
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceResolving the inner versus outer component of the 5f radial wavefunction in the early actinides
Nathan M. Alcock abc,
Myron S. Huzanabc,
Timothy G. Burrowd,
Myrtille O. J. Y. Hunaultd,
Christelle Tamaine,
Matthieu Autilloe,
Thomas Dumas*e and
Michael L. Baker*abc
abc,
Myron S. Huzanabc,
Timothy G. Burrowd,
Myrtille O. J. Y. Hunaultd,
Christelle Tamaine,
Matthieu Autilloe,
Thomas Dumas*e and
Michael L. Baker*abc
aDepartment of Chemistry, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: michael.baker@manchester.ac.uk
bCentre for Radiochemistry Research, The University of Manchester, Oxford Road, Manchester, M13 9PL, UK
cThe University of Manchester at Harwell, Diamond Light Source, Harwell Campus, OX11 0DE, UK
dSynchrotron SOLEIL, L'Orme des Merisiers, Départementale 128, 91190, Saint-Aubin, France
eCEA, DES, ISEC, DMRC, Univ. Montpellier, Marcoule 30207, France. E-mail: Thomas.DUMAS@cea.fr
First published on 7th May 2026
Abstract
Actinide chemistry is largely defined by 5f orbital behaviour, but experimental measurements of their role in bonding are scarce, despite their importance for understanding chemical reactivity. Here, we demonstrate that M4-edge 3d4f resonant inelastic X-ray scattering can discern the averaged 5f radial wavefunction, revealing how 5f electron density varies with distance from the nucleus. This approach enables differential quantification of how the inner and outer components of the 5f radial wavefunction respond to ligand bonding. Applied to a series of actinide hexachloride complexes, [AnCl6]2− (An = U, Np, Pu), it is shown that bonding-induced expansion of the 5f radial wavefunction increases along the series. However, this expansion is non-uniform; the outer region expands more when going from U to Pu than the inner region, which remains constrained by increasing nuclear charge. This identified sensitivity is used to rationalise 5f covalency trends across the early actinides. The results provide needed experimental insight into the mechanisms by which early actinide 5f orbitals engage in covalent bonding.
Introduction
The physical and chemical properties of the actinides are influenced by the extent of 5f orbital involvement in chemical bonding.1–3 Actinide coordination chemistry research is rapidly expanding, revealing rich and varied chemistry including an increasing diversity of transuranium studies,4–12 multiple bonds,13–15 wide-ranging redox behaviour,16–18 and catalysis.19,20 In much of this work, the degree of covalency present in actinide ligand bonds underpins the interpretation of physicochemical properties. The proposed presence of 5f metal–ligand covalency dates back to work by G. T. Seaborg and co-workers who rationalized ion-exchange behavior in early actinides as evidence of covalent mixing between actinide 5f and Cl 3p orbitals.21 This notion was supported further by Streitwieser and colleagues following the seminal discovery of the actinocenes.22 In more recent years, actinide computational chemistry studies have exposed deeper questions concerning 5f covalency, since different bonding analyses provide contrasting perspectives on both the degree of covalency present and the mechanism by which its presence occurs.23–26There is, therefore, a pressing need for high-quality experimental data to evaluate 5f bonding behavior. Experimental measures of 5f orbital engagement in bonding are required to correlate with physical properties, validate computational predictions, and develop improved models. There is a paucity of spectroscopic studies that have evaluated early actinide bonding trends. Electron paramagnetic resonance (EPR)27 and nuclear magnetic resonance (NMR)28–30 have been applied in cases where ligand ions are nuclear spin active, however studies into covalent contributions to bonding have thus far been limited to U and Th complexes. Chlorine ligand K-edge X-ray absorption near edge structure (XANES) has provided important insight into trends in actinide 5f and 6d contributions to bonding.31–35 This includes a study of [AnCl6]2− complexes (An = Th, U, Np, Pu) by Batista, Clark, Kozimor, Yang and co-workers that provides unique evidence of a 5f orbital covalency contribution to Cl bonding.34 Additionally, ligand K-edge XANES has also been proven as a probe of actinide carbon and actinide oxygen ligand bonding.36–41
Resonant inelastic X-ray scattering at actinide M4,5 edges (3d4f RIXS) is being increasingly applied as a probe of 5f electronic structure and bonding.42–52
Dipole selection rules enable resonant excitation into states of unoccupied 5f character, making 3d4f RIXS a promising route to exclusively evaluate 5f contributions to actinide bonding. There are clear advantages to 3d4f RIXS since it can be applied to actinide compounds with any ligand atom identity, nuclear spin, actinide oxidation state, or coordination symmetry. However, 3d4f RIXS is considerably less mature than established spectroscopies, with many remaining questions concerning the method's sensitivity to 5f bonding.
In the present study, we report M4-edge 3d4f RIXS on [AnCl6]2− complexes (An = U, Np, Pu) and show how the method carries a previously unidentified sensitivity to the distribution of the averaged 5f radial wavefunction, and hence, provides previously lacking insight into the influence of 5f orbitals in bonding. It is demonstrated that 3d4f RIXS can be analyzed to determine how bonding differentially influences the outer versus inner components of the 5f radial wavefunction. RIXS fine structure associated with 4f–5f spin-exchange is a unique probe of the inner component of the 5f radial wavefunction, while fine structure associated with 5f inter-electron repulsion is sensitive to the outer component of the 5f radial wavefunction. It is shown that this differential sensitivity to the distribution of the 5f radial wavefunction provides new insights into early actinide bonding trends. M4-edge 3d4f RIXS analysis shows that 5f radial expansion in going from an An4+ free ion to a [AnCl6]2− complex increases along the actinide series from U to Np and to Pu. However, this expansion becomes increasingly non-uniform. The outer component of the 5f radial wavefunction undergoes covalent expansion, due to proximity with the ligand orbitals, while the inner component expands less, due to increasing nuclear charge (Zeff) on traversing the series.
Results and discussion
Experimental RIXS data
The 3d4f RIXS measurements were conducted at the M4-edge (3d3/2 → 5f) and the Mβ (4f5/2 → 3d3/2) emission line (see SI for additional details on 3d4f RIXS spectroscopy). The M4 absorption profile is obtained by taking a constant emission energy cut through the two-dimensional RIXS plane, known as high-energy resolution fluorescence detection (HERFD). The Mβ emission profile can be resolved from a vertical cut through the RIXS plane at the resonant excitation energy, known as resonant X-ray emission spectroscopy (RXES). The M4 HERFD and Mβ RXES spectra for [UCl6]2−, [NpCl6]2−, and [PuCl6]2− are shown in Fig. 1. The HERFD spectra (Fig. 1(a)) are dominated by the resonant M4 absorption feature Ia, around 3725 eV (U), 3846 eV (Np), and 3967 eV (Pu), and exhibit additional fine structure at higher incident energies, labelled Ib. The two-peak HERFD fine structure for [PuCl6]2− is similar to that reported for PuO2.47,53 For [UCl6]2− only, an additional higher energy satellite feature is detected, labelled Ic. We previously assigned features Ib and Ic to originate from 5f inter-electron repulsion interactions.51,54 | ||
| Fig. 1 Experimental (solid colored) and simulated (dashed black) M4-edge HERFD (a) and Mβ RXES (b) spectra for [UCl6]2− (left), [NpCl6]2− (middle), and [PuCl6]2− (right), with inplots highlighting satellite features Ic and IIa. Simulated M4-edge 3d4f RIXS planes (c) are shown for [UCl6]2− (left), [NpCl6]2− (middle), and [PuCl6]2− (right), with the corresponding simulated HERFD and RXES cuts shown as diagonal and vertical red lines, respectively. | ||
The RXES spectra (Fig. 1(b)) are dominated by the actinide Mβ emission line (4f5/2 → 3d3/2) centered around 388 eV (U), 412 eV (Np), and 431 eV (Pu) energy transfer. Low energy transfer satellite features, labelled IIa, are detected for [UCl6]2−, [NpCl6]2−, and [PuCl6]2− at around 382.5 eV, 405 eV, and 424 eV energy transfer, respectively. An additional satellite feature is shown at higher transfer energies, labelled IIb. We previously assigned feature IIa for [UCl6]2− to originate from 4f–5f spin-exchange interactions.51 The energy position of satellite feature IIa relative to the maximum of the RXES increases from 6 eV in [UCl6]2−, to 6.3 eV in [NpCl6]2−, and 7.0 eV in [PuCl6]2−, along with a corresponding increase in broadness and intensity relative to the maximum of the RXES.
Computational analysis of RIXS
Uranium(IV) 3d4f RIXS fine structure carries a distinct sensitivity to 5f inter-electron repulsion and 4f–5f inter-shell spin-exchange.51 In a previous 3d4f RIXS study on [UX6]2− complexes (X = F, Cl, Br) we showed that 5f inter-electron repulsion and 4f–5f spin-exchange both decrease from [UF6]2− to [UCl6]2− and to [UBr6]2−. The trend follows the nephelauxetic series,55 reflecting reduced inter-electron repulsion and spin-exchange due to expansion of the 5f radial wavefunction as covalency increases in going from a hard to soft donor ligand. The 3d4f RIXS fine structure of uranium hexahalides is dominated by 5f inter-electron repulsion and 4f–5f inter-shell spin-exchange, which can be quantified via Slater integrals Fk5f5f and Gk4f5f, respectively.51 The extent by which bonding within a complex reduces Fk5f5f and Gk4f5f for an actinide ion can be expressed in terms of the nephelauxetic reduction parameter (β):55 e.g.
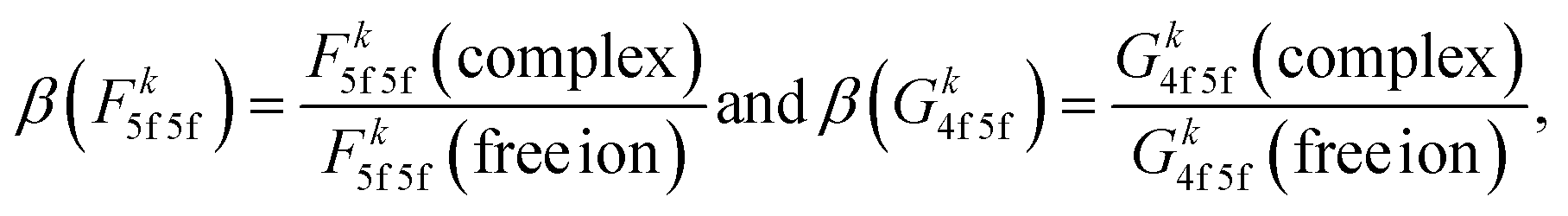 | (1) |
To predict the influence of bonding across the early actinides in [AnCl6]2− complexes (An = U, Np, Pu), we perform ligand field density functional theory (LFDFT) calculations implemented in the Amsterdam Density Functional (ADF) code.56,57 LFDFT utilizes an average of configuration (AOC) DFT method to define an active space via the selective occupation of frozen Kohn–Sham molecular orbitals. Slater integrals and ligand field parameters are calculated from first principles, including the Coulomb (Fk) and exchange (Gk) Slater integrals, relativistic spin–orbit coupling constants (ζnl), and the 5f ligand field splittings.58 All calculated ligand field parameters are listed in SI Tables S3 to S6. The octahedral ligand field transforms the 5f orbitals into three irreducible representations, t1u, t2u and a2u. The total calculated ligand field energy splitting does not exceed 0.365 eV, and there are only minor differences in the ligand field splitting for [UCl6]2−, [NpCl6]2−, and [PuCl6]2−. Calculated ligand field parameters describing 5f inter-electron repulsion (Fk5f5f) for the [AnCl6]2− complexes are shown in Table 1, expressed in the terms of the nephelauxetic reduction factor β (eqn (1)). The calculated nephelauxetic β(Fk5f5f) values (k = 2, 4, 6) in Table 1 show a sequential decrease in 5f inter-electron repulsion in going from [UCl6]2− to [NpCl6]2− and to [PuCl6]2− relative to their free ion values, indicating that 5f radial wavefunction expansion from free ion to complex increases across the series. The calculated 5f spin–orbit coupling constants are also reduced relative to their free-ion values due to the relativistic nephelauxetic effect.59 Relativistic β(ζ5f) decreases slightly, consistent with the trends identified in β(Fk5f5f), from [UCl6]2− to [NpCl6]2− and [PuCl6]2− with values of 0.84, 0.83 and 0.81, respectively (See SI Table S3).
| β(Fk5f5f) | [UCl6]2− | [NpCl6]2− | [PuCl6]2− |
|---|---|---|---|
| k = 2 | 0.80 | 0.77 | 0.72 |
| k = 4 | 0.79 | 0.76 | 0.71 |
| k = 6 | 0.79 | 0.75 | 0.71 |
To model the 3d4f RIXS, further LFDFT calculations are conducted for both RIXS intermediate (3d94f145fn+1) and final (3d104f135fn+1) state electron configurations and the resulting ligand field parameters are implemented in RIXS multiplet simulations.51 The calculated 5f ligand field splitting energies in the intermediate and final states are similar to the initial states for all three compounds and do not exceed 0.38 eV. The 5f ligand field splitting is therefore rather small relative to the measured RIXS energy resolution. LFDFT predicted parameters accurately reproduce the HERFD and RXES spectra, except the G04f5f values which, to obtain agreement with experiment, require a reduction to 62% of the LFDFT calculated value for [NpCl6]2− and 69% of the LFDFT calculated value for [PuCl6]2−, compared to a reduction to 54% of the LFDFT calculated value for [UCl6]2− (see Fig. S6 for LFDFT RIXS simulations excluding reduction of G04f5f).51,54 No other changes to the LFDFT parameters are required for good agreement with experiment. The simulated RIXS planes are shown in Fig. 1(c), and the corresponding HERFD and RXES simulations are compared with experiment in Fig. 1(a) and (b), respectively. Good reproduction of the general features is observed. The simulated HERFD captures the general shape of the spectra, including features Ia and Ib which result in an asymmetry in the HERFD shape for [NpCl6]2− and the double peak structure for [PuCl6]2−. To gain insight into the sensitivity of the RIXS spectra to the obtained LFDFT parameters, a series of systematic calculations were conducted, identifying significant sensitivity to 5f inter-electron repulsion, inter-shell spin-exchange, and spin–orbit coupling, and a very limited influence from 5f ligand field splitting (see SI Fig. S8 to S12). It was previously shown that RIXS fine-structure for [UCl6]2− has minimal sensitivity to 5f spin–orbit coupling.51 In contrast, the simulated RIXS fine structure for [NpCl6]2− and [PuCl6]2− shows a more significant sensitivity to 5f spin–orbit coupling (see SI Fig. S12). The relative energy and intensity of spectral features Ia and Ib for [NpCl6]2− and [PuCl6]2− are identified to depend on the magnitude of the Fk5f5f Slater integrals and 5f spin–orbit coupling constant, ζ5f. Varying these parameters away from those predicted by LFDFT worsens the agreement with experiment (See SI Fig. S9 and S12).
The experimental spectra, therefore, provide an independent confirmation for the LFDFT predicted values of Fk5f5f and ζ5f, confirming the predicted increase in covalency in moving across the series from U to Np and Pu. The simulated RXES, including satellite features IIa and IIb, are reproduced for all three complexes. The RXES satellites IIa and IIb are narrower than observed in experiment. Due to the different nature of the RXES satellite excitations relative to the main emission line, different line shape broadening can be expected. However, a constant line shape was used for the calculations of the RIXS planes, in order to describe electronic fine structure in the most transparent way. The simulated RXES show increasing intensity and energy separation of satellite IIa relative to the maximum of the RXES in going from [UCl6]2− to [NpCl6]2− to [PuCl6]2−, reproducing the experimental spectral trend (Fig. 2). The calculations confirm that satellite features IIa and IIb originate from 4f–5f spin-exchange interactions, and most strongly depend on G04f5f (see SI Fig. S11). The relative energy and intensity between the maximum of the RXES and IIa is therefore a direct probe of G04f5f.51,52
 | ||
| Fig. 2 Experimental (a) and simulated (b) Mβ RXES satellite feature IIa for [UCl6]2−, [NpCl6]2−, and [PuCl6]2−, plotted relative to the energy and intensity of the maximum of the RXES. | ||
The simulations reveal that the spectral features in the 3d4f RIXS of [AnCl6]2− are insensitive to the relatively small 5f ligand field splitting generated by the coordinated chloride ions (see further analysis in SI Section S3.2). The simulations reveal that 5f a2u, t2u, and t1u orbital character is strongly mixed over the full energy range of the absorption edge (see transition dipole tensor analysis, Fig. S7). Therefore, while symmetry-restricted mixing of metal and ligand orbitals will contribute to the reduction of inter-electron repulsion, 5f spin–orbit coupling and spin-exchange, the spectra are insensitive to the relative amounts of σ versus π covalency60 and [AnCl6]2− ligand field splitting.
Decomposition of the 5f radial wavefunction
Since the RIXS of all three compounds are found to carry a strong sensitivity to Fk5f5f and G04f5f, we focus on these Slater integrals for the [AnCl6]2− complexes in Table 2. For a given actinide, the nephelauxetic reduction parameters (β) express the amount the 5f radial wavefunction expands in going from free ion to [AnCl6]2− complex.| [UCl6]2− | [NpCl6]2− | [PuCl6]2− | |
|---|---|---|---|
| β(F25f5f) | 0.77 | 0.71 | 0.64 |
| β(F45f5f) | 0.76 | 0.70 | 0.64 |
| β(F65f5f) | 0.76 | 0.70 | 0.63 |
| β(G04f5f) | 0.45 | 0.50 | 0.53 |
The trend in β(Fk5f5f) when moving from An = U to Np to Pu indicates an increasing 5f radial expansion in response to bonding across the [AnCl6]2− series and, therefore, an increase in covalency. Conclusions from earlier Cl ligand K-edge XANES, DFT and relativistic multiconfigurational wavefunction theory studies on the same complexes identified that An 5f–Cl 3p covalency either increases slightly or does not significantly change across the series.25,34 A more recent DFT analysis of covalent bond formation in [UCl6]2−, [NpCl6]2− and [PuCl6]2−, applying a prebonding assessment of atomic orbital overlap versus atomic orbital energy matching, identify a small but non negligible An(5f)–Cl(3p) orbital overlap throughout the series.26 The computational analyses in these studies relate to symmetry-adapted mixing of Cl 3p and An 5f character (symmetry-restricted covalency). In contrast, 3d4f RIXS and LFDFT based determination of nephelauxetic β incorporates the effect of both average 5f radial wavefunction expansion (central-field covalency) and symmetry-restricted covalency. Since Cl K-edge XANES and theoretical calculations25,26,34 infer that symmetry-restricted covalency does not vary significantly along the early actinide series, we assign an increase in central-field covalency from U to Pu to explain the identified reductions in β(Fk5f5f). The β(G04f5f) values show an opposing trend relative to β(Fk5f5f) in moving from An = U to Np to Pu. This difference may seem counter intuitive, however the sensitivities of Fk5f5f and G04f5f to bonding inherently differ since G04f5f is linked to the 4f core sub-shell.51 The 4f core sub-shell has a contracted radial wavefunction and is unaffected by bonding (Fig. S13). Consequently, G04f5f is linked to the behaviour of the inner part of the 5f radial wavefunction. In contrast, Fk5f5f carries greater sensitivity to the behavior of the outer part of the 5f radial wavefunction. For example, the distinct radial sensitivities of F25f5f(r) and G04f5f(r) can be demonstrated by comparing them with their limiting values F25f5f(∞) and G04f5f(∞) alongside the associated 5f or 4f radial wavefunctions (Fig. 3). The F25f5f integral gains magnitude at radial distances greater than r ∼ 0.3 Å and reaches its terminal value (F25f5f(r)/F25f5f(∞) → 1) at large radial distances, showing the sensitivity of F25f5f to the outer component of the 5f radial wavefunction. In contrast, the G04f5f Slater integral gains magnitude at low radial distances and reaches its terminal value (G04f5f(r)/G04f5f(∞) → 1) by r ∼ 0.4 Å.
 | ||
| Fig. 3 (a) The averaged 5f radial wavefunction for [PuCl6]2− plotted with the normalized F25f5f(r)/F25f5f(∞) rolling integral, showing its strong sensitivity to the outer component of the 5f radial wavefunction. The rolling integral behaviour of Fk=4,65f5f are very similar to Fk=25f5f, see SI Fig. S14. (b) 4f and 5f radial wavefunctions for [PuCl6]2− plotted with the normalized G04f5f(r)/G04f5f(∞) rolling integral, showing its strong sensitivity to the inner component of the 5f radial wavefunction. Schematics showing cross-sections of a 5fx(x2−3y2) atomic orbital highlight the sensitivity the radial integrals have to the outer (a) and inner (b) components. | ||
A subtle sensitivity of different Slater integrals to inner versus outer component of radial wavefunctions has been identified in transition metal UV-Vis spectra61,62 and, more recently, high-resolution 2p3d valence-to-core RIXS.63,64 For transition metals, F23d3d is slightly more sensitive to the outer part of the 3d radial wavefunction than F43d3d.65 Consequently, a greater reduction from the free ion value of F23d3d is observed relative to F43d3d in covalent complexes.61 By contrast, a very distinct sensitivity of G04f5f to the inner components of the 5f radial wavefunction and F25f5f to the outer components enables deconvolution of the influence of bonding inside versus outside the 5f radial node.
On traversing the actinide series, radial contraction of the 5f orbitals is understood to occur due to imperfect screening of the increasing nuclear charge by successive addition of electrons in 5f orbitals.2 The consequence of imperfect screening of nuclear charge is the so-called actinide contraction, the sequential radial contraction of the atomic 5f radial wavefunction. The opposing trends in β values demonstrate that distortion of the 5f radial wavefunction increases from U to Pu, providing a partial counteraction of the actinide contraction, by maintaining a radially contracted electron density inside the 5f node, thereby permitting the outer component of the 5f radial wavefunction to expand for participation in covalent metal–ligand bonding.
The specific sensitivity of G04f5f to orbital behaviour at low radial distances provides an explanation for the required reduction of the LFDFT predicted G04f5f value to match experiment. The exclusion of semi-core correlation effects in the ab initio determination of ligand field parameters may lead to an underestimation of the nephelauxetic effect,66 and hence, an overestimation of Slater integral values. Since G04f5f depends on both the core 4f and the inner “semi-core” component of the 5f radial wavefunction, it follows that it will be more sensitive to these dynamic correlation effects than the outer “valence” component of the 5f radial wavefunction. Specific inclusion of semi-core correlation effects in the LFDFT model may, therefore, correct overestimation of G04f5f in the spectroscopic final state.
Conclusion
Our analysis reveals that 3d4f RIXS possesses exceptional sensitivity to the influence of bonding on the inner versus outer component of the actinide 5f radial wavefunction. This sensitivity is so distinct that it enables deconvolution of the response to bonding inside versus outside the 5f radial node. For [AnCl6]2− (An = U, Np, Pu), it is found that An–Cl bonding increasingly distorts the 5f radial wavefunction in going from U to Np and to Pu. Bonding induced expansion of the outer component of the 5f radial wavefunction increases from U to Pu whereas the inner component decreases, due to the increase in nuclear charge. This opposing trend in the behavior of the inner versus outer components of the 5f radial wavefunction contribute to partly counteract actinide contraction, aiding the availability of the 5f orbitals to participate in covalent metal–ligand bonding.There is a limited number of experimental methods available for quantifying trends in actinide electronic structure and bonding. The approach presented here is particularly versatile as it is applicable to actinide systems irrespective of ligand element, nuclear spin, oxidation state, and coordination symmetry. The identified 3d4f RIXS sensitivity to covalency and the 5f radial wavefunction significantly enhances our ability to establish structure–function relationships and physicochemical trends in actinide chemistry, and provides valuable experimental data to evaluate the predictive power of theoretical models.
Methods
Experimental
Caution! 237Np and 239Pu isotopes are high specific activity α-particle emitting radionuclides, and their use presents extreme hazards to human health. 238U isotopes are low specific-activity α-particle emitting radionuclides, and their use presents hazards to human health. This research was conducted in a radiological facility with appropriate analyses of these hazards and implementation of controls for the safe handling and manipulation of these toxic and radioactive materials.Synthesis of [(C2H5)4N]2PuCl6 (1) and [(C2H5)4N]2NpCl6 (2)
All manipulations involving radioactive materials were conducted at the ATALANTE facility (CEA-ISEC, France) under strictly controlled conditions. Due to the high radiotoxicity of neptunium and plutonium, all experiments were carried out in a negative-pressure glovebox under ambient air, with stringent operational protocols in place. All chemicals were used as received: nitric acid (HNO3, 65%) and hydrochloric acid (HCl, 37%) were obtained from Sigma-Aldrich, along with tetraethylammonium chloride (TEAC) and boron nitride (BN). Sodium hydroxide (NaOH) pellets were purchased from VWR and hydroxylamine hydrochloride (NH2OH HCl) from Merck.The syntheses of 1 and 2 were adapted from procedures previously reported for analogous actinide complexes of the type M2AnCl6, typically prepared via slow evaporation from aqueous solutions.67,68 For compound 1, the plutonium precursor was Pu(IV) in 1.5 M HNO3. 20 mg (V = 133 µL) was precipitated by the addition of concentrated NaOH. The resulting solid was washed three times with deionized water and subsequently dissolved in 4 M HCl (200 µL). After complete dissolution, two equivalents of TEAC (27.7 mg), pre-dissolved in 4 M HCl (100 µL) were added. Crystals of 1 suitable for single crystal X-ray diffraction (SC-XRD) were obtained by slow evaporation of the solution. For compound 2, the neptunium precursor was Np(V) in 3 M nitric acid. A 20 mg aliquot (V = 369 µL) was used. The reduction of Np(V) to Np(IV) was achieved by adding 100 equivalents of NH2OH·HCl followed by heating at 70 °C for several hours. The reduced Np(IV) species was then precipitated by addition of concentrated NaOH, was washed three times with water, and dissolved in 4 M HCl (200 µL) containing 4 equivalents of NH2OH·HCl. After complete dissolution, 2 equivalents of TEAC (27.7 mg) dissolved in 4 M HCl (100 µL) were added. Crystals of 2 suitable for SC-XRD were obtained by slow evaporation of the solution.
Single crystal X-ray diffraction
Single crystal X-ray diffraction (SC-XRD) measurements were performed using a Bruker D8 Quest diffractometer equipped with a PHOTON II charge-coupled device (CCD) detector and a Mo-target IµS microfocus radiation source (λ = 0.71073 Å). Data were collected at 200 K using an Oxford Cryosystems 800 series cryostream cooler. Individual crystals were mounted on a MiTeGen MicroMount and enclosed in MicroRT capillaries, which were sealed to the goniometer base using adhesive to prevent the release of radioactive material and ensure safe handling. Data were collected using ϕ and Ω scan strategies. Diffraction intensities were integrated using the SAINTPlus software package,69 and multi-scan absorption corrections were performed.70Unit cell parameters were refined from the complete dataset. Structural solution and refinement were carried out using SHELX-2017.71 Heavy atoms were located by direct methods, and the remaining atoms were identified through successive Fourier map analyses. All non-hydrogen atoms were refined anisotropically. Due to disorder in one of the TEA cations, hydrogen atoms on the ethyl chains could not be located from Fourier map analyses and were not included in the refinement. Hydrogen atoms bonded to the ordered TEA cation were placed in idealized positions using a riding model and assigned fixed isotropic displacement parameters.
Crystallographic data and refinement details for both structures (Fig. S15 and S16) are summarized in Table S11. The structures have been deposited on the Cambridge Crystallographic Data Centre (CCDC) structural database with deposition numbers 2466992 (1) and 2466993 (2). These data can be obtained free of charge via https://www.ccdc.cam.ac.uk/data_request/cif, or by emailing E-mail: data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Resonant inelastic X-ray scattering
For RIXS measurements, samples were prepared as pellets by finely grinding ca. 6 mg of single crystals with ca. 15 mg of BN. The pellets were mounted in special stainless steel sample holders enabling double confinement of the samples following the requirements of the French Nuclear Safety authorization at the MARS beamline. Further description of the sample holders can be found in the SI.Neptunium and plutonium M4-edge RIXS measurements were carried out at ambient temperature using the CX3 end-station of the MARS beamline72 at Synchrotron SOLEIL (Saint-Aubin, France). The storage ring operated in top-up mode at 2.5 GeV with an electron current of 500 mA. The incident photon energy was selected using a double-crystal monochromator (DCM) equipped with two Si(111) crystals. Vertical focusing and harmonic rejection were achieved using Si mirror strips positioned before and after the DCM at an incidence angle of 4 mrad. Energy calibration was performed using the L3 absorption edge of iodine in a KI pellet (4557 eV). The incident X-ray flux at 3700 eV was 1.9 × 109 per photons, with a beam size of 350 × 140 µm (horizontal × vertical, FWHM) at the sample position. The effects of photon-induced radiolysis for each sample were quantified before being mitigated by minimizing the scanning time per sample position on the pellets. The Mβ emission lines of Np (3435 eV) and Pu (3534 eV) were detected using a spherically bent Si(220) crystal analyzer (1 m bending radius) and a KETEK silicon drift detector arranged in a vertical Rowland circle geometry, with Bragg angles of 70.0° and 66.0°, respectively. Further details concerning the precise experimental scattering geometry for each measurement are found in the SI Section S1.1. Emission intensity was normalized to the incident flux. A He-filled chamber was used to minimize air scattering between the sample, analyzer, and detector. HERFD spectra were normalized to 1 in the post-edge region (3905–3940 eV for Np, 4015–4030 eV for Pu) using a linear fitting function. Background-subtracted RXES spectra were scaled so that their resonant intensity matched the maximum intensity of the HERFD spectra.
Uranium M4-edge RIXS measurements were performed at the European Synchrotron Research Facility (ESRF, Grenoble, France) as previously reported.51 HERFD and RXES spectra were normalized as above, using a HERFD post-edge region of 3820–3840 eV.
Computational
Electronic structure calculations were performed on [AnCl6]2− fragments (An = U, Np, Pu) based on single-crystal X-ray diffraction (XRD) crystallographic data.Ligand field density functional theory calculations
Average of configuration (AOC) DFT calculations were performed using the Amsterdam Density Functional (ADF) code implemented in the Amsterdam Modeling Suite (AMS, version 2023.104).56 The B3LYP functional was used for all calculations. Equivalent calculations were also conducted with the PBE0 function. There are negligible differences in LFDFT parameters between PBE0 and B3LYP; see SI Tables S7 and 8 for β values using PBE0 versus B3LYP. Scalar relativistic effects were treated with the zeroth order relativistic approximation (ZORA). Given known incompleteness errors originating in inner shells using ZORA,73 we also conducted calculations using X2C. LFDFT parameters obtained using X2C are very similar, and related RIXS simulations are nearly identical. SI Tables S9–S10 show that there are negligible differences in nephelauxetic β values using ZORA versus X2C.74,75 All calculations employed Slater type orbital (STO) triple-ζ-plus double polarisation all-electron basis sets from the ZORA/TZ2P database.76 The active spaces in the AOC calculations were defined as follows for An(IV) with ground state electron occupations of 5fn for An = U (n = 2), Np (n = 3), Pu (n = 4). For the ground state, each of the seven 5f MOs were fractionally populated with n/7 electrons, whilst for the intermediate and final states, the 5f MOs were fractionally populated with (n + 1)/7 electrons. The core holes were defined by fractionally occupying each actinide core 3d orbital with 9/5 electrons for the intermediate state and each core 4f orbital with 13/7 electrons in the final state. The AOC DFT results were treated by the ligand field density functional theory57,58 (LFDFT) implemented in ADF to obtain ligand field parameters for each of the initial (3d104f145fn), intermediate (3d94f145fn+1), and final (3d104f135fn+1) RIXS electronic configurations. Isosurface plots of the associated Kohn–Sham MOs are provided in SI Fig. S5 (isosurface level = 0.03). The plotted 4f and 5f radial wavefunctions are represented by radial distribution functions (RDFs) extracted from the AOC Kohn–Sham orbitals centered on the actinide ion and printed by inclusion of the debug keyword in the LFDFT input file.Ligand field multiplet simulations
Multiplet simulations of 3d4f RIXS planes were carried out using Quanty Version 0.6.77 The simulated RIXS planes include linewidth broadening to incorporate the effect of energy broadening due to core-hole lifetimes combined with spectrometer resolution. Lorentzian full-width half-maximum broadenings of 4.4 eV and 0.6 eV are applied to the incident energy (3d9) dimension and the energy transfer dimension (4f13), respectively. Additionally, a Gaussian full-width half-maximum broadening of 0.8 eV is applied to both incident and transfer energy dimensions.The selected broadenings were determined empirically. The Lorentzian broadenings applied are slightly larger than available core-hole lifetime broadening data,78 since they also incorporate instrumental broadening contributions to the linewidth. Improved fits can be obtained by including slightly different linewidths for each element, however, for simplicity and transparency, only one set of linewidth parameters is used for all compounds.
Hamiltonians for the initial, intermediate, and final electronic configurations of the 3d4f RIXS process were constructed using the results of LFDFT calculations, including 5f ligand field splitting, electron–electron interactions, and spin–orbit coupling. The influence of each term in the Hamiltonian was systematically investigated via an analysis of simulated RIXS fine structure to determine the relationship between the observed spectral features and the differences in electronic structure and bonding in going from [UCl6]2− to [NpCl6]2− and [PuCl6]2− (see SI S3.2 for further details).79,80 The G04f5f Slater integral required adjustment to obtain RIXS simulations that best fit the experimental results. For consistency, the other 4f–5f Slater integrals, including Fk4f5f (k = 2,4,6) and Gk4f5f (k = 2,4) are treated with the same scaling, although RIXS spectral simulations are less sensitive to these Slater integrals (SI Fig. S10 and S11), as previously established.51,52 Other Slater integrals determined from LFDFT analysis (including Fk5f5f, Fk3d5f and Gk3d5f) were found to be consistent with experiment. The 2D RIXS planes were calculated via a Green's theorem approach to the Kramers–Heisenberg equation, incorporating the experimental geometries81 (see SI S1.1 for further details).
Author contributions
MLB supervised and conceptualized the study. Np and Pu RIXS measurements were conducted by NMA, MSH, TGB, MLB, TD and MOJYH. NMA performed the data analysis and calculations under the supervision of MLB. Ligand field multiplet simulations were performed by NMA and MSH under the supervision of MLB. MA and CT synthesized compounds and determined their structures by SC-XRD under supervision of TD. The manuscript was written by NMA and MLB with review from all co-authors.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2466992 and 2466993 contain the supplementary crystallographic data for this paper.82a,bThe data supporting this article have been included as part of the supplementary information (SI). Further numerical research data supporting this publication are available from FigShare: https://doi.org/10.6084/m9.figshare.32114431. Supplementary information: includes further comments on RIXS experimental details, including instrument scattering geometry and radioactive sample confinement. Further details on ligand field DFT calculations, including AOC calculated active spaces and all calculated LFDFT and multiplet simulation parameters, and calculated radial wavefunctions for each complex. Ligand field multiplet simulations are included showing RIXS planes for [NpCl6]2− and [PuCl6]2− simulated with unscaled LFDFT parameters, a partial excitations analysis, as well as simulations showing the spectral dependence on 5f ligand field splitting, 5f spin–orbit coupling, and Slater integrals. A summary of the X-ray crystallographic analysis of the newly reported compounds is also included. See DOI: https://doi.org/10.1039/d6sc02413k.
Acknowledgements
We thank Synchrotron SOLEIL (20190807 and 20230706) and the European Synchrotron Radiation Facility (CH-6518) for the experimental time. This research was supported by the EPSRC (grant number EP/V029347/1 and EP/W029057/1). MLB was supported by the Analytical Chemistry Trust Fund and a Community for Analytical Measurement Science (CAMS) Fellowship. This work was supported by computing resources provided by the STFC Scientific Computing Department SCARF cluster and the University of Manchester Computational Shared Facility, and associated support services. We extend thanks to William Breton (https://orcid.org/0009-0002-6023-0679) at MARS for his technical assistance, as well as to Bruno Forgeat (SOLEIL) of the Radioprotection Services for their help.Notes and references
- S. T. Liddle, Angew. Chem., Int. Ed., 2015, 54, 8604–8641 CrossRef CAS PubMed.
- M. L. Neidig, D. L. Clark and R. L. Martin, Coord. Chem. Rev., 2013, 257, 394–406 CrossRef CAS.
- S. T. Liddle, Inorg. Chem., 2024, 63, 9366–9384 CrossRef CAS PubMed.
- J. Murillo, J. A. Seed, A. J. Wooles, M. S. Oakley, C. A. Goodwin, M. Gregson, D. Dan, N. F. Chilton, A. J. Gaunt and S. A. Kozimor, et al., J. Am. Chem. Soc., 2024, 146, 4098–4111 CrossRef CAS PubMed.
- C. A. Goodwin, M. T. Janicke, B. L. Scott and A. J. Gaunt, J. Am. Chem. Soc., 2021, 143, 20680–20696 CrossRef CAS PubMed.
- M. S. Dutkiewicz, C. A. Goodwin, M. Perfetti, A. J. Gaunt, J.-C. Griveau, E. Colineau, A. Kovács, A. J. Wooles, R. Caciuffo and O. Walter, et al., Nat. Chem., 2022, 14, 342–349 CrossRef CAS PubMed.
- T. N. Poe, H. Ramanantoanina, J. M. Sperling, H. B. Wineinger, B. M. Rotermund, J. Brannon, Z. Bai, B. Scheibe, N. Beck, B. N. Long, S. Justiniano, T. E. Albrecht-Schönzart and C.-B. Cristian, Nat. Chem., 2023, 15, 722–728 CrossRef CAS PubMed.
- J. E. Niklas, K. S. Otte, C. M. Studvick, S. Roy Chowdhury, B. Vlaisavljevich, J. Bacsa, F. Kleemiss, I. A. Popov and H. S. La Pierre, Nat. Chem., 2024, 16, 1490–1495 CrossRef CAS.
- D. R. Russo, A. N. Gaiser, A. N. Price, D.-C. Sergentu, J. N. Wacker, N. Katzer, A. A. Peterson, J. A. Branson, X. Yu, S. N. Kelly, E. T. Ouellette, J. Arnold, J. R. Long, W. W. Lukens Jr, S. J. Teat, R. J. Abergel, P. L. Arnold, J. Autschbach and S. G. Minasian, Science, 2025, 387, 974–978 CrossRef CAS.
- K. P. Carter, K. M. Shield, K. F. Smith, Z. R. Jones, J. N. Wacker, L. Arnedo-Sanchez, T. M. Mattox, L. M. Moreau, K. E. Knope, S. A. Kozimor, C. H. Booth and R. J. Abergel, Nature, 2021, 590, 85–88 CrossRef CAS PubMed.
- N. B. Beck, C. Celis-Barros, M. C. Martelles, J. M. Sperling, Z. Bai, J. P. Brannon, D. G. Martinez, Z. K. Huffman, B. N. Long, K. N. Mendoza, R. W. Merinsky, B. M. Rotermund, T. N. Poe and T. E. Albrecht, Nat. Commun., 2025, 16, 7759 CrossRef CAS PubMed.
- H. Zhang, A. Li, K. Li, Z. Wang, X. Xu, Y. Wang, M. V. Sheridan, H.-S. Hu, C. Xu, E. V. Alekseev, Z. Zhang, P. Yan, K. Cao, Z. Chai, T. E. Albrecht-Schönzart and S. Wang, Nature, 2023, 616, 482–487 CrossRef CAS PubMed.
- D. M. King, F. Tuna, E. J. McInnes, J. McMaster, W. Lewis, A. J. Blake and S. T. Liddle, Science, 2012, 337, 717–720 CrossRef CAS.
- H.-S. Hu, Y.-H. Qiu, X.-G. Xiong, W. E. Schwarz and J. Li, Chem. Sci., 2012, 3, 2786–2796 RSC.
- A. R. Fox and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 5716–5717 CrossRef CAS PubMed.
- C. L. Tracy, M. Lang, J. M. Pray, F. Zhang, D. Popov, C. Park, C. Trautmann, M. Bender, D. Severin, V. A. Skuratov and R. C. Ewing, Nat. Commun., 2015, 6, 6133 CrossRef CAS PubMed.
- C. Deng, J. Liang, R. Sun, Y. Wang, P.-X. Fu, B.-W. Wang, S. Gao and W. Huang, Nat. Commun., 2023, 14, 4657 CrossRef CAS.
- L. S. Natrajan, A. N. Swinburne, M. B. Andrews, S. Randall and S. L. Heath, Coord. Chem. Rev., 2014, 266–267, 171–193 CrossRef CAS.
- P. L. Arnold, T. Ochiai, F. Y. Lam, R. P. Kelly, M. L. Seymour and L. Maron, Nat. Chem., 2020, 12, 654–659 CrossRef CAS PubMed.
- X. Liu, Y. Xie, M. Hao, Y. Li, Z. Chen, H. Yang, G. I. Waterhouse, X. Wang and S. Ma, Nat. Commun., 2024, 15, 7736 CrossRef CAS.
- R. Diamond, K. Street Jr and G. T. Seaborg, J. Am. Chem. Soc., 1954, 76, 1461–1469 CrossRef CAS.
- A. Streitwieser Jr and U. Müller-Westerhoff, J. Am. Chem. Soc., 1968, 90, 7364 CrossRef.
- N. Kaltsoyannis and A. Kerridge, Nat. Rev. Chem., 2024, 8, 701–712 CrossRef CAS PubMed.
- N. Kaltsoyannis, Inorg. Chem., 2013, 52, 3407–3413 CrossRef CAS PubMed.
- D. C. Sergentu and J. Autschbach, Chem. Sci., 2022, 13, 3194–3207 RSC.
- A. Sen, A. Obeng, K. Stanistreet-Welsh and J. Autschbach, Inorg. Chem., 2025, 64, 23787–23803 CrossRef CAS.
- A. Formanuik, A.-M. Ariciu, F. Ortu, R. Beekmeyer, A. Kerridge, F. Tuna, E. J. McInnes and D. P. Mills, Nat. Chem., 2017, 9, 578–583 CrossRef CAS PubMed.
- J. Du, J. A. Seed, V. E. Berryman, N. Kaltsoyannis, R. W. Adams, D. Lee and S. T. Liddle, Nat. Commun., 2021, 12, 5649 CrossRef CAS PubMed.
- J. Du, J. Hurd, J. A. Seed, G. Balázs, M. Scheer, R. W. Adams, D. Lee and S. T. Liddle, J. Am. Chem. Soc., 2023, 145, 21766–21784 CrossRef CAS PubMed.
- T. W. Hayton and J. Autschbach, Acc. Chem. Res., 2025, 3259–3263 Search PubMed.
- S. A. Kozimor, P. Yang, E. R. Batista, K. S. Boland, C. J. Burns, D. L. Clark, S. D. Conradson, R. L. Martin, M. P. Wilkerson and L. E. Wolfsberg, J. Am. Chem. Soc., 2009, 131, 12125–12136 CrossRef CAS PubMed.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. D. Conradson, S. A. Kozimor, R. L. Martin, D. E. Schwarz, D. K. Shuh, G. L. Wagner, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2012, 134, 5586–5597 CrossRef CAS.
- L. P. Spencer, P. Yang, S. G. Minasian, R. E. Jilek, E. R. Batista, K. S. Boland, J. M. Boncella, S. D. Conradson, D. L. Clark, T. W. Hayton, S. A. Kozimor, R. L. Martin, M. M. MacInnes, A. C. Olson, B. L. Scott, D. K. Shuh and M. P. Wilkerson, J. Am. Chem. Soc., 2013, 135, 2279–2290 CrossRef CAS PubMed.
- J. Su, E. R. Batista, K. S. Boland, S. E. Bone, J. A. Bradley, S. K. Cary, D. L. Clark, S. D. Conradson, A. S. Ditter, N. Kaltsoyannis, J. M. Keith, A. Kerridge, S. A. Kozimor, M. W. Löble, R. L. Martin, S. G. Minasian, V. Mocko, H. S. La Pierre, G. T. Seidler, D. K. Shuh, M. P. Wilkerson, L. E. Wolfsberg and P. Yang, J. Am. Chem. Soc., 2018, 140, 17977–17984 CrossRef CAS PubMed.
- J. N. Cross, J. Su, E. R. Batista, S. K. Cary, W. J. Evans, S. A. Kozimor, V. Mocko, B. L. Scott, B. W. Stein, C. J. Windorff and P. Yang, J. Am. Chem. Soc., 2017, 139, 8667–8677 CrossRef CAS.
- S. G. Minasian, J. M. Keith, E. R. Batista, K. S. Boland, D. L. Clark, S. A. Kozimor, R. L. Martin, D. K. Shuh and T. Tyliszczak, Chem. Sci., 2014, 5, 351–359 RSC.
- J. A. Branson, P. W. Smith, J. Arnold and S. G. Minasian, Inorg. Chem., 2024, 63, 15557–15562 CrossRef CAS.
- Y. Zhang, W. Duan, Y. Yang, T. Jian, Y. Qiao, G. Ren, N. Zhang, L. Zheng, W. Yan and J. Wang, et al., Inorg. Chem., 2021, 61, 92–104 CrossRef.
- R. G. Denning, J. C. Green, T. E. Hutchings, C. Dallera, A. Tagliaferri, K. Giarda, N. B. Brookes and L. Braicovich, J. Chem. Phys., 2002, 117, 8008–8020 CrossRef CAS.
- Y. Qiao, G. Ganguly, C. H. Booth, J. A. Branson, A. S. Ditter, D. J. Lussier, L. M. Moreau, D. R. Russo, D.-C. Sergentu and D. K. Shuh, et al., Chem. Commun., 2021, 57, 9562–9565 RSC.
- D. E. Smiles, E. R. Batista, C. H. Booth, D. L. Clark, J. M. Keith, S. A. Kozimor, R. L. Martin, S. G. Minasian, D. K. Shuh, S. Chantal, E. Stieber and T. Tyliszczak, Chem. Sci., 2020, 11, 2796–2809 RSC.
- K. O. Kvashnina and S. M. Butorin, Chem. Commun., 2022, 58, 327–342 RSC.
- R. Caciuffo, G. H. Lander and G. van der Laan, Rev. Mod. Phys., 2023, 95, 015001 CrossRef CAS.
- K. O. Kvashnina, S. M. Butorin, P. Martin and P. Glatzel, Phys. Rev. Lett., 2013, 111, 253002 CrossRef CAS.
- T. Vitova, I. Pidchenko, D. Fellhauer, P. S. Bagus, Y. Joly, T. Pruessmann, S. Bahl, E. Gonzalez-Robles, J. Rothe, M. Altmaier, M. A. Denecke and H. Geckeis, Nat. Commun., 2017, 8, 16053 CrossRef CAS.
- D.-C. Sergentu, T. J. Duignan and J. Autschbach, J. Phys. Chem. Lett., 2018, 9, 5583–5591 CrossRef CAS.
- P. S. Bagus, B. Schacherl and T. Vitova, Inorg. Chem., 2021, 60, 16090–16102 CrossRef CAS PubMed.
- R. Polly, B. Schacherl, J. Rothe and T. Vitova, Inorg. Chem., 2021, 60, 18764–18776 CrossRef CAS.
- J. Tobin, S. Nowak, S. Yu, R. Alonso-Mori, T. Kroll, D. Nordlund, T. Weng and D. Sokaras, J. Phys.: Condens. Matter, 2022, 34, 505601 CrossRef CAS PubMed.
- C. Silva, L. Amidani, M. Retegan, S. Weiss, E. Bazarkina, T. Graubner, F. Kraus and K. Kvashnina, Nat. Commun., 2024, 15, 6861 CrossRef CAS PubMed.
- T. G. Burrow, N. M. Alcock, M. S. Huzan, M. A. Dunstan, J. A. Seed, B. Detlefs, P. Glatzel, M. O. J. Y. Hunault, J. Bendix, K. S. Pedersen and M. L. Baker, J. Am. Chem. Soc., 2024, 146, 22570–22582 CrossRef CAS.
- B. Schacherl, M. Tagliavini, H. Kaufmann-Heimeshoff, J. Göttlicher, M. Mazzanti, K. Popa, O. Walter, T. Pruessmann, C. Vollmer, A. Beck, R. S. K. Ekanayake, J. A. Branson, T. Neill, D. Fellhauer, C. Reitz, D. Schild, D. Brager, C. Cahill, C. Windorff, T. Sittel, H. Ramanantoanina, M. W. Haverkort and T. Vitova, Nat. Commun., 2025, 16, 1221 CrossRef CAS.
- K. O. Kvashnina, A. Y. Romanchuk, I. Pidchenko, L. Amidani, E. Gerber, A. Trigub, A. Rossberg, S. Weiss, K. Popa, O. Walter, R. Caciuffo, A. C. Scheinost, S. M. Butorin and S. N. Kalmykov, Angew. Chem., 2019, 131, 17722–17726 CrossRef.
- T. G. Burrow, N. M. Alcock, M. S. Huzan, M. A. Dunstan, J. A. Seed, B. Detlefs, P. Glatzel, M. O. J. Y. Hunault, J. Bendix and K. S. Pedersen, et al., J. Am. Chem. Soc., 2025, 147, 37876–37878 CrossRef CAS.
- C. K. Jørgensen, Orbitals in Atoms and Molecules, Academic Press, London, United Kingdom, 1962, p. 59 Search PubMed.
- G. te Velde, F. M. Bickelhaupt, E. J. Baerends, C. Fonseca Guerra, S. J. A. van Gisbergen, J. G. Snijders and T. Ziegler, J. Comput. Chem., 2001, 22, 931–967 CrossRef CAS.
- H. Ramanantoanina, W. Urland, F. Cimpoesu and C. Daul, Phys. Chem. Chem. Phys., 2013, 15, 13902–13910 RSC.
- M. Atanasov, C. Daul and C. Rauzy, Optical Spectra and Chemical Bonding in Inorganic Compounds: Special Volume dedicated to Professor Jørgensen I, 2004, pp. 97–125 Search PubMed.
- J. Jung, M. Atanasov and F. Neese, Inorg. Chem., 2017, 56, 8802–8816 CrossRef CAS PubMed.
- E. C. Wasinger, F. M. De Groot, B. Hedman, K. O. Hodgson and E. I. Solomon, J. Am. Chem. Soc., 2003, 125, 12894–12906 CrossRef CAS.
- J. Griffith and L. Orgel, J. Am. Chem. Soc., 1956, 4981–4983 CAS.
- J. Ferguson and D. Wood, Aust. J. Chem., 1970, 23, 861–871 CrossRef CAS.
- M. O. J. Y. Hunault, Y. Harada, J. Miyawaki, J. Wang, A. Meijerink, F. M. De Groot and M. M. Van Schooneveld, J. Phys. Chem. A, 2018, 122, 4399–4413 CrossRef CAS PubMed.
- A. W. Hahn, B. E. Van Kuiken, M. Al Samarai, M. Atanasov, T. Weyhermüller, Y.-T. Cui, J. Miyawaki, Y. Harada, A. Nicolaou and S. DeBeer, Inorg. Chem., 2017, 56, 8203–8211 CrossRef CAS PubMed.
- M. M. Gerloch and R. C. Slade, Ligand-field parameters, Cambridge University Press, London, 1973, pp. 200–228 Search PubMed.
- S. V. Rao, D. Maganas, K. Sivalingam, M. Atanasov and F. Neese, Inorg. Chem., 2024, 63, 24672–24684 CrossRef CAS.
- S. G. Minasian, K. S. Boland, R. K. Feller, A. J. Gaunt, S. A. Kozimor, I. May, S. D. Reilly, B. L. Scott and D. K. Shuh, Inorg. Chem., 2012, 51, 5728–5736 CrossRef CAS PubMed.
- M. Autillo and R. E. Wilson, Eur. J. Inorg. Chem., 2017, 2017, 4834–4839 CrossRef CAS.
- Bruker Analytical X-ray Systems, SAINTPlus, version 6.22, Madison, WI, 2001 Search PubMed.
- Bruker Analytical X-ray Systems, SADABS, version 2.03, Madison, WI, 2001 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. L. Solari, S. Schlutig, H. Hermange and B. Sitaud, J. Phys.: Conf. Ser., 2009, 190, 12042 CrossRef.
- D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2011, 7, 677–684 CrossRef CAS.
- E. van Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1994, 101, 9783–9792 CrossRef CAS.
- E. van Lenthe, A. Ehlers and E.-J. Baerends, J. Chem. Phys., 1999, 110, 8943–8953 CrossRef CAS.
- E. van Lenthe and E. J. Baerends, J. Comput. Chem., 2003, 24, 1142–1156 CrossRef CAS PubMed.
- M. W. Haverkort, M. Zwierzycki and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165113 CrossRef.
- J. Campbell and T. Papp, Atomic Data Nucl. Data Tables, 2001, 77, 1–56 CrossRef CAS.
- F. De Groot and A. Kotani, Core level spectroscopy of solids, CRC press, 2008 Search PubMed.
- S. M. Butorin, Inorg. Chem., 2020, 59, 16251–16264 CrossRef CAS.
- T. G. Burrow, M. O. J. Y. Hunault, F. Besnard, A. Juhin and C. Brouder, Phys. Rev. B, 2026, 113, 115131 CrossRef CAS.
- (a) CCDC 2466992: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nt3d0; (b) CCDC 2466993: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nt3f1.
| This journal is © The Royal Society of Chemistry 2026 |