DOI:
10.1039/D6SC02556K
(Edge Article)
Chem. Sci., 2026, Advance Article
Exploiting singlet fission in para-azaquinodimethane nanoaggregates with high energy triplets to trigger phototoxic reactive oxygen species
Received
27th March 2026
, Accepted 8th June 2026
First published on 9th June 2026
Abstract
In this study, aggregation-induced singlet fission (SF) was unveiled in water-dispersed organic nanoaggregates of push–pull para-azaquinodimethane (pAQM) derivatives exhibiting either asymmetrical (AsOMe and AsNMe2) or symmetrical (TPh and TPhOMe) molecular structures. Our ultrafast spectroscopic experiments revealed that SF occurs in a few to tens of picoseconds in these organic nanoparticles. The nanosecond and femtosecond transient absorption results showed quantitative SF for aggregates of three of the pAQMs, with triplet quantum yields of ca. 200% in water. In most cases, spherical nanoparticles with diameters of tens of nanometers were produced in an aqueous environment, and they could be successfully internalized within prostatic cancer and melanoma cells. The outstanding phototoxicity exhibited toward these cancer cells by the AsOMe nanoaggregates, with the highest production of ROS among the investigated compounds, was demonstrated to be due to their highest triplet energy (1.3 eV), which favours the energy transfer pathway to produce singlet oxygen. All the investigated molecules were also found to exhibit significant two-photon excited fluorescence, with their two-photon absorption cross sections being in trend with their symmetry and intramolecular charge transfer features. To the best of our knowledge, this is the first study highlighting the use of unconventional SF chromophores as new effective photosensitizers for two-photon triggered photodynamic therapy.
Introduction
Singlet fission (SF) is a downconversion process through which a high-energy singlet excited state (S1) couples to the ground state of a nearby chromophore (S0) to give two triplet excited states of roughly half the energy (T1 + T1).1–4 Thus, SF is a multiexciton generation phenomenon where two triplet excitons are produced upon absorption of a single photon, resulting in a maximum triplet quantum yield of 200%. SF is a spin-allowed triplet production mechanism as it proceeds through a peculiar biexcitonic intermediate, the correlated triplet pair or double triplet state 1(TT), where the two triplets are coupled together into an overall singlet transient species.5–8 SF has been generally observed in acene derivatives (e.g., pentacene and tetracene) which satisfy the critical thermodynamic condition required, namely E(S1) ≥ 2E(T1).9–22 However, acene-based materials show easy degradation in the presence of light and air as well as poor light absorption, which limits their potential for practical use. SF materials have been mainly investigated so far in the solar cell research field: (1) to overcome the Shockley–Queisser limit by reducing the thermalization losses in single-junction silicon solar cells or (2) to allow multiple charge extraction to electron acceptors in all-organic solar cells.23–27 The high yield (up to 200%) and ultrafast rate (in the picosecond time window) of triplet generation allowed by SF appears to be a highly promising alternative strategy to intersystem crossing for the design of effective all-organic, heavy-atom-free photosensitizers for photodynamic therapy (PDT).28 To date, the use of SF chromophores in PDT is largely unexplored in the literature and is limited to very few and very recent studies concerning acene derivatives.29–33 It has to be noted that, for implementation in photovoltaic devices and PDT, SF chromophores should show high triplet energy to be exploited as photosensitizers for lower band gap semiconductors (silicon 1.12 eV) or to produce singlet oxygen (0.98 eV).32,34–37
Our group has recently reported the synthesis, according to a green protocol, and photophysical study of new unconventional singlet fission chromophores38,39 exhibiting asymmetrical (AsOMe and AsNMe2 in Scheme 1) and symmetrical (TPh and TPhOMe in Scheme 1) quinoidal structures with different push–pull degrees due to the introduction of electron donor groups of different strengths (the weaker methoxy OMe and the stronger dimethylamino NMe2 substituents). In a previous study,39 such molecules were investigated in solution and thin films, where intermolecular SF was demonstrated. Time-resolved electron paramagnetic resonance (EPR) experiments highlighted the presence of the quintet correlated triplet pair intermediate 5(TT) in the solid state films, with significant implications for possible applications in quantum information science and technology.40,41 On the other hand, the SF-generated higher-energy triplet evidenced by the time-resolved optical study in films for AsOMe (1.3 eV), relative to the other molecules (1.1 eV for AsNMe2 and 1.2 eV for TPh/TPhOMe), makes the former particularly promising for SF silicon-matched photovoltaics. Triggered by these interesting results, we here inquire into the potential of these new unconventional SF chromophores as photosensitizers for reactive oxygen species (ROS) photoproduction. In the present investigation, we activated intermolecular SF upon aggregation by dispersing these aromatic molecules in a bad solvent such as water.42 We investigated the excited-state dynamics and the mechanism of triplet production in such water-dispersed organic nanoaggregates through nanosecond and femtosecond transient absorption spectroscopy. Through these advanced spectroscopic experiments, we evaluated the yield and rate of triplet generation. The dimensional and morphological features of the produced organic nanoaggregates were studied through Dynamic Light Scattering (DLS) and Scanning Electron Microscopy (SEM) measurements. Finally, the internalization of these organic nanoparticles within cancer cell lines (namely LN-CaP and MEL-501) was highlighted through fluorescence microscopy: dark and photoinduced cytotoxicity toward these tumour cells was evaluated through the MTT assay and quantified by the assessment of the IC50 values.43–45 Fluorimetric assays using specific probes46,47 were also carried out in a cell-free environment to test the photoproduction of selected ROS, i.e. singlet oxygen  and the oxygen radical anion (O2˙−), by the quinoidal organic nanoaggregates. To the best of our knowledge, the present study is the first attempt to employ unconventional SF chromophores, characterized by remarkable stability and high triplet energy, as novel photosensitizers for PDT.
and the oxygen radical anion (O2˙−), by the quinoidal organic nanoaggregates. To the best of our knowledge, the present study is the first attempt to employ unconventional SF chromophores, characterized by remarkable stability and high triplet energy, as novel photosensitizers for PDT.
 |
| | Scheme 1 Molecular structures of the investigated compounds. | |
Results and discussion
Linear and nonlinear optical properties
The para-azaquinodimethane (pAQM) derivatives were obtained following synthetic procedures previously reported.38,39 The water-dispersed nanoaggregates (NAs) were prepared through the reprecipitation method by injection into water of a concentrated solution of the dyes in dimethylsulphoxide (DMSO) or tetrahydrofuran (THF). The presence of water, being a bad solvent for these aromatic molecules, induces the formation of nanoaggregates.42,48 Absorption and emission spectra were recorded for the investigated compounds in dimethylsulphoxide (DMSO) solution and as nanoaggregates in water dispersion. Fig. 1 highlights the results obtained for AsOMe, while Fig. S1–S3 report in detail what was observed for all the pAQM derivatives. The absorption spectrum for the AsOMe chromophore in DMSO peaks at ca. 480 nm. When AsOMe was investigated as water-dispersed nanoaggregates, the absorption spectrum showed a main peak at ca. 450 nm assigned to H-aggregates and an apparent shoulder at ca. 510 nm likely due to J-aggregates. A similar behaviour was observed for the absorption of the other nanoaggregates. The emission spectra in water dispersion exhibited a large red shift relative to the DMSO solution spectra. Table S2 reports the absorption and emission spectral shifts observed for the four investigated compounds when passing from the monomer in DMSO solution to the J-aggregates in water. The smallest spectral shifts within the series are obtained for AsNMe2, both in absorption and emission.
 |
| | Fig. 1 Normalized absorption (a) and emission (b) spectra of AsOMe in DMSO solution (black) and of AsOMe nanoaggregates in water dispersion (red). | |
The fluorescence quantum yield measured for the compounds in the polar DMSO solvent was found to be low (Table 1), lower for the asymmetric (6.6% for AsOMe and 1.4% for AsNMe2) than for the symmetric derivatives (15.6% for TPh and 15.8% for TPhOMe). This may be due to the larger push–pull excited state degree of the asymmetric dipolar systems, as proven by their larger dipole moment difference between the excited and the ground state obtained in our previous study.39 When aggregates were formed in water, the fluorescence quantum yield was found to be significantly quenched relative to the value obtained in DMSO for the symmetric molecules (0.04% for TPh and 0.06% for TPhOMe) as well as for AsOMe (0.32%) and AsNMe2 (0.24%). Indeed, moving from the monomers in DMSO to the aggregates, 95% fluorescence quenching was observed for AsOMe, while 99.8% and 99.6% quenching for TPh and TPhOMe, respectively. On the other hand, a fluorescence quenching of only 83% was observed for AsNMe2 when moving from DMSO solution to water dispersion. The fluorescence kinetics in DMSO, recorded by nanosecond time-correlated single photon counting, exhibited decays longer than the Instrumental Response Function (IRF) for TPh and TPhOMe (Fig. S4), resulting in fluorescence lifetimes of 1.03 and 0.77 ns, respectively (Table S1); whereas the asymmetric compounds showed lifetimes shorter than the IRF and were evaluated from the fs-TA experiments (Table S1). The generally short fluorescence lifetimes implied fluorescence rate constants ranging from 1.4 × 108 to 2.1 × 108 s−1 for all the compounds in DMSO (Table S1). For the water-dispersed nanoaggregates, the fluorescence kinetics were generally found to resemble the IRF (Fig. S4) and no significant excitation/emission wavelength effect was observed on the fluorescence decay (Fig. S5).
Table 1 Aggregation effect on the fluorescence quantum yields
Two-photon excited fluorescence measurements were performed for the investigated molecules in solution upon exciting the samples with an amplified femtosecond laser at 800 nm in the NIR region. Fluorescence emission spectra resembling those recorded through steady state fluorescence techniques were recorded for the four dyes in toluene solution (Fig. 2), as generally more intense emission was detected in nonpolar media. A linear trend in the logarithmic plot of the fluorescence as a function of the laser power was found, exhibiting a slope of ca. 2 (Fig. 2). This result proves the biphotonic and nonlinear excitation of the sample fluorescence. Two-photon absorption cross sections at 800 nm were estimated through the comparative method by using rhodamine B in ethanol as a reference compound42,49,50 (Table in Fig. 2). Interestingly, higher cross sections were generally measured for the symmetric relative to the asymmetric compounds.51 Moreover, larger cross sections were found for the compounds bearing a stronger electron donor group when similar molecular structures are compared: a larger cross section was found for TPhOMe (113 GM) relative to TPh (29 GM), and for AsNMe2 (46 GM) relative to AsOMe (22 GM).52 Due to the remarkable fluorescence quenching observed for the pAQM compounds upon aggregation, it was not possible to obtain with our two-photon excited fluorescence set-up the cross sections for these nanoaggregates in water, which are generally expected to be enhanced relative to the isolated molecules.53–55
 |
| | Fig. 2 Upper graph: Normalized two-photon excited fluorescence emission spectra for the investigated compounds in toluene solution. Lower graph: Logarithmic plot of the fluorescence (area of the emission spectrum recorded upon femtosecond laser excitation at 800 nm) as a function of the laser power for TPhOMe in toluene solution. The table reports the two-photon absorption cross sections calculated for the investigated compounds in toluene using rhodamine B in ethanol as the reference compound. | |
Triplet properties
Nanosecond transient absorption measurements were carried out on DMSO solutions and water-dispersed aggregates of the four compounds. In DMSO, a significant transient signal was revealed for AsOMe, TPh, and TPhOMe (Fig. 3a and S6) characterized by a broad excited-state absorption peaked at ca. 610–630 nm and a negative ground-state bleaching band below 550 nm, while no significant absorption was detected for AsNMe2. This transient exhibited a lifetime strongly dependent upon the presence of molecular oxygen, going from hundreds of nanoseconds in air-equilibrated solution to tens of microseconds in nitrogen-purged solution (Table 2). This evidence, together with the effective production of this species in sensitization experiments,39 allowed such a transient to be assigned to the lowest excited triplet state T1. The sensitization experiments, combined with relative actinometry measurements, enabled the triplet yield for such pAQM compounds in DMSO solution to be evaluated at around 10% (Table 2). Interestingly, a transient species characterized by a similar absorption spectrum (Fig. 3b and S7) and by a lifetime of ca. 1 µs (Table 2) was also revealed in water dispersion for all the investigated compounds, suggesting that efficient triplet production occurs in the organic nanoparticles. Detailed sensitization experiments carried out in a previous study39 allowed the triplet energy to be estimated at ca. 1.3 eV for AsOMe, ca. 1.1 eV for AsNMe2, and ca. 1.2 eV for TPh/TPhOMe. These experimental results (Table 3), together with the computational predictions about the excited singlet and triplet state energies, demonstrated SF to be thermodynamically allowed for these molecules: −0.05 eV < ΔETT–S = 2ET − ES < +0.20 eV.
 |
| | Fig. 3 Nanosecond transient absorption spectra of AsOMe in DMSO solution (a) and of AsOMe nanoaggregates in water dispersion (b). | |
Table 2 Triplet properties of the investigated compounds in DMSO solution and as water-dispersed nanoaggregates
| From fs-TA. |
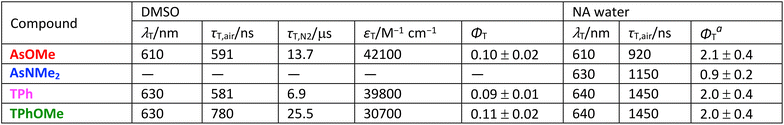 |
Table 3 Experimental energies for the excited states (ES singlet and ET triplet).39
| ΔETT–S = 2ET − ES. |
 |
Femtosecond transient absorption
The mechanism of triplet production for the investigated compounds in DMSO solution and in water-dispersed nanoaggregates was investigated via femtosecond transient absorption measurements. In DMSO (Fig. S8–S11), the singlet excited-state absorption spectrum, which peaked at ca. 700 nm, was found to evolve in hundreds of picoseconds (310 ps for AsOMe, 75 ps for AsNMe2, 810 ps for TPh, and 700 ps for TPhOMe) to give the triplet excited-state absorption at ca. 610–630 nm (revealed as a residual transient characterized by an infinite lifetime, Inf, in the ultrafast experiments and spectral features resembling those obtained for the triplet in the ns-TA experiments). This dynamics is compatible with intersystem crossing for the investigated push–pull molecules in solution (Table 4).39 When the excited-state dynamics was investigated in water-dispersed aggregates, a faster triplet formation was observed in all cases. As shown in Fig. 4 for AsOMe upon 500 nm excitation as a representative example, the Global Analysis revealed the presence of four exponential components: the first exhibiting a spectral shape resembling the singlet excited-state absorption and the other components exhibiting triplet-like Evolution Associated Spectra (EAS). The first transient was assigned to the S1 state decay occurring in ca. 700 fs (Table 4). The Inf component was assigned to the T1 state. The second and third exponential components, with lifetimes typical of a singlet excited state and a triplet-like absorption spectrum (while still retaining some S1 absorption spectral features), were associated with the correlated triplet pair intermediates between S1 and T1: the spin entangled spatially correlated 1(TT) and the spin entangled but spatially decorrelated 1(T⋯T). Similar results were obtained for all the investigated compounds as water-dispersed nanoaggregates upon 500 nm excitation (Fig. S16–S19): in the case of AsOMe, TPh, and TPhOMe, a 600–800 fs lifetime was revealed for S1 and a ca. 9 ps lifetime for 1(TT) (Table 4). Relatively slower excited state dynamics were instead observed for AsNMe2 (8 ps lifetime for S1 and 400 ps for 1(TT), Table 4). Our results demonstrate the occurrence of intermolecular SF triggered by aggregation in the organic nanoparticles produced in water for the investigated molecules. The significant intramolecular charge transfer degree of the pAQM push–pull compounds may play a role in favoring SF, thus accelerating the dynamics of triplet production via virtual intramolecular charge transfer states.56–60 The femtosecond transient absorption experiments were also performed for the nanoaggregates by using a 400 nm pump. At this wavelength, the absorbance of the nanoaggregates produced by the reprecipitation method was lower than that at 500 nm, resulting in a lower signal-to-noise ratio of the transient absorption data (Fig. S12–S15). While the 500 nm pump likely leads to the preferential J-aggregate excitation, particularly in the case of AsOMe, excitation at 400 nm results in a more important photoselection of the H-aggregates is realized. Relatively slower SF dynamics occur when a 400 nm laser excitation is employed: the correlated triplet pair is formed in tens of picoseconds for aggregates of AsOMe, TPh, and TPhOMe, while appearing in hundreds of femtoseconds for the same samples at 500 nm (Table 4). It can also be noticed that the kinetics recorded in correspondence with the J-aggregate ground state bleaching (540 nm for AsOMe and 560–580 nm for the other chromophores) do not show the conventional regular decay of the negative signal to zero with time. On the other hand, they exhibit the typical features expected for the SF dynamics: ground state bleach enhancement as the intermolecular SF proceeds due to the consumption of a second ground state belonging to a nearby chromophore in the nanoaggregates (Fig. S20). These experimental observations altogether suggest that SF occurs within the J-aggregates of the investigated compounds. Upon 400 nm excitation, it may be possible that an energy transfer from the H- to the J-aggregates occurs, followed by SF.38 Indeed, the prevalence of H-aggregates in water dispersion relative to the more important production of J-aggregates in films led to slower SF in the aqueous nanoaggregates (occurring in 5–27 ps) than previously observed in the films (occurring in 1–2 ps) upon the same 400 nm laser excitation.38,39,42
Table 4 Femtosecond transient absorption results for all the investigated compounds in DMSO and as water-dispersed nanoaggregates: τTA lifetimes resulting from Global Analysis and relative assignment. Measurements were performed at 400 nm with a 300 µJ cm−2 laser fluence and at 500 nm with a 190 µJ cm−2 laser fluence
 |
| | Fig. 4 Left: Results of Global Analysis of the femtosecond transient absorption data for AsOMe water-dispersed nanoaggregates obtained upon photoexcitation at 500 nm with a 65 µJ cm−2 laser fluence in terms of Evolution Associated Spectra (EAS). Right: Population dynamics of the excited singlet and triplet states for AsOMe nanoaggregates in water. | |
Detailed analysis of the femtosecond transient absorption data allowed an estimate of the triplet yield to be obtained for the water-dispersed nanoaggregates of these pAQM compounds (see Fig. 4 and the relative SI section).61–64 Interestingly, upon 400 nm excitation the triplet yield was found to be ca. 200% for AsOMe, TPh, and TPhOMe, confirming multiexciton triplet generation for aggregates of these molecules (Table 2). On the other hand, a triplet yield of ca. 90% was obtained for AsNMe2 upon 400 nm excitation, in agreement with the slowest SF observed in water. This result also seems to be consistent with the smallest fluorescence quenching experienced by this compound among the investigated molecules upon aggregation. Such an experimental body of evidence suggests less effective aggregation for AsNMe2 in water relative to the analogous investigated compounds under our experimental conditions. This may be due to its less hydrophobic and more dipolar molecular structure, because of the asymmetric backbone and the presence of the strongest electron donor NMe2 group.39
Experiments with a 500 nm pump were also performed at two different excitation laser fluences of 190 µJ cm−2 and 65 µJ cm−2 for the case of AsOMe and TPhOMe nanoaggregates. The results obtained from global fitting of these data, in terms of Evolution Associated Spectra and lifetimes, are shown in Fig. S21. It is apparent that the spectral shape of the first transient species with a lifetime of ca. 600–700 fs assigned to the singlet excited state S1 is different when derived at the two distinct laser powers. This may be an indication of some singlet–singlet annhilation occurring under our experimental conditions and opening a deactivation pathway competitive with SF.65 Actually, the triplet quantum yields obtained for the case of AsOMe nanoaggregates from these data were 90 ± 20% at 190 µJ cm−2 (number of excited chromophores per cm3: 2.9 × 1014) and 220 ± 40% at 65 µJ cm−2 (number of excited chromophores per cm3: 1.0 × 1014), as detailed in the corresponding section of the SI. These results suggest that, when excited with a higher laser fluence some singlet excited states might be deactivated via singlet–singlet annhilation and thus not be available for the SF. However, at a lower laser fluence, SF occurs quantitatively for the investigated AsOMe nanoaggregates. Due to the lower absorbance at 400 nm, the femtosecond transient absorption results obtained with a 400 nm pump (number of excited chromophores per cm3: 1.2 × 1014) were indeed not significantly affected by singlet–singlet annhilation processes, as also suggested by the S1 EAS shape obtained from Global Analysis and the 210 ± 40% triplet yield estimated under these experimental conditions for the AsOMe nanoaggregates with the same method.
Dimensional and morphological characterization of the nanoaggregates
The dimensional features of the nanoaggregates prepared for the photophysical and photobiological study (upon injection of a concentrated DMSO solution into water, DMSO![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water V/V 1:99) were investigated through Dynamic Light Scattering (DLS) experiments. The obtained size distributions (Fig. S52) revealed two main populations for the AsOMe, TPh, and TPhOMe nanoaggregates: one characterized by diameters of tens of nanometers (30–70 nm) and another characterized by diameters of hundreds of nanometers (250–480 nm). The details are reported in Table S5. In the case of AsNMe2, a different behavior was observed, with a single peak in the size distribution at 47 nm. The smaller nanoaggregates obtained in this case may be due to the more dipolar and less hydrophobic molecular structure of AsNMe2. When the nanoaggregates were prepared upon injecting into water a concentrated tetrahydrofuran (THF) solution, also with the help of probe sonication, a single size population peaking around 100–200 nm was revealed by the DLS measurements (Fig. 5 and S53). In fact, THF is a solvent in which the pAQM compounds were more readily soluble than in DMSO, thus determining a more homogenous size distribution of the nanoparticles. Unfortunately, THF shows lower biocompatibility than DMSO, and so it was considered not suitable for the biological study.
:water V/V 1:99) were investigated through Dynamic Light Scattering (DLS) experiments. The obtained size distributions (Fig. S52) revealed two main populations for the AsOMe, TPh, and TPhOMe nanoaggregates: one characterized by diameters of tens of nanometers (30–70 nm) and another characterized by diameters of hundreds of nanometers (250–480 nm). The details are reported in Table S5. In the case of AsNMe2, a different behavior was observed, with a single peak in the size distribution at 47 nm. The smaller nanoaggregates obtained in this case may be due to the more dipolar and less hydrophobic molecular structure of AsNMe2. When the nanoaggregates were prepared upon injecting into water a concentrated tetrahydrofuran (THF) solution, also with the help of probe sonication, a single size population peaking around 100–200 nm was revealed by the DLS measurements (Fig. 5 and S53). In fact, THF is a solvent in which the pAQM compounds were more readily soluble than in DMSO, thus determining a more homogenous size distribution of the nanoparticles. Unfortunately, THF shows lower biocompatibility than DMSO, and so it was considered not suitable for the biological study.
 |
| | Fig. 5 Dynamic Light Scattering (DLS) results showing the size distribution of the nanoaggregates for all the compounds prepared upon injection of a concentrated THF solution into water. | |
Information about the dimensional as well as the morphological features of the produced nanoparticles was gained through Scanning Electron Microscopy (SEM) images acquired on the water-dispersed nanoaggregates deposited on silicon supports upon drop casting (Fig. S54–S57) at low voltage (1 kV). A few representative images obtained for aggregates of the four investigated molecules are reported in Fig. 6. These images highlight the formation of generally spherical nanoparticles with sizes of tens of nanometers (Fig. S58). The presence of larger clusters produced by agglomeration of such nanoparticles was also highlighted for AsOMe, TPh, and TPhOMe, while more isolated objects were generally observed for the more hydrophilic AsNMe2. While the size distributions obtained by analyzing the SEM images reflect the dimensions of the individual dry nanoparticles (30–60 nm), the ones obtained by DLS experiments show the ensemble behaviour of each water-dispersed sample, which also takes into account the larger aggregates of nanoparticles and the associated hydration shell (130–250 nm).
 |
| | Fig. 6 SEM images at 250 K magnification of water-dispersed nanoaggregates. | |
Phototoxicity toward cancer cells
The size of the produced nanoaggregates was revealed to be compatible with their inclusion within cells. Indeed, fluorescence microscopy showed that all the pAQM nanoparticles could be successfully internalized within prostatic cancer (LN-CaP) and melanoma (MEL-501) cells (Fig. 7, S59 and S61). In particular, bright red fluorescence due to the nanoaggregates was observed in the cytoplasmic and perinuclear cell regions. This could be unambiguously highlighted when the cells were co-stained with the blue-emitting DAPI dye as a fluorescent nuclear marker.
 |
| | Fig. 7 Representative fluorescence microscopy images of fixed LN-CaP cells. Cells stained with DAPI (blue, DAPI filter) and 10 µM pAQM compounds (red, TRITC filter) and merged images (image magnification: 60×). | |
A dark cytotoxic effect was observed toward the cancer cells, especially by the nanoaggregates of the symmetrical pAQM derivatives (Fig. 8, S60 and S62) with IC50 (half-maximal inhibitory concentration) values on the order of a few micromolar. However, it is noteworthy that more significant and remarkable toxicity was observed upon irradiating the cells containing the nanoaggregates. In particular, IC50 was more than halved for TPh and TPhOMe in the presence of light, while an even more apparent effect was observed for the asymmetric compounds. This phototoxicity was found to be particularly important when AsOMe nanoaggregates were considered (Fig. 8, S60 and S62). In fact, for this molecule, an IC50 higher than 10 µM was determined in the dark, while it was reduced by more than one order of magnitude when AsOMe nanoaggregates were exposed to light, reaching a value of just 0.42 µM. This finding is especially appealing in view of a practical application in photodynamic therapy, where an appropriate concentration has to be chosen for the photosensitizers to avoid toxic effects in the absence of an external stimulus and induce an effective response when triggered by light. The origin of the observed phototoxicity toward both prostatic cancer (LN-CaP) and melanoma (MEL-501) cells was investigated through experiments aimed at searching for ROS using the DCFH2-DA assay.46 As a rule, ROS were found to be generated in the investigated tumour cells upon irradiation above the control levels (untreated cells exposed to light with no pAQM compounds) when solutions of the dyes at 1 or 10 µM concentrations were considered (Fig. 9 and S63). Particularly, ROS production upon irradiation of AsOMe nanoaggregates was found to stand out against the outcomes of the other dyes, with ROS levels reaching almost 500% and 250% of their relative controls in LN-CaP and MEL-501 cells, respectively.
 |
| | Fig. 8 Upper panel: dark cytotoxicity and phototoxicity of the pAQM compounds (at 1 µM) on LN-CaP cells with a 28 minute-irradiation time (corresponding to 1.80 J cm−2). Both dark cytotoxicity and phototoxicity are expressed as the mean of two independent experiments with four replicas each ± SD. 100% corresponds to the control mean values. Lower table: dark cytotoxicity and phototoxicity of the investigated nanoaggregates on Ln-CAP cells at 28-minute irradiation time, expressed as IC50 (half-maximal inhibitory concentration) values. | |
 |
| | Fig. 9 Upper graph: ROS production induced by the pAQM compounds on LN-CaP cells with a 28 minute-irradiation time (1.80 J cm−2). ROS production is expressed as mean of two independent experiments of four replicas each ± SD. 100% corresponds to control mean values. Lower graph: area of the emission spectra of the SOSG singlet oxygen probe as a function of irradiation time under 458 nm light (1.07 mW cm−2) for water-dispersed aggregates of the pAQM compounds (produced by THF injection into water upon sonication) in the presence of SOSG and for the fluorescent probe alone as a control. | |
These results obtained in the cellular environment were understood in depth through fluorimetric tests performed for the pAQM aggregates in cell-free water dispersion in the presence of two other fluorescent probes for the detection of specific ROS species: Singlet Oxygen Sensor Green (SOSG) for singlet oxygen47,66 (Fig. S64 and S66) and Dihydroethidium (DHE) for the superoxide anion (Fig. S68).67 When the DHE probe was used, larger photoinduced superoxide anion generation was generally observed for aggregates of the symmetrical derivatives relative to the asymmetrical derivatives (Fig. S69). On the other hand, when the SOSG probe was employed, the largest singlet-oxygen production was revealed in the case of AsOMe nanoaggregates (Fig. 9 and S65). In these experiments, AsOMe nanoaggregates were found to be more effective singlet-oxygen photosensitizers than 9,10-bis(phenylethynyl)anthracene (BPEA) aggregates, one of the few literature examples of SF-enhanced photosensitizers for photodynamic therapy.31 The BPEA nanoaggregates were prepared under the same experimental conditions and using stock solutions with the same concentration as for the investigated compounds, resulting in nanoparticles with a similar size distribution (Fig. S53). As for the other less effective pAQM aggregates, a relatively larger singlet oxygen production capability was found for TPh than for TPhOMe, followed by AsNMe2 (as shown by zooming in on the obtained results in Fig. S65 and S67). Interestingly, the capability of singlet oxygen generation of these pAQM molecules is in trend with their triplet energy (1.3 eV for AsOMe, 1.2 eV for TPh and TPhOMe, and 1.1 eV for AsNMe2).39 Our results suggest that the main pathway leading to the exceptional phototoxicity of AsOMe nanoparticles toward cancer cells is the energy transfer to molecular oxygen, leading to remarkable singlet oxygen generation, which is especially favoured for this molecule by its highest SF-generated triplet energy among the investigated chromophores.
Conclusions
In this investigation, the photophysical and photobiological properties of water-dispersed organic nanoaggregates of four para-azaquinodimethane push–pull molecules, characterized by symmetrical (TPh and TPhOMe) and asymmetrical (AsOMe and AsNMe2) molecular structures and by electron donor groups of different strengths (OMe and NMe2), were investigated in depth. Our femtosecond and nanosecond transient absorption experiments proved ultrafast aggregation-induced intermolecular singlet fission. The triplet yield was found to be ca. 200% for the TPh, TPhOMe, and AsOMe nanoaggregates, while being only 90% for AsNMe2. All these molecules proved to give significant two-photon excited fluorescence, with their two-photon absorption cross sections found to be enhanced for the structures characterized by higher symmetry and charge transfer degree. Isolated spherical nanoparticles with diameters of 30–60 nm were observed through scanning electron microscopy, with size distributions peaking at 100–200 nm resulting from the dynamic light scattering experiments, which also take into account the larger clusters and the hydration shell associated with the nanoaggregates. These dimensional features allowed successful inclusion of the produced organic nanoparticles in the cytoplasmic and perinuclear regions of prostatic cancer and melanoma cells, as highlighted through fluorescence microscopy. Remarkable phototoxicity toward tumour cells was exhibited by the pAQM nanoaggregates, particularly relevant in the case of AsOMe, which was instead the least cytotoxic when kept in the dark. These features make AsOMe nanoaggregates the best candidates as photosensitizers for photodynamic therapy. Specifically, their phototoxicity was proven to be due to an excellent photoinduced intracellular reactive oxygen species production. Fluorimetric tests performed by employing fluorescent probes for the specific detection of singlet oxygen and superoxide anion demonstrated that the exceptional phototoxicity exhibited by the AsOMe nanoparticles, greater than that observed for literature photosensitizers for photodynamic therapy, is due to their ability to produce singlet oxygen via energy transfer. Such behaviour is connected to its remarkable capacity to trigger via singlet fission, the highest energy triplets (1.3 eV) among the chromophores investigated here. To the best of our knowledge, this is the first study highlighting the use of unconventional singlet fission chromophores as new effective photosensitizers for two-photon-triggered photodynamic therapy.
Author contributions
Enrico Sorbelli: formal analysis, data curation, investigation, methodology, visualization, writing – review and editing; Alessio Cesaretti: formal analysis, data curation, investigation, methodology, visualization, writing – review and editing; Martina Alebardi: formal analysis, data curation, investigation, writing – review and editing; Roberto Cantoni: formal analysis, data curation, investigation, writing – review and editing; Matilde Carloni: formal analysis, data curation, investigation; Cristina Munzone: methodology, validation; Alessandro Grasso: methodology, validation; Alessandro Di Michele: formal analysis, data curation, writing – review and editing; Eleonora Calzoni: formal analysis, data curation, investigation, writing – review and editing; Cosimo Gianluca Fortuna: funding acquisition, resources; Anna Spalletti: conceptualization, investigation, supervision, writing – review and editing; Carmela Bonaccorso: conceptualization, investigation, supervision, writing – review and editing; Benedetta Carlotti: conceptualization, data curation, formal analysis, funding acquisition, investigation, supervision, writing – original draft.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6sc02556k.
Acknowledgements
This work was funded by the European Union – NextGenerationEU under the Italian Ministry of University and Research (MUR) National Innovation Ecosystem grant ECS00000041 – VITALITY (at Perugia) and grant ECS00000022 – SAMOTHRACE (at Catania). B. C. acknowledges MUR financial support under the PRIN 2022 program, grant no. 2022RRFJC4. C. B. acknowledges the University of Catania for the funding received under the “PIAno di inCEntivi per la Ricerca di Ateneo 2020/2022 Pia.Ce.Ri.” – Linea di intervento 3 Starting Grant – Project SiFiChrom (51722152225).
Notes and references
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Annu. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS.
- N. Monahan and X.-Y. Zhu, Annu. Rev. Phys. Chem., 2015, 66, 601–618 CrossRef CAS.
- R. M. Young and M. R. Wasielewski, Acc. Chem. Res., 2020, 53, 1957–1968 CrossRef CAS PubMed.
- H. Kim and P. M. Zimmerman, Phys. Chem. Chem. Phys., 2018, 20, 30083–30094 RSC.
- K. Miyata, F. S. Conrad-Burton, F. L. Geyer and X.-Y. Zhu, Chem. Rev., 2019, 119, 4261–4292 CrossRef CAS.
- A. D. Chien, A. R. Molina, N. Abeyasinghe, O. P. Varnavski, T. Goodson and P. M. Zimmerman, J. Phys. Chem. C, 2015, 119, 28258–28268 CrossRef CAS.
- L. Mencaroni, M. Alebardi, F. Elisei, I. Škorić, A. Spalletti and B. Carlotti, Phys. Chem. Chem. Phys., 2023, 25, 21089–21099 RSC.
- T. Ullrich, D. Munz and D. M. Guldi, Chem. Soc. Rev., 2021, 50, 3485–3518 RSC.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, S. R. Reddy, B. T. Phelan, M. D. Krzyaniak, M. K. Volland, P. B. Coto, R. M. Young, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Chem, 2018, 4, 1092–1111 CAS.
- B. S. Basel, J. Zirzlmeier, C. Hetzer, B. T. Phelan, M. D. Krzyaniak, S. R. Reddy, P. B. Coto, N. E. Horwitz, R. M. Young, F. J. White, F. Hampel, T. Clark, M. Thoss, R. R. Tykwinski, M. R. Wasielewski and D. M. Guldi, Nat. Commun., 2017, 8, 15171 CrossRef CAS PubMed.
- D. Guzmán, I. Papadopoulos, G. Lavarda, P. R. Rami, R. R. Tykwinski, M. S. Rodríguez-Morgade, D. M. Guldi and T. Torres, Angew. Chem., Int. Ed., 2021, 60, 1474–1481 CrossRef.
- J. Kim, H. T. Teo, Y. Hong, Y. C. Liau, D. Yim, Y. Han, J. Oh, H. Kim, C. Chi and D. Kim, J. Am. Chem. Soc., 2023, 145, 19812–19823 CrossRef CAS PubMed.
- I. Papadopoulos, J. Zirzlmeier, C. Hetzer, Y. J. Bae, M. D. Krzyaniak, M. R. Wasielewski, T. Clark, R. R. Tykwinski and D. M. Guldi, J. Am. Chem. Soc., 2019, 141, 6191–6203 CrossRef CAS PubMed.
- R. Ringström, F. Edhborg, Z. W. Schroeder, L. Chen, M. J. Ferguson, R. R. Tykwinski and B. Albinsson, Chem. Sci., 2022, 13, 4944–4954 RSC.
- C. Zeiser, L. Moretti, D. Lepple, G. Cerullo, M. Maiuri and K. Broch, Angew. Chem., Int. Ed., 2020, 59, 19966–19973 CrossRef CAS PubMed.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- N. V. Korovina, J. Joy, X. Feng, C. Feltenberger, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2018, 140, 10179–10190 CrossRef CAS.
- O. J. Ogunyemi, C. Rani, O. Varnavski, R. Wajahath, M. Norscia, B. Malaikal, Y. Qiu, S. Sun, Y. Gu, S. Mukamel, M. R. Wasielewski and T. I. Goodson, J. Phys. Chem. C, 2024, 128, 20287–20296 CrossRef CAS.
- M. W. B. Wilson, A. Rao, K. Johnson, S. Gélinas, R. di Pietro, J. Clark and R. H. Friend, J. Am. Chem. Soc., 2013, 135, 16680–16688 CrossRef CAS.
- C. K. Yong, A. J. Musser, S. L. Bayliss, S. Lukman, H. Tamura, O. Bubnova, R. K. Hallani, A. Meneau, R. Resel, M. Maruyama, S. Hotta, L. M. Herz, D. Beljonne, J. E. Anthony, J. Clark and H. Sirringhaus, Nat. Commun., 2017, 8, 15953 CrossRef CAS.
- S. Lukman, A. J. Musser, K. Chen, S. Athanasopoulos, C. K. Yong, Z. Zeng, Q. Ye, C. Chi, J. M. Hodgkiss, J. Wu, R. H. Friend and N. C. Greenham, Adv. Funct. Mater., 2015, 25, 5452–5461 CrossRef CAS.
- J. Lee, P. Jadhav, P. D. Reusswig, S. R. Yost, N. J. Thompson, D. N. Congreve, E. Hontz, T. Van Voorhis and M. A. Baldo, Acc. Chem. Res., 2013, 46, 1300–1311 CrossRef CAS PubMed.
- J. Xia, S. N. Sanders, W. Cheng, J. Z. Low, J. Liu, L. M. Campos and T. Sun, Adv. Mater., 2017, 29, 1601652 CrossRef.
- M. Einzinger, T. Wu, J. F. Kompalla, H. L. Smith, C. F. Perkinson, L. Nienhaus, S. Wieghold, D. N. Congreve, A. Kahn, M. G. Bawendi and M. A. Baldo, Nature, 2019, 571, 90–94 CrossRef CAS.
- C. J. Lee, A. Sharma, N. A. Panjwani, I. M. Etchells, E. M. Gholizadeh, J. M. White, P. E. Shaw, P. L. Burn, J. Behrends, A. Rao and D. Jones, Adv. Opt. Mater., 2024, 12, 2301539 CrossRef CAS.
- H. Kim, B. Keller, R. Ho-Wu, N. Abeyasinghe, R. J. Vázquez, T. I. Goodson and P. M. Zimmerman, J. Am. Chem. Soc., 2018, 140, 7760–7763 CrossRef CAS PubMed.
- T. Tsuneda and T. Taketsugu, Sci. Rep., 2024, 14, 829 CrossRef CAS.
- T. Wang, H. Liu, X. Wang, L. Tang, J. Zhou, X. Song, L. Lv, W. Chen, Y. Chen and X. Li, ACS Catal., 2023, 13, 13902–13911 CrossRef CAS.
- R. Zhang, G. Cheng, M. Li, H. Lv and J. Li, Part. Part. Syst. Char., 2023, 40, 2200174 CrossRef CAS.
- Y. Liu, J. Li, S. Gong, Y. Yu, Z.-H. Zhu, C. Ji, Z. Zhao, X. Chen, G. Feng and B. Z. Tang, ACS Mater. Lett., 2024, 6, 896–907 CrossRef CAS.
- B. Manna and A. Nandi, J. Photochem. Photobiol., A, 2021, 412, 113251 CrossRef CAS.
- D. Chen, J. Shao, T. Zhang, K. Xu, C. Liang, Y. Cai, Y. Guo, P. Chen, X.-Z. Mou and X. Dong, Nano Lett., 2024, 24, 7524–7533 CrossRef CAS PubMed.
- O. Millington, S. Montanaro, A. Leventis, A. Sharma, S. A. Dowland, N. Sawhney, K. J. Fallon, W. Zeng, D. G. Congrave, A. J. Musser, A. Rao and H. Bronstein, J. Am. Chem. Soc., 2023, 145, 2499–2510 CrossRef CAS.
- L. Mencaroni, B. Carlotti, F. Elisei, A. Marrocchi and A. Spalletti, Chem. Sci., 2022, 13, 2071–2078 RSC.
- L. Mencaroni, A. Zaykov, B. Carlotti, F. Elisei, G. Bastien, R. Germani, Z. Havlas, A. Spalletti and J. Michl, Chem. Sci., 2025, 16(33), 15129–15140 RSC.
- D. J. Valentine, D. Manawadu and W. Barford, Phys. Rev. B, 2020, 102, 125107 CrossRef CAS.
- M. Alebardi, C. Munzone, E. Sorbelli, A. Grasso, L. Mencaroni, F. Elisei, C. G. Fortuna, A. Spalletti, C. Bonaccorso and B. Carlotti, Adv. Funct. Mater., 2024, 34, 2403706 CrossRef CAS.
- M. Alebardi, A. Carella, A. Grasso, E. Sorbelli, F. Lazzarin, C. Munzone, C. G. Fortuna, F. Elisei, A. Spalletti, C. Bonaccorso, M. D. Valentin and B. Carlotti, Angew. Chem., Int. Ed., 2026, 65(6), e20838 CrossRef CAS.
- R. M. Jacobberger, Y. Qiu, M. L. Williams, M. D. Krzyaniak and M. R. Wasielewski, J. Am. Chem. Soc., 2022, 144, 2276–2283 CrossRef CAS.
- K. E. Smyser and J. D. Eaves, Sci. Rep., 2020, 10, 18480 CrossRef CAS PubMed.
- R. Cantoni, M. Alebardi, E. M. D’Amato, C. Popli, Y. Patil, P. Mancini, A. Di Michele, A. Spalletti, R. Misra, T. Goodson and B. Carlotti, Adv. Funct. Mater., 2026, 36(17), e17911 CrossRef CAS.
- T. Bianconi, A. Cesaretti, P. Mancini, N. Montegiove, E. Calzoni, A. Ekbote, R. Misra and B. Carlotti, J. Phys. Chem. B, 2023, 127, 1385–1398 CrossRef CAS PubMed.
- P. Mancini, C. Montanari, P. Venturi, K. Yadav, T. Bianconi, E. Calzoni, A. Cesaretti, R. Misra and B. Carlotti, Chem. Commun., 2025, 61, 19628–19631 RSC.
- E. Calzoni, A. Cesaretti, N. Montegiove, M. L. Valicenti, F. Morena, R. Misra, B. Carlotti and S. Martino, Nanomaterials, 2025, 15, 894 CrossRef CAS PubMed.
- R. Bresolí-Obach, L. Busto-Moner, C. Muller, M. Reina and S. Nonell, Photochem. Photobiol., 2018, 94, 1143–1150 CrossRef PubMed.
- X. Ragàs, A. Jiménez-Banzo, D. Sánchez-García, X. Batllori and S. Nonell, Chem. Commun., 2009, 2920 RSC.
- E. Kurek, P. Mancini, J.-B. Verlhac, B. Carlotti and M. Blanchard-Desce, Chem. Commun., 2026, 62(15), 4570–4574 RSC.
- C. Xu and W. W. Webb, J. Opt. Soc. Am. B, 1996, 13, 481 CrossRef.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef PubMed.
- F. Ricci, B. Carlotti, B. Keller, C. Bonaccorso, C. G. Fortuna, T. Goodson, F. Elisei and A. Spalletti, J. Phys. Chem. C, 2017, 121, 3987–4001 CrossRef.
- B. Carlotti, Z. Cai, H. Kim, V. Sharapov, I. K. Madu, D. Zhao, W. Chen, P. M. Zimmerman, L. Yu and T. I. Goodson, Chem. Mater., 2018, 30, 4263–4276 CrossRef.
- G. D'Avino, F. Terenziani and A. Painelli, J. Phys. Chem. B, 2006, 110, 25590–25592 CrossRef PubMed.
- V. Parthasarathy, S. Fery-Forgues, E. Campioli, G. Recher, F. Terenziani and M. Blanchard-Desce, Small, 2011, 7, 3219–3229 CrossRef PubMed.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742–15755 Search PubMed.
- S. Masoomi-Godarzi, C. R. Hall, B. Zhang, M. A. Gregory, J. M. White, W. W. H. Wong, K. P. Ghiggino, T. A. Smith and D. J. Jones, J. Phys. Chem. C, 2020, 124, 11574–11585 Search PubMed.
- C. Lin, Y. Qi, P. J. Brown, M. L. Williams, J. R. Palmer, M. Myong, X. Zhao, R. M. Young and M. R. Wasielewski, J. Phys. Chem. Lett., 2023, 14, 2573–2579 CrossRef PubMed.
- M. Chen, N. E. Powers-Riggs, A. F. Coleman, R. M. Young and M. R. Wasielewski, J. Phys. Chem. C, 2020, 124, 2791–2798 CrossRef.
- D. H. Arias, J. L. Ryerson, J. D. Cook, N. H. Damrauer and J. C. Johnson, Chem. Sci., 2016, 7, 1185–1191 RSC.
- J. Lee, J. Yi, H. Hong, Y. J. Lee, F. Fang, S. Lee, W. B. Lee, S. Yun, K.-B. Park, J.-I. Park, D. Kim and W. Kim, Adv. Funct. Mater., 2025, 35, 2423223 CrossRef.
- E. A. Margulies, C. E. Miller, Y. Wu, L. Ma, G. C. Schatz, R. M. Young and M. R. Wasielewski, Nat. Chem., 2016, 8, 1120–1125 CrossRef PubMed.
- B. Carlotti, I. K. Madu, H. Kim, Z. Cai, H. Jiang, A. K. Muthike, L. Yu, P. M. Zimmerman and T. Goodson, Chem. Sci., 2020, 11, 8757–8770 RSC.
- A. K. Muthike, B. Carlotti, I. K. Madu, H. Jiang, H. Kim, Q. Wu, L. Yu, P. M. Zimmerman and T. Goodson, J. Phys. Chem. B, 2021, 125, 5114–5131 CrossRef PubMed.
- L. Mencaroni, A. Zaykov, B. Carlotti, F. Elisei, G. Bastien, R. Germani, Z. Havlas, A. Spalletti and J. Michl, Chem. Sci., 2025, 16, 15129–15140 RSC.
- E. Fron, G. Schweitzer, J. Jacob, A. Van Vooren, D. Beljonne, K. Müllen, J. Hofkens, M. Van der Auweraer and F. C. De Schryver, ChemPhysChem, 2007, 8, 1386–1393 CrossRef PubMed.
- H. Lin, Y. Shen, D. Chen, L. Lin, B. C. Wilson, B. Li and S. Xie, J. Fluoresc., 2013, 23, 41–47 CrossRef CAS PubMed.
- R. R. Nazarewicz, A. Bikineyeva and S. I. Dikalov, J. Biomol. Screen, 2013, 18, 498–503 CrossRef PubMed.
Footnote |
| † Equally contributed as first authors. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Martina Alebardi
a,
Martina Alebardi and the oxygen radical anion (O2˙−), by the quinoidal organic nanoaggregates. To the best of our knowledge, the present study is the first attempt to employ unconventional SF chromophores, characterized by remarkable stability and high triplet energy, as novel photosensitizers for PDT.
and the oxygen radical anion (O2˙−), by the quinoidal organic nanoaggregates. To the best of our knowledge, the present study is the first attempt to employ unconventional SF chromophores, characterized by remarkable stability and high triplet energy, as novel photosensitizers for PDT.












