DOI:
10.1039/D6SC02823C
(Review Article)
Chem. Sci., 2026, Advance Article
The electron buffer effect for advanced electrocatalysis
Received
6th April 2026
, Accepted 11th June 2026
First published on 15th June 2026
Abstract
The electronic structure of catalysts often undergoes irreversible transformation during electrocatalysis at applied potential, significantly impacting their activity and stability. Recent studies have revealed the critical role of the electron buffer effect in enhancing electrocatalytic performance. Conceptually analogous to a pH buffer, this effect involves the dynamic regulation of electron density at active sites through reversible electron transfer with a functional support. It helps stabilize the optimal valence states of active sites, mitigates over-oxidation or over-reduction, and optimizes the adsorption/desorption behavior of key reaction intermediates. These insights underscore the need for a deeper atomic-level understanding of dynamic electronic structure design. This review systematically elaborates the fundamental mechanisms and distinctive features of the electron buffer effect, categorizes the relevant buffer materials into metal-based and nonmetal-based systems, and highlights their crucial roles in key electrocatalytic reactions. Finally, current challenges and future prospects concerning the precise manipulation and characterization of electron buffer effects are discussed, providing guidance for the rational design of advanced materials in highly efficient electrocatalysis.
1. Introduction
The global energy landscape remains heavily dependent on traditional fossil fuels, which drive the socioeconomic development.1,2 However, this persistent reliance intensifies the dual crisis of energy shortage and environmental pollution, underscoring the urgent need to accelerate the global energy transition and achieve carbon neutrality.3–5 In this context, the utilization of renewable electricity to drive chemical transformation reactions has emerged as a cutting-edge and pivotal research direction in chemistry and materials science.6 Electrocatalysis holds immense promise for storing intermittent renewable energy (e.g., solar and wind) in the form of chemical bonds and converting it into value-added fuels and chemicals.7,8 Currently, key techniques including water splitting for hydrogen production, fuel cells, CO2 conversion, and ammonia production are garnering widespread interest and involve critical electrocatalytic reactions such as the hydrogen/oxygen evolution reaction (HER/OER), oxygen reduction reaction (ORR), CO2 reduction reaction (CO2RR), and nitrogen/nitrate reduction reaction (NRR/NO3RR).9–13 Nevertheless, the widespread implementation of these electrocatalytic processes is still hindered by inherent challenges, including sluggish reaction kinetics, high overpotential, and limited energy conversion efficiency.14,15 Consequently, the fundamental understanding of catalytic mechanisms, coupled with the rational design of highly efficient electrocatalysts that exhibit superior activity and stability, is essential to propel these electrochemical reactions towards large-scale practical application.
Early research in electrocatalysis predominantly focused on modulating the geometric structure of catalysts, such as particle size, morphology, and exposed specific surface area.16,17 However, these strategies faced inherent limitations, as the intrinsic activity of an electrocatalyst is governed not merely by the number of active sites, but more critically by its electronic structure. Specifically, the electronic density of states near the Fermi level, particularly the d-band center in transition metals, determines the degree of orbital overlap between reactant molecules and the catalyst, thereby precisely regulating the adsorption strength of reaction intermediates. In 1978, Tauster et al. reported a pronounced suppression of H2 and CO chemisorption on Pt/TiO2, following high-temperature reduction (HTR), leading to the pioneering concept of the strong metal–support interaction (SMSI) mechanism.18 The efficient electron transfer from the support to the metal was found to fundamentally alter the electronic density of the metal surface. Since then, the SMSI effect has been observed across various supported catalysts induced by diverse methods (e.g., reduction, oxidation, and photo-treatment).19 This discovery provided profound insight into how an “inert” support governs the electronic structure and adsorption behavior of a metal catalyst through the electronic interaction effect. Nonetheless, according to the Sabatier principle, an ideal catalyst should exhibit moderate adsorption strength toward reaction intermediates to facilitate their rapid generation and facile desorption. Furthermore, under harsh electrocatalytic conditions, active sites are susceptible to irreversible oxidation or reduction, leading to rapid catalyst deactivation. Hence, developing effective strategies to precisely regulate and maintain the optimal electronic structure of active sites has become a paramount objective in catalyst design.
With the development of nanotechnology and carbon-based materials, the SMSI-like effect has been observed in some novel carbon structures, such as sulfur-doped carbon and graphene.20,21 Inspired by the concept of a buffer solution resisting pH changes, the more advanced electron buffer effect was proposed to describe the reversible electron exchange between metal nanoparticles and highly conductive carbon supports. In this mechanism, the conductive carbon support serves as an “electron reservoir”, dynamically donating or accepting electrons in response to variations in external potential or reaction conditions, which has been recently validated in the thermocatalytic processes for ethylene glycol and ammonia synthesis.22,23 Owing to the delocalized π electron system, carbon supports can rapidly respond to environmental changes and promptly modulate the electronic structure of metal centers.24 By dynamically mitigating the over-oxidation or over-reduction of the metal sites, the induced electron buffer effect effectively stabilizes the optimal valence state of active sites, leading to favorable intermediate adsorption and enhanced catalytic performance.25 Currently, this concept has expanded beyond carbon materials to other functional compounds (e.g., CeO2)26 and from heterostructures to atomic-level doped architectures.27,28 In view of these superior features, inducing the electron buffer effect to precisely regulate the electronic structure of the active sites has emerged as the key strategy for improving the electrocatalytic performance. However, the efficacy of this effect is highly dependent on the integrity of interfaces between components, which may become unstable in severe electrochemical environments due to elemental dissolution, phase transformation, and undesirable surface reconstruction. Therefore, a timely review that systematically outlines the mechanisms, applications and challenges of the electron buffer effect in electrocatalysis is urgently needed.
In this review, we highlight the fundamental mechanisms and origins of the electron buffer effect and establish its integral connection to electrocatalytic processes (Fig. 1). We systematically classify the materials capable of inducing the electron buffer effect into two groups, including metal-based components and nonmetal-based components. More importantly, this review categorizes and elaborates on the pivotal role of the electron buffer effect across major important electrocatalytic reactions (e.g., the OER, HER, ORR, CO2RR, NRR, and NO3RR), highlighting its contribution to boosting catalytic activity and stability by optimizing the adsorption/desorption behavior of key intermediates and stabilizing the valence states of active sites. Finally, we discuss the prevailing challenges and future prospects in the precise manipulation and real-time characterization of electron buffer effects, aiming to inspire the rational design of high-performance electrocatalysts.
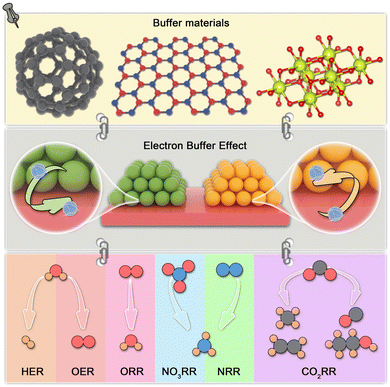 |
| | Fig. 1 Schematic illustration of the electron buffer effect for electrocatalysis. | |
2. Fundamentals of the electron buffer effect in electrocatalysis
The rational design of advanced electrocatalysts necessitates a profound understanding of interfacial electronic effects, among which the electron buffer effect has emerged as a pivotal strategy for enhancing catalytic performance. Distinct from conventional strategies that focus solely on the intrinsic properties of active sites, this effect enables dynamic regulation of the electronic structure, allowing real-time adaptation to the demanding electrochemical environment. Hence, a concise analysis of the mechanism and unique features of the electron buffer effect serves as a starting point for this review (Fig. 2).
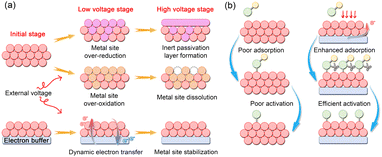 |
| | Fig. 2 Schematic illustration of the electron buffer effect toward (a) modulation of the valence state and (b) intermediate adsorption behaviors. | |
2.1. Mechanism
The core mechanism of the electron buffer effect lies in the ability of a material with a specific electron structure to function as a dynamic “electron reservoir”, reversibly injecting or extracting electrons into adjacent active sites in response to fluctuations in external potential or the reaction microenvironment. Physically, this is not a static electron distribution. This behavior originates from the electronic interactions in a built-in electron channel at the interface between active sites and the buffer component. A promising buffer component can undergo repeated redox cycles (filling/emptying of its frontier orbitals or bands) with minimal structural degradation or significant shift in its intrinsic buffering potential, thereby sustaining its function. For instance, in highly oxidizing environments, the active sites are prone to over-oxidation, forming high-valence soluble species, such as RuO2 in the acidic OER process.29 In such cases, the buffer component functions as the electron donor, rapidly supplying electrons to stabilize the oxidation states of metal sites and thereby preserving stability of the catalyst. Conversely, in many cathodic reactions, the active sites may become over-reduced, which diminishes their capacity for reactant adsorption and activation. Here, the electron buffer effect can promptly snatch excess electrons from the active sites, maintaining a moderately electron-deficient state that favors intermediate adsorption.30 Therefore, the electron buffer effect holds great promise for advancing electrocatalytic systems.
2.2. Multifunctional roles in catalysis
Compared to traditional single-component catalysts or the synergistic effect, the electron buffer effect is not a static electronic modification, but a dynamic and continuous equilibrium process capable of real-time adaptation to the reaction environment. Moreover, this effect operates spontaneously during the electrocatalytic process, preventing the active sites from deviating from their optimal electronic state, rather than repairing them after deactivation, thereby fundamentally enhancing catalytic stability. Beyond regulating the valence state of active sites for stability optimization, the electron buffer effect also exhibits several other distinctive capabilities. First, by finely tuning the electron density of active sites, it induces shifts in the d-band center position, which effectively modulates the adsorption energy of key intermediates and significantly reduces the overpotential.31 Second, the built-in electron channels established by this effect facilitate rapid electron shuttling across the surface and interface of the catalyst, lowering the charge transfer resistance and accelerating reaction kinetics.26 Third, in multiple-step reactions involving multiple intermediates, the electron buffer effect can disrupt linear scaling relationships among the key intermediates by altering the adsorption behavior of the key intermediates.32 Therefore, the deliberate incorporation of the electron buffer effect opens a novel avenue for designing catalytic materials that surpass the performance limitations of conventional catalysts.
3. In situ characterization techniques
In situ characterization techniques are indispensable for unravelling the dynamic evolution of catalysts under operating conditions, which allows real-time monitoring of changes in the valence state, local coordination environment, and reaction intermediates, providing direct evidence for the electron buffer effect. In this section, we summarize several in situ characteristic techniques, including in situ X-ray absorption near-edge spectroscopy/extended X-ray adsorption fine structure (XANES/EXAFS), in situ X-ray photoelectron spectroscopy (XPS), in situ Fourier-transform infrared spectroscopy (FTIR), and in situ Raman spectroscopy.
3.1. In situ XANES and EXAFS
In situ XANES and EXAFS can monitor the dynamic valence state and coordination structure changes of both active sites and buffer components.28,32,33 Peng et al. developed an inter-doped tungsten–ruthenium oxide [(Ru–W)Ox] catalyst and used in situ XANES and EXAFS techniques to monitor the dynamic changes in Ru oxidation states under operating conditions.33 For (Ru–W)Ox, starting from the open-circuit voltage (OCV) to 1.4 V vs. RHE, the average Ru valence state slightly decreased from +3.7 to +3.5, which corresponds to a pre-catalytic redox process (Fig. 3a). As the potential rose from 1.4 V to 1.6 V vs. RHE, the Ru valence increased from +3.5 to +4.1, indicating the formation of higher-valent Ru sites that are active for the OER. Remarkably, when the potential exceeded 1.8 V vs. RHE, the Ru oxidation state decreased again from +4.1 back to approximately +3.7. This reversible decrease demonstrated that the high-valent W species donate electrons back to the Ru sites and prevent their over-oxidation to soluble RuO42− species. In contrast, the RuOx catalyst showed a continuous and irreversible increase in the Ru valence state with increasing potential, leading to structural degradation and Ru dissolution (Fig. 3a). Besides, in situ EXAFS analysis indicated that (Ru–W)Ox possessed a minor increase in the Ru–O coordination number (CN) while RuOx exhibits a significant decrease in the CN, which is attributed to the structural damage (Fig. 3b). Hence, (Ru–W)Ox enables a reversible valence oscillation of Ru sites and stabilizes the Ru–O coordination structure, thereby enhancing its stability.
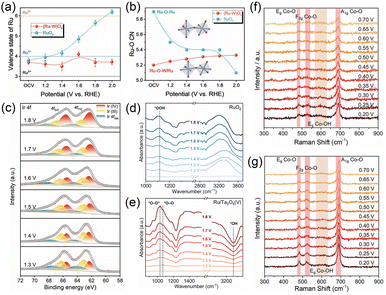 |
| | Fig. 3 (a) The valence states and (b) the Ru–O coordination numbers of RuOx and (Ru–W)Ox at different potentials. Reproduced with permission.33 Copyright 2023, Wiley-VCH. (c) XPS spectra of Ir–Ce SSO after operating at different potentials. Reproduced with permission.35 Copyright 2024, Wiley-VCH. In situ FTIR spectra of (d) RuO2 and (e) Ru/Ta2O5. Reproduced with permission.36 Copyright 2025, Royal Society of Chemistry. In situ Raman spectra of (f) CoO and (g) P–Ce SAs@CoO. Reproduced with permission.38 Copyright 2023, Wiley-VCH. | |
3.2. In situ XPS
In situ XPS can also quantitatively track the valence state of active sites under operating conditions.26,34,35 For example, Guo et al. fabricated an iridium–cerium substitution solid solution oxide (Ir–Ce SSO) for efficient OER and used XPS to investigate the valence state change of Ir species in Ir–Ce SSO.35 After operating at different potentials, the XPS spectra revealed that the amount of Ir3+ species in Ir–Ce SSO decreases slowly, just from 51.8% to 35.5% (Fig. 3c). This result suggested that CeO2 with the electron buffer effect effectively inhibits the over-oxidation and dissolution of Ir in Ir–Ce SSO. Hence, the Ir–Ce SSO showed great stability in a PEMWE device for 100 h at 500 mA cm−2. Besides, the binding energy shifts of active sites and buffer components can also be observed with the change of applied potential, which can directly reflect the electron buffer effect during the electrocatalytic process. Therefore, in situ XPS can provide intuitive evidence for dynamic changes in the charge state through the changes of binding energies and peak area.
3.3. In situ FTIR
By probing the vibrational adsorption modes of chemical bonds on the catalyst surface, in situ FTIR allows real-time determination of reaction intermediates and their adsorption configurations, which can be used to analyze the impact of the electron buffer effect on the catalytic process and mechanism.36,37 Liu et al. synthesized a Ru/Ta2O5 catalyst with an abundant 5d electron buffering interface.36 The anchoring of Ru on the surface of Ta2O5 modulated the electron cloud density around Ru sites via Ta 5d electrons, optimizing the reaction pathway from the adsorbate evolution mechanism (AEM) to the oxide path mechanism (OPM), which was confirmed by the in situ FTIR technique. RuO2 exhibited a peak near 1120 cm−1 at high potentials, attributed to *OOH species, suggesting that it proceeds via the AEM pathway at high potentials (Fig. 3d). In contrast, for Ru/Ta2O5, distinct absorption peaks at 1033 cm−1 and 1067 cm−1 appeared at potentials above 1.3 V vs. RHE, which are assigned to adsorbed *O–O* and *O–O intermediates, indicating that the catalyst follows the OPM pathway where direct O–O coupling releases O2 without forming *OOH intermediates (Fig. 3e). Hence, in situ FTIR can effectively reflect the influence of the electron buffer effect on regulating the reaction intermediates.
3.4. In situ Raman spectroscopy
In situ Raman spectroscopy can not only detect the reaction intermediates but also reflect the structural transformation during electrocatalytic processes.30,38,39 Fu et al. prepared an atomically dispersed Ce on CoO (P–Ce SAs@CoO) catalyst for the OER.38 As shown in in situ Raman spectra of P–Ce SAs@CoO and CoO, three characteristic Co–O vibrational modes of the CoO phase were observed at 482 cm−1, 521 cm−1, and 690 cm−1, along with a broad peak between 580 cm−1 and 620 cm−1 assigned to Co–OH species formed by OH− adsorption at low potential. As the applied potential increased, CoO showed a rapid attenuation of the Co–O peak intensities beyond 0.45 V vs. Ag/AgCl, suggesting the surface reconstruction, lattice oxygen evolution, and dissolution–redeposition processes of the catalysts during the OER (Fig. 3f). However, P–Ce SAs@CoO preserved the Co–O vibrational signals even at high potentials, indicating that the introduction of Ce SAs significantly strengthens the Co–O bond and suppresses its breakage during the OER (Fig. 3g). Normalized intensity analysis of the A1g peak (690 cm−1) further confirmed that the Ce–O–Co unit site remains stable under harsh oxidative conditions. The in situ Raman spectroscopy results revealed that Ce acts as an electronic protector that prevents the structural degradation of CoO, thereby enhancing both activity and stability for the oxygen evolution reaction.
4. Material platforms for electron buffering
As emphasized earlier, the realization of the electron buffer effect hinges critically on the inherent electronic properties of the buffer component. A systematic understanding of such materials is vital for the rational design of high-performance electrocatalysts. This section comprehensively introduces materials capable of inducing the electron buffer effect, which are primarily categorized into two groups including metal-based and nonmetal-based systems.
4.1. Metal-based components
The key feature of these metal-based buffer components is the reversible multi-electron redox capability of their metal centers, which allows them to function as localized electron exchange reservoirs. As the most representative metal-based material, CeO2 exhibits the significant electron buffering capability owing to the Ce3+/Ce4+ redox couple and the formation/annihilation of oxygen vacancies within its stable fluorite crystal structure.26,35,40–43 The shielding effect of 5s/5p orbitals, progressive filling of the 4f orbital, and the presence of abundant electronic energy levels endow Ce with a highly adaptable pathway for electronic structure optimization. For instance, Wang et al. developed a scalable strategy to achieve dynamic electron backflow across heterogeneous interfaces by constructing a CeO2/Fe–Co(OH)2 heterojunction.26 Driven by the built-in electric field at the interfaces between CeO2 and Co(OH)2, the resulting high-valence Coδ+ species synergistically activate lattice oxygen with high-spin Fe3+, significantly enhancing catalytic activity (Fig. 4a). Moreover, the higher work function of Fe–CoO2 compared with CeO2 provides the driving force for the electron backflow from CeO2 to Fe–CoO2, thereby suppressing the over-oxidation of Co and improving structural stability (Fig. 4b). Besides, the d-band center of CeO2/Fe–Co(OH)2 lied between those of Fe–Co(OH)2 and CeO2/Co(OH)2, indicating the optimized adsorption strength for intermediates. CeO2/Fe–Co(OH)2 exhibited an overpotential of 189 mV at 10 mA cm−2 and stability for 800 h at 1000 mA cm−2 and outperformed Fe–Co(OH)2 (220 mV at 10 mA cm−2 and 100 h at 1000 mA cm−2). This reversible electron shuttling between Co2+ → Ce3+/4+ and Ce3+/4+ → Co3+/4+, which is induced by the electron buffer effect of CeO2, dynamically modulates the valence state of Co active sites, thereby enhancing both electrocatalytic activity and operational stability. Beyond Ce-based systems, other variable-valence metals characterized by weak electron affinities and strong ionization tendencies can also serve as effective electron buffer components, which mitigate the localized charge imbalance and polarization to achieve a more uniform electron density distribution.27,28,36 Li et al. introduced Cr into intermetallic PtFe alloy nanoparticles to modulate the electronic structure of Pt shells.28 In the L10-PtFe/C system, partial oxidation of the Pt shell increases surface charge density, inducing electrostatic repulsion and lattice expansion (Fig. 4c). Encouragingly, the incorporation of Cr as an electron buffer with electron-donating ability effectively suppresses the surface polarization of Pt shells by relieving the increase in the valence state and the tensile strain at increasing potential (Fig. 4d and e). L10-Cr–PtFe/C showed a moderate d-band center position (−2.487 eV) compared to that of L10-PtFe/C (−2.541 eV) and pure Pt (−2.382 eV), indicating the optimized adsorption of surface oxygen species. This mechanism significantly enhances the electrocatalytic activity and simultaneously inhibits the dissolution of both Pt and Fe. L10-Cr–PtFe/C possessed a half-wave potential of 0.952 V and exhibited stability for 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ADT cycles with 2.9% degradation, which are superior to those of L10-PtFe/C (almost 0.92 V and 30000 ADT cycles with 20.2% degradation). Therefore, in designing advanced electrocatalysts with electron buffer functionality, the strategic utilization of the multi-valence metal-based buffer components, coupled with precise electron density modulation, offers a powerful approach to simultaneously boost electrocatalytic activity and long-term stability.
000 ADT cycles with 2.9% degradation, which are superior to those of L10-PtFe/C (almost 0.92 V and 30000 ADT cycles with 20.2% degradation). Therefore, in designing advanced electrocatalysts with electron buffer functionality, the strategic utilization of the multi-valence metal-based buffer components, coupled with precise electron density modulation, offers a powerful approach to simultaneously boost electrocatalytic activity and long-term stability.
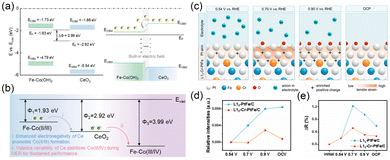 |
| | Fig. 4 (a) Schematic diagram of band structures for Fe–Co(OH)2, CeO2, and CeO2/Fe–Co(OH)2. (b) The proposed work functions of CeO2/Fe–Co(OH)2 during the OER. Reproduced with permission.26 Copyright 2025, Royal Society of Chemistry. (c) Schematic diagram of electrochemical processes on L10-Cr–PtFe/C at different potentials. In situ potential-dependent relative (d) white-line intensity and (e) radial distances of L10-Cr–PtFe/C and L10-PtFe/C. Reproduced with permission.28 Copyright 2024, American Chemical Society. | |
4.2. Nonmetal-based components
Nonmetal-based materials offer a distinct and powerful platform for electron buffering, operating not through valence state changes in metal cations but via defect engineering and delocalized electron systems. The spherical structure and highly delocalized three-dimensional π–electron system of fullerene (C60) grant it high electron affinity, and its low-energy lowest unoccupied molecular orbital (LUMO) acts as a dedicated electron reservoir, enabling it to function as an electron buffer component to reversibly accept or donate electrons in response to the surrounding electronic environment.34,44–46 Zhang et al. developed a C60-buffered Ru–RuO2 heterostructure (Ru–RuO2/C60−x), in which the Ru-based active species reversibly transition between Ru and RuO2 during the HER and OER.34 When the applied potential decreased from open-circuit-potential (OCP) to −0.4 V, C60−x donated electrons to RuO2 (Ru4+), inducing the generation of more Ru0 species (Fig. 5a and b), which is beneficial for lowering the adsorption energy barrier of *H intermediates. Conversely, during the OER process, C60−x withdrew the electrons from Ru0 species to facilitate the regeneration of Ru4+ species, thereby enhancing the OER activity (Fig. 5a and c). The Bader charge results showed that C60−x tended to transfer more negative charge (0.68|e|) to the surface Ru atoms compared with carbon nanotubes (0.01|e|) and graphene (0.13|e|), effectively alleviating the full oxidation of Ru to RuO2 and stabilizing the desired Ru/RuO2 heterostructure. This C60−x-mediated dynamic interfacial reconstruction between Ru0 and Ru4+ enables the catalyst to exhibit excellent bifunctional electrocatalytic activity for both the HER and OER (Fig. 5d). Ru–RuO2/C60−x exhibits overpotentials of 7 and 198 mV at 10 mA cm−2 for the HER and OER, which are lower than those of RuO2 (129 and 265 mV). Furthermore, the strong intermolecular interactions among C60 molecules facilitate the formation of thin and highly dispersible C60 crystals. Recently, the two-dimension (2D) C60 network assembled with covalently bonded C60 cages within a plane has been successfully exfoliated from bulk crystals through micromechanical cleavage and the intercalation-based exfoliation method.32,47,48 Yang et al. synthesized few-layer 2D-C60 networks using an acid etching-liquid exfoliation method and integrated them with Ru nanoparticles to construct Ru NPs/2D-C60.49 The Bader charge analysis revealed that under the electron buffer effect of C60 with an electron-withdrawing ability, Ru loses few electrons (from 0.63|e| to 0.52|e|), increasing the electron density of *H and *OH species (from 0.17|e| and 0.22|e| to 0.38|e| and 0.41|e|, respectively), thereby accelerating their desorption and ultimately improving the HER activity. Ru NPs/2D-C60 exhibits an overpotential of 24 mV at 10 mA cm−2, which is lower than that of Ru NPs (74 mV). Beyond pristine C60, fullerenol (C60(OH)x) functionalized with hydroxyl groups not only offers abundant O sites to anchor metal species (e.g., Ru, Ni, and Fe) but also retains the inherent advantages of C60, making it another effective regulator of the interfacial electronic structure in electrocatalysis.50,51
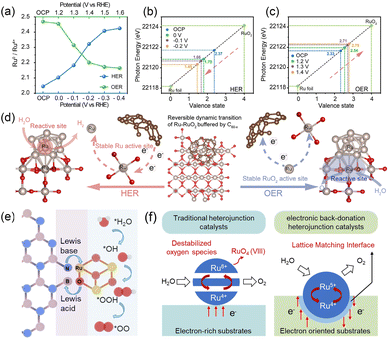 |
| | Fig. 5 (a) The ratio of Ru0/Ru4+ in Ru–RuO2/C60−x against applied potential in the HER and OER. The valence state of Ru in Ru–RuO2/C60−x against applied potential in (b) HER and (c) OER. (d) Schematic illustration of the reversible dynamic transition of Ru–RuO2/C60−x during the HER and OER. Reproduced with permission.34 Copyright 2025, Wiley-VCH. (e) Schematic illustration of electron transport between RuO2 and BNNSs. (f) Schematic illustration of charge compensation and transition processes on the different heterojunctions. Reproduced with permission.54 Copyright 2024, Wiley-VCH. | |
Apart from carbon-based materials, hexagonal boron nitride (BN) has also emerged as a promising electron buffer support for modulating the electron density of active sites. The B and N sites in BN act as electron-deficient and electron-rich centers, respectively, creating a polarized B–N bond that enables flexible regulation of the electronic structure of supported catalysts.52 However, conventional hexagonal BN is an electrical insulator with poor conductivity, which limits efficient directional electron transfer. Constructing atomically thin BN nanosheets has proven effective in facilitating rapid electron tunneling in the direction perpendicular to the exposed plane.53 Peng et al. designed defect-rich ultrathin BN nanosheets as the support for RuO2 (RuO2/BNNS).54 In this hybrid, N sites serve as Lewis base sites that donate electrons to Ru, suppressing its over-oxidation, while B sites function as Lewis acid sites that accept electrons from O, thereby stabilizing the lattice oxygen species (Fig. 5e). Hence, there is a bidirectional electron transfer between Ru and BNNSs. Leveraging this mechanism, RuO2/BNNS displayed remarkable activity and stability compared to traditional heterojunction catalysts. Normally, as the applied potential increases, the sluggish replenishment of surface charges leads to electron loss from surface oxygen, triggering oxygen release and structural collapse (Fig. 5f). In contrast, the local charge cycling induced by BNNSs effectively retained the electrocatalytic stability of RuO2/BNNS with slight fluctuations of the Ru oxidation state around 3.92. Moreover, the polarized B sites are susceptible to nucleophilic attacks, which facilitates the deprotonation of *OH and *OOH intermediates on Ru sites. Therefore, RuO2/BNNS possessed an overpotential of 180 mV at 10 mA cm−2, which is lower than that of RuO2 (270 mV). In addition to structural design, introducing interstitial oxygen can also induce an electron buffer effect to regulate the electron localization behavior.37 Guo et al. designed a CrMnFeNiPt high entropy alloy (HEA) catalyst, in which the strong oxygen affinity of Cr facilitated the incorporation of oxygen as an electron buffer with a strong electron-withdrawing ability.37 The presence of interstitial oxygen modulated the electron density of adjacent metal atoms, reducing their d-band centers and increasing their vacancy formation energies, thereby enhancing the electrocatalytic performance. In summary, nonmetal-based electron-buffering materials represent a highly promising direction for further electrocatalyst design.
4.3. Selection principle
Due to the different buffering mechanisms of nonmetal-based and metal-based buffer materials, their roles in electrocatalytic reactions are also different. Taking C60 as an example of nonmetal-based materials, its three-dimensional π–electron system enables reversible acceptance/release of multiple electrons, stabilizing the valence state of adjacent metal active centers and preventing their over-oxidation or over-reduction. The cage structure of C60 exhibits high thermal stability and chemical inertness, and it serves as a stable support to offer flexible integration approaches (van der Waals forces, π–π stacking, or chemical bonding) to form heterojunctions with various materials (e.g., single atoms and metal phthalocyanines). Moreover, acting as an electron transfer mediator, C60 can induce strong electron coupling at heterojunction interfaces, effectively reducing interfacial charge transfer resistance and accelerating reaction kinetics. However, the carbon sites in C60 are often catalytically inert and cannot effectively provide additional active sites. Its large molecular size may introduce a steric hindrance effect that impedes the adsorption of reactants at the active sites. In addition, nonmetal-based buffer materials would suffer from irreversible structural changes after exposure to an oxidative environment for a long time, which significantly compromises the efficiency of the electron buffer effect.
As for metal-based buffer materials, CeO2, as a representative material, can accept electrons from adjacent metal–oxygen bonds, thereby effectively stabilizing the integrated metal oxides (e.g., RuO2) against structural collapse caused by excessive delocalization of lattice oxygen electrons at high potential. This enables long-term stable operation under highly oxidizing conditions, even at large current densities. Besides, CeO2 provides abundant oxygen vacancies that originated from the Ce3+/Ce4+ redox transition, which not only act as active sites to promote water dissociation but also facilitate the optimization of reaction intermediate adsorption. CeO2 exhibits excellent structural stability, retaining good chemical resistance in both acidic and alkaline electrolytes, along with high-temperature durability. However, CeO2 is a wide-bandgap semiconductor with intrinsically low electronic conductivity. In electrocatalysis, this shortcoming directly restricts the electron transport rate at the bulk phase and interfaces, increases the overpotential of the electrochemical reaction, and thus constitutes a major bottleneck for improving efficiency. Both nonmetal-based and metal-based buffer materials possess their advantages and disadvantages. Therefore, the selection of an electron buffer material is not fixed but should be rationally guided by the specific requirements of the catalytic reaction and the nature of the active component.
5. Applications of the electron buffer effect in various electrocatalytic reactions
Based on the systematically established material platforms capable of inducing the electronic buffer effect, the strategic application of this mechanism to specific electrocatalytic reactions is essential for addressing their respective bottlenecks. It is noteworthy that while the physical nature of the electronic buffer effect is universal, its operational behavior in different reaction environments exhibits significant variations. These differences stem from the distinct thermodynamic energy barrier and unique kinetic pathway inherent to each reaction. This section delves into the role of the electron buffer effect across five key electrocatalytic reactions (OER, HER, ORR, CO2RR, and NRR/NO3RR), focusing on how it enhances electrocatalytic activity and stability under distinct electrocatalytic conditions (Table 1).
Table 1 The electrocatalytic performance of the electrocatalysts for various reactions with/without buffer components
| OER |
| Catalysts |
Electrolyte |
η (mV)@j (mA cm−2) |
Tafel slope (mV dec−1) |
Stability (h)@j (mA cm−2) |
Reference |
| (Ru–W)Ox |
0.5 M H2SO4 |
170@10 |
46.2 |
300@10 |
Adv. Mater., 2023, 35, 2305939 |
| RuOx |
240@10 |
78.3 |
75@10 |
| Re0.06Ru0.94O2 |
0.1 M HClO4 |
190@10 |
45.5 |
200@10 |
Nat. Commun., 2023, 14, 354 |
| RuO2 |
258@10 |
50.3 |
20@10 |
| Ir NCs/C60NET |
0.5 M H2SO4 |
237@10 |
41 |
600@10 |
J. Am. Chem. Soc., 2025, 147, 20600–20611 |
| Ir NCs/graphene |
259@10 |
50 |
29@10 |
| Ir–Ce SSO |
0.5 M H2SO4 |
238@10 |
65.9 |
100@10 |
Adv. Funct. Mater., 2024, 34, 2400809 |
| Com-IrO2 |
297@10 |
77.5 |
40@10 |
| Ce–Mn2O3 |
160 g per L H2SO4 |
329@10 |
112.3 |
N/A |
ACS Appl. Mater. Interfaces, 2025, 17, 47047–47056 |
| Mn2O3 |
364@10 |
138.6 |
N/A |
| CeO2/Fe–Co(OH)2 |
1 M KOH |
189@10 |
39 |
800@1000 |
Energy Environ. Sci., 2025, 18, 7188–7202 |
| Fe–Co(OH)2 |
228@10 |
52.9 |
100@1000 |
| IrOx/CeO2 |
0.5 M H2SO4 |
220@10 |
63 |
300@10 |
Nano Energy, 2022, 104, 107960 |
| c-IrOx |
290@10 |
78 |
30@10 |
| CeO2/FeOOH |
1 M KOH |
261@10 |
37.12 |
100@1000 |
Energy Environ. Mater., 2025, e70136 |
| FeOOH |
281@10 |
49.63 |
40@1000 |
| Mn/Ru-NC |
0.5 M H2SO4 |
180@10 |
46.5 |
80@10 |
Chem. Eng. J., 2024, 497, 154724 |
| Ru-NC |
280@10 |
56.1 |
40@10 |
| Ni3Fe LDH/NiFe2O4/Pt–Tm |
1 M KOH |
224@10 |
54.81 |
N/A |
J. Mater. Chem. A, 2024, 12, 17574–17585 |
| Ni3Fe LDH/NiFe2O4/Pt |
259@10 |
62.61 |
N/A |
| Ru/Ta2O5 |
0.1 M HClO4 |
272@10 |
64.5 |
190@10 |
J. Mater. Chem. A, 2025, 13, 23998–24004 |
| RuO2 |
303@10 |
100.3 |
10@10 |
| Ta–RuO2 |
0.1 M HClO4 |
201@10 |
55 |
280@10 |
Adv. Energy Mater., 2025, 15, 2403388 |
| R–RuO2 |
218@10 |
62 |
100@10 |
| Ru–RuO2/C60−x |
1 M KOH |
194@10 |
113.25 |
200@65 |
Angew. Chem., Int. Ed., 2025, 64, e202503608 |
| RuO2 |
265@10 |
174.93 |
N/A |
| Ru3Cr1Srx |
0.1 M HClO4 |
214@10 |
40.6 |
300@10 |
Nano Lett., 2024, 24, 10899–10907 |
| C–RuO2 |
270@10 |
52.2 |
50@10 |
| RuO2/BNNS |
0.5 M H2SO4 |
180@10 |
63.3 |
350@10 |
Angew. Chem., Int. Ed., 2024, 63, e202402018 |
| RuO2 |
270@10 |
136.9 |
N/A |
| P–Ce SAs@CoO |
1 M KOH |
261@10 |
75 |
30@10 |
Adv. Mater., 2023, 35, 2302462 |
| CoO |
310@10 |
80.2 |
5@10 |
| HER |
| Catalysts |
Electrolyte |
η (mV)@j (mA cm−2) |
Tafel slope (mV dec−1) |
Stability (h)@j (mA cm−2) |
Reference |
| Pt1–MoL–Mo2C |
1 M KOH |
12@10 |
17 |
192@80 |
Adv. Mater., 2025, 37, 2502989 |
| Pt1–Mo2C |
47@10 |
34 |
N/A |
| RCO/TF |
1 M KOH |
17@10 |
42 |
N/A |
J. Mater. Chem. A, 2025, 13, 14964–14971 |
| RuO2/TF |
27@10 |
47 |
N/A |
| Ru–RuO2/C60−x |
1 M KOH |
7@10 |
19.2 |
200@250 |
Angew. Chem., Int. Ed., 2025, 64, e202503608 |
| RuO2 |
129@10 |
63.22 |
N/A |
| Ru NPs/2D-C60 |
1 M KOH |
24@10 |
41 |
200@10 |
Small, 2025, 21, 2506131 |
| Ru NPs |
74@10 |
105 |
50@10 |
| ORR |
| Catalysts |
Electrolyte |
Half-wave potential (V) |
Stability@CV (cycles) |
Reference |
| L12-Pt3Co/Ti-a-NPC |
0.1 M HClO4 |
0.936 |
3 mV decay@3000 |
Adv. Funct. Mater., 2024, 34, 2406347 |
| L12-Pt3Co/a-NPC |
0.926 |
5 mV decay@3000 |
| L10-Cr–PtFe/C |
0.1 M HClO4 |
0.952 |
2.9% decay@30000 |
J. Am. Chem. Soc., 2024, 146, 2033–2042 |
| L10-PtFe/C |
∼0.92 |
20.2% decay@30000 |
| FePc/Eu2O3 |
0.1 M KOH |
0.939 |
9 mV decay@20000 |
Adv. Funct. Mater., 2025, 35, 2425138 |
| FePc |
0.906 |
22 mV decay@20000 |
| Mn SAs/Fe3C NPs@NPC |
0.1 M KOH |
0.88 |
No decay@10000 |
Nano Res., 2022, 15, 7976–7985 |
| Fe3C NPs@NPC |
∼0.8 |
N/A |
| FeRu-DACs |
0.5 M H2SO4 |
∼0.83 |
15 mV decay@20000 |
Angew. Chem., Int. Ed., 2025, 64, e202508141 |
| Fe–N–C |
∼0.79 |
N/A |
| Fe–N3S/SNC |
1 M KOH |
0.924 |
1 mV decay@5000 |
Adv. Energy Mater., 2026, e71096 |
| Fe–N4/NC |
0.9 |
N/A |
| CO2RR |
| Catalysts |
Electrolyte |
E (V) |
Faraday efficiency |
Reference |
| CuO–C60 |
1 M KOH |
−1.6 |
61%@C2+ products |
Adv. Energy Mater., 2023, 13, 2204346 |
| CuO |
32%@C2+ products |
| G–CuxO-2 h |
1 M KOH+ 0.5 M KHCO3 |
−0.8 |
81%@formic acid |
Appl. Catal., B, 2019, 259, 118044 |
| CuxO-2 h |
29.7%@formic acid |
| CuCeO–Ov |
1 M KOH + 0.1 M KHCO3 |
−1.3 |
51.7%@C2H4 |
Appl. Catal., B, 2026, 395, 126871 |
| CuO–Ov |
33.5%@C2H4 |
| C60/Cu(OH)F |
0.1 M KHCO3 |
−1.5 |
76.9%@C2+ products |
Chem. Commun., 2025, 61, 1681–1684 |
| Cu(OH)F |
45.3%@C2+ products |
| c-Cu2O–C60 |
1 M KOH+ 0.1 M KHCO3 |
−1.2 |
60.4%@C2+ products |
ACS Nano, 2025, 19, 41658–41668 |
| c-Cu2O |
−1.4 |
46.6%@C2+ products |
| NRR/NO3RR |
| Catalysts |
Electrolyte |
Faraday efficiency@E (V) |
NH3 yield@E (V) |
Reference |
| STRs |
0.1 M HCl |
24.56%@−0.3 |
21.54 µg h−1 mg−1@−0.3 |
Adv. Sci., 2019, 6, 1901627 |
| Te NWs |
8.77%@−0.3 |
6.15 µg h−1 mg−1@−0.3 |
| Ag0–CuAgOx |
0.1 M KOH + 0.01 M KNO3 |
91.5%@−0.6 |
1920 µg h−1 cm−2@−0.6 |
Adv. Energy Mater., 2025, 15, 2405534 |
| Cu nanosheets |
∼25%@−0.6 |
∼400 µg h−1 cm−2@−0.6 |
| Cu2O–InSA |
0.5 M Na2SO4 + 0.1 M KNO3 |
99.36%@−0.8 |
23.37 mg h−1 mg−1@−0.8 |
Angew. Chem., Int. Ed., 2026, 65, e20730 |
| Cu2O |
∼80%@−0.8 |
∼15 mg h−1 mg−1@−0.8 |
| Fe(OH)2/Fe@CNTs |
1 M KOH + 0.1 M KNO3 |
95.1%@−0.4 |
0.67 mmol h−1 cm−2@−0.4 |
Adv. Funct. Mater., 2025, 35, 2501079 |
| Fe(OH)2@CNTs |
∼75%@−0.4 |
∼0.45 mmol h−1 cm−2@−0.4 |
| Fe@CNTs |
∼92%@−0.4 |
∼0.38 mmol h−1 cm−2@−0.4 |
5.1. OER
Under highly oxidizing conditions, electrocatalysts (e.g., Ru and Co-based materials) often suffer from inevitable over-oxidation during the OER process, leading to structural degradation and diminished stability.55,56 However, high-valence metal sites typically exhibit superior catalytic activity due to the more empty d-orbitals that preferably adsorb intermediates.57 Moreover, the OER process is constrained by the linear scaling relationships among oxygen-contained intermediates, which fundamentally limits further enhancement of the catalytic activity.58
In this context, the electron buffer effect has emerged as a pivotal regulatory mechanism to address these issues by modulating the electronic structure of active sites.33,39,59–63 Qiao et al. introduced Re dopants into RuO2 to simultaneously boost OER activity and stability.59 Leveraging the multivalent nature of Re and its strong metal–oxygen bonding capability, the authors effectively induced dynamic charge transfer at the Ru sites. In situ XANES analysis revealed a progressive increase in the oxidation state of Re (from +6.33 to +6.67) during the pre-catalytic process with the increasing potential. As the potential exceeded 1.3 V, the oxidation state of Re decreased from +6.67 to +6.29 due to the electron acceptance from Ru, facilitating the generation of high-valence Ru sites to boost electrocatalytic activity. At even higher overpotential (>1.5 V), the Re sites (from +6.29 to +6.53) donated electrons back to Ru, preventing their over-oxidation and thereby preserving the structural integrity of RuO2 (Fig. 6a). This reversible electron exchange enabled Re dopants to function as an effective electron reservoir, dynamically activating and stabilizing the Ru sites and significantly promoting the OER performance of RuO2 (Fig. 6b). Re0.06Ru0.94O2 showed an overpotential of 190 mV at 10 mA cm−2 and stability for 200 h at 10 mA cm−2, outperforming RuO2 (258 mV and 20 h at 10 mA cm−2). Beyond valence state regulation, the electron buffer effect can also optimize the adsorption behaviors of OER intermediates by tuning the electronic structure of active sites. Chen et al. employed a 2D C60 network to precisely modulate the electronic structure of Ir nanoclusters (Ir NCs/C60NET).32 Ir NCs/C60NET exhibited an overpotential of 237 mV at 10 mA cm−2 and great stability over 600 h at 10 mA cm−2. Owing to the unique electron buffer effect of C60NET, Ir NCs/C60NET showed a higher anodic peak (Ir3+ → Ir4+) compared to Ir NCs/graphene, indicating the enhanced resistance to oxidation of Ir3+ (Fig. 6c). Besides, the buffering by C60NET significantly suppressed the increase in reduction current for Ir NCs/C60NET, demonstrating that electrons from Ir NCs were partially captured by C60NET rather than being fully transferred to the electrode. Bader charge analysis further confirmed the strong electron-withdrawing and electron-donating ability of C60NET, which dynamically modulate the electron density of the Ir NCs (Fig. 6d). Besides, the in situ Ir L3-edge XANES quantitatively analyzed the electron buffer strength of C60NET. Ir NCs/C60NET showed a lower increase rate [∂µ(E)/∂η] of 0.6 for the intensities of the white line peaks relative to the overpotential than that of Ir NCs/graphene (0.84) from the open-circuit voltage (OCV) to 1.65 V vs. RHE, indicating that the oxidation state of Ir in Ir NCs/C60NET can be retarded with the electron buffer effect of C60NET. More importantly, during the OER process, the electron buffer effect of C60NET increased the electron density on Ir nanoclusters while decreasing it on reaction intermediates, thereby promoting the formation of *OOH. This electronic redistribution narrowed the free energy difference between ΔG*OOH and ΔG*OH, breaking the conventional linear scaling relationship (ΔG*OOH − ΔG*OH = 3.2 ± 0.2 eV) for single-site catalysts and consequently reducing the energy barrier of the rate-determining step (RDS) (Fig. 6e). Besides, a P–Ce SAs@CoO catalyst with Ce(4f)–O(2p)–Co(3d) gradient orbital coupling was designed to optimize the adsorption energies of oxygen-containing intermediates.38 The introduction of Ce into CoO induced surface charge redistribution. Ce acts as an electron donor with a higher charge than Co, which alleviates Co self-oxidation. Moreover, the RDS of the OER switches from *O to *OOH in CoO to *OH to *O in P–Ce SAs@CoO with the decreased energy barrier of RDS, and the scaling relation (ΔG*OOH − ΔG*OH) of P–Ce SAs@CoO reduces from 3.3 to 3.25 eV, indicating enhanced OER performance. These findings underscore the potential of the electron buffer effect in rationally modulating the thermodynamics of surface electrocatalytic reactions.
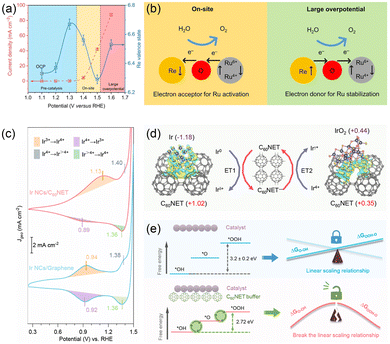 |
| | Fig. 6 (a) The change in the Re valence state and OER current density as a function of applied potential. (b) Schematic illustration of dynamic electron transfer between Re and Ru sites. Reproduced with permission.59 Copyright 2023, Springer Nature. (c) The cyclic voltammetry (CV) curves of Ir NCs/C60NET and Ir NCs/graphene. (d) The Bader charge values for Ir–C60NET and IrO2–C60NET systems. (e) Schematic illustration of conventional and C60NET-buffered correlations among OER intermediates. Reproduced with permission.32 Copyright 2025, American Chemical Society. | |
5.2. HER
The HER proceeds through a two-electron transfer pathway comprising the Volmer step followed by either the Heyrovsky or Tafel step.64 While optimizing the adsorption strength of the *H intermediate at active sites to align with the Sabatier principle has been widely demonstrated to boost HER activity, the critical initial steps of H2O adsorption and activation remain frequently overlooked in mechanistic studies.65,66 Given that the electron density of active sites significantly influences the adsorption and activation of H2O, the introduction of the electronic buffer effect holds promise for enhancing these processes.49,67 For instance, Ma et al. constructed a Mo nanolayer as an electron buffer between Pt and Mo2C (Pt1@MoL/Mo2C) via a carburization method under a reductive atmosphere.68 Bader charge analysis revealed a strong electron interaction between Pt sites and Mo2C (0.76|e|), as well as with the interfacial Mo (0.49|e|). Upon inserting a single Mo atomic layer between Pt and Mo2C, the Bader charge on Pt decreased to 0.35|e| and further dropped to 0.04|e| when the Mo nanolayer was increased to three atomic layers (Fig. 7a). The Mo nanolayer effectively weakened the electron interaction between Pt and Mo2C substrate by mitigating the electron withdrawal from the Mo2C matrix. Moreover, the electron buffer effect primarily originated from the second and third Mo layers, which remained nearly charge-neutral (Fig. 7b). Density functional theory (DFT) calculations indicated that the Mo nanolayer not only improved H2O accumulation and adsorption, but also promoted H2O activation and dissociation into *H and *OH. Besides, the electron buffer effect rendered Pt sites in a near-zero valence state with a free-atom-like d state, facilitating the adsorption/desorption of dissociated *H on Pt sites. As a result, the synergistic interaction between the Mo nanolayer and Pt sites enabled Pt1@MoL/Mo2C with an optimal HER performance (Fig. 7c). Pt1–MoL–Mo2C showed an HER overpotential of 12 mV at 10 mA cm−2, which is lower than that of Pt1–Mo2C (47 mV). In addition to water activation, maintaining active sites in a desirable valence state is equally crucial for precise modulation of the adsorption affinity for key reaction intermediates during the HER. For instance, Wang et al. introduced a Cu dopant as an electron buffer to regulate the electronic structure of RuO2 supported on titanium felt (RCO/TF) during the HER.69 Owing to the multivalence nature of Cu (+2/+1/0) at HER potential, the dynamically adaptive electron buffer effect facilitates the generation of an active Ru0/Ru4+ surface while suppressing the excessive reduction of Ru species. Post-HER characterization confirmed that the Ru4+/Ru0 ratio in RCO/TF remained nearly unchanged, whereas RuO2/TF showed an obvious increase in the proportion of Ru0 (Fig. 7d). The resulting partial oxygen-deficient Ru0-rich surface on the Cu-doped RuO2 showed a more favorable hydrogen adsorption energy than single Ru and RuO2, delivering enhanced HER activity with an overpotential of 17 mV at 10 mA cm−2. Therefore, the electronic buffer effect exhibits multiple functions in HER electrocatalysis, though its underlying mechanisms warrant further systematic investigation.
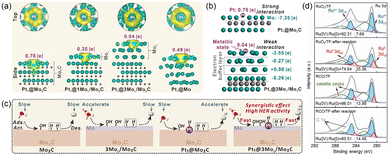 |
| | Fig. 7 (a) Charge density distribution and Bader charge of the Pt1 site in different structures. (b) Bader charge of the Mo nanolayer in Pt1@Mo2C and Pt1@3MoL/Mo2C. (c) Schematic illustration of the HER process on different surface structures. Reproduced with permission.68 Copyright 2025, Wiley-VCH. (d) The Ru 3d XPS spectra of RuO2/TF, RuO2/TF-after reaction, RCO/TF, and RCO/TF-after reaction. Reproduced with permission.69 Copyright 2025, Royal Society of Chemistry. | |
5.3. ORR
As a multi-electron transfer process that proceeds in the reverse direction of the OER, the ORR also presents a fundamental challenge in achieving the balanced adsorption of oxygen-contained intermediates at active sites.70,71 Although Pt-based materials remain benchmark ORR catalysts, the integration of an electron buffer component offers a promising strategy to not only reduce material cost but also finely tune the d-band center of Pt sites, thereby optimizing the adsorption behavior of key reaction intermediates.28,31,72,73 Cheng et al. designed a Ti single atom-modified nitrogen-doped porous carbon (Ti-a-NPC) to balance the O2 activation and *OH desorption on ultra-small L12-Pt3Co nanoparticles.31 Functioning as an electron buffer with electron-withdrawing ability, the Ti atoms trapped electrons and modulated the electron transfer from L12-Pt3Co to the N atoms, thus regulating the local electron density at the Pt sites (Fig. 8a–c). The resulting charge rearrangement on Ti atoms formed an electron channel bridging the support and L12-Pt3Co (Fig. 8d). This interaction significantly shifted the d-band center of Pt sites, thereby tuning the adsorption energy for oxygen-contained intermediates. The optimized O bonding strength of L12-Pt3Co/Ti-a-NPC that was buffered by Ti single atoms promoted O2 activation and switched the RDS to the *OH removal, thereby enhancing the intrinsic ORR activity (Fig. 8e). L12-Pt3Co/Ti-a-NPC showed a higher half-wave potential of 0.936 V, compared to L12-Pt3Co/a-NPC (0.926 V). In addition to Pt-based materials, other metal-based materials integrated with electron buffer components can also effectively regulate the adsorption behavior of reaction intermediates, thereby enhancing electrocatalytic performance.74–78 Luo et al. designed an Fe single atom catalyst (Fe–N3S/SNC) with a broken D4h symmetric structure by substituting a N atom in the Fe–N4 moiety with a S atom, resulting in an Fe–N3S coordination.74 The S atom, with lower electronegativity than N, acts as an electron buffer that dynamically retains a stable electron filling in the eg orbital of Fe during the ORR. Compared with the symmetric Fe–N4 site, the Fe–N3S site preserves an eg filling value close to 1 throughout the ORR. This stability originates from the electron buffer capability of S, which can reversibly accept or donate electrons to compensate for the charge density variation on Fe. Consequently, the energy barrier for electron transfer between t2g and eg orbitals is lowered, facilitating the formation of *OOH and the desorption of *OH. Thus, the Fe–N3S/SNC catalyst achieves an outstanding half-wave potential of 0.924 V vs. RHE in alkaline OER. Moreover, the electron buffer effect can also be extended to dual-atom configuration to regulate the behavior of reaction intermediates for the ORR. Xiang et al. implanted isovalent Ru ions into the secondary coordination structure of an Fe–N4 site, forming FeRu dual-atom catalysts (FeRu-DACs).75 The Ru center with highly similar d-orbital characteristics to Fe serves as an electron buffer site that dynamically neutralizes the electronic polarization induced by the adsorption of oxygen-containing intermediates. Therefore, Ru lowers the Fe d-band center by only 0.27 eV, shifting the RDS from *OH desorption to *OOH to *O conversion and reducing the energy barrier. Besides, FeRu-DACs achieve a peak power density of 1.73 W cm−2 in H2–O2 fuel cells and retain over 97% of Fe sites after prolonged stability tests, which is a 22-fold improvement over Fe–N–C. The stabilization of FeRu-DACs during the ORR process is attributed to the reversible electron transfer between Fe and Ru sites. Notably, the electron buffer effect contributes significantly to both catalytic activity and stability, as further demonstrated in rare-earth-based systems. The f-band in rare-earth materials can serve as an electron buffer to preserve covalency by compensating for electron loss, thereby boosting the electrocatalytic performance of active centers.79,80 Fu et al. constructed an f–p–d gradient orbital coupling configuration by integrating Fe phthalocyanine with Eu2O3 (FePc/Eu2O3) on carbon nanotubes to achieve an efficient and stable ORR electrocatalysis.80 Owing to the intrinsically higher energy level of Eu 4f orbitals relative to the lower Hubbard band (LHB) in Fe–O*, the adsorbed oxygen intermediates preferentially extracted electrons from the Eu f-band rather than the Fe d-orbitals (Fig. 8f). This electron buffer effect from the Eu f-band with electron-donating ability maintained the covalency and high-spin d-orbital occupancy of Fe–N4 sites, significantly achieving the enhanced ORR stability. Furthermore, the f–p–d gradient orbital coupling facilitated O–O bond cleavage in *OOH and shifted the RDS of FePc/Eu2O3 from *OOH → *O to *O → *OH, thereby boosting the ORR activity (Fig. 8g). Compared with FePc (0.906 V and 20000 cycles with 22 mV degradation), FePc/Eu2O3 exhibited a higher half-wave potential of 0.939 V and greater stability over 20000 cycles with 9 mV degradation. In summary, the electron buffer effect serves as a versatile electronic regulation strategy that concurrently enhances both the activity and stability of ORR electrocatalysts.
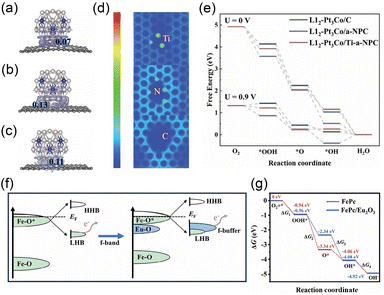 |
| | Fig. 8 The charge density difference distribution of L12-Pt3Co supported on (a) C, (b) a-NPC, and (c) Ti-a-NPC. (d) Electron localization function diagram and (e) free energy diagram of L12-Pt3Co/Ti-a-NPC, L12-Pt3Co/a-NPC, and L12-Pt3Co/C. Reproduced with permission.31 Copyright 2024, Wiley-VCH. (f) Schematic illustrations of the proposed f–p–d gradient orbital coupling effect on ORR activity and stability. (g) Free energy diagram of FePc and FePc/Eu2O3. Reproduced with permission.80 Copyright 2025, Wiley-VCH. | |
5.4. CO2RR
The electrochemical CO2RR faces challenges in product selectivity, primarily due to the diverse CO2 adsorption configurations and complex reaction pathway, along with severe competition from the HER.81–86 HER-inert Cu-based materials serve as the most promising candidates for steering the product distribution of the CO2RR.87 However, Cu+ sites are prone to reduction to Cu0 during the CO2RR process, particularly at high negative potentials. As an effective strategy for regulating the valence states, the electron buffer effect has demonstrated significant applicability in Cu-based systems.88,89 Yan et al. introduced an electron buffer effect by decorating CuxO on graphene oxide (G–CuxO).88 During the electrocatalytic process, Cu2+ species act as a sacrificial agent, continuously replenishing Cu+ active sites from the bulk to the surface of CuxO, thereby enhancing catalytic stability. Meanwhile, the stabilized Cu+ sites effectively weaken the adsorption strength of *COOH intermediates, leading to improved selectivity toward HCOOH. The synthesized G–CuxO-2 h showed higher Faraday efficiency (FE) (81%) at −0.8 V toward HCOOH than CuxO-2 h (29.7%). Currently, the conversion of CO2 into high-value multicarbon (C2+) products holds immense economic and environmental interest, yet it faces a key challenge due to the difficulty in facilitating C–C coupling. It has been reported that the coupling between *CHO and *CO intermediates is thermodynamically and kinetically favorable.90 Cu-based materials with Cu0–Cu+ active sites are considered promising for facilitating the generation of *CHO intermediates, which are crucial for the subsequent formation of C2+ products.91,92 Zhang et al. synthesized a CuO–C60 composite with in situ formed Cu0–Cu+ dual active sites after electroreduction.89 The charge density difference analysis confirmed that the generation and stabilization of Cu+ sites originated from the electron transfer from Cu0 sites to C60, which possesses a strong electron-withdrawing ability (Fig. 9a). In contrast to the maintained valence state distribution of Cu in the CuO–C60 structure, the Cu species in pure CuO were completely reduced to Cu0 within 10 min of electrolysis (Fig. 9b and c). Moreover, in situ attenuated total reflection FTIR (ATR-FTIR) verified the presence of *CHO and coupled *CO–CHO intermediates on CuO–C60, indicating the preferential coupling between *CO and *CHO over *CO dimerization at the Cu0–Cu+ dual sites (Fig. 9d). Compared with CuO (32%), CuO–C60 showed a higher FE of 61% for C2+ products. In addition, the FE ratio of C2+ products to CO for CuO–C60 was also higher than that for CuO, especially at high reduction potentials, further demonstrating the crucial role of the electron buffer effect in promoting C–C coupling (Fig. 9e). Yuan et al. developed a cubic Cu2O–C60 catalyst for the CO2RR.93 The C60 molecules act as an electron buffer, dynamically accepting and donating electrons during the CO2RR, thereby preventing over-reduction of Cu+ to Cu0. This electron buffer effect maintains abundant Cu+/Cu0 grain boundaries on the surface of the catalyst. The in situ Raman spectra of c-Cu2O–C60 showed both the presence of Raman bands of the atop-bound (2085–2125 cm−1) and bridge-bound (1785–1876 cm−1) *CO species, which are essential for efficient C–C coupling, while only weak *COatop adsorption was observed on the c-Cu2O surface. Hence, C60 acts as a molecular electron reservoir that preserves the active Cu+/Cu0 interface, promotes *CO dimerization, and dramatically enhances both C2+ selectivity and long-term durability. The c-Cu2O–C60 catalyst achieved FEs for C2+ products of 60.4% in the H-cell and 65.6% in the flow cell and exhibited stable operation for 100 h at −1.2 V vs. RHE without obvious activity loss. Moreover, Wu et al. synthesized a Ce-doped CuO catalyst enriched with oxygen vacancies (CuCeO–Ov).94 In the synthesized gradient orbital coupling Ov–[Ce 4f–O 2p–Cu 3d] unit, the Ce 4f orbitals serve as an electron buffer, while the Ov act as an electron supply station, enhancing Cu–O covalency and stabilizing the Cuδ+ (1<δ < 2) state during the CO2RR. The in situ FTIR spectra were used to identify key reaction intermediates during the CO2RR. Compared to pristine CuO–Ov, CuCeO–Ov exhibited significantly stronger infrared bands at ∼1557 cm−1 (*COCHO of the C–C coupling), ∼1772 cm−1 (*CHO), and 1735 cm−1 (*C2H4), indicating that the electron buffer effect from Ce and Ov promoted the formation and stabilization of *CHO and facilitated its coupling with *CO to generate *COCHO. Moreover, in a CuO catalyst with oxygen vacancies, the Bader charge of the C atom in adsorbed *CHO is 1.65|e|. After Ce doping, this value increases to 1.82|e|, which facilitates electron transfer to the adsorbed *CHO and strengthens the Cu–C bond. Besides, the energy barrier for coupling *CO and *CHO to generate *COCHO of CuCeO–Ov is lower than that of CuO–Ov, which is consistent with the higher FE of C2H4 (51.7% at −1.3 V vs. RHE). In summary, the electron buffer effect plays an important role in the modulation of metal valence states and the adsorption behavior of key intermediates, thereby governing CO2RR product selectivity.
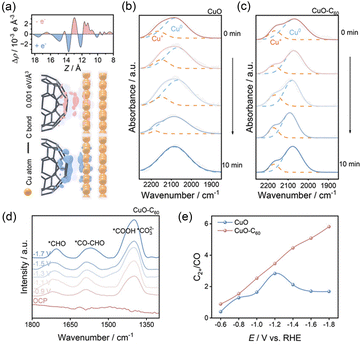 |
| | Fig. 9 (a) The charge density difference and planar-averaged electron density difference for C60–Cu systems. In situ ATR-FTIR spectra of (b) CuO and (c) CuO–C60 at different times. (d) In situ ATR-FTIR spectra of CuO–C60 at different potentials. (e) The FE ratios of C2+ products to CO of CuO and CuO–C60 at different potentials. Reproduced with permission.89 Copyright 2023, Wiley-VCH. | |
5.5. Ammonia synthesis
Ammonia production from the electrochemical NRR presents a sustainable and efficient route that circumvents the energy-intensive conditions and high-pressure H2 requirements of the conventional Haber–Bosch method.95,96 However, the extreme inertness of the N2 molecule severely limits NRR activity, making efficient N2 adsorption and activation a critical challenge.97–99 TeO2, which exhibits inert HER activity, demonstrates considerable N2 activation capability due to its exposed lone-pair electrons that facilitate N–N bond elongation (Fig. 10a). Nevertheless, O ligands in TeO2 cannot buffer the oxidation state variation of Te during the NRR. To address this limitation, Li et al. introduced Se into TeO2 to enhance the hybridization between Te 5p and O 2p orbitals, increasing the DOS near the Fermi level (Fig. 10b).100 The incorporation of Se promoted efficient charge transfer from Te sites to N2, significantly reducing the adsorption energy of N2 (Fig. 10c), leading to higher FE (24.56%) and NH3 yield (21.54 g h−1 mg−1) at −0.3 V. Moreover, the resulting electron-donating SeO ligands near Te sites served as electron reservoirs to effectively buffer the oxidation state of Te, which is critical for π-backdonation interaction between Te sites and N2. This electronic modulation also shifted the RDS from the protonation of *NNH to that of *N2, lowering the energy barrier to 0.98 eV (Fig. 10d). Given the persistent inefficiency in cleaving the highly stable triple bond of N2, the NO3RR has emerged as a thermodynamically more favorable alternative for ammonia production, owing to the high solubility and lower activation barrier of nitrate.101–103 In one representative study, Li et al. constructed heterovalent Fe(OH)2/Fe pair sites on carbon nanotubes (Fe(OH)2/Fe@CNTs) to drive efficient NO3RR.104 The d-band center for Fe(OH)2/Fe (1.104 eV) shows an upward shift toward the Fermi level compared to Fe (0.622 eV) alone and a downward shift compared to Fe(OH)2 (3.478 eV) alone, tuning adsorption energies of N-containing intermediates. The Fe(OH)2/Fe structure provided both a sufficient proton source and moderate *NO3 adsorption affinity, facilitating continuous hydrogenation steps during the NO3RR. Compared to single Fe and Fe(OH)2, the Fe(OH)2/Fe configuration delivered the lowest energy barrier for the RDS, indicating superior nitrate-to-ammonia efficiency (Fig. 10e). Fe(OH)2/Fe@CNTs showed higher FE (95.1%) and NH3 yield (0.67 mmol h−1 cm−2) at −0.4 V by comparing with Fe(OH)2@CNTs (almost 75% and 0.45 mmol h−1 cm−2) and Fe@CNTs (almost 92% and 0.38 mmol h−1 cm−2). Crucially, the electron transfer between Fe and Fe(OH)2 induced the electron buffer effect that dynamically stabilized the Fe(OH)2/Fe sites when exposed to air and during electroreduction, thereby retaining both catalytic activity and structural integrity throughout the NO3RR process. Besides, Wang et al. developed a heterostructured catalyst composed of metallic Ag clusters embedded in oxidized CuAgOx nanosheets (Ag0–CuAgOx).105 The Ag0 acts as an electron buffer, dynamically accepting electrons from Cu+ to generate electron-deficient Cu sites, thus modulating the Cu d-band center (from −2.82 eV to −2.54 eV relative to the Fermi level) with the optimized adsorption/activation energy barriers of *NO2 and *NO intermediates, steering the reaction selectivity toward NH3 rather than nitrite or H2. As shown in the in situ FTIR spectra, the peak that is assigned to *NO2 emerges at −0.4 V vs. RHE on Ag0–CuAgOx, while Cu nanosheets showed a *NO2 peak at a more negative potential of −0.8 V vs. RHE, demonstrating the enhanced kinetics of nitrate-to-nitrite conversion on Ag0–CuAgOx. Moreover, compared to the Cu nanosheets, as the potential becomes more negative, a much wider redshift for the band position of *NO2 and *NO on Ag0–CuAgOx can be observed, indicating the higher stark tuning rate and stronger electric field of Ag0–CuAgOx with the electron-deficient Cu sites, which can strengthen the activation of *NO2 and *NO and facilitate the subsequent hydrogenation to NH3. In addition, the DFT results showed that the Cu2O–Ag model possesses a lower energy barrier of the RDS (*NO to *NOH) than that of Cu and Cu2O. Hence, Ag0–CuAgOx exhibited a high FE of 91.5% and an NH3 yield rate of 1920 µg h−1 cm−2 at −0.6 V vs. RHE. These studies collectively demonstrate that introducing electron buffer components can effectively stabilize the oxidation state of active metals, lower the energy barrier of the RDS, and enhance the selectivity and stability toward valuable ammonia production. Although the electron buffer effect has demonstrated preliminary potential in stabilizing metal sites and modulating the adsorption behavior of N-contained intermediates, its precise regulatory mechanisms governing complex reaction pathways and product selectivity in electrocatalytic ammonia synthesis require further exploration.
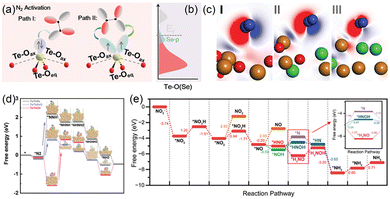 |
| | Fig. 10 (a) The N2 activation on TeO2 through backbonding interactions. (b) Schematic band diagrams of TeO2 after introduction of Se. (c) Charge density difference of N2-adsorbed TeO2, TeSeO, and TeSe. (d) Schematic illustration of the free energy diagram for Te/TeSe, Te/TeSeO, and Te/TeO2. Reproduced with permission.100 Copyright 2019, Wiley-VCH. (e) Free energy diagram of the NO3RR on Fe(OH)2/Fe via different pathways. Reproduced with permission.104 Copyright 2025, Wiley-VCH. | |
6. Summary and outlook
In summary, precise modulation of the electronic structure of active sites through the introduction of an electron buffer effect has emerged as a pivotal strategy for enhancing electrocatalytic performance. This approach offers considerable potential for boosting activity, improving stability, and steering reaction pathways, thereby offering a novel avenue for overcoming the performance limitations of conventional electrocatalysts. Current research on the electronic buffer effect is undergoing a paradigm evolution, progressing from phenomenological description to mechanistic understanding and further advancing toward rational design. Although this review has summarized key progress in this field, there are still several serious challenges that guide future research directions (Fig. 11).
 |
| | Fig. 11 Schematic illustration of outlook and challenges for the electron buffer effect. | |
6.1. Establishing evaluation criteria for buffering strength
To quantitatively assess electron buffering capability, a multi-dimensional framework that integrates complementary theoretical and experimental descriptors is essential. Theoretically, a key metric is the interfacial electron transfer energy barrier, which quantifies the thermodynamic driving force for electron transfer between the buffer component and the active sites. A lower interfacial electron transfer energy barrier corresponds to a more facile and reversible buffering process in the catalyst. Concurrently, the projected Density of States (pDOS) analysis of the catalyst with and without the buffer component can reveal the extent of electronic modulation. The shifts in the d-band center and changes in electron occupancy near the Fermi level serve as intrinsic descriptors directly correlated with adsorption energetics. Experimentally, the multi-potential step stability test can determine the operational potential window of the buffer component by monitoring activity retention (e.g., current or potential decay) under progressively oxidizing or reducing conditions. A wider stable potential window indicates greater applicability. Furthermore, in situ X-ray absorption spectroscopy (XAS) can track the valence state of the active metal as a function of applied potential (E). The d(valence state)/dE reflects the potential-dependent sensitivity of the metal center. Therefore, a smaller d(valence state)/dE over the relevant potential range signifies stronger buffering strength, as it indicates effective suppression of over-oxidation or over-reduction. Thus, a robust evaluation of buffering strength necessitates the integrated application of these complementary descriptors, which bridge theoretical insight with experimental validation to enable systematic comparison across different buffer systems.
6.2. Rational design of novel buffer materials
Based on first-principles calculations and high-throughput screening, it is feasible to identify novel buffer materials featuring broader potential windows and higher electron affinity. For instance, MXenes (Mn+1XnTx, where M is a transition metal, X is C and/or N, and T is surface functional groups) represent an innovative class of catalyst supports for electrocatalysis. Their two-dimensional layered architecture, coupled with high electrical conductivity, intrinsic hydrophilicity, tunable interlayer spacing, and abundant terminal functional groups (e.g., –O, –OH, and –F), render them a highly attractive platform. The conductive backbone of transition metal carbides/nitrides ensures efficient charge transport, while the terminal functional groups can serve as electron donors or acceptors. This dual functionality allows for flexible modulation of the electronic structure of the supported active components, making MXenes promising candidates for advanced electron buffer materials. Furthermore, introducing multiple variable-valence metals as dopants into the active component not only simplifies the synthetic process, but also effectively induces the electron buffer effect by leveraging their multivalent characteristics. Among these, high-valent d0 metal ions (e.g., W6+, Mo6+, and Nb5+) are promising candidates due to their fully empty d-orbitals, which allow the high-valent d0 metal ions to function as electron reservoirs. During the initial stage of oxidation reactions, these empty d-orbitals accept electrons and promote the formation of high-valent active centers. Subsequently, by donating electrons back, high-valent d0 metal ions can mitigate over-oxidation of the active sites during prolonged operation. Thus, engineering with high-valent d0 metal dopants directly addresses the interfacial instability issue in conventional supported catalysts, while simultaneously enhancing the overall buffering capacity.
6.3. Advanced in situ characterization and simulation techniques
Current investigations on the electron buffer effect in catalysts predominantly rely on the static electronic structure analysis conducted before and after reactions. In situ characterization techniques and dynamic theoretical simulations based on first-principles calculations will be crucial for advancing fundamental research on the electron buffer effect. By employing in situ XANES, the dynamic valence state evolution of active sites in response to the changes of potential or time can be tracked during catalytic processes, which can be used to quantify the buffering strength, allowing for the direct assessment of the electron buffer effect. In addition, in situ EXAFS can provide insights into the dynamic evolution of the coordination structure of active sites, reflecting changes in the local electronic structure and geometric configuration around metal atoms. In situ XPS allows for the quantitative tracking of valence state distributions in both metal-based buffer materials and active sites, thereby directly revealing the dynamic electron redistribution processes at their interface. Furthermore, in situ infrared and Raman spectroscopy can analyze the evolution of reaction intermediates on the surface of a catalyst. A systematic correlation of these findings with the valence state evolution of active sites enables a direct investigation into the mechanistic role of the electron buffer effect in steering the reaction pathway. Furthermore, constant-potential density functional theory (DFT) combined with ab initio molecular dynamics (AIMD) simulations enables the accurate modeling of dynamic interfacial evolution between the buffer component and active sites under an applied electric field. Analyses of the density of states (DOS), band structure, and differential charge density can directly reveal the electron transfer pathways between buffer components and active sites. Additionally, comparing the adsorption energies of key intermediates with and without the buffer components can elucidate how the electron buffer effect modulates the reaction pathway.
6.4. Expansion and integration strategies for electrocatalytic application
During electrocatalysis, the local pH near the electrode surface can deviate substantially from the bulk value, particularly at high current densities where rapid proton consumption or generation occurs. Such local pH changes may significantly affect the redox behavior of metal-based electron buffer components. Most studies focus on bulk electrolyte pH effects on catalytic activity or product selectivity rather than on the reversible electron transfer dynamics of the buffer component itself. Future investigations on the electron buffer effect should also focus on the microenvironment changes on the surface of the electrode, which can guide the design of robust electrocatalysts for practical high-current-density operation. The electron buffer effect has been extensively applied in the fields of the OER, HER, and ORR. Future research should emphasize its underexplored role in the CO2RR and NRR/NO3RR, where its influence on reaction pathways and product selectivity remains unclear. A mechanistic understanding of these systems is urgently needed. Extending this concept to organic oxidation reactions, particularly the urea oxidation reaction (UOR) and 5-hydroxymethylfurfural oxidation reaction (HMFOR) that proceed via in situ formation of high-valence metal oxyhydroxide species (e.g., NiOOH and CoOOH), represents a promising avenue for further research.106–108 The buffer component in these organic oxidation reactions can act as an electron acceptor, facilitating the oxidation of metal centers and lowering the kinetic barrier for generating active high-valence species. Moreover, as reported by Du et al., NiOOH can undergo over-oxidation to form OER-favored species (e.g., NiO(OH)2 and NiOO2), particularly under conditions of high applied potential or when the concentration of organic molecules adsorbed on the catalyst surface is insufficient.109 Acting as a reversible electron donor, the buffer component can supply electrons to the metal center when its valence state increases excessively. This mechanism ensures that the catalyst remains in its optimal oxidation state for efficient organic molecule activation. Besides, through fine modulation of electron density of active sites, the buffer component tunes the adsorption/desorption behavior of key intermediates, balances surface coverages of organic molecules and OH−, and promotes selective bond activation to direct the reaction toward desired products, for example, favoring C–N bond cleavage over N–H bond cleavage in the UOR for higher Faraday efficiency of NOx− products. Hence, an effective electron buffer component for these organic oxidation reactions should meet two key criteria: intrinsic OER inertness to suppress the competition of the OER and efficient electron storage/release capacity that can dynamically regulate the valence states of active sites. Based on the abovementioned potential for regulation in catalytic reactions, translating the electron buffer effect into practical applications still requires overcoming a series of challenges, necessitating a focused research pathway that moves from scalable material synthesis to practical device validation. To address scalable manufacturing, future work should develop facile synthesis routes for incorporating variable-valence metals into the bulk phase of active components. This approach inherently bypasses the need for complex interfacial control, which is often difficult to scale up. Besides, to validate long-term durability, device-level integration must be assessed by fabricating membrane electrode assemblies and performing accelerated stress tests under operating conditions (e.g., potential cycling). Finally, system-level optimization requires integrating the electron buffer effect with electrode architectures designed for efficient mass and charge transport, supported by economic analysis to assess practical feasibility. This roadmap aims to translate the concept of the electron buffer effect from precisely defined materials into robust and scalable electrochemical devices.
Overcoming the existing bottlenecks hinges on the deep integration of multi-scale theoretical simulations, artificial intelligence, and advanced in situ characterization techniques. By synergistically employing these tools, the accurate prediction and rational design of the electron buffer effect will be ultimately achieved. Through such mechanistic insights and material innovations, the electron buffer effect is expected to drive rapid advancement in electrocatalysis and plays a significant role in energy conversion, chemical production and environmental remediation.
Author contributions
All of the authors contributed to the literature search and writing and editing of this review.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22571275, 21975223, and 52473034), Natural Science Foundation of Zhejiang Province (LZ22B030002 and LQN26B030005), Jinhua Major Project (2024-3-001), Jinhua Key Project (2026-3-098), and Open Research Fund of Key Laboratory of the Ministry of Education for Advanced Catalysis Materials and Zhejiang Key Laboratory for Reactive Chemistry on Solid Surfaces, Zhejiang Normal University (KLMEACM202501).
References
- S. Chu and A. Majumdar, Nature, 2012, 488, 294–303 Search PubMed.
- N. S. Lewis, Science, 2016, 351, aad1920 CrossRef PubMed.
- K. Kawashima, R. A. Marquez, L. A. Smith, R. R. Vaidyula, O. A. Carrasco-Jaim, Z. Wang, Y. J. Son, C. Cao and C. B. Mullins, Chem. Rev., 2023, 123, 12795–13208 CrossRef CAS.
- M. B. Ross, P. De Luna, Y. Li, C. T. Dinh, D. Kim, P. Yang and E. H. Sargent, Nat. Catal., 2019, 2, 648–658 CrossRef CAS.
- H. Ding, H. Liu, W. Chu, C. Wu and Y. Xie, Chem. Rev., 2021, 121, 13174–13212 CrossRef CAS PubMed.
- M. F. Lagadec and A. Grimaud, Nat. Mater., 2020, 19, 1140–1150 Search PubMed.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X. Lin, H. Xu, L. Wang and D. Liu, Science, 2023, 380, 609–616 CrossRef CAS.
- T. Wang, X. Cao and L. Jiao, Angew. Chem., Int. Ed., 2022, 61, e202213328 Search PubMed.
- L. Quan, H. Jiang, G. Mei, Y. Sun and B. You, Chem. Rev., 2024, 124, 3694–3812 Search PubMed.
- L. Yan, P. Li, Q. Zhu, A. Kumar, K. Sun, S. Tian and X. Sun, Chem, 2023, 9, 280–342 Search PubMed.
- H. Wang, X. Kang and B. Han, Chem. Soc. Rev., 2025, 54, 10156–10244 Search PubMed.
- Y. Ren, C. Yu, X. Tan, H. Huang, Q. Wei and J. Qiu, Energy Environ. Sci., 2021, 14, 1176–1193 Search PubMed.
- Y. Xiong, Y. Wang, J. Zhou, F. Liu, F. Hao and Z. Fan, Adv. Mater., 2024, 36, 2304021 CrossRef CAS.
- H. Chem, X. Liang, Y. Liu, X. Ai, T. Asefa and X. Zou, Adv. Mater., 2020, 32, 2002435 CrossRef.
- C. Hu, R. Paul, Q. Dai and L. Dai, Chem. Soc. Rev., 2021, 50, 11785–11843 RSC.
- R. W. Zhang, J. L. Sun, Y. Z. Chen, Q. Shen, C. Ding, S. Zhang and J. Zhang, Coord. Chem. Rev., 2025, 543, 216958 CrossRef CAS.
- H. Wang, L. Chen, H. Pang, S. Kaskel and Q. Xu, Chem. Soc. Rev., 2020, 49, 1414–1448 RSC.
- S. J. Tauster, Acc. Chem. Res., 1987, 20, 389–394 Search PubMed.
- T. Pu, W. Zhang and M. Zhu, Angew. Chem., Int. Ed., 2023, 62, e202212278 CrossRef CAS.
- P. Yin, Q. Yan and H. Liang, Angew. Chem., Int. Ed., 2023, 62, e202302819 CrossRef CAS PubMed.
- S. Lyu, C. Guo, J. Wang, Z. Li, B. Yang, L. Lei, L. Wang, J. Xiao, T. Zhang and Y. Hou, Nat. Commun., 2022, 13, 6171 CrossRef CAS PubMed.
- J. Zheng, L. Huang, C. Cui, Z. Chen, X. Liu, X. Duan, X. Cao, T. Yang, H. Zhu, K. Shi, P. Du, S. Ying, C. Zhu, Y. Yao, G. Guo, Y. Yuan, S. Xie and L. Zheng, Science, 2022, 376, 288–292 CrossRef CAS.
- Y. Zhang, X. Peng, H. Tian, B. Yang, Z. Chen, J. Li, T. Zhang, M. Zhang, X. Liang, Z. Yu, Y. Zhou, L. Zheng, X. Wang, J. Zheng, Y. Tang, C. T. Au, L. Jiang and S. Xie, Nat. Chem., 2024, 16, 1781–1787 CrossRef CAS PubMed.
- M. Zhang, Y. Luo, X. Peng, S. Zhang, L. Zhang, C. Xu, Y. Zhou, L. Zheng, J. Ni, X. Wang, L. Jiang and J. Li, Adv. Funct. Mater., 2025, 35, 2415651 CrossRef CAS.
- Y. Zi, C. Zhang, J. Zhao, Y. Cheng, J. Yuan and J. Hu, Small, 2024, 20, 2406657 Search PubMed.
- Z. Li, J. Yi, Y. Tang, Z. Zhang, C. Li, R. Bao and J. Wang, Energy Environ. Sci., 2025, 18, 7188–7202 Search PubMed.
- Z. Liu, H. Liu, T. Xue, K. Zhou, C. Li, Y. Shen, X. Su, Z. Wu, H. Li, H. Li and C. Li, Nano Lett., 2024, 24, 10899–10907 CrossRef CAS PubMed.
- X. Liu, Y. Wang, J. Liang, S. Li, S. Zhang, D. Su, Z. Cai, Y. Huang, L. Elbaz and Q. Li, J. Am. Chem. Soc., 2024, 146, 2033–2042 CrossRef CAS PubMed.
- F. Qian, D. Cao, S. Chen, Y. Yuan, K. Chen, P. J. Chimtali, H. Liu, W. Jiang, B. Sheng, L. Yi, J. Huang, C. Hu, H. Lei, X. Wu, Z. Wen, Q. Chen and L. Song, Nat. Commun., 2025, 16, 6894 CrossRef CAS.
- Z. Zhao, Y. Zhao, W. Wang, X. Xin, Y. Jiao, A. D. Abell, C. S. Law and A. Santos, Adv. Sci., 2026, 13, e14558 CrossRef CAS.
- Z. Wang, W. Wu, H. Jiang, S. Chen, R. Chen, Y. Zhu, Y. Xiao, H. Lv, J. Zhong and N. Cheng, Adv. Funct. Mater., 2024, 34, 2406347 CrossRef CAS.
- X. Chen, H. Ma, X. Wang, H. Jin, Y. Wu, S. Wang, Y. Xiao, R. Jiang, Y. Da, L. Fan, Y. Sun, S. Xi, Y. Lum, Q. He, H. Li, D. Liu, S. Yang and W. Chen, J. Am. Chem. Soc., 2025, 147, 20600–20611 CrossRef CAS PubMed.
- L. Deng, S. Huang, Z. Lin, Y. Zhang, C. Zhang, Y. Hao, S. Liu, C. Kuo, H. Chen, J. Peng, J. Wang and S. Peng, Adv. Mater., 2023, 35, 2305939 CrossRef CAS PubMed.
- Y. Wang, J. Wang, J. Xu, X. Qu, L. Lin, L. Zhao, Q. Liu, Y. Wei, X. Li, Q. Ma, J. Zhang, W. Fan, B. Wu, X. Kong, J. Huang, Y. Wang, Y. Ye, Y. Feng and F. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202503608 Search PubMed.
- Z. Dong, C. Zhou, W. Chen, F. Lin, H. Luo, Z. Sun, Q. Huang, R. Zeng, Y. Tan, Z. Xiao, H. Huang, K. Wang, M. Luo, F. Lv and S. Guo, Adv. Funct. Mater., 2024, 34, 2400809 CrossRef CAS.
- J. Shang, X. Zhang, Y. Che, X. Qin, Y. Zhang, J. Zhang, B. Li, C. Yang, Y. Jiang, X. Lin and Q. Liu, J. Mater. Chem. A, 2025, 13, 23998–24004 Search PubMed.
- X. Zou, X. Zhao, B. Pang, H. Ma, K. Zeng, S. Zhi and H. Guo, Adv. Mater., 2024, 36, 2412954 Search PubMed.
- M. Li, X. Wang, K. Liu, H. Sun, D. Sun, K. Huang, Y. Tang, W. Xing, H. Li and G. Fu, Adv. Mater., 2023, 35, 2302462 CrossRef CAS.
- X. Wang, Z. Li, H. Jang, C. Chen, S. Liu, L. Wang, M. Kim, J. Cho, Q. Qin and X. Liu, Adv. Energy Mater., 2025, 15, 2403388 CrossRef CAS.
- W. Gou, Z. Xia, X. Tian, Q. Xue, F. Ye, S. Dai, M. Zhang, R. Si, Y. Zou, Y. Ma, J. Ho and Y. Qu, Nano Energy, 2022, 104, 107960 CrossRef CAS.
- H. Yue, S. Huo, X. Wang, M. Wang, H. Sun, Y. Chen, Y. Dai, M. Liu and J. Zou, Mater. Today, 2025, 89, 44–56 CrossRef CAS.
- D. Kim, S. Jo, J. I. Jeon, J. I. Sohn and J. Hong, Energy Environ. Mater., 2025, e70136 Search PubMed.
- Y. Zhi, Z. Li, Y. Tang, J. Peng, Z. Zhang, C. Li, R. Bao, G. Chen, J. Yi, J. Chen, J. Wang and J. Wang, Nano Energy, 2025, 134, 110565 CrossRef CAS.
- J. Chen, M. Aliasgar, F. B. Zamudio, T. Zhang, Y. Zhao, X. Lian, L. Wen, H. Yang, W. Sun, S. M. Kozlov, W. Chen and L. Wang, Nat. Commun., 2023, 14, 1711 CrossRef CAS.
- Y. H. Choi, J. Lee, A. Parija, J. S. Cho, S. V. Verkhoturov, M. Al-Hashimi, L. Fang and S. Banerjee, ACS Catal., 2016, 6, 6246–6254 CrossRef CAS.
- R. Gao, Q. Dai, F. Du, D. Yan and L. Dai, J. Am. Chem. Soc., 2019, 141, 11658–11666 Search PubMed.
- L. Hou, X. Cui, B. Guan, S. Wang, R. Li, Y. Liu, D. Zhu and J. Zheng, Nature, 2022, 606, 507–510 CrossRef CAS PubMed.
- E. Meirzadeh, A. M. Evans, M. Rezaee, M. Milich, C. J. Dionne, T. P. Darlington, S. Bao, A. K. Bartholomew, T. Handa, D. J. Rizzo, R. A. Wiscons, M. Reza, A. Zangiabadi, N. Fardian-Melamed, A. C. Crowther, P. J. Schuck, D. N. Basov, X. Zhu, A. Giri, P. E. Hopkins, P. Kim, M. L. Steigerwald, J. Yang, C. Nuckolls and X. Roy, Nature, 2023, 613, 71–76 CrossRef CAS PubMed.
- X. Wang, X. Chen, R. Lv, H. Ma, Y. Yao, H. Jin, S. Qiao, Y. Sun, D. Liu, L. Song, P. Du, W. Chen, Y. Lu and S. Yang, Small, 2025, 21, 2506131 CrossRef CAS.
- Q. Tang, L. Wang, S. Zhang, P. Xue, Y. Zhang, H. Li and D. Zhu, J. Mater. Chem. A, 2025, 13, 5229–5237 RSC.
- Y. Li, T. Xu, Q. Huang, L. Zhu, Y. Yan, P. Peng and F. Li, ACS Catal., 2023, 13, 7597–7605 CrossRef CAS.
- Q. Li, X. Wang, Z. Xie, X. Peng, L. Guo, X. Yu, X. Yang, Z. Lu, X. Zhang and L. Li, Appl. Catal., B, 2022, 350, 121020 CrossRef.
- Y. Lu, B. Li, N. Xu, Z. Zhou, Y. Xiao, Y. Jiang, T. Li, S. Hu, Y. Gong and Y. Cao, Nat. Commun., 2023, 14, 6965 CrossRef CAS PubMed.
- Y. Hao, S. Hung, C. Tian, L. Wang, Y. Chen, S. Zhao, K. Peng, C. Zhang, Y. Zhang, C. Kuo, H. Chen and S. Peng, Angew. Chem., Int. Ed., 2024, 63, e202402018 Search PubMed.
- L. Wang, S. Hung, S. Zhao, Y. Wang, S. Bi, S. Li, J. Ma, C. Zhang, Y. Zhang, L. Li, T. Chen, H. Chen, F. Hu, Y. Wu and S. Peng, Nat. Commun., 2025, 16, 3502 Search PubMed.
- J. Huang, C. N. Borca, T. Huthwelker, N. S. Yuezbasi, D. Baster, M. El Kazzi, C. W. Schneider, T. J. Schmidt and E. Fabbri, Nat. Commun., 2024, 15, 3067 Search PubMed.
- H. Wang, T. Zhai, Y. Wu, T. Zhou, B. Zhou, C. Shang and Z. Guo, Adv. Sci., 2023, 10, 2301706 Search PubMed.
- Z. Huang, J. Song, S. Dou, X. Li, J. Wang and X. Wang, Matter, 2019, 1, 1494–1518 Search PubMed.
- H. Jin, X. Liu, P. An, C. Tang, H. Yu, Q. Zhang, H. Peng, L. Gu, Y. Zheng, T. Song, K. Davey, U. Paik, J. Dong and S. Qiao, Nat. Commun., 2023, 14, 354 Search PubMed.
- L. Jiang, J. Chen, Y. Zhou, J. Liu, F. Liu, Z. Zhang, J. Qin, Y. Lai and J. Li, ACS Appl. Mater. Interfaces, 2025, 17, 47047–47056 CrossRef CAS PubMed.
- Y. Wang, K. Wei, Y. Song, A. A. Obisanya, H. Li, J. Wang, H. Li and F. Gao, Chem. Eng. J., 2024, 497, 154724 Search PubMed.
- X. Yang, S. Li, Y. Zhang, F. Qiu, Y. Sun, W. Ning, Q. Tao, W. Li and S. Miao, J. Mater. Chem. A, 2024, 12, 17574–17585 Search PubMed.
- H. Jin, S. Choi, G. J. Bang, T. Kwon, H. S. Kim, S. J. Lee, Y. Hong, D. W. Lee, H. S. Park, H. Baik, Y. Jung, S. J. Yoo and K. Lee, Energy Environ. Sci., 2022, 15, 1119–1130 Search PubMed.
- M. N. Hossain, L. Zhang, R. Neagu and S. Sun, Chem. Soc. Rev., 2025, 54, 3323–3386 Search PubMed.
- C. Xu, H. Yu, H. Huang, S. Li, Y. Cao, W. Peng, Y. Li, H. Ke, S. Xu, H. Mo, C. Wu, H. Wang, Y. Zhang, X. Li and W. Chen, Angew. Chem., Int. Ed., 2025, 64, e202504667 CrossRef CAS.
- M. Jiang, J. Xu, Q. Zhou, Y. Chen, P. Munroe, L. Li, Z. Xie, Y. Wu and S. Peng, Angew. Chem., Int. Ed., 2025, 64, e202510259 Search PubMed.
- M. Maji, S. Dutta, R. Jena, A. Dey, T. K. Maji, S. K. Pati and S. Bhattacharyya, Angew. Chem., Int. Ed., 2024, 63, e202403697 Search PubMed.
- L. Xu, Y. Xu, B. Xia, B. Guo, K. M. Kamal, B. Likozar, X. Li, F. Dong, S. Li and Y. Ma, Adv. Mater., 2025, 37, 2502989 Search PubMed.
- X. Wang, T. Liu, J. Wang, B. Xia, Y. Wang, Z. Li, Z. Zhang and F. Wang, J. Mater. Chem. A, 2025, 13, 14964–14971 RSC.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Norskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- Y. Pan, M. Li, W. Mi, M. Wang, J. Li, Y. Zhao, X. Ma, B. Wang, W. Zhu, Z. Cui, H. Yin and Y. Liu, Nano Res., 2022, 15, 7976–7985 Search PubMed.
- M. Zhou, H.-L. Wang and S. Guo, Chem. Soc. Rev., 2016, 45, 1273–1307 RSC.
- X. Mou, Z. Zhang, Z. Zhao, B. Liu, X. Wang, C. Yang, L. Shen, Y. Zhang, L. Zhao, W. Qu and Z. Wang, Int. J. Hydrogen Energy, 2025, 182, 151698 CrossRef CAS.
- Y. Xie, Y. Feng, S. Pan, Q. Xu, Z. Yang, J. Zhang, X. Fu and F. Luo, Adv. Energy Mater., 2026, e71096 CrossRef.
- H. Yu, C. Li, Y. Lei and Z. Xiang, Angew. Chem., Int. Ed., 2025, 64, e202508141 CrossRef CAS PubMed.
- N. Li, K. Guo, S. Lu, L. Bao, Z. Yu and X. Lu, Chem. Commun., 2024, 60, 11964–11967 RSC.
- R. Jin, C. Guo, Y. Si, Y. Liu, L. Teng, F. Fu, Y. Jian, X. Dong and T. G. Cherkasova, J. Mater. Chem. A, 2026, 14, 14499–14512 RSC.
- Y. Liu, Z. Wang, S. Cao, F. Liu, Q. Tan, Y. Chen and W. Wang, J. Mater. Chem. A, 2026, 14, 20394–20404 RSC.
- M. Li, X. Wang, K. Liu, Z. Zhu, H. Guo, M. Li, H. Du, D. Sun, H. Li, K. Huang, Y. Tang and G. Fu, Adv. Energy Mater., 2023, 13, 2301162 Search PubMed.
- R. Cheng, X. He, M. Jiang, X. Shao, W. Tang, B. Ran, H. Li and C. Fu, Adv. Funct. Mater., 2025, 35, 2425138 CrossRef CAS.
- T. Yan, X. Chen, L. Kumari, J. Lin, M. Li, Q. Fan, H. Chi, T. J. Meyer, S. Zhang and X. Ma, Chem. Rev., 2023, 123, 10530–10583 CrossRef CAS PubMed.
- J. He, J. Hua, Z. Wang, Y. Xia, C. Shao and K. Dai, Small, 2025, 21, 2503852 CrossRef CAS PubMed.
- Y. Xiao, J. Chen, G. Chen, X. Geng, L. Fan, Y. Ding, M. Wang, H. Jin, S. Xi, X. Chen and W. Chen, Small, 2026, 22, e14087 CrossRef CAS PubMed.
- W. Zhang, Y. Wang, L. Cui, M. Wang and K. Yu, J. Mater. Chem. A, 2026, 14, 17316–17327 RSC.
- S. Wen, J. Wen, M. Zhao, Z. Huang, L. Gao and Z. He, J. Environ. Chem. Eng., 2026, 14, 123311 CrossRef CAS.
- X. Xiong, X. Wu, Y. Cheng, D. Yu, X. Xu, Y. Cheng, F. Wu and X. Wei, Chem. Commun., 2025, 61, 1681–1684 RSC.
- X. He, L. Lin, X. Li, M. Zhu, Q. Zhang, S. Xie, B. Mei, F. Sun, Z. Jiang, J. Cheng and Y. Wang, Nat. Commun., 2024, 15, 9923 CrossRef CAS.
- W. Ni, C. Li, X. Zang, M. Xu, S. Huo, M. Liu, Z. Yang and Y. Yan, Appl. Catal., B, 2019, 259, 118044 CrossRef CAS.
- B. Zhao, F. Chen, C. Cheng, L. Li, C. Liu and B. Zhang, Adv. Energy Mater., 2023, 13, 2204346 CrossRef CAS.
- X. Zhi, A. Vasileff, Y. Zheng, Y. Jiao and S. Qiao, Energy Environ. Sci., 2021, 14, 3912–3930 RSC.
- H. Xiao, W. A. Goddard III, T. Cheng and Y. Liu, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 6685–6688 CrossRef CAS PubMed.
- E. Bertheussen, A. Verdaguer-Casadevall, D. Ravasio, J. H. Montoya, D. B. Trimarco, C. Roy, S. Meier, J. Wendland, J. K. Norskov, I. E. L. Stephens and I. Chorkendorff, Angew. Chem., Int. Ed., 2016, 55, 1450–1454 CrossRef CAS.
- N. Chen, X. Deng, Z. Chen, P. Du, X. Liu, J. Liu, B. Hong, R. Qin, J. Zheng, J. Li, S. Xie and Y. Yuan, ACS Nano, 2025, 19, 41658–41668 Search PubMed.
- J. Wang, J. Li, F. Wang, X. Zhang, S. Liu, S. Niu, L. Chen and X. Wu, Appl. Catal., B, 2026, 395, 126871 CrossRef CAS.
- Y. Pang, C. Su, G. Jia, L. Xu and Z. Shao, Chem. Soc. Rev., 2021, 50, 12744–12787 RSC.
- L. Jiang, X. Zhi, X. Bai and Y. Jiao, J. Am. Chem. Soc., 2025, 147, 16935–16947 CrossRef CAS PubMed.
- T. Zhang, Z. Che, Y. Song, R. Yao, J. Li, Y. Sun and G. Liu, Angew. Chem., Int. Ed., 2025, 64, e202514028 CrossRef CAS.
- H. Cai, J. Wang, Y. Guo and Q. Pan, Appl. Surf. Sci., 2026, 720, 165232 CrossRef CAS.
- C. He, L. Wang and W. Zhang, Appl. Surf. Sci., 2026, 726, 165867 CrossRef CAS.
- G. Zhang, H. Xu, Y. Li, C. Xiang, Q. Ji, H. Liu, J. Qu and J. Li, Adv. Sci., 2019, 6, 1901627 CrossRef CAS PubMed.
- Z. Kou, D. Shi, B. Yang, Z. Li, Q. Zhang, J. Lu, T. Zhang, L. Lei, Y. Li, L. Dai and Y. Hou, Chem. Soc. Rev., 2025, 54, 10796–10844 RSC.
- H. Zhang, K. Fang, J. Yang, H. Chen, J. Ning, H. Wang and Y. Hu, Coord. Chem. Rev., 2024, 506, 215723 CrossRef CAS.
- G. Li, X. Wang, H. Pang and H. Ma, Adv. Sci., 2026, 13, e21317 CrossRef CAS PubMed.
- R. Xia, W. Wang, Y. Zhou, Q. Guan, Y. Liu and W. Li, Adv. Funct. Mater., 2025, 35, 2501079 CrossRef.
- H. Huang, Y. Zhang, W. Chen, J. Chen, X. Zou, J. Lv, X. Chen, Z. Shen, Z. Ge, L. Guo, Y. Yao and Y. Wang, Adv. Energy Mater., 2025, 15, 2405534 Search PubMed.
- J. Li, C. Yin, S. Wang, B. Zhang and L. Feng, Chem. Sci., 2024, 15, 13659–13667 RSC.
- C. Yin, F. Yang, S. Wang and L. Feng, Adv. Energy Mater., 2025, 15, e04064 CrossRef.
- C. Yin, F. Yang, T. Liu, S. Wang and L. Feng, Adv. Energy Mater., 2026, 16, e70876 CrossRef.
- R. Luo, Y. Li, L. Xing, R. Zhong, Z. Qian, G. Yin, Y. Wang and L. Du, Appl. Catal., B, 2022, 311, 121357 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *acd,
Xiwen Huangd,
Lin Xuc,
Xiaofeng Lu
*acd,
Xiwen Huangd,
Lin Xuc,
Xiaofeng Lu









