
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced interfacial charge transport motivated by Bi2Te3–SbPO4@NC heterointerface for superior Zn2+ and NH4+ storage system
Zhiyuan Zhaa,
Ruinan Chena,
Tong Zhou*bd,
Daohong Zhang *ac and
Qiufan Wang*ac
*ac and
Qiufan Wang*ac
aKey Laboratory of Catalysis and Energy Materials Chemistry of Ministry of Education, Hubei Key Laboratory of Catalysis and Materials Science, Hubei R&D Center of Hyperbranched Polymers Synthesis and Applications, South-Central Minzu University, Wuhan 430074, China. E-mail: daohong.zhang@scuec.edu.cn; ygdf@mail.scuec.edu.cn
bHubei Key Laboratory of Energy Storage and Power Battery, School of New Energy, Hubei University of Automotive Technology, Shiyan 442002, China. E-mail: zhoutong1018@huat.edu.cn
cGuangdong Provincial Laboratory of Chemistry and Fine Chemical Engineering Jieyang Center, Jieyang 515200, China
dKey Laboratory of Advanced Energy Materials Chemistry, Nankai University, Tianjin 300071, China
First published on 22nd May 2026
Abstract
Aqueous energy storage technologies hold great promise in the renewable energy field. Transition metal dichalcogenides (TMDs) have garnered significant attention as prospective cathode materials. However, their development has been hampered by sluggish ionic transport kinetics and structural instability during repeated ion insertion/extraction processes. Herein, a heterostructured Bi2Te3–SbPO4 composite anchored on nitrogen-doped carbon frameworks (Bi2Te3–SbPO4@NC) is designed to address these challenges. The formation of a built-in electric field at the heterojunction interface, in conjunction with the conductive NC framework, promotes uniform material distribution and effectively mitigates volume expansion during Zn2+ and NH4+ intercalation/de-intercalation. Density functional theory (DFT) calculations reveal that the Bi2Te3–SbPO4@NC heterointerface exhibits enhanced adsorption energies for both Zn2+ (−1.687 eV) and NH4+ (−2.296 eV) compared to pristine Bi2Te3. These synergistic effects collectively contribute to outstanding dual-ion storage capabilities, as evidenced by the high specific capacity of 295 mA h g−1 at 1 A g−1 for zinc-ion storage and 325 mA h g−1 at 0.5 A g−1 for ammonium-ion storage. Remarkably, the hybrid device achieves exceptional long-term cycling stability with 99.1% capacity retention after 9000 cycles for Zn2+ and 84.66% retention after 2000 cycles for NH4+. This work provides fundamental insights into heterointerface engineering for developing high-performance dual-ion battery systems.
Introduction
The depletion of traditional fossil fuels, climate warming, and environmental pollution have driven the development of renewable energy technologies. Aqueous energy batteries have emerged as an attractive candidate due to their eco-friendliness, intrinsic safety and ultrafast charge/discharge capability.1–7 Among various systems, dual-ion batteries represent a promising direction due to their ability to utilize multiple charge carriers (e.g., Zn2+ and NH4+). However, such systems face challenges including sluggish reaction kinetics and poor structural stability under the intercalation of different ionic species.Transition metal dichalcogenides (TMDs), such as bismuth telluride (Bi2Te3), have been widely investigated as cathode candidates for aqueous rechargeable batteries due to their layered structures and tunable electrochemical properties.8–11 While their widespread application is constrained by sluggish ion diffusion kinetics and structural instability during repeated charge–discharge cycles, primarily attributable to intrinsic lattice limitations and interfacial instability.12–15 Although heterojunction engineering has demonstrated potential in optimizing charge transfer dynamics,16,17 the rational design of heterointerfaces with balanced lattice compatibility and robust built-in electric fields remains a significant challenge, particularly in dual-ion storage systems involving diverse charge carriers.
Recent advances highlight the pivotal role of interface modulation in enhancing battery performance.18,19 Heterostructures with work function differences can create spontaneous interfacial electric fields, while lattice matching at phase boundaries alleviates mechanical strain during ion intercalation.20,21 Nevertheless, current strategies often neglect the synergistic requirements of dual-ion coordination chemistry and long-term structural durability. To address these limitations, we introduce a heterojunction engineering strategy via the integration of bismuth telluride (Bi2Te3) and antimony phosphate (SbPO4) anchored on nitrogen-doped carbon frameworks (Bi2Te3–SbPO4@NC). This architecture leverages the complementary attributes of the constituent phases. Bi2Te3 provides a stable conductive matrix, while SbPO4 serves as a structural stabilizer and interface modifier. Its lattice compatibility with Bi2Te3 not only creates a strong built-in electric field but also provides a coherent and durable mechanical support to buffer the volume change of Bi2Te3.22–24 The nitrogen-doped carbon substrate further enhances electron transport and mitigates volume fluctuations.25,26
Theoretical calculations and experimental assessments confirm that the designed heterointerface establishes an efficient charge redistribution network. The work function difference between Bi2Te3 and SbPO4 induces a strong built-in electric field, significantly lowering the energy barriers for Zn2+ and NH4+ migration. Simultaneously, their lattice coherence minimizes interfacial strain, preserving structural integrity during repetitive ion insertion/extraction. Moreover, the hybrid configuration demonstrates exceptional dual-ion storage performance, including high reversible capacities of 295 mA h g−1 at 1 A g−1 for Zn2+ and 325 mA h g−1 at 0.5 A g−1 for NH4+, along with exceptional long-term cycling stability (99.1% retention after 9000 cycles for Zn2+ and 84.66% after 2000 cycles for NH4+). Furthermore, a hybrid-ion battery assembled in a 15 M CH3COONH4 + 2 M ZnSO4 aqueous electrolyte maintains 78.27% capacity retention over 8000 cycles under a high current density of 5 A g−1. This work provides fundamental insights into heterointerface design for high-energy-density, long-lifespan dual-ion storage devices.
Results and discussion
The role of Bi2Te3–SbPO4 heterointerface in charge transfer and ion storage processes was studied by the first-principles calculations based on density functional theory (DFT). Fig. 1a–c present the optimized crystal structure models of Bi2Te3, SbPO4 and Bi2Te3–SbPO4, respectively. Compared with pure Bi2Te3 material, Bi2Te3–SbPO4 exhibits enhanced electrical conductivity, with the interface offering abundant active sites and efficient pathway for Zn2+ and NH4+ transport (Fig. 1d). The charge density difference analysis reveals a significant charge redistribution at the Bi2Te3–SbPO4 heterojunction interface, indicating pronounced electron transfer from SbPO4 to Bi2Te3 (Fig. 1e–h). Yellow and blue regions represent charge accumulation and depletion, respectively. Furthermore, the Bi2Te3–SbPO4 system demonstrates higher adsorption energies for Zn2+ (−1.687 eV) and NH4+ (−2.296 eV) compared to the pure Bi2Te3 model, suggesting that the heterointerface is more conducive to the storage of Zn2+ and NH4+.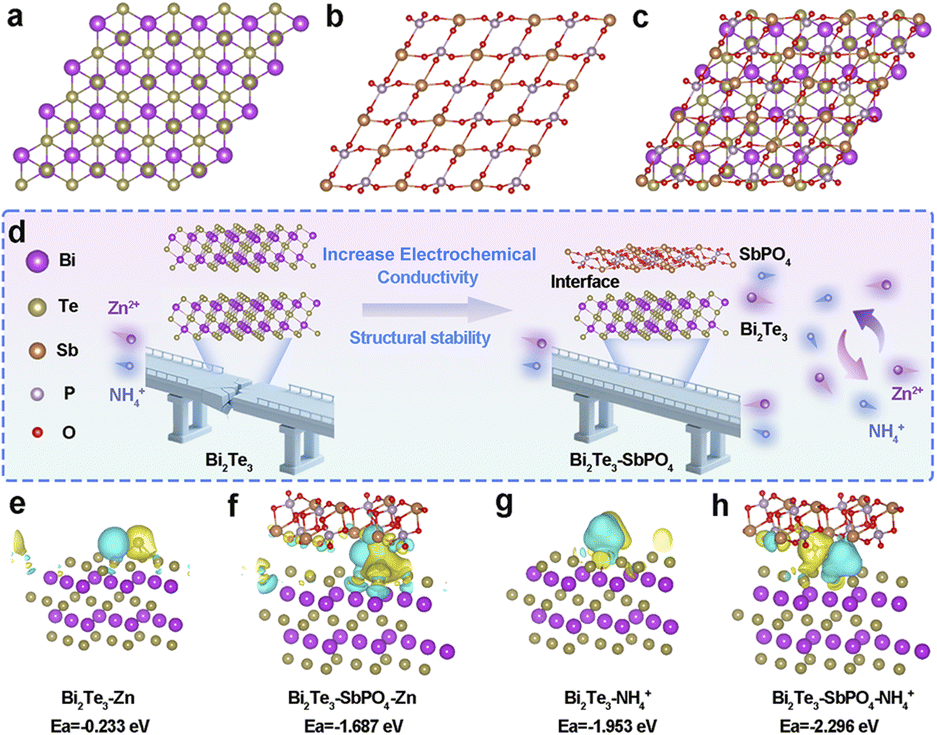 | ||
| Fig. 1 Theoretical simulation models of (a) Bi2Te3, (b) SbPO4 and (c) Bi2Te3–SbPO4. Schematic illustration of the optimization strategies of Bi2Te3–SbPO4 electrode and (d) the feasibility analyses of NH4+/Zn2+ co-storage chemistry in Bi2Te3–SbPO4. Zn2+ and NH4+ adsorption energy of the proposed configurations, yellow and blue regions represent the charge accumulation and the charge depletion (e–h). | ||
Fig. 2a shows the synthesis process of Bi2Te3–SbPO4@NC nanorods. Detailed synthesis information is provided in the Experimental Section. The phase composition and crystalline structure of the samples were characterized by X-ray diffraction (XRD). As shown in Fig. 2b and S1, the diffraction peaks of the composite match well with Bi2Te3 (PDF#15-0863) and SbPO4 (PDF#71-2275), confirming the successful construction of Bi2Te3–SbPO4@NC heterojunction. X-ray photoelectron spectroscopy (XPS) verified the presence of Bi, Te, Sb, P, O and C elements in the Bi2Te3–SbPO4@NC sample (Fig. S2). In the high-resolution Bi 4f spectrum (Fig. 2c), the peaks at 159.4 eV and164.7 eV are assigned to Bi 4f7/2 and Bi 4f5/2, respectively. In the Te 3d spectrum (Fig. 2d), two distinct peaks at 572.2 eV (Te 3d5/2) and 586.3 eV (Te 3d3/2) correspond to Te–Bi and Te–O bonding environments.27 Notably, in both the Bi 4f and Te 3d spectra, the binding energies exhibit a positive shift in the heterojunction material, which can be attributed to the difference in work function between Bi2Te3 and SbPO4 (Fig. S3). This drives electron transfer from Bi2Te3 to SbPO4, resulting in the formation of a built-in electric field. Additionally, the presence of Sb, P, O signals in the heterojunction (Fig. S4) further confirms the successful incorporation of SbPO4.28 Morphological and internal structure of the prepared materials were characterized by field emission scanning electron microscopy (SEM) and transmission electron microscopy (TEM). The Bi-BTC precursor exhibits uniform nanorod morphology (Fig. S5), and the resulting Bi2Te3@NC maintains the one-dimensional structure well after tellurization process (Figure S6-7). In contrast to pristine Bi2Te3@NC, the heterojunction material displays porous nanorods with larger particles encapsulated within a carbon shell (Fig. 2e and f). The porous architecture effectively mitigates mechanical fracture during ions insertion/(de)insertion processes, thereby facilitating ion transport (Fig. S8). The high-resolution TEM (HRTEM) images (Fig. 2g–k) reveal the interplanar spacings of 0.32 nm and 0.235 nm, corresponding to the (015) plane of Bi2Te3 and the (002) plane of SbPO4, respectively. The selected area electron diffraction (SAED) pattern in Fig. S9 confirms the polycrystalline nature of the material. Furthermore, the high-angle annular dark-field (HAADF) image and corresponding energy dispersive spectrometer (EDS) elemental mappings demonstrate the uniform distribution of Bi, Te, Sb, P and O elements (Fig. 2l–q). These results confirm the successful synthesis of Bi2Te3–SbPO4@NC heterojunction nanorods. Compared to Bi2Te3@NC, heterojunction materials enhances ion adsorption and effectively accommodates volume changes during repeated charge–discharge cycles. Since SbPO4 is electrochemically inert (Fig. S10), we only compare the electrochemical performance of Bi2Te3@NC and Bi2Te3–SbPO4@NC in the following.
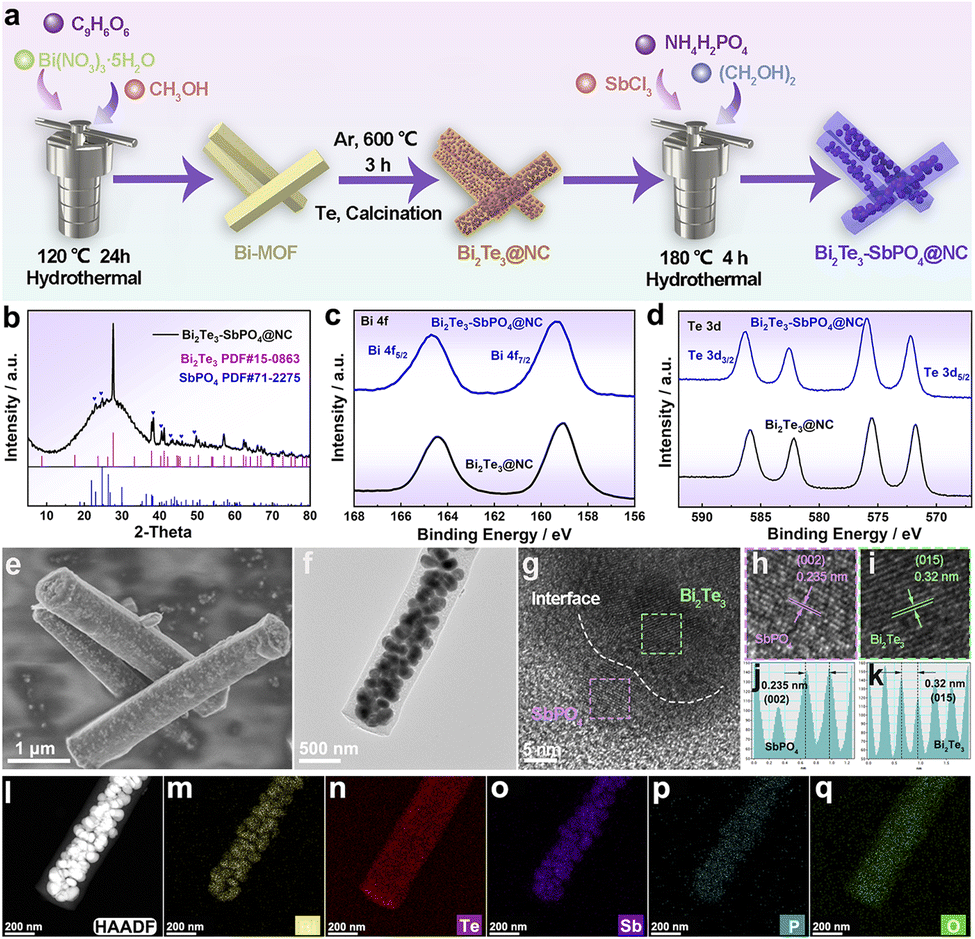 | ||
| Fig. 2 (a) Schematic of the Bi2Te3–SbPO4@NC heterojunction fabrication process. (b) XRD pattern. XPS spectra of (c) Bi 4f, (d) Te 3d. (e) SEM image of Bi2Te3–SbPO4@NC. (f) TEM image of Bi2Te3–SbPO4@NC. (g–i) HRTEM image. (j and k) Lattice distance. (l) The HAADF image of Bi2Te3–SbPO4@NC and (m–q) the corresponding element mappings. | ||
The electrochemical properties of Bi2Te3–SbPO4@NC and pristine Bi2Te3@NC electrode were systematically studied in 2 M ZnSO4 electrolyte. The cyclic voltammetry (CV) curve of the heterojunction exhibits minimal variation compared to those of Bi2Te3@NC (Fig. 3a and S11), retaining a pair of well-defined and reversible redox peaks, indicating similar redox behavior. Fig. 3b shows the galvanostatic charge–discharge (GCD) profiles of the heterojunction at various current densities. Reversible capacities of 330, 318, 295, 280, 259 mA h g−1 are achieved at current densities of 0.5, 0.8, 1, 3, 5 A g−1, respectively (Fig. 3c). When the current density is restored to 0.5 A g−1, the electrode recovers its capacity, exhibiting excellent rate capability (Fig. S12). To further evaluate and compare the electrochemical performance, key parameters including coulombic efficiency (CE), capacity retention, and specific capacity are plotted in a visual radar chart (Fig. 3d). The Bi2Te3–SbPO4@NC heterojunction outperforms previously reported transition metal compounds in zinc ion storage performance.29–34 The long-term electrochemical stability was assessed, the Bi2Te3–SbPO4@NC delivers an initial capacity of 300 mA h g−1 at 0.5 A g−1 and maintains the capacity over 100 cycles, exhibiting superior cycling stability relative to pristine Bi2Te3@NC (Fig. 3e). Even under high current densities of 5 A g−1 for 1100 cycles (Fig. 3f) and 10 A g−1 for 9000 cycles (Fig. 3g), the heterojunction demonstrates significantly enhanced capacity retention and exceptional cycle durability. These results underscore the outstanding rate capability and remarkable structural stability of the Bi2Te3–SbPO4@NC heterojunction. The outstanding electrochemical performance of Bi2Te3–SbPO4@NC heterojunction primarily is attributed to the strong built-in electric field at the interface of Bi2Te3 and SbPO4, which effectively promotes charge transfer and mitigates volume expansion during cycling. The reaction kinetics of the samples were investigated in Fig. S13. To fully elucidate the energy storage mechanism of Bi2Te3–SbPO4@NC, the phase evolution during the charge–discharge process was analyzed by ex-situ XRD, XPS and SEM. GCD test was conducted at 0.8 A g−1 (Fig. 3h). The XRD pattern in Fig. 3i clearly shows the characteristic peaks of Bi2Te3 (PDF#15-0863) and SbPO4 (PDF#71-2275), with no additional diffraction peaks observed throughout the cycling process. It is worth noting that at 2θ = 27.8°, the (015) crystal plane of Bi2Te3 gradually moves to a lower angle upon discharge to 0.1 V (Fig. 3j), and reverts to its original position when charge to 1.6 V, indicating the reversible Zn2+/H+ insertion/extraction. XPS analysis provides further insights into electrochemical behavior. The Bi 4f spectrum shows negligible changes in the Bi–O and Bi–Te bonding environments during the redox process (Fig. 3k). In the Te XPS profile, the Te–Te bond disappears during cycling, while the Te–O and Te–Bi peaks remain largely unchanged (Fig. 3l), further highlighting the structural stability of the heterojunction. In the Zn 2p XPS spectrum (Fig. 3m), two obvious Zn signals are detected at 1022.58 eV and 1045.6 eV in the fully discharged state, and these signals are weaker significantly in the fully charged state, confirming the reversible insertion/de-insertion of Zn2+. To verify the H+ intercalation, CV curves were compared in ZnSO4/DMSO and ZnSO4/H2O electrolytes, respectively (Fig. S14 and 15). The higher CV area observed in the ZnSO4/H2O electrolyte compared to that in ZnSO4/DMSO electrolyte suggests a synergistic Zn2+/H+ co-insertion mechanism.35,36 The morphological evolution of Bi2Te3–SbPO4@NC and Bi2Te3@NC was examined by SEM. In the discharge state, obvious Zn4SO4(OH)6·xH2O (ZSH) nanoflakes are observed on both electrodes (Fig. 3n and p). However, in the fully charged state, residual ZSH deposits persist on Bi2Te3@NC, whereas they are nearly absent on Bi2Te3–SbPO4@NC (Fig. 3o and q), indicating enhanced reaction reversibility in the heterojunction. Based on the above studies, a schematic illustration summarizing the structural evolution during electrochemical intercalation is proposed to depict the Zn2+/H+ storage behavior within the Bi2Te3–SbPO4@NC heterojunction host (Fig. 3r).37
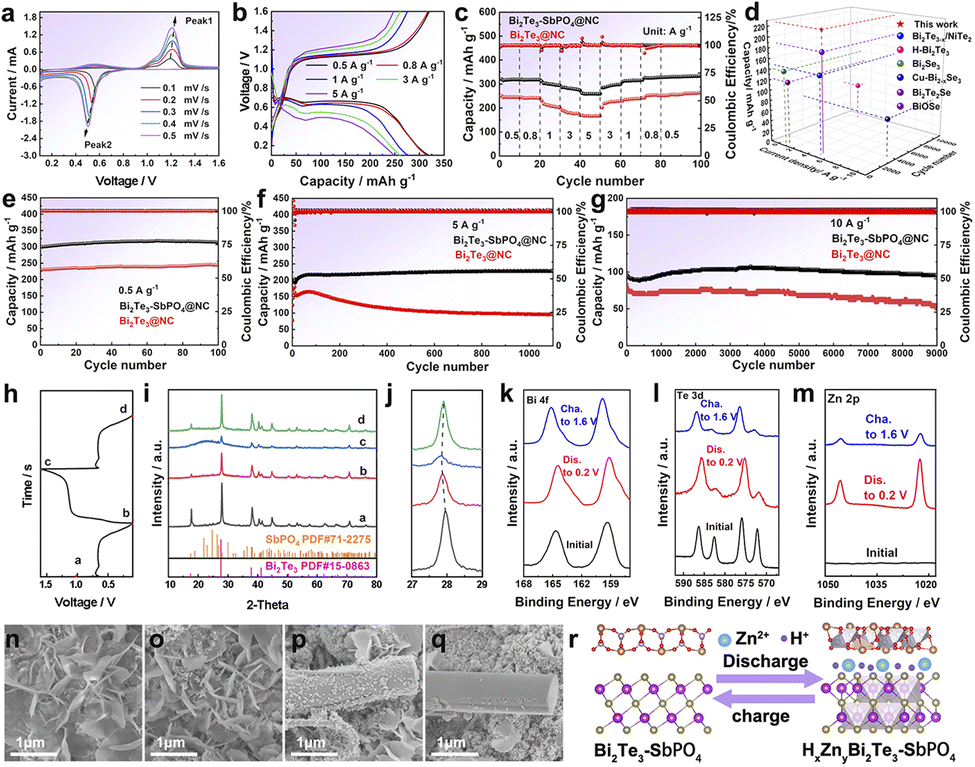 | ||
| Fig. 3 The electrochemical properties of Bi2Te3–SbPO4@NC: (a) CV curves from 0.1 to 1.6 V at 0.1–0.5 mV s−1. (b) The GCD curves under 0.5–5 A g−1. (c) Rate performance of Bi2Te3–SbPO4@NC and Bi2Te3@NC. (d) Capacity comparison of Bi2Te3–SbPO4@NC with state-of-the-art Zn2+ storage cathode. (e–g) Cycling performance of Bi2Te3@NC and Bi2Te3–SbPO4@NC. (h–j) Ex situ XRD result obtained from the heterojunction cathodes at different states. (k) Bi 4f, (l) Te 3d, (m) Zn 2p XPS obtained from the Bi2Te3–SbPO4@NC heterojunction cathodes. SEM images of (n-o) Bi2Te3@NC and (p and q) Bi2Te3–SbPO4@NC at the fully discharged-charged states, respectively. (r) Schematic diagram presents the charge storage mechanism in the heterojunction cathode. | ||
The Bi2Te3–SbPO4@NC material exhibits excellent electrochemical performance in ZIBs, prompting further investigation into its behavior in ammonium-ion batteries (AIBs). We tested the electrochemical performance of Bi2Te3–SbPO4@NC heterojunction materials in 1 M and 5 M CH3COONH4 as electrolytes (Fig. S16). It can be found that the electrochemical performance increases with the increase of concentration. In the 15 M CH3COONH4 as electrolyte, it shows better electrochemical performance. This is due to the fact that acetate ions are involved in the formation of solvated NH4+ in high concentration electrolyte, which promotes the adsorption of solvated NH4+ clusters on the electrode surface.38 The electrochemical properties of Bi2Te3 and Bi2Te3–SbPO4@NC were studied by using 15 M CH3COONH4 as electrolyte. From Fig. 4a–b, the heterojunction electrode displays stronger CV redox peak and more obvious charge–discharge platform, indicating enhanced NH4+ storage capability. The CV and GCD curves of Bi2Te3–SbPO4@NC and Bi2Te3@NC are shown in Fig. 4c, d and S17, respectively. In impedance test (Fig. 4e), the Bi2Te3–SbPO4@NC delivers a lower charge transfer resistance, delivering the enhanced charge transport efficiency. Rate capability of the samples was also evaluated (Fig. 4f). The Bi2Te3–SbPO4@NC electrode exhibits superior rate performance and reversibility with a recoverable capacity of 350 mA h g−1. The Bi2Te3–SbPO4@NC can maintain a high capacity retention of 84.66% when cycled for 2000 times at 3 A g−1 (Fig. 4g). The energy storage kinetics of the sample in 15 M CH3COONH4 is evaluated (Fig. S18 and 19).
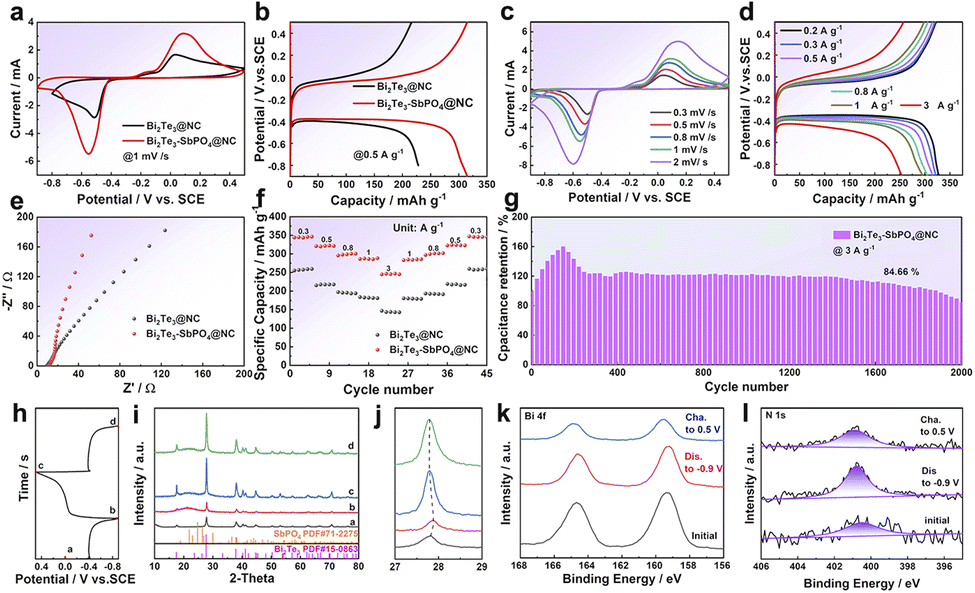 | ||
| Fig. 4 Comparison of the electrochemical performance of Bi2Te3 and Bi2Te3–SbPO4@NC electrodes in 15 M CH3COONH4: (a) CV curve at 1 mV s−1; (b) GCD curve at 0.5 A g−1; The electrochemical properties of Bi2Te3–SbPO4@NC: (c) CV curves, (d) GCD curve. (e) EIS spectra. (f) The rate performance of Bi2Te3 and Bi2Te3–SbPO4@NC electrodes. (g) The cycling performance of Bi2Te3–SbPO4@NC electrode at 3 A g−1. (h) GCD curve. (i and j) Ex-situ XRD patterns, (k) Bi 4f XPS spectra, and (l) N 1s XPS spectra of Bi2Te3–SbPO4@NC. | ||
To explore the NH4+ storage mechanism in Bi2Te3–SbPO4@NC, ex-situ XRD and XPS tests were carried out. The charge–discharge test was conducted at 0.5 A g−1 (Fig. 4h). XRD test revealed no formation of new phase after fully charge–discharge process (Fig. 4i). By enlarging the XRD spectrum near 27.8°, it can be observed that the peak moves to a low angle during the discharge process, which is attributed to the insertion process of NH4+. During the subsequent charge process, the peak returns to a high angle, corresponding to the extraction of NH4+ (Fig. 4j). The chemical states of Bi, Te, N, and O in charge–discharge state were studied by ex-situ XPS spectroscopy (Fig. 4k and S20). In the three-dimensional XPS spectra of Bi 4f and Te 3d, it can be found that the Bi–Te peak moves to the direction of high binding energy during charging, and then does not return to the initial state during discharging, indicating that there is a certain degree of irreversible reaction. In the N 1s spectrum (Fig. 4l), a strong –N–H– peak appears in the fully discharged state, which proves the insertion of NH4+. However, upon fitting and integration of the N 1s spectra, the charged state shows no significant extra area compared to the pristine material, indicating that the N signal mainly comes from the NC framework itself rather than from residual NH4+. Therefore, the insertion and extraction of NH4+ are essentially reversible. In the O 1s signal (Fig. S21), the enhancement of the O–H bond in the fully discharged state illustrated that the coinsertion of H+ was accompanied by the embedding of NH4+. The above results prove the reaction mechanism of NH4+ insertion/extraction in Bi2Te3–SbPO4@NC.
Fig. 5a shows the electron flow mechanism at the heterogeneous interface within the Bi2Te3–SbPO4@NC heterojunction during the charge and discharge process. During charging, electrons transfer from Bi2Te3 to SbPO4, while during discharging, the direction reverses with electrons moving from SbPO4 back to Bi2Te3. The electronic properties of both Bi2Te3 and Bi2Te3–SbPO4 were investigated using density functional theory (DFT) calculations. The computed band structures and partial density of states (PDOS) are presented in Fig. 5b and c. Obviously, the pristine Bi2Te3 exhibits typical semiconductor behavior with a band gap of 0.95 eV. In contrast, the Bi2Te3–SbPO4 heterostructure displays additional occupied states near the Fermi level, indicating that interface coupling enhances the local electron concentration and improves electronic conductivity. Subsequently, the electrochemistry performance of Bi2Te3–SbPO4@NC was evaluated in a 15 M CH3COONH4 +2 M ZnSO4 aqueous electrolyte by assembling coin cells. As shown in the CV curves (Fig. 5d), the heterojunction electrode exhibits well-defined redox peak. Even at a high current density of 5 A g−1, Bi2Te3–SbPO4@NC achieves a capacity retention of 78.27% after 8000 cycles (Fig. 5e), with negligible changes in the GCD profiles (Fig. S22), showing exceptional cycling stability. This work demonstrates the feasibility of designing a bi-functional electrode for Zn2+ and NH4+ storage.
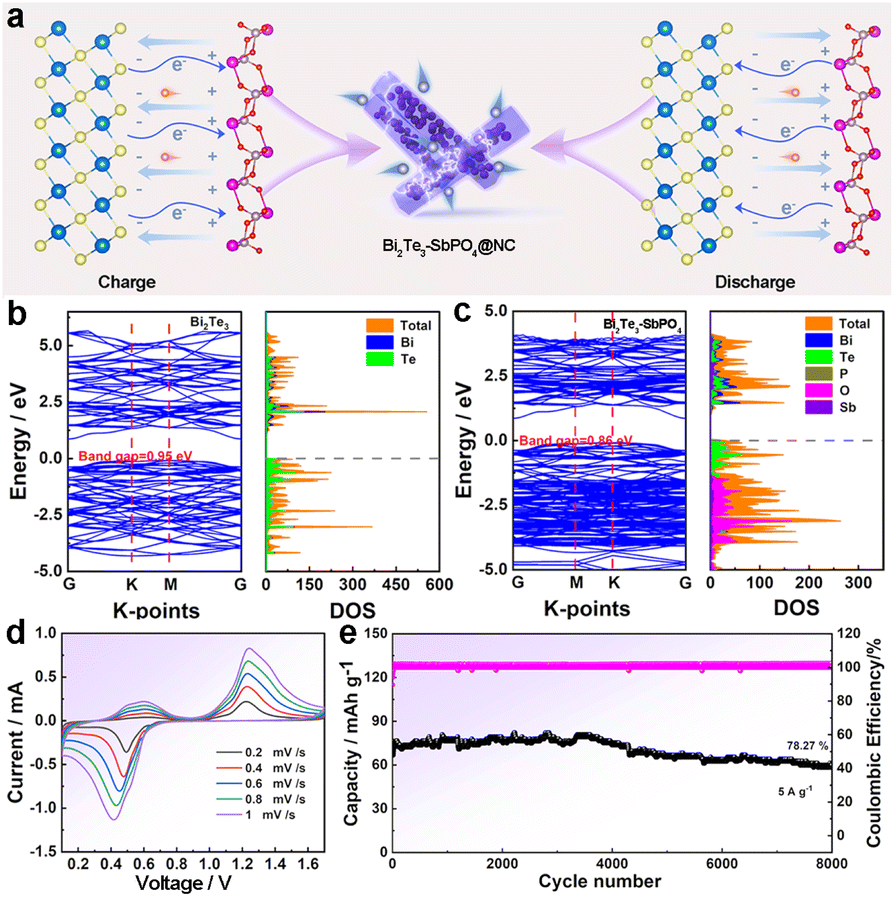 | ||
| Fig. 5 (a) During the charge and discharge process, the electrons flow at the heterogeneous interface. (b and c) Energy band structure and density of state (DOS) of Bi2Te3 and Bi2Te3–SbPO4. (d) CV curves of Bi2Te3–SbPO4@NC at 15 M CH3COONH4 +2 M ZnSO4 hybrid-ion electrolyte. (e) The cycling performance of Bi2Te3–SbPO4@NC electrode at 5 A g−1. | ||
Conclusions
In summary, theoretical calculation and experimental validation demonstrate that the designed heterogeneous interface establishes an efficient charge redistribution network. The work function difference between Bi2Te3 and SbPO4 induces a strong built-in electric field, which significantly lowers the energy barrier of Zn2+ and NH4+ insertion. Concurrently, their lattice coherence minimizes the interfacial strain, thereby preserving structural integrity during repeated ion insertion/extraction processes. As a result, the electrode delivers a high specific capacity of 295 mA h g−1 (1 A g−1) in Zn2+ storage and 325 mA h g−1 (0.5 A g−1) in NH4+ storage. Moreover, the hybrid structure maintains 99.1% of its capacity after 9000 cycles for Zn2+ storage and 84.66% capacity after 2000 cycles for NH4+ storage. Furthermore, a hybrid-ion battery was assembled in a 15 M CH3COONH4 + 2 M ZnSO4 aqueous electrolyte. Under a high current density of 5 A g−1, the material maintains 78.27% capacity retention over 8000 cycles. This work demonstrates the feasibility of designing a bi-functional electrode for Zn2+ and NH4+ storage.Author contributions
Zhiyuan Zha: data curation, investigation, methodology, writing – original draft. Ruinan Chen: data curation, investigation. Tong Zhou: software. Daohong Zhang: supervision, funding. Qiufan Wang: writing – review & editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available on request from the corresponding author, upon reasonable request.Supplementary information: experimental details, XRD, SEM and TEM images, XPS, and electrochemical performance. See DOI: https://doi.org/10.1039/d6sc02844f.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (51702369, 22509055), Hubei Provincial Young Scientific and Technological Talent Cultivation Program (2025DJA042), and Fundamental Research Funds for the Central Universities of South-Central Minzu University (CZD24001) (https://www.sciencedirect.com/science/article/pii/S2405829724004987?via=ihub)).Notes and references
- M. C. Tang, Q. Liu, X. H. Zou, B. Zhang and L. An, Adv. Mater., 2025, 37, 2501361 CrossRef CAS PubMed.
- C.-C. Kao, C. Ye, J. Hao, Y. Chen, S.-J. Zhang and S.-Z. Qiao, Adv. Energy Mater., 2025, 15, 2501201 CrossRef CAS.
- D. D. Ling, G. F. Tian, D. H. Zhang and Q. F. Wang, Nano Lett., 2025, 25, 2684 CrossRef CAS PubMed.
- M. Kim, J. Lee and J. W. Choi, Adv. Energy Mater., 2025, 15, e04692 CrossRef CAS.
- M. M. Shi and X. Y. Zhang, Adv. Mater., 2025, 37, 2415676 CrossRef CAS PubMed.
- Y. J. Zhao, Y. Y. Wang, J. Z. Li, J. W. Xiong, Q. Li, K. Abdalla, Y. Zhao, Z. Cai and X. M. Sun, eScience, 2025, 5, 100331 CrossRef.
- L. Zhang, J. Xiao, X. L. Xiao, W. L. Xin, Y. H. Geng, Z. C. Yan and Z. Q. Zhu, eScience, 2024, 4, 100205 CrossRef.
- Z. Y. Zha, R. N. Chen, D. H. Zhang and Q. F. Wang, Nano Lett., 2026, 26, 260–268, DOI:10.1021/acs.nanolett.5c05017.
- Q. F. Wang, Z. H. Zhao, S. F. Chen, T. Zhou and D. H. Zhang, Adv. Funct. Mater., 2025, e12019 Search PubMed.
- P. De, P. Lazar, M. Otyepka and M. Pumera, Adv. Sci., 2025, 12, e07255 CrossRef CAS PubMed.
- S. D. Liu, H. L. Zhang, X. Peng, J. M. Chen, L. Kang, X. Yin, Y. Yusuke and B. Ding, ACS Nano, 2025, 19, 13591–13636 CrossRef CAS PubMed.
- L. Wen, Q. X. Zhang, J. J. Shi, F. Wang, S. L. Wang, Z. W. Chen, Y. Yue and Y. H. Gao, ACS Nano, 2024, 18, 5981–5990 CAS.
- O. C. Kenneth and N. J. Megan, J. Am. Chem. Soc., 2025, 147, 4643–4653 CrossRef PubMed.
- S. J. Zhang, M. Fang, F. Wang, L. Wen, Q. Wang, J. A. Dai, P. B. Gui, X. G. Ren, Z. L. Chen, W. Zeng, Z. X. Huang, Y. Yue and S. L. Wang, Chem. Eng. J., 2024, 496, 153980 CrossRef CAS.
- T. R. Gong, L. Gao, L. F. Kang, M. L. Shi, G. Hou, S. H. Zhang, D. H. Meng, J. T. Li and W. Su, Adv. Sci., 2024, 11, 2403845 CrossRef CAS PubMed.
- X. J. Wang, J. Q. Qiao, L. W. Guo, M. Feng, P. Wang and Z. M. Liu, Adv. Funct. Mater., 2025, 35, 2422017 CrossRef CAS.
- S. C. Liu, Q. Zhou, D. Wen, C. Y. Wu, Y. Q. Pan, X. Liu, Z. Huang and N. Li, ACS Catal., 2024, 14, 8105–8115 CrossRef CAS.
- B. T. Zhang, Y. X. Huang, S. Y. Gao, N. Zhang, Y. Mei, Y. T. Huang, T. F. Ding, X. Hu, L. Li, F. Wu and R. J. Chen, J. Energy Chem., 2025, 107, 908–918 CrossRef CAS.
- J. J. Feng, Z. H. Li, L. W. Zhao, Y. J. Zong, H. Sun, B. Liu, L. L. Li, C. L. Wang and H. C. Wang, Adv. Funct. Mater., 2025, 35, 2504803 CrossRef CAS.
- W. T. Qu, Y. Cai, B. H. Chen and M. Zhang, Energy Environ. Mater., 2024, 7, e12645 CrossRef CAS.
- M. L. Chen, M. Zhou, Q. Y. Wang, C. Xu, S. Wang, J. Ning, T. Q. Wang, K. L. Wang and K. Jiang, Adv. Funct. Mater., 2025, 35, 2414032 CrossRef CAS.
- X. Q. Song, D. Tian, Y. Qiu, X. Sun, B. Jiang, C. H. Zhao, Y. Zhang, L. S. Fan and N. Q. Zhang, Adv. Funct. Mater., 2022, 32, 2109413 CrossRef CAS.
- H. Q. Wang, L. Gou, W. F. Jing, D. An, Y. Li, M. Wang, N. Li, S. L. Hu and Y.-B. He, Mater. Adv., 2020, 1, 206–214 RSC.
- J. Zhang, J. Y. Yu, Q. Lang, Y. X. Yang, J. L. Che, F. X. Wang, L. Q. Luo, J. H. Ye, Z. Y. Liu, L. Chen, G. Wang and Y. P. Wu, ACS Energy Lett., 2025, 10, 1013–1021 CrossRef CAS.
- L. L. Zhao, J. Y. Wang, Y. M. Wu, P. F. Wang, Z. L. Liu, J. Shu, T. F. Yi and Q. B. Zhang, J. Energy Chem., 2025, 111, 249–273 CrossRef CAS.
- Y. M. Qin, S. H. Zhao, H. D. Wu, C. X. Sun, J. Xu, M. Yang, R. D. Shi and H. F. Bao, J. Energy Storage, 2025, 134, 118300 CrossRef CAS.
- Y. X. Hou, P. F. Ma, F. Long, M. Y. Liu, Y. F. Zheng, L. Sun, J. J. Shi, K. Niu, J. Su, Y. N. Ma and Y. H. Gao, ACS Nano, 2024, 18, 27358–27371 CrossRef CAS PubMed.
- L. J. Li, F. Li and T. H. Li, Inorg. Chem. Commun., 2023, 156, 1111128 Search PubMed.
- W. H. Zhang, S. S. Zhang, F. F. Chen, R. Ma, L. L. Ai, M. J. Xu, D. Z. Jia, N. N. Guo and L. X. Wang, ACS Appl. Energy Mater., 2025, 8, 7530–7537 CrossRef CAS.
- Z. J. Tang, W. S. Chen, Z. L. Deng, Z. Y. Zhu, H. Y. Meng, N. Ju, F. Ye, Y. P. Du, Y. P. Wu and L. F. Hu, Sci. China Mater., 2026, 69, 1969–1977, DOI:10.1007/s40843-025-3599-5.
- Y. Y. Chen, S. T. Wang, Y. Y. Wang, Z. M. Fang, B. B. Liu, Y. Mi, J. T. Bai, B. B. Wang and G. Wang, J. Energy Chem., 2025, 105, 130–141 CrossRef CAS.
- Y. Zong, H. C. Chen, J. S. Wang, M. H. Wu, Y. Chen, L. Y. Wang, X. L. Huang, H. W. He, X. Ning, Z. C. Bai, W. Wen, D. M. Zhu, X. C. Ren, N. N. Wang and S. X. Dou, Adv. Mater., 2023, 35, 2306269 CrossRef CAS PubMed.
- H. N. Yue, M. W. Han, X. N. Li, T. Song, Y. Pei, X. Y. Wang, X. W. Wu, T. F. Duan and B. Long, J. Colloid Interface Sci., 2023, 651, 558–566 CrossRef CAS PubMed.
- B. B. Liu, S. T. Wang, K. M. Yan, Y. Y. Chen, Y. H. Ren, H. H. Xia, R. Y. Chen, J. F. Yan and G. Wang, Chem. Eng. J., 2025, 506, 160246 CrossRef CAS.
- Q. H. Zhao, A. Song, W. G. Zhao, R. Z. Qin, S. X. Ding, X. Chen, Y. L. Song, L. Y. Yang, H. Lin, S. N. Li and F. Pan, Angew. Chem., Int. Ed., 2021, 133, 4215 CrossRef.
- D. D. Ling, Q. Wang, G. F. Tian, H. Yu, D. H. Zhang and Q. F. Wang, ACS Nano, 2023, 17, 25222–25233 CrossRef CAS PubMed.
- G. F. Zeng, Q. Sun, S. Horta, S. Wang, X. Lu, C. Y. Zhang, J. Li, J. S. Li, L. J. Ci, Y. H. Tian, M. Ibáñez and A. Cabot, Adv. Mater., 2024, 36, 2305128 CrossRef CAS.
- J. M. Meng, Y. Song, J. Wang, P. Hei, C. Liu, M. X. Li, Y. L. Lina and X. X. Liu, Chem. Sci., 2024, 15, 220–229 RSC.
| This journal is © The Royal Society of Chemistry 2026 |