
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAgBF4-catalyzed insertion of unactivated alkynes into C–F bonds of acyl fluorides
Tomoya Emmeia and
Mamoru Tobisu *ab
*ab
aDepartment of Applied Chemistry, Graduate School of Engineering, The University of Osaka, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), The University of Osaka, Osaka 565-0871, Japan
First published on 13th May 2026
Abstract
We report an AgBF4-catalyzed intermolecular C–F insertion of unactivated alkynes into acyl fluorides, providing a general route to monofluoroalkenes. This transformation enables both C–F bond cleavage and reformation without activated coupling partners, driven by cooperative Ag+/BF4− catalysis, where BF4− functions as a fluoride shuttle. The reaction exhibits broad substrate scope, accommodating aromatic and aliphatic acyl fluorides with high Z-selectivity. Given that monofluoroalkenes are valuable amide bioisosteres, this method enables direct access to bio-relevant motifs from carboxylic acid derivatives and supports late-stage monofluoroalkene installation in complex molecules.
Introduction
Given its highest electronegativity and ability to form the strongest bond with carbon, fluorine has been widely employed as a substituent to strategically modulate the physicochemical properties of target molecules.1 Consequently, the development of methods for synthesizing organofluorine compounds has remained a central theme in contemporary organic chemistry. Significant progress has been achieved in fluorination reactions of non-fluorinated compounds,2 enabling access to a diverse array of fluorinated compounds. Leveraging such organofluorine compounds as building blocks for synthesizing more complex derivatives would represent a powerful synthetic strategy. In this context, the insertion of organic fragments into C–F bonds (hereafter referred to as C–F insertion) is particularly attractive, as it enables the direct transformation of fluorinated building blocks into structurally elaborated organofluorine derivatives without the need for costly, toxic, or unstable external fluorinating reagents (Scheme 1A). Despite its conceptual appeal, C–F insertion remains exceptionally challenging because it requires a single reaction system capable of orchestrating both C–F bond cleavage and C–F bond formation—each of which is intrinsically demanding. Although a number of C–F bond cleavage reactions have been reported, these are largely limited to substitution processes in which fluorine is expelled as a leaving group. In contrast, examples of C–F insertion are scarce and remain restricted in both substrate scope and the diversity of insertable fragments.3 To date, only four classes of fluorinated substrates have been shown to participate in C–F insertion reactions: acyl fluorides,4 benzyl and allyl (or propargyl) fluorides,5 and difluorocyclopropanes.6 Among these, acyl fluorides are particularly attractive because they are readily accessible from the corresponding carboxylic acids, isolable by column chromatography, and bench-stable (Scheme 1B).7 Nevertheless, C–F insertion reactions of acyl fluorides suffer from two major limitations. First, the scope of viable insertion partners is narrowly confined to activated substrates, including activated alkynes,4a,b tetrafluoroethylene,4c 1,1-difluoroalkenes,4e,f and benzofuran.4d Consequently, C–F insertion with simple, unactivated alkenes or alkynes remains unexplored, except for a few notable intramolecular reactions (vide infra).8,9 Second, unlike intramolecular reactions,8,9 intermolecular C–F insertion has thus far been limited to aromatic acyl fluorides, with aliphatic analogues being inapplicable. Herein, we report a catalytic solution to these challenges through the development of an intermolecular C–F insertion of unactivated alkynes into acyl fluorides, enabling a broadly applicable and conceptually distinct approach to C–F bond functionalization (Scheme 1C). Notably, it enables rapid access to structurally diverse monofluoroalkenes, which are of particular interest as potential amide bioisosteres (Scheme 1D).10 | ||
| Scheme 1 C–F Insertion: background and this work. | ||
Results and discussion
We began our investigation by screening catalysts for the C–F insertion of unactivated alkenes and alkynes with acyl fluorides. Acyl fluorides were selected for two key reasons: (1) they are easily accessible from the corresponding carboxylic acids and exhibit high chemical stability (isolable by chromatography), making them convenient fluorine-containing building blocks;7 and (2) prior studies by Le8 and our group9 demonstrated that C–F insertion of unactivated alkenes and alkynes can occur when tethered to acyl or carbamoyl fluorides. Central to these transformations is the pivotal role of BF4− as a fluoride shuttle, which facilitates both fluoride elimination and recombination. We hypothesized that this fluoride shuttle strategy could be extended to intermolecular C–F insertion — a significant challenge, given the difficulty of suppressing undesired side reactions, such as alkene/alkyne oligomerization, in the absence of the entropic advantage inherent to intramolecular reactions.Building on our prior success in intramolecular C–F insertion,9 we examined the reaction of acyl fluoride 1a with unactivated alkyne 2a using Rh(cod)2BF4 as the catalyst in 1,1,2,2-tetrachloroethane at 140 °C for 24 h. Under these conditions, the desired insertion product 3aa was formed in 22% yield (Table 1, Entry 1). Improved yield of 3aa were observed when other BF4 salts were employed, including Cu(CH3CN)4BF4 (53%, Entry 2), Ph3CBF4 (61%, Entry 3) and AgBF4 (72%, Entry 4). In contrast, the use of BF3·OEt2 resulted in a diminished yield (52%, Entry 5), while HBF4 afforded the product in only 20% yield (Entry 6). Based on these results, AgBF4 was identified as the optimal catalyst. In all cases, the product was formed stereoselectively as the Z isomer. This selectivity is attributed to isomerization of the initially formed, kinetically favored E isomer to the more thermodynamically stable Z isomer under the reaction conditions (see the SI for details).
|
|||
|---|---|---|---|
| Entry | Cat. | NMR yield [%] | |
| 3aa (Z/E) | Recovered 1a | ||
| 1 | Rh(cod)2BF4/dppbz | 22 (>20/1) | 21 |
| 2 | Cu(CH3CN)4BF4 | 53 (9/1) | 16 |
| 3 | Ph3CBF4 | 61 (19/1) | 1 |
| 4 | AgBF4 | 72 (>20/1) | 3 |
| 5 | BF3·OEt2 | 52 (>20/1) | 2 |
| 6 | HBF4 | 20 (Z only) | 18 |

Having optimized the catalyst and reaction conditions, we next explored the scope of this C–F insertion reaction (Scheme 2). Benzoyl fluorides bearing diverse functional groups, including alkyl (1c), alkoxy (1a), halides (1d), and acetals (1e), underwent C–F insertion smoothly upon increasing the catalyst loading to 20 mol%, affording the corresponding alkenyl fluorides 3aa–3ea. Acyl fluorides derived from naphthalene (1g, 1h) and heteroarene (1l–1n) frameworks successfully participated in this reaction. Sterically hindered ortho-substituted benzoyl fluorides 1i and 1j proved compatible as well. Notably, aliphatic acyl fluorides were also viable substrates, which represents a significant contrast to previously reported intermolecular C–F insertion reactions.4 Acyl fluorides with primary (1o), secondary (1p), and tertiary (1q) alkyl groups all participated successfully, delivering the insertion products 3oa–3qa with excellent Z-selectivity. Moreover, acyl fluorides derived from α,β-unsaturated carboxylic acids (e.g., 1r) were also suitable. Regarding the alkyne component, phenylacetylene derivatives bearing fluorides (2b, 2e), chlorides (2c), bromides (2d, 2f![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 11), and alkoxy groups (2g) were well-tolerated. The compatibility of halide functionalities is particularly advantageous, offering a handle for subsequent structural modifications. A current limitation of this methodology is the incompatibility with aliphatic alkynes and internal alkynes. A current limitation of this methodology is its incompatibility with aliphatic and internal alkynes. Notably, in the reaction with diphenylacetylene under the standard conditions, the desired C–F insertion product was not observed; instead, cyclized indenone derivatives were formed. This outcome is consistent with the intermediacy of a vinyl cation species (see Scheme 4), which undergoes rapid intramolecular SEAr cyclization, in preference to trapping by BF4− (see the SI for details).
11), and alkoxy groups (2g) were well-tolerated. The compatibility of halide functionalities is particularly advantageous, offering a handle for subsequent structural modifications. A current limitation of this methodology is the incompatibility with aliphatic alkynes and internal alkynes. A current limitation of this methodology is its incompatibility with aliphatic and internal alkynes. Notably, in the reaction with diphenylacetylene under the standard conditions, the desired C–F insertion product was not observed; instead, cyclized indenone derivatives were formed. This outcome is consistent with the intermediacy of a vinyl cation species (see Scheme 4), which undergoes rapid intramolecular SEAr cyclization, in preference to trapping by BF4− (see the SI for details).
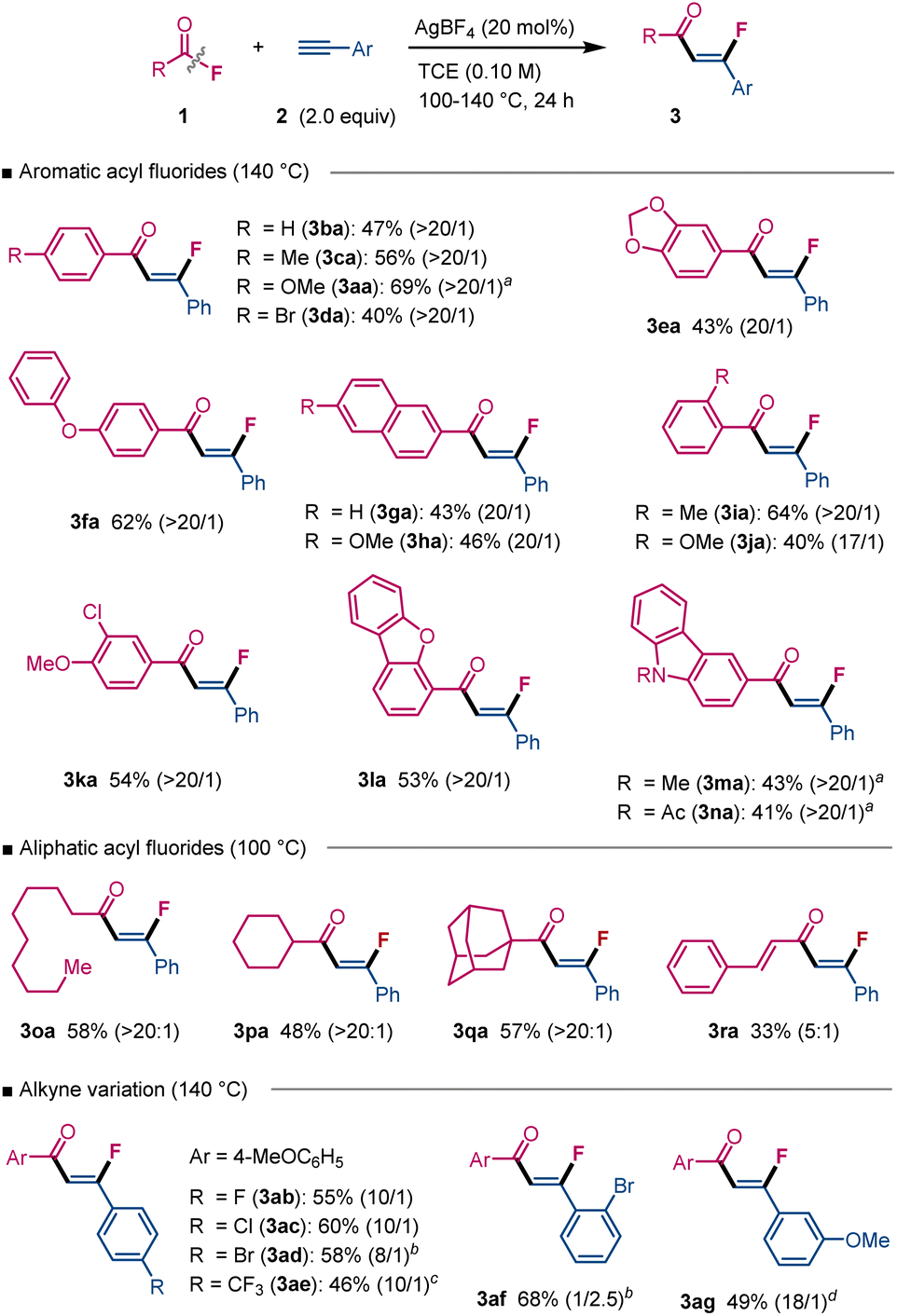 | ||
| Scheme 2 Reaction scope. aWith AgBF4 (15 mol%). bWith alkyne (3 equiv). cWith AgBF4 (30 mol%) for 48 h. dAt 120 °C. | ||
Several experiments were conducted to gain insights into the reaction mechanism. First, acyl fluoride 1a was reacted with anisole instead of an alkyne under Ag-catalyzed conditions, yielding acylated anisole 4 (Scheme 3A). This result suggests that 1a undergoes electrophilic activation by AgBF4, enabling nucleophilic attack by an external nucleophile. Next, the effect of the counteranions in Ag(I) salts was examined (Scheme 3B). While BF4− facilitated the C–F insertion, SbF6− and OTf− were completely ineffective, highlighting the essential role of BF4−. Notably, under AgSbF6-catalyzed conditions, the C–F insertion product 3aa was obtained in 55% yield when BF4− was added externally. When PF6− was used, a catalytic amount of 3aa (9%) was generated, suggesting that the fluoride ion affinity (FIA)12 of the parent Lewis acids must fall within an optimal range to act as a fluoride shuttle (vide infra). During the C–F insertion reactions, 10–20% of alkynyl ketone 5 was consistently detected as a side product. This observation led us to consider a plausible reaction pathway, as outlined in Scheme 3C. In this scenario, AgBF4 promotes acyl substitution of 1 with terminal alkyne 2 to generate 5 along with HF. The HF could then add across 5 to produce the C–F insertion product 3. To examine the feasibility of this reaction pathway, we designed the experiment shown in Scheme 3D. The AgBF4-catalyzed reaction of 1a with 2a was conducted in the presence of alkynyl ketone 5ba, which is a side product from the reaction of a different acyl fluoride, 1b. If the proposed HF addition pathway were operative, the HF adduct of 5ba (i.e., 3ba) would be formed. However, only 3aa (the C–F insertion product from 1a) and its side product 5aa were observed, while 3ba was not detected and 5ba was quantitatively recovered. These results rule out a pathway involving alkynyl ketone 5 as an intermediate.
 | ||
| Scheme 3 Mechanistic studies. | ||
Based on these mechanistic experiments, we propose the mechanism depicted in Scheme 4. The carbonyl oxygen in acyl fluoride 1 initially coordinates to the Ag(I) cation, forming intermediate A. This interaction is supported by 13C and 19F NMR analysis of a mixture of 1a/AgBF4 (see SI for details). The resulting electrophilically activated carbonyl carbon in A undergoes nucleophilic attack by alkyne 2, generating vinyl cation intermediate B. The Ag center then abstracts the neighboring fluoride, leading to acylated vinyl cation C, accompanied by AgF. An alternative pathway involving the generation of an acylinium cation through direct fluoride abstraction from 1 by AgBF4 cannot be excluded at this stage. Vinyl cation C subsequently captures fluoride from BF4−, furnishing the C–F insertion product 3 alongside BF3.13 The reaction between AgF and BF3 regenerates AgBF4,14 thereby ensuring catalytic turnover. This step was confirmed experimentally (see SI for details). The observed side product 5 likely arises from deprotonation of C. This proposed mechanism implies that the counteranion must exhibit balanced reactivity profile: it should efficiently donate fluoride to C, while its parent Lewis acid must possess sufficient FIA to regenerate Ag+ via fluoride abstraction from AgF. The FIA of the corresponding Lewis acids follows the order SbF5 > PF5 > BF3, indicating that fluoride-donating ability of the counteranions increases in the reverse order (SbF6− < PF6− < BF4−). The success of AgBF4 in promoting this C–F insertion is thus attributed to the relatively high fluoride-donating ability of BF4−, combined with the moderate Lewis acidity of BF3.
 | ||
| Scheme 4 Proposed mechanism. | ||
The present C–F insertion reaction provides a straightforward approach to access a diverse range of monofluoroalkenes from readily available acyl fluorides and simple alkynes. This transformation is particularly valuable for the synthesis of bio-relevant molecules, as monofluoroalkenes are recognized bioisosteres for amides10 (Scheme 5). Given that carboxylic acids are prevalent functional groups in bioactive compounds, our C–F insertion strategy enables the installation of a monofluoroalkene moiety directly from carboxylic acid derivatives. This utility is demonstrated through the late-stage modification of complex molecules such as cholic acid, gemfibrozil and adapalene.
 | ||
| Scheme 5 Late-stage introduction of fluoroalkene moieties into bio-relevant molecules. | ||
Conclusions
In summary, we have developed AgBF4-catalyzed intermolecular insertion of unactivated alkynes into acyl fluorides. Both Ag+ and BF4− play essential roles in facilitating the capture and release of fluoride. While the synthesis of organofluorine compounds has traditionally focused on fluorination of non-fluorinated substrates, the concept of C–F insertion provides an alternative approach that repurposes existing organofluorine compounds as starting materials to generate more valuable derivatives. Further investigations to expand this strategy are currently ongoing in our laboratory.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
Additional data and NMR spectra can be found in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6sc02480g.Acknowledgements
This work was supported by JSPS KAKENHI Grant Number 24H02207 (M.T.) in Transformative Research Areas (A) JP24A202 Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP). We thank Ms. Sakura Takahashi and Mr Kota Shintaku for assistance with NMR experiments. We also thank the Instrumental Analysis Center, Faculty of Engineering, The University of Osaka, for assistance with HRMS.Notes and references
- Selected reviews: (a) K. Müller, C. Faeh and F. Diederich, Science, 2007, 317, 1881–1886 CrossRef PubMed; (b) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320–330 RSC; (d) C. Ni and J. Hu, Chem. Soc. Rev., 2016, 45, 5441–5454 RSC.
- Selected reviews: (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed; (c) S. Fustero, D. M. Sedgwick, R. Román and P. Barrio, Chem. Commun., 2018, 54, 9706–9725 RSC.
- Reviews: (a) D.-S. Deng, S.-Q. Tang, Y.-T. Yuan, D.-X. Xie, Z.-Y. Zhu, Y.-M. Huang and Y.-L. Liu, Chin. Chem. Lett., 2024, 35, 109417 CrossRef CAS; (b) A. Garg, A. Haswell and M. N. Hopkinson, Chem.–Eur. J., 2024, 30, e202304229 CrossRef CAS PubMed; (c) Y. Zeng, X. Yan and Y. Xia, Tetrahedron Chem, 2024, 12, 100109 CrossRef CAS.
- (a) H. Fujimoto, T. Kodama, M. Yamanaka and M. Tobisu, J. Am. Chem. Soc., 2020, 142, 17323–17328 CrossRef CAS PubMed; (b) H. Fujimoto, S. Yamamura, N. Takenaka and M. Tobisu, Synthesis, 2023, 55, 899–906 CrossRef CAS; (c) N. Ishida, H. Iwamoto, D. E. Sunagawa, M. Ohashi and S. Ogoshi, Synthesis, 2021, 53, 3137–3143 CrossRef CAS; (d) X. Yu, Q.-Y. Meng, C. G. Daniliuc and A. Studer, J. Am. Chem. Soc., 2022, 144, 7072–7079 CrossRef CAS PubMed; (e) X. Yu, A. Maity and A. Studer, Angew. Chem., Int. Ed., 2023, 62, e202310288 CrossRef CAS PubMed; (f) Y. Mao, Y. Liu, X. Wang, S. Ni, Y. Pan and Y. Wang, Chin. Chem. Lett., 2024, 35, 109443 CrossRef CAS.
- (a) F. Wang, Y. Nishimoto and M. Yasuda, J. Am. Chem. Soc., 2021, 143, 20616–20621 CrossRef CAS PubMed; (b) A. Garg, N. J. Gerwien, C. Fasting, A. Charlton and M. N. Hopkinson, Angew. Chem., Int. Ed., 2023, 62, e202302860 CrossRef CAS PubMed; (c) Y.-B. Wang and M. Chen, J. Am. Chem. Soc., 2025, 147, 25061–25071 CrossRef CAS PubMed; (d) H. Luo, X. Cai, Z. Li, J. Hu and T. Wu, ACS Catal., 2025, 15, 12148–12156 CrossRef CAS; (e) J. Duran, V. G. Moldoveanu, C. Barroso, G. A. Pereira, B. Limburg and X. Companyó, Angew. Chem., Int. Ed., 2026, 65, e20513 CrossRef CAS PubMed; (f) X. Cai, T. Shi, W. Wen, M. Cen and T. Wu, Org. Lett., 2025, 27, 13449–13455 CrossRef CAS PubMed.
- (a) D. Li, C. Shen, Z. Si and L. Liu, Angew. Chem., Int. Ed., 2023, 62, e202310283 CrossRef CAS PubMed; (b) Y. Zhu, J. Jia, X. Song, C. Gong and Y. Xia, Chem. Sci., 2024, 15, 13800–13806 RSC; (c) Y. Zeng, H. Gao, Z.-T. Jiang, Y. Zhu, J. Chen, H. Zhang, G. Lu and Y. Xia, Nat. Commun., 2024, 15, 4317 CrossRef CAS PubMed.
- (a) Y. Ogiwara and N. Sakai, Angew. Chem., Int. Ed., 2020, 59, 574–594 CrossRef CAS PubMed; (b) T. Tian, Q. Chen, Z. Li and Y. Nishihara, Synthesis, 2022, 54, 3667–3697 CrossRef CAS; (c) N. Blanchard and V. Bizet, Angew. Chem., Int. Ed., 2019, 58, 6814–6817 CrossRef CAS PubMed.
- (a) E. A. McKnight, R. Arora, E. Pradhan, Y. H. Fujisato, A. J. Ajayi, M. Lautens, T. Zeng and C. M. Le, J. Am. Chem. Soc., 2023, 145, 11012–11018 CrossRef CAS PubMed; (b) A. McKnight, Y. H. Fujisato, N. Khanal and C. M. Le, Org. Lett., 2025, 27, 1322–1326 CrossRef CAS PubMed.
- T. Yoshida, M. Ohta, T. Emmei, T. Kodama and M. Tobisu, Angew. Chem., Int. Ed., 2023, 62, e202303657 CrossRef CAS PubMed.
- S. Kumari, A. V. Carmona, A. K. Tiwari and P. C. Trippier, J. Med. Chem., 2020, 63, 12290–12358 CrossRef CAS PubMed.
- When the alkyne 2f was used, the E isomer was formed preferentially. DFT calculations indicate that, unlike other products, E-3af is more thermodynamically stable than Z-3af, which is consistent with the experimental results. See SI for details.
- P. Erdmann, J. Leitner, J. Schwarz and L. Greb, ChemPhysChem, 2020, 21, 987–994 CrossRef CAS PubMed.
- (a) A. J. Cresswell, S. G. Davies, P. M. Roberts and J. E. Thomson, Chem. Rev., 2015, 115, 566–611 CrossRef CAS PubMed; (b) R. Guo, X. Qi, H. Xiang, P. Geaneotes, R. Wang, P. Liu and Y.-M. Wang, Angew. Chem., Int. Ed., 2020, 59, 16651–16660 CrossRef CAS PubMed.
- G. A. Olah and H. W. Quinn, J. Inorg. Nucl. Chem., 1960, 14, 295–296 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |