuous electrolyte corrosion.67,68 Li3N typically possesses high ionic conductivity, enhancing interfacial ion migration capability, which is beneficial for promoting fast Li-ion transport within the SEI.45,69 Inorganic components like LiF, Li2O, and Li3N are key building blocks for a uniform, dense, and stable high-performance SEI.44 X-ray photoelectron spectroscopy (XPS) and time-of-flight secondary ion mass spectrometry (ToF-SIMS) depth profiling have repeatedly confirmed a significant increase in inorganic content within the SEI formed on ferroelectric material-modified electrode surfaces.70
(3) Enhancing the mechanical stability of the SEI. An inorganic-rich, uniform SEI typically exhibits better mechanical strength, allowing it to better accommodate volume changes during Li deposition/stripping, reducing fracture risks, and thereby improving cycling stability.71 The high modulus of inorganic components helps resist Li dendrite penetration.
Research shows that using a separator modified with ferroelectric BTO nanoparticles results in a significantly higher inorganic content in the SEI on the cycled Li metal surface compared to the control group, along with markedly enhanced density and flatness of the Li deposition morphology.55 These findings, together with the mechanistic hypothesis outlined above, highlight the promising potential of the FD strategy, while recognizing that direct evidence is still needed.
It is important to recognize that the proposed sequence of “FD-induced anion polarization, followed by preferential reduction, then inorganic-rich SEI formation” remains a working hypothesis. Most existing evidence is indirect, based primarily on in situ XPS and ToF-SIMS analysis of the final SEI compositions.55,70 These preliminary data support the hypothesis, but further in situ characterization studies (e.g., in situ PFM, cryo-TEM coupled with EELS) are required to directly visualize the dynamic processes of anion polarization and reduction at the ferroelectric interface.
3.4. Pre-adsorbed anion-type FDs enable targeted SEI design
The aforementioned strategy primarily relies on inherent anions or additives in the electrolyte. To further enhance the precision and flexibility of SEI design, we propose a more advanced concept: pre-adsorbed anion-type FDs. The core of this strategy lies in pre-adsorbing or fixing specific types or combinations of anions onto or near the surface region of ferroelectric materials through physical or chemical methods before battery assembly.72 These pre-adsorbed anions become “advantageous precursors” for SEI construction and, combined with the polarization and activation effects of FDs, enable targeted introduction of SEI components and precise regulation of the microstructure (Fig. 6). The working mechanisms of this strategy include the followings.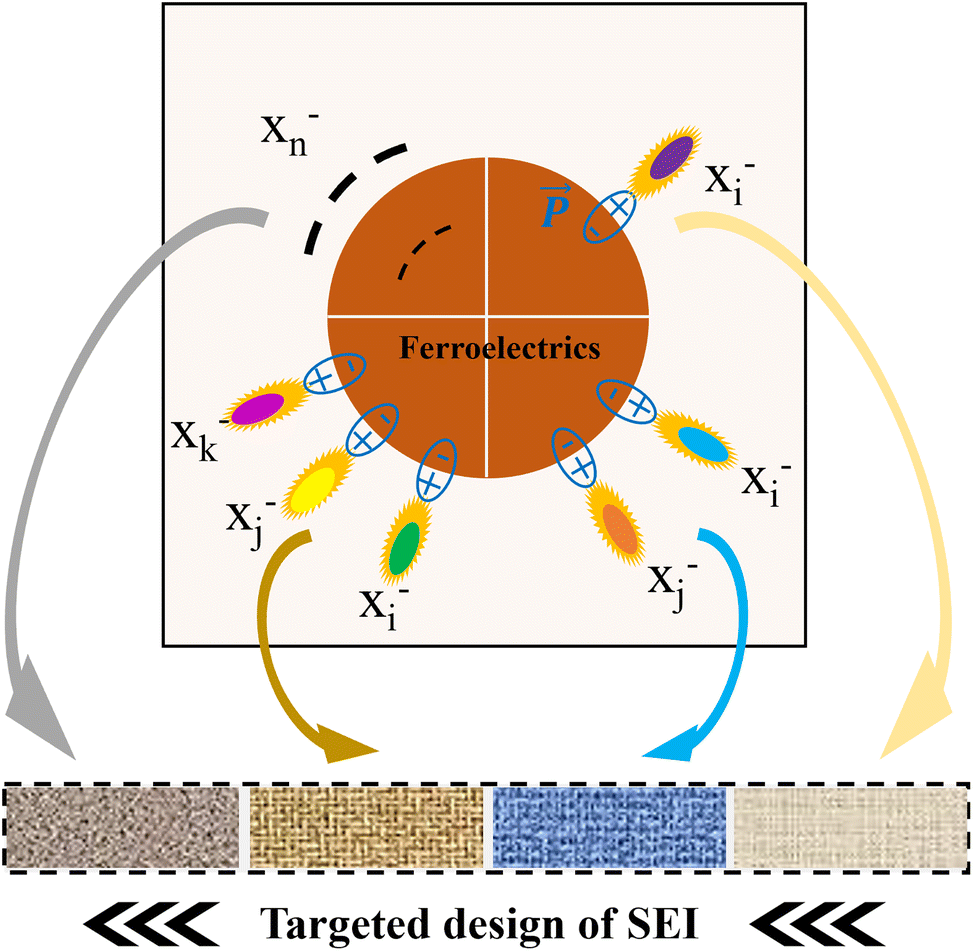 | ||
| Fig. 6 Schematic illustration of how pre-adsorbed anion-type ferroelectrics achieve targeted design of the SEI by polarizing various types and combinations of anions. | ||
(1) Pre-adsorption and enrichment. Target anions (denoted as X−) are made to preferentially occupy the surface or interface layer of ferroelectric materials through surface modification (e.g., grafting positively charged functional groups), simple solution immersion, or ultrasonic-stimulated adsorption. This is equivalent to pre-setting the “building blocks” of the SEI at the interface.
(2) Ferroelectric field polarization and activation. After battery assembly, the strong local electric field of the FDs exerts intense polarization on the pre-adsorbed X−, significantly lowering its reduction decomposition energy barrier, causing it to be preferentially reduced over other electrolyte components at the beginning of electrochemical cycling.
(3) Targeted decomposition and SEI construction. The activated X− is reduced at the electrode surface, and its decomposition products are precisely introduced into the SEI. By selecting different types/combinations of X−, we can design the chemical composition of the SEI as needed, akin to “building blocks”.
The main advantage of this strategy is that it transforms SEI formation from relying on the random, competitive reduction of bulk electrolyte components to a directed, selective reduction dominated by interface-predefined “templates”. It greatly reduces the uncertainty in the SEI formation process, offering the possibility of controllable fabrication and performance prediction of the SEI. Of course, realizing this concept also faces challenges, such as the stability of pre-adsorbed anions, their binding strength with ferroelectric materials, and the feasibility for large-scale production. Nonetheless, this undoubtedly opens up an imaginative new direction for future LMB interface engineering.
4. Summary and outlook
Before summarizing the key contributions of this perspective, it is helpful to briefly position the proposed FD strategy within the broader landscape of Li metal anode stabilization. Numerous approaches have been developed, including electrolyte additives (e.g., FEC and LiNO3),73,74 artificial SEI layers (e.g., LiF, Al2O3, and polymers),75–77 3D porous hosts,13,78 and separator modifications.79 These strategies are largely passive in nature, aimed at suppressing side reactions or guiding Li deposition by providing a static physical or chemical barrier. In contrast, FD-based strategies provide active tuning capabilities, exemplified by an ever-present built-in polarization field and the versatile choice of pre-adsorbed species, among others. However, FD engineering is not a universal replacement, which may be most effective when combined with other approaches. For example, a ferroelectric coating on a 3D host could synergistically homogenize the electric field.70,80 We emphasize that the FD strategy is still at an early stage, and further research is needed to address material stability and integration challenges (discussed below). With this context in mind, we now summarize the key mechanisms and future directions.We emphasize that initial chemical corrosion is by no means a negligible transient process. Instead, by inducing initial surface tips, heterogeneous SEI formation, and unstable chemical composition, it initiates a vicious cycle leading to rampant Li dendrite growth and battery failure. Traditional “patchwork” optimization cannot solve this problem. To this end, we outline a new research path: FD engineering. By introducing a ferroelectric layer with spontaneous polarization at the Li metal interface, we can achieve multi-level regulation from a physical field perspective.
(1) Homogenizing ion and electric fields. Surface-bound charges can bidirectionally regulate anions and Li+ distribution, achieving homogenization of interfacial ion flux and suppressing the tip effect.
(2) Regulating the solvation structure. A high dielectric environment and local electric field can guide anions to preferentially enter the inner solvation sheath.
(3) Driving precise SEI design. Polarization and activation of anions lower their decomposition energy barriers and guide the formation of a uniform, dense, inorganic-rich, highly stable SEI.
(4) Targeted construction of an advanced SEI. The innovative pre-adsorbed anion strategy achieves on-demand design and controllable fabrication of SEI chemical components.
This strategy pushes interface research from “passive protection” towards “active design” and “precise programming”, demonstrating vast application prospects. However, advancing this strategy towards practical applications still faces numerous challenges and opportunities.
(1) Material and structural innovation. There is an urgent need to develop ferroelectric coating technologies compatible with battery manufacturing processes and to explore ultrathin, highly flexible, high-polarization-strength ferroelectric materials (e.g., two-dimensional ferroelectrics and ferroelectric polymer nanofibers). Optimizing the loading and distribution of the ferroelectric phase, as well as its interfacial contact with Li metal, is crucial. Particularly, attention must be paid to the long-term chemical and electrochemical stability of ferroelectric materials in the battery environment. For oxide ceramics like BTO and PZT, concerns include chemical reduction of Ti4+ (to Ti3+ or lower), dissolution of Ba2+/Pb2+ into the electrolyte (potentially contaminating the cathode), mechanical detachment of particles during Li volume changes (leading to localized blocking layers or increased resistance), and unknown polarization retention over repeated cycling. A promising direction for future research is to improve the chemical and electrochemical stability of ferroelectrics via bulk doping or surface modification, without compromising their high polarization. Additionally, polymer ferroelectrics (e.g., P(VDF-TrFE)) and 2D ferroelectrics (e.g., In2Se3) offer more compliant and potentially more stable alternatives.51,81 These challenges are surmountable but must be explicitly addressed in future experimental work.
(2) In-depth mechanistic exploration. It is imperative to advance in situ/operando characterization techniques (e.g., in situ piezoresponse force microscopy (PFM), Kelvin probe force microscopy (KPFM), and cryogenic transmission electron microscopy (cryo-TEM)) to track in real-time the dynamic processes of solvation structure evolution, anion activation, SEI formation, and Li deposition under the influence of the ferroelectric interface, providing direct evidence for mechanistic models. Theoretical calculations (e.g., density functional theory and molecular dynamics) need to further reveal the microscopic mechanisms by which the ferroelectric field affects anion polarization and reduction pathways, especially simulations for pre-adsorbed anion systems.
(3) Full-cell integration verification. Future research must validate the long-term cycling stability, safety performance, and cost-effectiveness of this strategy in Ah-level pouch cells, assess its compatibility with high-capacity cathodes, and evaluate performance under wide temperature ranges, high-rate conditions, and calendar aging coupling.
(4) Multi-field coupling effects. Beyond the electrostatic field, the coupling of the piezoelectric effects of ferroelectric materials with internal mechanical stresses in batteries and the coupling of pyroelectric effects with temperature-field should be explored to achieve more intelligent multifunctional interface regulation. For example, stresses generated by volume changes of active materials during cycling could be harnessed to induce piezoelectric potentials for self-driven interfacial ion flow regulation.
In conclusion, interface design based on FDs represents a paradigm shift in fundamentally addressing LMB interface issues. By deeply understanding the failure root cause of chemical corrosion and utilizing the unique physical properties of ferroelectric materials for precise intervention, we have the potential to break the existing performance bottlenecks. Particularly, the introduction of the pre-adsorbed anion-type FD concept elevates SEI research to a new height. Through interdisciplinary collaborative efforts, harnessing this powerful “physical field” tool, we may ultimately overcome the core challenges of the Li metal anode, ushering in a new era of high-energy-density, high-safety secondary batteries.
Author contributions
B. X. proposed the mechanisms and wrote the original draft; W. W. conceived and supervised the project; W. W., Z. X., C. Z., and L. Z. funded the project; all authors discussed the results and commented on the paper. All authors read and approved the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We would like to acknowledge financial support from the Foundation for Outstanding Research Groups of the National Natural Science Foundation of China (Grant No. 72088101), the National Natural Science Foundation of China (Grant No. 52477228, 52407257 and 52377220), and the Key Project of Xiangjiang Laboratory (No. 25XJ02001).References
- J. Liu, Z. Bao, Y. Cui, E. J. Dufek, J. B. Goodenough, P. Khalifah, Q. Li, B. Y. Liaw, P. Liu, A. Manthiram, Y. S. Meng, V. R. Subramanian, M. F. Toney, V. V. Viswanathan, M. S. Whittingham, J. Xiao, W. Xu, J. Yang, X.-Q. Yang and J.-G. Zhang, Nat. Energy, 2019, 4, 180–186 CrossRef CAS.
- M. He, L. G. Hector, F. Dai, F. Xu, S. Kolluri, N. Hardin and M. Cai, Nat. Energy, 2024, 9, 1199–1205 CrossRef.
- H. Wan, J. Xu and C. Wang, Nat. Rev. Chem., 2024, 8, 30–44 CrossRef CAS.
- X.-B. Cheng, R. Zhang, C.-Z. Zhao and Q. Zhang, Chem. Rev., 2017, 117, 10403–10473 CrossRef CAS.
- X. Zhang, A. Wang, X. Liu and J. Luo, Acc. Chem. Res., 2019, 52, 3223–3232 CrossRef CAS PubMed.
- C. Fang, J. Li, M. Zhang, Y. Zhang, F. Yang, J. Z. Lee, M.-H. Lee, J. Alvarado, M. A. Schroeder, Y. Yang, B. Lu, N. Williams, M. Ceja, L. Yang, M. Cai, J. Gu, K. Xu, X. Wang and Y. S. Meng, Nature, 2019, 572, 511–515 CrossRef CAS PubMed.
- Y. Zhou, X. Zhang, Y. Ding, J. Bae, X. Guo, Y. Zhao and G. Yu, Adv. Mater., 2020, 32, 2003920 CrossRef CAS PubMed.
- J. Zhi, S. Li, M. Han and P. Chen, Sci. Adv., 2020, 6, eabb1342 CrossRef CAS PubMed.
- C. Yang, H. Xie, W. Ping, K. Fu, B. Liu, J. Rao, J. Dai, C. Wang, G. Pastel and L. Hu, Adv. Mater., 2018, 31, 1804815 CrossRef PubMed.
- H. Gao, X. Ai, H. Wang, W. Li, P. Wei, Y. Cheng, S. Gui, H. Yang, Y. Yang and M. S. Wang, Nat. Commun., 2022, 13, 5050 CrossRef CAS PubMed.
- J.-i. Yamaki, S.-i. Tobishima, K. Hayashi, K. Saito, Y. Nemoto and M. Arakawa, J. Power Sources, 1998, 74, 219–227 CrossRef CAS.
- D. Lin, Y. Liu and Y. Cui, Nat. Nanotechnol., 2017, 12, 194–206 CrossRef CAS PubMed.
- D. J. Yoo, A. Elabd, S. Choi, Y. Cho, J. Kim, S. J. Lee, S. H. Choi, T. w. Kwon, K. Char, K. J. Kim, A. Coskun and J. W. Choi, Adv. Mater., 2019, 31, 1901645 CrossRef.
- J. Chen, J. Zhao, L. Lei, P. Li, J. Chen, Y. Zhang, Y. Wang, Y. Ma and D. Wang, Nano Lett., 2020, 20, 3403–3410 CrossRef CAS PubMed.
- J. Qian, W. A. Henderson, W. Xu, P. Bhattacharya, M. Engelhard, O. Borodin and J.-G. Zhang, Nat. Commun., 2015, 6, 6362 CrossRef CAS.
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, Nat. Commun., 2013, 4, 1481 CrossRef.
- Y. Liu, Y.-K. Tzeng, D. Lin, A. Pei, H. Lu, N. A. Melosh, Z.-X. Shen, S. Chu and Y. Cui, Joule, 2018, 2, 1595–1609 CrossRef CAS.
- J. Zhao, L. Liao, F. Shi, T. Lei, G. Chen, A. Pei, J. Sun, K. Yan, G. Zhou, J. Xie, C. Liu, Y. Li, Z. Liang, Z. Bao and Y. Cui, J. Am. Chem. Soc., 2017, 139, 11550–11558 CrossRef CAS PubMed.
- A. Kolesnikov, M. Kolek, J. F. Dohmann, F. Horsthemke, M. Börner, P. Bieker, M. Winter and M. C. Stan, Adv. Energy Mater., 2020, 10, 2000017 CrossRef CAS.
- D. Lin, Y. Liu, Y. Li, Y. Li, A. Pei, J. Xie, W. Huang and Y. Cui, Nat. Chem., 2019, 11, 382–389 CrossRef CAS.
- W. Zhang, P. Sayavong, X. Xiao, S. T. Oyakhire, S. B. Shuchi, R. A. Vilá, D. T. Boyle, S. C. Kim, M. S. Kim, S. E. Holmes, Y. Ye, D. Li, S. F. Bent and Y. Cui, Nature, 2024, 626, 306–312 CrossRef CAS PubMed.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- F. Baakes, D. Witt and U. Krewer, Chem. Sci., 2023, 14, 13783–13798 RSC.
- C. Jin, Y. Huang, L. Li, G. Wei, H. Li, Q. Shang, Z. Ju, G. Lu, J. Zheng, O. Sheng and X. Tao, Nat. Commun., 2023, 14, 8269 CrossRef CAS.
- C.-X. Bi, Y.-J. Zhu, X.-Y. Li, J. Ma, X.-Q. Zhang, M. Zhao, B.-Q. Li and J.-Q. Huang, J. Am. Chem. Soc., 2025, 147, 34632–34640 CrossRef CAS.
- J. Kim, I. Jung, K. Lee, I. Hwang, M. Kim, E. Park, D. Park, H. Park, Y. Jeon, G. Park, H. C. Ahn, J. Oh, J. Park, A. Coskun and J. W. Choi, Adv. Energy Mater., 2025, e04147 CrossRef.
- S.-H. Yu, X. Huang, J. D. Brock and H. D. Abruña, J. Am. Chem. Soc., 2019, 141, 8441–8449 CrossRef CAS PubMed.
- Q. Wang, B. Liu, Y. Shen, J. Wu, Z. Zhao, C. Zhong and W. Hu, Adv. Sci., 2021, 8, 2101111 CrossRef CAS.
- C. Xie, D. Yan, H. Li, S. Du, W. Chen, Y. Wang, Y. Zou, R. Chen and S. Wang, ACS Catal., 2020, 10, 11082–11098 CrossRef CAS.
- W. Li, D. Wang, Y. Zhang, L. Tao, T. Wang, Y. Zou, Y. Wang, R. Chen and S. Wang, Adv. Mater., 2020, 32, 1907879 CrossRef CAS PubMed.
- Y. Zhang, J. Liu, Y. Xu, C. Xie, S. Wang and X. Yao, Chem. Soc. Rev., 2024, 53, 10620–10659 RSC.
- C. Xie, D. Yan, W. Chen, Y. Zou, R. Chen, S. Zang, Y. Wang, X. Yao and S. Wang, Mater. Today, 2019, 31, 47–68 CrossRef CAS.
- L. Enze, J. Phys. D: Appl. Phys., 1987, 20, 1609 CrossRef.
- P. Liu, B. Chen, C. Liang, W. Yao, Y. Cui, S. Hu, P. Zou, H. Zhang, H. J. Fan and C. Yang, Adv. Mater., 2021, 33, 2007377 CrossRef CAS PubMed.
- M. Liu, Y. Pang, B. Zhang, P. De Luna, O. Voznyy, J. Xu, X. Zheng, C. T. Dinh, F. Fan, C. Cao, F. P. G. de Arquer, T. S. Safaei, A. Mepham, A. Klinkova, E. Kumacheva, T. Filleter, D. Sinton, S. O. Kelley and E. H. Sargent, Nature, 2016, 537, 382–386 CrossRef CAS PubMed.
- S. Liu, X. Xia, Y. Zhong, S. Deng, Z. Yao, L. Zhang, X. B. Cheng, X. Wang, Q. Zhang and J. Tu, Adv. Energy Mater., 2017, 8, 1702322 CrossRef.
- C. Monroe and J. Newman, J. Electrochem. Soc., 2003, 150, A1377 CrossRef CAS.
- E. Wang, S. Ge, W. Li, B. Fu, F. Zhou and W. Chen, Matter, 2025, 8, 102368 CrossRef CAS.
- Q. Zhao, S. Stalin and L. A. Archer, Joule, 2021, 5, 1119–1142 CrossRef CAS.
- H. Wu, H. Jia, C. Wang, J.-G. Zhang and W. Xu, Adv. Energy Mater., 2021, 11, 2003092 CrossRef CAS.
- M. D. Tikekar, S. Choudhury, Z. Tu and L. A. Archer, Nat. Energy, 2016, 1, 16114 CrossRef CAS.
- Y. Xu, H. Jia, P. Gao, D. E. Galvez-Aranda, S. P. Beltran, X. Cao, P. M. L. Le, J. Liu, M. H. Engelhard, S. Li, G. Ren, J. M. Seminario, P. B. Balbuena, J.-G. Zhang, W. Xu and C. Wang, Nat. Energy, 2023, 8, 1345–1354 CrossRef CAS.
- C. Jiang, Q. Jia, M. Tang, K. Fan, Y. Chen, M. Sun, S. Xu, Y. Wu, C. Zhang, J. Ma, C. Wang and W. Hu, Angew. Chem., Int. Ed., 2021, 60, 10871–10879 CrossRef CAS PubMed.
- S. Xu, S. Xu, X. Guo, J. Xiong, Z. Wei, S. Zhu, J. Xu, S. Gong, P. Shi, S. Guo and Y. Min, Adv. Funct. Mater., 2025, 35, 2500335 CrossRef CAS.
- J. Pokharel, A. Cresce, B. Pant, M. Y. Yang, A. Gurung, W. He, A. Baniya, B. S. Lamsal, Z. Yang, S. Gent, X. Xian, Y. Cao, W. A. Goddard, K. Xu and Y. Zhou, Nat. Commun., 2024, 15, 3085 CrossRef CAS PubMed.
- A. Kushima, K. P. So, C. Su, P. Bai, N. Kuriyama, T. Maebashi, Y. Fujiwara, M. Z. Bazant and J. Li, Nano Energy, 2017, 32, 271–279 CrossRef CAS.
- Z. Chang, H. Yang, Y. Qiao, X. Zhu, P. He and H. Zhou, Adv. Mater., 2022, 34, 2201339 CrossRef CAS.
- Z. Piao, R. Gao, Y. Liu, G. Zhou and H.-M. Cheng, Adv. Mater., 2023, 35, 2206009 CrossRef CAS.
- A. Chen, J. Wang, W. Yang, L. Wang, L. Zhou, Y. Qiao, P. He and H. Zhou, Nat. Commun., 2025, 16, 11400 CrossRef CAS.
- E. Peled and S. Menkin, J. Electrochem. Soc., 2017, 164, A1703 CrossRef CAS.
- V. K. T. Prateek and R. K. Gupta, Chem. Rev., 2016, 116, 4260–4317 CrossRef CAS PubMed.
- M. Acosta, N. Novak, V. Rojas, S. Patel, R. Vaish, J. Koruza, G. Rossetti Jr and J. Rödel, Applied Physics Reviews, 2017, 4, 041305 CrossRef.
- B. Jiang, J. Iocozzia, L. Zhao, H. Zhang, Y. W. Harn, Y. Chen and Z. Lin, Chem. Soc. Rev., 2019, 48, 1194–1228 RSC.
- L. Chen, J.-T. Ren and Z.-Y. Yuan, Adv. Energy Mater., 2023, 13, 2203720 CrossRef CAS.
- B. Xu, L. Ma, W. Wang, H. Zhu, Y. Zhang, C. Liang, L. Zhou, L. Wang, Y. Zhang, L. Chen, C. Zhang and W. Wei, Adv. Mater., 2024, 36, 2311938 CrossRef CAS PubMed.
- W. Zhao, K. Xu, Y. Zhang, Q. Dong, N. Zhang, H. Wang, R. Yi, Y. Tang, Y. Shen and L. Chen, Chem. Sci., 2026, 17, 5145–5153 RSC.
- S. Xia, Y. Zhao, J. Yan, J. Yu and B. Ding, ACS Nano, 2021, 15, 3161–3170 CrossRef CAS.
- Z. Peng, J. Song, L. Huai, H. Jia, B. Xiao, L. Zou, G. Zhu, A. Martinez, S. Roy, V. Murugesan, H. Lee, X. Ren, Q. Li, B. Liu, X. Li, D. Wang, W. Xu and J.-G. Zhang, Adv. Energy Mater., 2019, 9, 1901764 CrossRef CAS.
- A. Pei, G. Zheng, F. Shi, Y. Li and Y. Cui, Nano Lett., 2017, 17, 1132–1139 CrossRef CAS PubMed.
- D. Wang, W. Zhang, W. Zheng, X. Cui, T. Rojo and Q. Zhang, Adv. Sci., 2017, 4, 1600168 CrossRef PubMed.
- T. Abe, H. Fukuda, Y. Iriyama and Z. Ogumi, J. Electrochem. Soc., 2004, 151, A1120 CrossRef CAS.
- K. Xu, Y. Lam, S. S. Zhang, T. R. Jow and T. B. Curtis, J. Phys. Chem. C, 2007, 111, 7411–7421 CrossRef CAS.
- G. Li, Z. Niu, Y. Wu, F. Wu, P. Yang and Z. Chen, Energy Storage Mater., 2026, 84, 104778 CrossRef.
- C. Liu, Z. Jiang, Y. Zhang, W. Xie, J. Zou, S. Wu, M. Sun and Y. Li, Chem. Sci., 2025, 16, 7847–7857 RSC.
- Y. Liu, X. Tao, Y. Wang, C. Jiang, C. Ma, O. Sheng, G. Lu and X. W. Lou, Science, 2022, 375, 739–745 CrossRef CAS PubMed.
- M. Chen, J. Zheng, Y. Liu, O. Sheng, Z. Ju, G. Lu, T. Liu, Y. Wang, J. Nai, Q. Wang and X. Tao, Adv. Funct. Mater., 2021, 31, 2102228 CrossRef CAS.
- G. M. Hobold, C. Wang, K. Steinberg, Y. Li and B. M. Gallant, Nat. Energy, 2024, 9, 580–591 CrossRef CAS.
- H. Zeng, K. Yu, J. Li, M. Yuan, J. Wang, Q. Wang, A. Lai, Y. Jiang, X. Yan, G. Zhang, H. Xu, J. Wang, W. Huang, C. Wang, Y. Deng and S.-S. Chi, ACS Nano, 2024, 18, 1969–1981 CrossRef CAS.
- X. Zhang, Q. Su, G. Du, B. Xu, S. Wang, Z. Chen, L. Wang, W. Huang and H. Pang, Angew. Chem., Int. Ed., 2023, 135, e202304947 CrossRef.
- B. Xu, C. Zhang, W. Wang, H. Zhu, L. Ma, M. Wang, C. Liang, L. Zhou, L. Wang, L. Chen, D. G. Ivey and W. Wei, Angew. Chem., Int. Ed., 2025, 64, e202416565 CrossRef CAS PubMed.
- Z. Hao, G. Li, C. Zheng, X. Liu, S. Wu, H. Li, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2024, 63, e202407064 CrossRef CAS PubMed.
- B. Xu, Y. Wu, R. Ke, K. Yan, C. Liang, L. Chen, X. Chen, B. Han, C. Zhang and W. Wei, ResearchGate, 2026 DOI:10.13140/RG.2.2.32148.56962.
- Q. Zhao, N. W. Utomo, A. L. Kocen, S. Jin, Y. Deng, V. X. Zhu, S. Moganty, G. W. Coates and L. A. Archer, Angew. Chem., Int. Ed., 2022, 61, e202116214 CrossRef CAS PubMed.
- J.-H. Song, J.-T. Yeon, J.-Y. Jang, J.-G. Han, S.-M. Lee and N.-S. Choi, J. Electrochem. Soc., 2013, 160, A873 CrossRef CAS.
- X. Q. Zhang, X. Chen, R. Xu, X. B. Cheng, H. J. Peng, R. Zhang, J. Q. Huang and Q. Zhang, Angew. Chem., Int. Ed., 2017, 56, 14207–14211 CrossRef CAS PubMed.
- E. Kazyak, K. N. Wood and N. P. Dasgupta, Chem. Mater., 2015, 27, 6457–6462 CrossRef CAS.
- B. Zhu, Y. Jin, X. Hu, Q. Zheng, S. Zhang, Q. Wang and J. Zhu, Adv. Mater., 2016, 29, 1603755 CrossRef PubMed.
- Z. Sun, S. Jin, H. Jin, Z. Du, Y. Zhu, A. Cao, H. Ji and L. J. Wan, Adv. Mater., 2018, 30, 1800884 CrossRef PubMed.
- L. Zhao, Z. Wu, Z. Wang, Z. Bai, W. Sun and K. Sun, ACS Nano, 2022, 16, 20891–20901 CrossRef CAS PubMed.
- Y. Guo, R. Wang, C. Cui, R. Xiong, Y. Wei, T. Zhai and H. Li, Nano Lett., 2020, 20, 7680–7687 CrossRef CAS PubMed.
- S. Lin, G. Zhang, Q. Lai, J. Fu, W. Zhu and H. Zeng, Adv. Funct. Mater., 2023, 33, 2304139 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |