
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceZerovalent transition-metal inverse-sandwich complexes of a diborataanthracene dianion
Alexander Gerstnerab,
Merle Arrowsmith ab,
Maximilian Dietzab,
Cornelius Mihmab and
Holger Braunschweig*ab
ab,
Maximilian Dietzab,
Cornelius Mihmab and
Holger Braunschweig*ab
aInstitute for Inorganic Chemistry, Julius-Maximilians-Universität Würzburg, Am Hubland, Würzburg 97074, Germany. E-mail: h.braunschweig@uni-wuerzburg.de
bInstitute for Sustainable Chemistry & Catalysis with Boron, Julius-Maximilians-Universität Würzburg, Am Hubland, Germany
First published on 22nd May 2026
Abstract
The 9,10-dihydro-9,10-diboraanthracene (9,10-DHDBA) 2,3,6,7,9,10-Me6-9,10-DHDBA (2) reacted with 1 equiv. [(MeCN)3Cr(CO)3] to yield the unsymmetrical half-sandwich complex [(η6-2)Cr(CO)3] (2-Cr), in which the Cr(CO)3 unit binds exclusively to one DHDBA benzo ring. Reactions of 2 with 2 equiv. [(MeCN)3M(CO)3] (M = Cr, Mo, W) yielded the centrosymmetric slipped inverse-sandwich complexes [{µ-(η6,η′6-2)}{M(CO)3}2] (2-M2), in which each M(CO)3 unit binds to one of the DHDBA benzo rings, one above and one below the DHDBA plane. The twofold reduction of 2 with Li sand afforded the corresponding diborataanthracene (DBA) dianion, isolated as the C4B2-bound inverse-sandwich dilithio complex [{µ-(η6,η6-2)}{Li(thf)2}2] (3). The latter reacted with 2 equiv. [(MeCN)3Cr(CO)3] (M = Cr, Mo) to yield the first stable zerovalent transition-metal inverse-sandwich complexes of a [9,10-DBA]2− dianion, complexes [{µ-(η6,η6-2)}{M(CO)3Li(thf)3}2] (3-M2). All new compounds were characterised by multinuclear NMR, UV-vis and IR spectroscopy, and their solid-state structures ascertained by single-crystal X-ray diffraction analyses. Furthermore, DFT calculations provide insight into the electronic structure of the 9,10-DHDBA and [9,10-DBA]2− fused ring systems, as well as metal-(DH)DBA bonding in the chromium complexes 2-Cr, 2-Cr2 and 3-Cr2.
Introduction
More than seven decades after the landmark isolations of ferrocene, [(η5-C5H5)2Fe], and the first bis(benzene) complex, [(η6-C6H6)2Cr],1 sandwich complexes (or metallocenes), in which the metal centre is wedged between two π carbocycles, have become ubiquitous in organometallic chemistry, and are still finding new applications in small-molecule activation,2 catalysis,3 materials design,4 and even pharmaceutical compounds.5 While sandwich complexes are now known for nearly all the metals of the periodic table, inverse-sandwich complexes, in which the π carbocycle is wedged between two metal centres (Fig. 1), are less well explored. The formation of stable inverse-sandwich complexes requires a particularly electron-rich, often highly reduced π ligand, capable of coordinating one metal centre on each side, such as the 6π-aromatic cyclobutadiene dianion [C4H4]2−,6 the 8π-antiaromatic benzene dianion [C6H6]2−,7 the 10π-aromatic benzene tetraanion [C6H6]4−,8 or the 10π-aromatic cyclooctatetraene (COT) dianion [C8H8]2−.9 As metal⋯arene bonding in these complexes is mostly ionic in nature, the vast majority of inverse-sandwich complexes involve the more Lewis-acidic s- and f-block metals, kinetic stabilisation being provided by bulky ligand spheres shielding the metal centres and the highly reactive reduced arene.10 | ||
| Fig. 1 Examples of known non-d0 TM inverse-sandwich complexes. Dipp = 2,6-diisopropylphenyl; Tipp = 2,4,6-triisopropylphenyl; Xyl = 2,6-dimethylphenyl. | ||
Excluding triple-decker complexes, in which the metal centres are additionally sandwiched by another arene (usually cyclopentadienyl (Cp−) or [COT]2−),11 and complexes with only partial metal⋯arene binding, there are only a handful of non-d0-metal inverse-sandwich complexes. These include some β-diketiminate-stabilised V(I), Nb(III) and Cr(I) complexes (I, Fig. 1),12 two amidinate-chelated Cr(I) derivatives (II),13 and a series of monovalent Cr(I), Mn(I) and Fe(I) terphenyl complexes (III),14 all with neutral benzene or toluene as the η6,η6-bridging ligand. Importantly, there are no known solely CO-bearing transition-metal (TM) inverse-sandwich complexes of organic π carbocycles of the form [µ-(η6,η6-arene){TM(CO)n}2], as the TM(CO)n fragments are usually too electron-rich and insufficiently stabilised by the CO ligands.
The replacement of one or more endocyclic CR units by isoelectronic [BL] or [BR]− units (L = neutral donor, R = anionic substituent) results in a dramatic increase in the electron-donating and π-acceptor ability of the resulting π boracycle compared to its all-carbon analogue, as the incorporation of the electropositive boron atom(s) raises the energy levels of the π-symmetry HOMOs while lowering that of the LUMO.15 Thus, monoanionic boratabenzenes, [C5H5BR]−, although isoelectronic to benzene, are much stronger donors, on a par with the Cp− ligand,16 enabling the isolation of a wide array of d- and f-block (half)-sandwich and triple-decker complexes.17 Whereas benzene only forms sandwich and half-sandwich complexes with Cr(0), Fontaine reported the formation of the Cr(CO)3 boratabenzene inverse-sandwich anion IV (Fig. 2) as a byproduct in the synthesis of the piano-stool complex [{η6-(2-SiMe3-C5H4BCl)}Cr(CO)3].18 Similarly, Herberich showed that a 4π-antiaromatic borole enables the isolation of the Mn(CO)3 inverse-sandwich complex V.19 Finally, borole dianions have been shown to be able to displace isoelectronic Cp− ligands,20 thus confirming their stronger donor ability.
 | ||
| Fig. 2 TM(CO)n half- and inverse-sandwich complexes of π (di)boracycles. | ||
The incorporation of two boron atoms into aromatic scaffolds generates even more powerful π ligands. Thus, we have shown that a neutral cyclic alkyl(amino)carbene (CAAC)-stabilised 1,4-diborabenzene (1,4-DBB) ligand is capable of stabilising zerovalent half-sandwich complexes of groups 6 (VI-M, M = Cr, Mo, W),21 8 (M(CO)2, M = Fe, Ru),22 and 10 (M(CO), M = Ni).23 Similarly, neutral diboraacenes like an N-heterocyclic carbene (NHC)-stabilised 2,3-diboranaphthalene and a CAAC-stabilised 9,10-diboraanthracene (9,10-DBA) have enabled the isolation of the zerovalent group 6 half-sandwich complexes VII-M and VIII-M (M = Cr, Mo, W), respectively.24,25 However, no inverse-sandwich formation has ever been observed with these neutral diboraacenes. Our attention therefore turned to dianionic diboraacenes, which are obtained by the twofold reduction of neutral dihydrodiboraacene precursors,26 and are known to form sandwich and triple-decker complexes with a small number of d- and f-block metals.27 Herein, we report the synthesis of the mono- and bimetallic zerovalent group 6 complexes of a neutral 9,10-dihydro-9,10-diboraanthracene (9,10-DHDBA) and the first TM inverse-sandwich complexes of its 9,10-diborataanthracene dianion ([9,10-DBA]2−).
Results and discussion
Synthesis of (DH)DBA group 6 complexes
The dibromo precursor 1 was synthesised in a manner analogous to its non-methylated analogue29 by heating a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.2 mixture of 1,2-bis(trimethylsilyl)-4,5-dimethylbenzene and BBr3 in hexane in a sealed thick-walled flask at 120 °C for 3 days (Scheme 1a).† Subsequent methylation with MeMgBr yielded 2,3,6,7,9,10-Me6-9,10-DHDBA (2) in near-quantitative yield (Scheme 1b). The 11B NMR shifts in C6D6 of 1 and 2 at 62.5 and 67.2 ppm, respectively, as well as their structural parameters (see Fig. S61 and S62 in the SI) are in agreement with those reported for their non-methylated analogues.28,29 It is noteworthy that in THF, the 11B NMR resonance of compound 2 is upfield-shifted to 58.3 ppm, presumably owing to slow reversible THF coordination to the Lewis-acidic boron centres on the NMR timescale.
:2.2 mixture of 1,2-bis(trimethylsilyl)-4,5-dimethylbenzene and BBr3 in hexane in a sealed thick-walled flask at 120 °C for 3 days (Scheme 1a).† Subsequent methylation with MeMgBr yielded 2,3,6,7,9,10-Me6-9,10-DHDBA (2) in near-quantitative yield (Scheme 1b). The 11B NMR shifts in C6D6 of 1 and 2 at 62.5 and 67.2 ppm, respectively, as well as their structural parameters (see Fig. S61 and S62 in the SI) are in agreement with those reported for their non-methylated analogues.28,29 It is noteworthy that in THF, the 11B NMR resonance of compound 2 is upfield-shifted to 58.3 ppm, presumably owing to slow reversible THF coordination to the Lewis-acidic boron centres on the NMR timescale.
 | ||
| Scheme 1 Synthesis and two-electron-reduction of 2. | ||
The cyclovoltammogram of 2 in THF shows a reversible reduction wave at E1/2 = −2.06 V and an irreversible one at Epc = −2.71 V (see Fig. S61 in the SI). The chemical two-electron reduction of 2 with a large excess of lithium sand in d8-THF resulted in an instant colour change to pink, then dark red, and the appearance of a new broad 11B NMR resonance at 22.0 ppm, identical to that of the parent [9,10-DBA]2− and the [9,10-Me2-9,10-DBA]2− dilithio complexes (Scheme 1c).26a,30 The UV-vis spectrum of 3 in THF shows a major absorption at 436 nm with a shoulder at 415 nm, as well as two very broad, low-intensity bands, overlapping in the 500–600 nm region (Fig. 3c). These are slightly red-shifted compared to those of the parent [9,10-DBA][Li(thf)2]2 complex (λmax = 420 nm, λ2 = 400 nm, λ3 = 500 nm).26a While single crystals of complex 3 suitable for X-ray diffraction analysis could be isolated (see X-ray crystallographic analyses), the compound was not indefinitely stable in the solid state.
 | ||
| Fig. 3 (a–c) Overlays of the UV-vis spectra of the DHDBA and DBA metal complexes presented herein. | ||
The 1:1 reaction of [(MeCN)3Cr(CO)3] and 2 in THF at rt led to the formation of the monometallic half–sandwich complex 2-Cr, isolated as a red crystalline solid in 72% yield (Scheme 2a). In d8-THF the broad 11B NMR resonance of 2-Cr at 46.3 ppm is significantly upfield-shifted compared to that of 2 (δ11B = 58.3 ppm). In benzene, however, in which 2-Cr is only poorly soluble, its 11B NMR resonance shifts back downfield to 64.9 ppm, 6 ppm upfield of the known unsymmetrical Cr(CO)3 complex of 9,10-Me2-9,10-DHDBA (δ11B = 71 ppm in C6D6).31 The larger upfield-shift of the 11B NMR resonance of 2-Cr upon switching from benzene to THF (Δδ ≈ −19 ppm) compared to 2 (Δδ ≈ −9 ppm) suggests that the boron centres in 2-Cr bind THF more strongly and are therefore more Lewis-acidic.
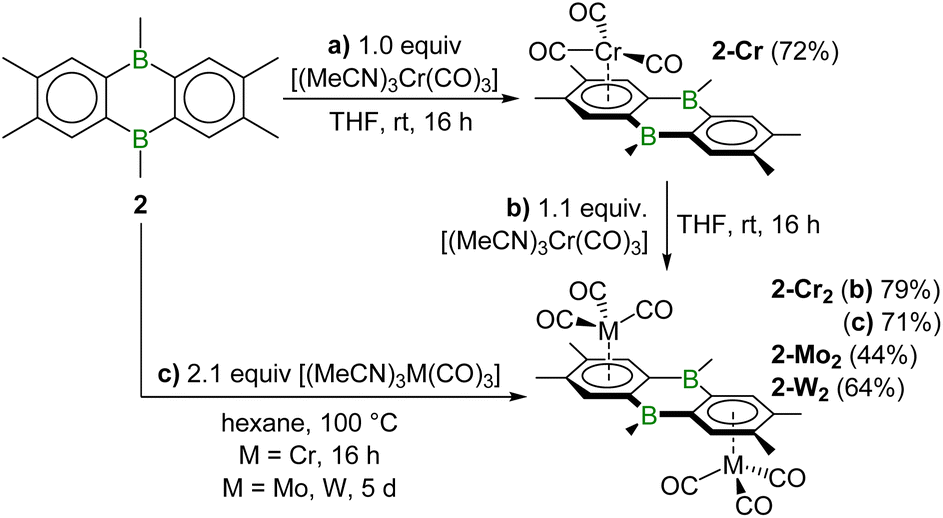 | ||
| Scheme 2 Synthesis of mono- and bimetallic group 6 complexes of 2. | ||
In the 1H NMR spectrum, two 2H singlets for the benzo protons, one of which is upfield-shifted to 5.83 ppm due to chromium coordination, reflect the unsymmetrical nature of 2-Cr. The 13C{1H} NMR spectrum shows a single CO resonance at 234.5 ppm, ca. 22 ppm downfield-shifted from those of its 9,10-Me2-DHDBA analogue (δ13C = 212.8, 212.2 ppm),31 indicating that the Cr(CO)3 moiety rotates freely in solution. It is noteworthy that the coordination to the 9,10-DHDBA benzo ring is unique to the group 6 metals, as all other low-valent TMs (Fe0, Ru0, CoI, RhI, Ni0, Pd0, Pt0) preferentially coordinate to the central 1,4-dibora-2,5-cyclohexadiene ring.31,32
The addition of a further equivalent of [(MeCN)3Cr(CO)3] to 2-Cr yielded the centrosymmetric slipped inverse-sandwich complex 2-Cr2, isolated as a red crystalline solid in 79% yield (Scheme 2b). The coordination of the second Cr(CO)3 unit results in a small downfield shift of the 11B NMR resonance to 49.1 ppm (THF). A single upfield-shifted benzo singlet (4H) at 5.86 ppm confirms the symmetrical nature of the complex. 2-Cr2 could also be obtained directly from the 2:1 reaction of [(MeCN)3Cr(CO)3] and 2 in hexane at 100 °C in a pressurised Schlenk flask (Scheme 2c). Attempted 1:1 reactions of the heavier group 6 precursors [(MeCN)3M(CO)3] (M = Mo, W) and 2 did not afford the corresponding mononuclear complexes 2-M but only a 1:1 mixture of bimetallic 2-M2 and unreacted 2, as determined by NMR spectroscopy. These could be accessed selectively by 2:1 reactions in hexane at 100 °C in pressurised Schlenk flasks, yielding 2-Mo2 and 2-W2 as red crystalline solids in moderate yields of 44% and 64%, respectively (Scheme 2c). Both show an 11B NMR resonance around 45 ppm, upfield-shifted from that of 2-Cr2 (δ11B = 49.1 ppm). The 13C NMR CO resonance of 2-M2 experiences a marked upfield shift upon moving down group 6 (δ13C = 234.6 (2-Cr2), 221.8 (2-Mo2), 212.4 (2-W2) ppm), with similar chemical shifts to those observed in the [(η6-mesitylene)M(CO)3] series.33 The solid-state IR spectra of 2-M2 all display a sharp C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching band around 1936–1944 cm−1 and a very broad unresolved band covering the 1800–1875 cm−1 range. Both are shifted to slightly lower wavenumbers compared to the parent benzene complexes [(η6-C6H6)M(CO)3] (ν(CO)solid–state = 1965–1971, 1845–1875 cm−1),34 indicating slightly weaker C–O bonds. This suggests that the benzo rings of the 9,10-DHDBA ligand are slightly better overall donors than benzene.
O stretching band around 1936–1944 cm−1 and a very broad unresolved band covering the 1800–1875 cm−1 range. Both are shifted to slightly lower wavenumbers compared to the parent benzene complexes [(η6-C6H6)M(CO)3] (ν(CO)solid–state = 1965–1971, 1845–1875 cm−1),34 indicating slightly weaker C–O bonds. This suggests that the benzo rings of the 9,10-DHDBA ligand are slightly better overall donors than benzene.
All four 9,10-DHDBA complexes are red. The UV-vis spectra of 2-Cr and 2-M2 (M = Cr, Mo, W) all show an absorption maximum around λmax = 330–350 nm, as well as two very broad, overlapping, medium-intensity bands in the λ2 = 415–430 nm and λ3 = 470–500 nm regions, the latter accounting for their red colour. Comparison of the mono- and bimetallic complexes 2-Cr (λ = 333, 423, 481 nm) and 2-Cr2 (λ = 349, 431, 497 nm) shows a small redshift for all three bands in the latter (Fig. 3a). Comparison of the three 2-M2 complexes (Fig. 3b), shows a small blueshift upon moving from 2-Cr2 (λ = 349, 431, 497 nm) to 2-Mo2 (λ = 336, 417, 464 nm), and a very minor redshift of λ2 and λ3 upon moving from 2-Mo2 to 2-W2 (λ = 334, 422, 469 nm).
Siebert reported that the reduction of his 9,10-Me2-9,10-DHDBA analogue of 2-Cr with potassium did not lead to the desired shift of the Cr(CO)3 unit to the central C4B2 ring but rather to decomposition.31 We therefore decided to attempt the synthesis of inverse-sandwich complexes starting from the already doubly reduced complex 3. The addition of 2 equiv. [(MeCN)3Cr(CO)3] (M = Cr, Mo, W) to a freshly prepared THF solution of 3 at rt led to the instant formation of dark suspensions. The 11B NMR spectra of the reaction mixture showed the formation of several unidentified sp3-boron-containing species, as well as a broad resonance at 12 ppm, present in the Cr- and Mo-based reactions, but not in the W-based one. After filtration, removal of the solvent and washing of the solid residues with various solvents, 3-Cr2 and 3-Mo2 were isolated as orange and yellow solids, respectively, in rather poor yields of 14% and 29%, respectively (Scheme 3). It is noteworthy that once formed, 3-Cr2 and 3-Mo2 were indefinitely stable in solution at rt. In the case of the W-based reaction, no equivalent 3-W2 complex was observed, and no other product could be identified. Carrying out the reactions at −78 °C yielded no improvement in their selectivity. To our knowledge, these are the first examples of zerovalent TM inverse-sandwich complexes of a diborataanthracene ligand. The only other known [9,10-DBA]2− complexes beyond the alkali metals are the dilanthanide triple-decker complexes recently reported by Morena-Pineda and Roesky.27b
 | ||
| Scheme 3 Synthesis of zerovalent inverse-sandwich group 6 complexes of 3. | ||
The 11B NMR resonance of 3-Cr2 and 3-Mo2 at ca. 12 ppm is 10 ppm upfield-shifted compared to 3 (δ11B = 22 ppm), indicating a significant increase in electron density at the boron centres from interaction with the electron-rich Cr(0) centres (see DFT calculations). The centrosymmetric nature of 3-M2 was attested by the single 1H NMR benzo singlet (4H) at 7.45 ppm. In solution the Cr(CO)3 units rotate freely and the [Li(thf)3]+ units fluctuate between all three CO ligands on the NMR timescale, as only one 13C NMR carbonyl resonance is observed (δ11C = 242.0 (3-Cr2), 234.6 (3-Mo2) ppm). The solid-state IR spectra reflect the presence of the Li-bound CO ligands through a series of stretching bands spanning the 1711–1935 cm−1 region, with the bands above 1850 cm−1 pertaining to the terminal CO ligands and those below 1800 cm−1 to those of the weaker Li-bound CO bonds. As expected from the differences in colour, the UV-vis spectra of 3-M2 (M = Cr, orange; M = Mo, yellow) differ greatly from that of their precursor 3 (red). Thus, 3-M2 show only low-intensity absorptions around 440 nm, where 3 displays its λmax, and lack any bands in the 500–600 nm region (Fig. 3c). Both their absorption maxima (3-Cr2 λ = 308, 418 nm; 3-Mo2 λ = 296, 344 nm) are significantly blueshifted from those of 2-M2. Furthermore, a blueshift is observed upon moving from 3-Cr2 to the heavier 3-Mo2, as already noted between 2-Cr2 and 2-Mo2.
X-ray crystallographic analyses
 (see edge labels below);
(see edge labels below);  between the metal centre and the mean plane of the ring it coordinates to;
between the metal centre and the mean plane of the ring it coordinates to;  between the metal centre and the carbon atoms of the e/e′ (and d/d′) edges of the ring it coordinates to;
between the metal centre and the carbon atoms of the e/e′ (and d/d′) edges of the ring it coordinates to;  ;
;  ; |ωmax| = absolute value of the largest endocyclic torsion angle of the central B2C4 ring; φ = angle between the mean planes of the benzo and C4B2 rings
; |ωmax| = absolute value of the largest endocyclic torsion angle of the central B2C4 ring; φ = angle between the mean planes of the benzo and C4B2 rings
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 11B NMR |
|
|
|
|
|
|
|
|
|
|ωmax| | φ/φ′ | |
a Within the metal-containing complex moiety.b within the metal-free complex moiety.c  .d .d  .e values provided for both molecules present in the asymmetric unit.f different avg. values measured for the e and e′ edge.g for terminal CO ligands.h for Li-bound CO ligands. .e values provided for both molecules present in the asymmetric unit.f different avg. values measured for the e and e′ edge.g for terminal CO ligands.h for Li-bound CO ligands. |
||||||||||||
| 2 | 58.3 | 1.565(2) | 1.419(2) | 1.404(2) | 1.395(2) | 1.401(2) | — | — | — | — | 10.6(2) | ca. 4.0 |
| 3 | 22.0 | 1.531(3) | 1.472(3) | 1.438(3) | 1.370(3) | 1.441(3) | ca. 1.87 | 2.385(4)c | — | — | 1.1(3) | ca. 0.9 |
| 2-Cr | 46.3 | 1.573(4)a | 1.433(4)a | 1.434(4)a | 1.406(4)a | 1.427(4)a | ca. 1.72 | 2.193(3)c | 1.847(3) | 1.156(4) | 8.4(4) | ca. 5.2a |
| 1.557(4)b | 1.421(4)b | 1.400(4)b | 1.396(4)b | 1.392(4)b | 2.263(3)d | ca. 5.0b | ||||||
| 2-Cr2 | 49.1 | 1.559(3) | 1.451(3) | 1.415(3) | 1.421(3) | 1.405(3) | ca. 1.71 | 2.201(2)c | 1.853(3) | 1.151(3) | 2.5(3) | ca. 9.3 |
| 2.263(2)d | ||||||||||||
| 2-Mo2 | 45.0 | 1.562(4) | 1.450(4) | 1.428(4) | 1.403(4) | 1.434(4) | ca. 1.88 | 2.316(3)c | 1.984(3) | 1.147(4) | 6.3(4) | ca. 9.4 |
| 2.400(3)d | ||||||||||||
| 2-W2 | 44.4 | 1.560(7) | 1.444(6) | 1.425(7) | 1.415(7) | 1.423(7) | ca. 1.87 | 2.311(2)c | 1.964(7) | 1.144(9) | 8.9(6) | ca. 11.8 |
| 2.390(6)d | ||||||||||||
| 3-Cr2e | 11.9 | 1.551(3) | 1.457(2) | 1.449(3) | 1.355(3) | 1.449(2) | ca. 1.78 | 2.287(2) | 1.816(2)g | 1.173(3)g | 2.0(2) | ca. 2.3 |
| 2.369(2)f | 1.779(2)h | 1.191(3)h | ca. 0.8f | |||||||||
| 3-Mo2e | 12.0 | 1.557(9) | 1.463(8) | 1.448(9) | 1.357(9) | 1.443(9) | ca. 1.95 | 2.424(6) | 1.931(7)g | 1.173(9)g | 1.6(8) | ca. 2.2 |
| 2.500(6)f | 1.889(10)h | 1.197(10)h | ca. 0.7f | |||||||||










 | ||
| Fig. 4 Crystallographically determined solid-state structures of 2-Cr, 2-Cr2 and 2-Mo2. Top: views from above the DHDBA plane, highlighting the various conformations of the M(CO)3 moieties with respect to the DHDBA framework and to each other. Below: truncated views (methyl groups omitted) along the B⋯B axis, highlighting the bend in the DHDBA framework. Thermal displacement ellipsoids set at 50% probability. Hydrogen atoms omitted for clarity. | ||
In contrast to 2-Cr, the three bimetallic complexes 2-M2 are centrosymmetric, with one M(CO)3 fragment η6-coordinated to each benzo ring, one above and one below the 9,10-DHDBA plane, in what may be called a slipped inverse-sandwich coordination mode (Fig. 4 and Table 1, see Fig. S68 in the SI for 2-W2). Whereas the central C4B2 ring in 2-Cr2 is virtually planar (|ωmax| = 2.5(3)°), it is slightly twisted in the two heavier analogues (2-Mo2 6.3(4)°, 2-W2 8.9(6)°). Furthermore, all three complexes display a significant bend in the overall 9,10-DHDBA framework, the angle φ formed by the C6 and C4B2 mean planes ranging from 9.3° to 11.8°. The endocyclic C–C and B–C bond lengths of all three dinuclear complexes are similar within the error of the measurement. The M⋯C6 distance increases significantly from 2-Cr2 (ca. 1.71 Å) to 2-Mo2 (ca. 1.88 Å) but shows no further increase for 2-W2 (ca. 1.87 Å). This is in line with the much lower difference between the hexacoordinate metallic radii of W and Mo, than of Mo and Cr (Cr 1.285, Mo 1.402, W 1.410 Å),36 owed to the f-orbital contraction in W. These M⋯C6 distances are all slightly shorter than those found in the [(η6-C6H6)M(CO)3] series (Cr 1.72, Mo 1.91, W 1.90 Å),37 suggesting stronger metal⋯arene bonding. Complexes 2-M2 display the same slightly unsymmetrical positioning of the metal centres above the C6 rings as 2-Cr, the M–Cd bonds being ca. 3% longer than the M–Ce bonds, owing to the influence of the fused electron-poor C4B2 ring (see DFT calculations). Whereas the M(CO)3 fragments in 2-Cr2 are staggered with respect to the benzo rings, this time with one CO ligand each pointing towards the central C4B2 ring, those in 2-Mo2 and 2-W2 are nearly eclipsed. These conformational differences may be at the origin of the differing degrees of distortion from planarity observed in their C4B2 rings (vide supra). Finally, the C–O bond lengths of all three complexes are statistically similar, in line with their very similar IR C–O stretching bands (vide supra).
 | ||
| Fig. 5 Left: crystallographically determined solid-state structure of 3. Right: truncated view of 3 (methyl groups and THF ligands omitted) along the B⋯B axis, highlighting the planarity of the DBA framework. Thermal displacement ellipsoids set at 50% probability. Hydrogen atoms omitted for clarity. | ||
 | ||
| Fig. 6 Top: crystallographically determined solid-state structure of 3-Cr2. Bottom left: view of 3-Cr2 (THF ligands omitted) from above the DBA plane, highlighting the eclipsed conformation of the Cr(CO)3 moieties. Bottom right: truncated view of 3-Cr2 along the B⋯B axis (methyl groups and THF omitted), highlighting the planarity of the DBA framework. Thermal displacement ellipsoids set at 50% probability. Hydrogen atoms omitted for clarity. | ||
DFT calculations
| C4B2 ring | C6 rings | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| NICS | WBI | NICS | WBI | ||||||
| (0/1) | (±1)zz | a | e | (0/1) | (±1)zz | b | c | d | |
| a The slight bend in the C4B2 ring results in different NICS(1)zz and NICS(−1)zz values. | |||||||||
| C6H6 | — | — | — | — | −8.9/−11.0 | −29.1 | 1.41 | ||
| 2 | 10.3/2.4 | 19.7, 13.0a | 1.06 | 1.33 | −4.8/−8.8 | −20.5 | 1.33 | 1.39 | 1.39 |
| [2]2− | −8.2/−11.4 | −32.2 | 1.33 | 1.21 | −4.6/−7.4 | −17.5 | 1.16 | 1.53 | 1.20 |
For the two benzo rings the aromaticity decreases slightly upon reduction to the dianion, as seen in their slightly less negative NICS(±1)zz value (2 –20.5 ppm; [2]2− −17.5 ppm). In contrast, the central C4B2 ring switches from antiaromatic in 2, with an average positive NICS(±1)zz value of +16.6 ppm, to highly aromatic in [2]2−, with a NICS(±1)zz value of −32.3 ppm, more negative than that of benzene. It is noteworthy that in compound 2 the substantial difference between the NICS(−1)zz (19.7 ppm) and NICS(+1)zz (13.0 ppm) values of the C4B2 ring is owed to its slightly bent geometry (see X-ray crystallographic analyses). For [2]2− the NICS(−1)zz and NICS(+1)zz values are identical within the error of the calculation, in line with the planarisation of the C4B2 ring upon twofold reduction. For comparison, the central ring of the literature-known [9,10-Mes2-9,10-DBA]2− dianion (Mes = 2,4,6-trimethylphenyl), for which only the NICS(0) and NICS(1) values are reported, (−9.0/−13.1 ppm),40 is slightly more aromatic than that of [2]2− (−8.2/−11.4 ppm). Moreover, the aromatisation of the C4B2 ring is also visible in its B–C Wiberg bond indices (WBIs), which increase from 1.06 in 2 (single bonds) to 1.33 in [2]2− (partial double bonds), concomitant with a slight decrease in its C–C WBIs, from 1.33 to 1.21, respectively (Table 2).41 At the same time the benzo rings lose some of their aromatic character, as seen in the wider range of C–C WBIs ([2]2− 1.16–1.53, 2 1.33–1.39).
The aromatisation of the C4B2 ring is also apparent in the frontier MOs. Whereas the π-symmetry HOMOs of 2 are all delocalised over the two benzo rings, the HOMO (which corresponds to the LUMO of 2), HOMO−1, and HOMO−4 of [2]2− show π delocalisation over the 1,4-diboratabenzene ring akin to that of benzene (Fig. 7).
 | ||
| Fig. 7 Plots of the frontier MOs of [2]2− involving π delocalisation over the C4B2 ring, energy levels in parentheses. Isovalues: 0.04. | ||
The optimised structures of 2-Cr and 2-Cr2 in the solid-state conformations reproduce the slightly off-centre position of the Cr atoms, which are closer to the e than d edge(s) (Cr–Cd 2.229, Cr–Ce 2.184 Å). A look at the natural bond orbital (NBO)42 charges of the benzo rings of 2 (Fig. 8a) shows that the e-edge carbon atoms are significantly more negative (−0.39) than the d-edge ones (−0.02) due to the polarisation of the benzo π electron density towards the electron-deficient boron atoms (+0.98) of the central ring. This polarisation is also visible in the unsymmetrical π-electron distribution of the HOMO−9 and HOMO−12 (Fig. 8b and c), resulting in tighter binding of the Cr centres to the e edges in 2-Cr and 2-Cr2.
 | ||
| Fig. 8 (a) Optimised structure of 2 at the ωB97X-D4-def2-SVP level of theory, with NBO charges of the d and e carbon and boron atoms in red. Atom colours: carbon = grey, boron = green. Hydrogen atoms omitted for clarity. (b and c) Plots of the HOMO−9 and HOMO−12 of 2. Isovalues: 0.04. | ||
 | ||
| Fig. 9 Plots of the frontier MOs of [3′-Cr2]2− involved in Cr⋯C4B2 bonding. Energy levels in parentheses. Isovalues: 0.04. | ||
An intrinsic bonding orbital (IBO)43 analysis shows three IBOs involved in Cr⋯C4B2 bonding (Fig. 10). IBO-1 and IBO-2 involve σ donation from essentially the B1–C4 (68% contribution) and C1–C2 (75%) π bonds into the two chromium dxz orbitals (6%, respectively), while IBO-3 involves donation from the B2–C3 π bond (68%) into the chromium dyz orbitals (8%). It is noteworthy that the contribution of each boron 2pz orbital to these three IBOs (ca. 24%) is significantly lower than that of each carbon one (ca. 40% for C1/C2, ca. 50% for C3/C4). The rather low chromium contributions (≤4% per Cr) to these IBOs suggest that the Cr0←[2]2−→Cr0 interaction is mainly electrostatic in nature. Interestingly, an atoms-in-molecules (AIM)44 analysis provides bond critical points (BCP) only for the Cr–C bonds, and none for the Cr⋯B interactions (see Fig. S76 in the SI).
 | ||
| Fig. 10 (a) Optimised structure of [3′-Cr2]2− at the ωB97X-D4-def2-SVP level of theory. The hydrogen atoms and Li(thf)3 moieties have been omitted for clarity. (b–d) IBOs of [3′-Cr2]2− involved in Cr⋯C4B2 bonding. Main orbital contributions to IBO-1: 21% 2pz(B1), 47% 2pz(C4), 4% 2pz(C15), 4% 2pz(C3), 2% 2pz(C2), 3% 3dxz(Cr2), 2% 3dxz(Cr1); to IBO-2: 4% 2pz(B1), 40% 2pz(C1), 35% 2pz(C2), 2% 2pz(B2), 1% 3dxz(Cr1), 1% 3dxz(Cr2); to IBO-3: 2% 2pz(C2), 22% 2pz(B2), 46% 2pz(C3), 3% 2pz(C18), 3% 2pz(C4), 4% 3dyz(Cr1), 4% 3dyz(Cr2). Isovalues: 0.04. | ||
Conclusions
We have shown that the neutral 9,10-DHDBA 2 and its doubly reduced [9,10-DBA]2− dilithio inverse-sandwich complex 3 are suitable precursors for the complexation of group 6 carbonyls. While compound 2 forms a stable, unsymmetrical half-sandwich complex (2-Cr) by coordination of Cr(CO)3 to one of the benzo rings, the analogous Mo and W complexes are not accessible. DFT calculations show that coordination to the central C4B2 ring is disfavoured by 22 kcal mol−1. In contrast, all three group 6 metals form stable centrosymmetric slipped inverse-sandwich complexes (2-M2, M = Cr, Mo, W) by coordination of one M(CO)3 to each benzo ring, one above and one below the DHDBA plane. These all display a slightly bent DHDBA framework, with the metal centres coordinating more tightly to the ring junctions, where π density accumulates due to the attraction exerted by the central electrophilic boron atoms.Twofold reduction of 2 with lithium yields the inverse-sandwich complex 3, in which the two lithium cations coordinate to either side of the now aromatic central 1,4-diborabenzene ring. While 3 is only stable in solution, its reaction with [M(CO)3(NCMe)3] (M = Cr, Mo) yielded the first stable zerovalent TM inverse-sandwich complexes of a diborataanthracene ligand (3-M2, M = Cr, Mo), albeit in poor yields. SCXRD analyses confirm that the two M(CO)3 units bind in an η6 fashion to the central C4B2 ring of the DBA dianion, while the [Li(thf)3]+ countercations bind to one CO ligand of each M(CO)3 unit. Finally, DFT calculations show that M(CO)3 bonding to the central 1,4-diborabenzene ring in 3-M2 is mainly electrostatic in nature, without formal Cr–B bonding.
Author contributions
AG designed the study, conducted the experimental work and data analysis, and wrote the SI. MA carried out computations and wrote the manuscript. AG, MD and CM carried out SCXRD experiments and refined the data. HB conceived the project, and provided funding and supervision. All authors were involved in the finalisation of manuscript and SI.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2538786, 2538787, 2538788, 2538789, 2538790, 2538791, 2538792, 2538793 and 2538794 contain the supplementary crystallographic data for this paper.45a–iThe data supporting the findings are included in the supplementary information (SI). Supplementary information: synthetic procedures, NMR, IR, and UV-vis spectra, cyclovoltamograms, X-ray crystallographic and computational details, Cartesian coordinates of calculated structures. See DOI: https://doi.org/10.1039/d6sc02501c.
Acknowledgements
The authors gratefully acknowledge funding from the Deutsche Forschungsgesellschaft (DFG grants BR1149/30-1 466754611) and a Fonds der Chemischen Industrie (FCI) Kékulé stipend for AG.Notes and references
- (a) T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS; (b) E. O. Fischer and W. U. Hafner, Z. Naturforsch., B, 1955, 10, 665–668 CrossRef.
- (a) U. Rosenthal, Angew. Chem., Int. Ed., 2018, 57, 14718–14735 CrossRef CAS PubMed; (b) L. Becker and U. Rosenthal, Coord. Chem. Rev., 2017, 345, 137–149 CrossRef CAS; (c) S. Schäfer, S. Kaufmann, E. S. Rösch and P. W. Roesky, Chem. Soc. Rev., 2023, 52, 4006–4045 RSC; (d) P. J. Chirik, Organometallics, 2010, 29, 1500–1517 CrossRef CAS.
- (a) A. Desgranges, F. D'Agosto and C. Boisson, ChemPlusChem, 2024, 89, e202400262 CrossRef CAS PubMed; (b) J. Okuda, J. Organomet. Chem., 2023, 1000 Search PubMed; (c) H. G. Alt, Metallocene Complexes as Catalysts for Olefin Polymerization, Coord. Chem. Rev., 2006, 250, 1–272 CrossRef CAS.
- (a) T. Wang and D. Astruc, Coord. Chem. Rev., 2025, 524, 216300 CrossRef CAS; (b) M. V. Kharlamova and C. Kramberger, Nanomat, 2023, 13, 774 CrossRef CAS PubMed; (c) A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS; (d) H. Gu, R. Ciganda, S. Gatard, F. Lu, P. Zhao, J. Ruiz and D. Astruc, J. Organomet. Chem., 2016, 821, 54–61 CrossRef CAS; (e) D. Astruc, C. Ornelas and J. Ruiz, Chem. Eur J., 2009, 15, 8936–8944 CrossRef CAS PubMed.
- (a) A. Kalamatianou, C. Ludwig, S. Zhong, K. Cariou and G. Gasser, Chem. Soc. Rev., 2025, 54, 3930–3961 RSC; (b) I. Kostova, Molecules, 2024, 29, 824 CrossRef CAS PubMed; (c) M. M. Santos, P. Bastos, I. Catela, K. Zalewska and L. C. Branco, Mini-Rev. Med. Chem., 2017, 17, 771–784 CrossRef CAS PubMed.
- (a) D. Patel, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Nat. Commun., 2013, 4, 2323 CrossRef PubMed; (b) C. Yu, B. Wu, Z. Yang, H. Chen, W.-X. Zhang and Z. Xi, Bull. Chem. Soc. Jpn., 2020, 93, 1314–1318 CrossRef; (c) A. Sekiguchi, T. Matsuo and H. Watanabe, J. Am. Chem. Soc., 2000, 122, 5652–5653 CrossRef CAS.
- (a) D. Jędrzkiewicz, M. Morasch, O. P. E. Townrow, B. Rösch, J. Langer, Z. Mathe and S. Harder, Chem. Sci., 2025, 16, 17793–17802 RSC; (b) Y. Wang, R. Sun, J. Liang, Y. Zhang, B. Tan, C. Deng, Y.-H. Wang, B.-W. Wang, S. Gao and W. Huang, J. Am. Chem. Soc., 2025, 147, 7741–7748 CrossRef CAS PubMed; (c) F. Delano IV and S. Demir, Angew. Chem., Int. Ed., 2025, 64, e202417217 CrossRef PubMed; (d) C. A. Gould, J. Marbey, V. Vieru, D. A. Marchiori, R. D. Britt, L. F. Chibotaru, S. Hill and J. R. Long, Nat. Chem., 2021, 13, 1001–1005 CrossRef CAS PubMed; (e) P. L. Arnold, S. M. Mansell, L. Maron and D. McKay, Nat. Chem., 2012, 4, 668–674 CrossRef CAS PubMed; (f) P. L. Diaconescu, P. L. Arnold, T. A. Baker, D. J. Mindiola and C. C. Cummins, J. Am. Chem. Soc., 2000, 122, 6108–6109 CrossRef CAS.
- (a) M. Liu, Y.-C. Chen, A. Mondal, H. Wang, M.-L. Tong, R. A. Layfield and F.-S. Guo, J. Am. Chem. Soc., 2025, 147, 11359–11367 CrossRef CAS PubMed; (b) K. R. McClain, A. H. Vincent, A. Rajabi, D. X. Ngo, K. R. Meihaus, F. Furche, B. G. Harvey and J. R. Long, J. Am. Chem. Soc., 2024, 146, 32708–32716 CrossRef CAS PubMed; (c) Y. Wang, Y. Zhang, J. Liang, B. Tan, C. Deng and W. Huang, Chem. Sci., 2024, 15, 10669 RSC; (d) W. Huang, F. Dulong, T. Wu, S. I. Khan, J. T. Miller, T. Cantat and P. L. Diaconescu, Nat. Commun., 2013, 4, 1448 CrossRef PubMed; (e) S. Krieck, H. Görls, L. Yu, M. Reiher and M. Westerhausen, J. Am. Chem. Soc., 2009, 131, 2977–2985 CrossRef CAS PubMed.
- (a) M. S. Hill, M. F. Mahon, A. S. S. Wilson, C. Dinoi, L. Maron and E. Richards, Chem. Commun., 2019, 55, 5732–5735 RSC; (b) P. L. Diaconescu and C. C. Cummins, J. Am. Chem. Soc., 2002, 124, 7660–7661 CrossRef CAS PubMed; (c) H. Schumann, J. Winterfeld, L. Esser and G. Kociok-Köhn, Angew Chem. Int. Ed. Engl., 1993, 32, 1209–1210 CrossRef.
- S. T. Liddle, Coord. Chem. Rev., 2015, 293–294, 211–227 CrossRef CAS.
- V. Beck and D. O'Hare, J. Organomet. Chem., 2004, 689, 3920–3938 CrossRef CAS.
- (a) T. L. Gianetti, G. Nocton, S. G. Minasian, N. C. Tomson, A. L. D. Kilcoyne, S. A. Kozimor, D. K. Shuh, T. Tyliszczak, R. G. Bergman and J. Arnold, J. Am. Chem. Soc., 2013, 135, 3224–3236 CrossRef CAS PubMed; (b) Y.-C. Tsai, P.-Y. Wang, S.-A. Chen and J.-M. Chen, J. Am. Chem. Soc., 2007, 129, 8066–8067 CrossRef CAS PubMed; (c) W. H. Monillas, G. P. A. Yap and K. H. Theopold, Angew. Chem., Int. Ed., 2007, 46, 6692–6694 CrossRef CAS PubMed; (d) Y.-C. Tsai, P.-Y. Wang, K.-M. Lin, S.-A. Chen and J.-M. Chen, Chem. Commun., 2008, 205–207 RSC.
- Y.-S. Huang, G.-T. Huang, Y.-L. Liu, J.-S. K. Yu and Y.-C. Tsai, Angew. Chem., Int. Ed., 2017, 56, 15427–15431 CrossRef CAS PubMed.
- C. Ni, B. D. Ellis, J. C. Fettinger, G. J. Long and P. P. Power, Chem. Commun., 2008, 1014–1016 RSC.
- (a) G. C. Bazan, W. D. Cotter, Z. J. A. Komon, R. A. Lee and R. J. Lachicotte, J. Am. Chem. Soc., 2000, 122, 1371–1380 CrossRef CAS; (b) G. E. Herberich and D. Söhnen, J. Organomet. Chem., 1983, 254, 143–147 CrossRef CAS; (c) D. W. Clack and K. D. Warren, Inorg. Chem., 1979, 18, 513–519 CrossRef CAS.
- (a) R. Wang, C.-S. Lee and Z. Lu, J. Organomet. Chem., 2023, 984, 122564 CrossRef CAS; (b) K. Rojales, M. Tamizmani, T. A. Bartholome and C. D. Martin, Dalton Trans., 2022, 51, 17216–17223 RSC; (c) J. He, F. Rauch, M. Finze and T. B. Marder, Chem. Sci., 2021, 12, 128–147 RSC; (d) S. K. Mellerup and S. Wang, Trends Chem., 2019, 1, 77–89 CrossRef CAS; (e) E. von Grotthuss, A. John, T. Kaese and M. Wagner, Asian J. Org. Chem., 2018, 7, 37–53 CrossRef CAS.
- (a) P. Cui and Y. Chen, Coord. Chem. Rev., 2016, 314, 2–13 CrossRef CAS; (b) A. J. Ashe, III, Organometallics, 2009, 28, 4236–4248 Search PubMed; (c) G. C. Fu, Adv. Organomet. Chem., 2001, 47, 101–119 CrossRef CAS; (d) G. E. Herberich and H. Ohst, Adv. Organomet. Chem., 1986, 25, 199–236 CrossRef CAS.
- V. Pérez, S. S. Barnes and F.-G. Fontaine, Eur. J. Inorg. Chem., 2014, 5698–5702 CrossRef.
- G. E. Herberich, J. Hengesbach, G. Huttner, A. Frank and U. Schubert, J. Organomet. Chem., 1983, 246, 141–149 CrossRef CAS.
- (a) T. Heitkemper, J. Sarcevic and C. P. Sindlinger, J. Am. Chem. Soc., 2020, 142, 21304–21309 CrossRef CAS PubMed; (b) G. E. Herberich, I. Hausmann, B. Hessner and M. Negele, J. Organomet. Chem., 1989, 362, 259–264 CrossRef CAS.
- J. Böhnke, H. Braunschweig, J. O. C. Jiménez-Halla, I. Krummenacher and T. E. Stennett, J. Am. Chem. Soc., 2018, 140, 848–853 Search PubMed.
- M. Dietz, M. Arrowsmith, S. Reichl, L. I. Lugo-Fuentes, J. O. C. Jiménez-Halla, M. Scheer and H. Braunschweig, Angew. Chem., Int. Ed., 2022, 61, e202206840 CrossRef CAS PubMed.
- M. Dietz, M. Arrowsmith, L. Endres, V. Paprocki, B. Engels and H. Braunschweig, J. Am. Chem. Soc., 2023, 145, 22222–22231 CrossRef CAS PubMed.
- T. Zhao, Y. Dai, P. Cui, C.-H. Tung and L. Kong, J. Am. Chem. Soc., 2025, 147, 40113–40119 Search PubMed.
- M. Dietz, M. Arrowsmith and H. Braunschweig, Dalton Trans., 2024, 53, 449–453 RSC.
- (a) A. Lorbach, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2010, 29, 5762–5765 CrossRef CAS; (b) C. Balzereit, H.-J. Winkler, W. Massa and A. Berndt, Angew Chem. Int. Ed. Engl., 1994, 33, 2306–2308 CrossRef; (c) G. E. Herberich, B. Hessner and M. Hostalek, Angew Chem. Int. Ed. Engl., 1986, 25, 642–643 CrossRef.
- (a) M. Tamizmani, J. R. Tidwell, E. W. Reinheimer, B. M. Lindley and C. D. Martin, Inorg. Chem., 2023, 62, 7150–7154 CrossRef CAS PubMed; (b) C. Uhlmann, L. Münzfeld, A. Hauser, T.-T. Ruan, S. Kumar Kuppusamy, C. Jin, M. Ruben, K. Fink, E. Moreno-Pineda and P. W. Roesky, Angew. Chem., Int. Ed., 2024, 63, e202401372 CrossRef CAS PubMed; (c) G. E. Herberich and B. Heßner, Chem. Ber., 1982, 115, 3115–3127 CrossRef CAS.
- P. Müller, S. Hucka, H. Köppel, H. Pritzkowa and W. Siebert, Z. Naturforsch., 1995, 50b, 1476–1484 Search PubMed.
- E. Januszewski, A. Lorbach, R. Grewal, M. Bolte, J. W. Bats, H.-W. Lerner and M. Wagner, Chem. Eur J., 2011, 17, 12696–12705 CrossRef CAS PubMed.
- E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, Angew. Chem., Int. Ed., 2018, 57, 16491–16495 CrossRef CAS PubMed.
- P. Müller, B. Gangnus, H. Pritzkow, H. Schulz, M. Stephan and W. Siebert, J. Organomet. Chem., 1995, 487, 235–243 Search PubMed.
- (a) H. Schulz, H. Pritzkow and W. Siebert, Chem. Ber., 1991, 124, 2203–2207 Search PubMed; (b) P. Müller, H. Pritzkow and W. Siebert, J. Organomet. Chem., 1996, 524, 42–47 CrossRef.
- B. E. Mann, Chem. Commun., 1971, 976–977 RSC.
- R. D. Fischer, Chem. Ber., 1960, 93, 165–175 Search PubMed.
- F. Hanic and O. S. Mills, J. Organomet. Chem., 1968, 11, 151–158 CrossRef CAS.
- M. Trömel, Z. Naturforsch., 2000, 55b, 243–247 Search PubMed.
- (a) M. F. Bailey and L. F. Dahl, Inorg. Chem., 1965, 4, 1314–1319 CrossRef CAS; (b) H.-B. Bürgi, A. Raselli, D. Braga and F. Grepioni, Acta Crystallogr., Sect. B:Struct. Sci., 1992, 48, 428–437 CrossRef; (c) J. M. Oh, S. J. Geib and N. J. Cooper, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1998, 54, 581–583 CrossRef.
- (a) E. von Grotthuss, M. Diefenbach, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Angew. Chem., Int. Ed., 2016, 55, 14067–14071 CrossRef CAS PubMed; (b) E. von Grotthuss, S. E. Prey, M. Bolte, H.-W. Lerner and M. Wagner, J. Am. Chem. Soc., 2019, 141, 6082–6091 CrossRef CAS PubMed.
- (a) Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. von Ragué Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed; (b) A. Stanger, J. Org. Chem., 2006, 71, 883–893 CrossRef CAS PubMed.
- C. Hoffend, M. Diefenbach, E. Januszewski, M. Bolte, H.-W. Lerner, M. C. Holthausen and M. Wagner, Dalton Trans., 2013, 42, 13826–13837 RSC.
- K. B. Wiberg, Tetrahedron, 1968, 24, 1083–1096 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- G. Knizia, J. Chem. Theory Comput., 2013, 9, 4834–4843 CrossRef CAS PubMed.
- R. F. W. Bader, International Series of Monographs on Chemistry, Oxford University Press: Oxford, UK, 1990, Vol. 22 Search PubMed.
- (a) CCDC 2538786: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tb4; (b) CCDC 2538787: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tc5; (c) CCDC 2538788: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6td6; (d) CCDC 2538789: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tf7; (e) CCDC 2538790: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tg8; (f) CCDC 2538791: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6th9; (g) CCDC 2538792: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tjb; (h) CCDC 2538793: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tkc; (i) CCDC 2538794: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2r6tld.
Footnote |
| † There is one important difference: whereas 9,10-Br2-9,10-DHDBA only forms under static reduced pressure (i.e. evacuation of the headspace of the flask prior to heating), 1 only forms under elevated pressure. Under static reduced pressure, the reaction yields 1,2-bis(dibromoboryl)-4,5-dimethylbenzene instead. |
| This journal is © The Royal Society of Chemistry 2026 |