
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDe novo construction of C–O axial chirality via cobalt-catalyzed atroposelective C–H activation/annulation
Yanbo Zhang†
a,
Yichao Xie†a,
Baicheng Guoa,
Yingchao Dou *a,
Dandan Yang*a and
Jun-Long Niu*ab
*a,
Dandan Yang*a and
Jun-Long Niu*ab
aCollege of Chemistry, Pingyuan Laboratory, Zhengzhou University, Zhengzhou, 450001, China. E-mail: yingchaodou@zzu.edu.cn; yangdandan@zzu.edu.cn; niujunlong@zzu.edu.cn
bState Key Laboratory of Coking Coal Resources Green Exploitation, Zhengzhou University, Zhengzhou, 450001, China
First published on 6th May 2026
Abstract
The catalytic asymmetric synthesis of axially chiral compounds has attracted growing attention over the past decades. C–O axially chiral compounds represent a distinctive class of atropisomers characterized by a unique dual-axial architecture. Their atroposelective construction remains a formidable challenge due to inherent low rotational barriers and high conformational flexibility. Current catalytic approaches rely predominantly on enantioselective desymmetrization of preformed diaryl ethers. Here, we report an unprecedented de novo synthetic strategy, enabling the construction of a new class of C–O axially chiral aryl-heterocyclic ethers. This protocol is achieved by an earth-abundant cobalt-catalyzed atroposelective C–H activation/annulation of arylamides with phenylethynyl ethers, which concurrently builds the heterocycle and the C–O chiral axis in a single step. Notably, a traceless directing group strategy is successfully implemented, featuring smooth in situ cleavage. Furthermore, a series of C–O atropisomers bearing multiple stereogenic elements are prepared with excellent stereoselectivity. Racemization experiments reveal high configurational stability of the products. Derivatization studies afford a versatile library of non-biaryl C–O axially chiral scaffolds, extending the applicability of this strategy.
Introduction
Developing efficient strategies for the construction of diverse chiral entities has long been a central pursuit in chemical research.1–10 In contrast to the extensively studied point chirality, axial chirality arises from restricted rotation around a stereogenic axis. Given the widespread application of atropisomeric frameworks in pharmaceuticals and advanced materials, substantial efforts have been devoted to their asymmetric construction. Distinct from well-established mono-axial chiral systems (Fig. 1a), such as C–C, C–N, C–B, and N–N atropisomers, the distinctive C–O axially chiral architecture features a unique dual-axial chirality,11–15 wherein two contiguous C–O axes are bonded to a central oxygen atom. This structural motif is not only prevalent in bioactive molecules and natural products,16–23 but also serves as a privileged scaffold in functional molecules24 and phosphine ligands25–28 (Fig. 1b). Despite their significance, the asymmetric construction of C–O atropisomers remains largely underdeveloped, and is hindered by several intrinsic challenges: (1) the inherently low rotational barriers associated with two contiguous C–O axes endow C–O axially chiral compounds with flexible atropisomerism,29 triggering facile racemization via concerted rotation of both axes; (2) simultaneous stereocontrol over two C–O axes complicates the achievement of high atroposelectivity compared to mono-axial chirality molecules; (3) the oxygen bridge between the two C–O axes weakens spatial and electronic interactions of substituents, resulting in enhanced conformational freedom. Consequently, these factors collectively compromise the configurational stability and synthetic accessibility of dual-axial scaffolds, making their catalytic asymmetric synthesis a considerable challenge. | ||
| Fig. 1 Background and project synopsis for the synthesis of C–O atropisomers. (a) Axially chiral skeletons and synthetic challenges in C–O axially chiral compounds. (b) Selected bioactive molecule and ligand featuring C–O axis. (c) Previous catalytic strategies for synthesizing C–O atropisomers. (d) This work: de novo construction of C–O axially chiral aryl-isoquinolinol ethers via cobalt-catalyzed C–H activation/annulation. | ||
Since the pioneering investigations of C–O atropisomers by Fuji,30 the catalytic asymmetric construction of such architectures has gained growing attention in the synthetic community and represents a significant frontier in modern stereochemistry.11–15,31,32 Several elegant catalytic enantioselective approaches toward C–O atropisomers have been achieved (Fig. 1c). However, current strategies rely mostly on atroposelective desymmetrization via enzymatic,33,34 organocatalytic35–43 or transition-metal-catalyzed functionalizations,44–48 and focus predominantly on the synthesis of C–O axially chiral diaryl ether frameworks. The exclusive construction of axially chiral naphthoquinone-aryl ethers was achieved by Gustafson via a dynamic kinetic resolution approach.49 Given the significance of privileged C–O axial structures, it is highly desirable to develop novel synthetic strategies that could enable the efficient construction of C–O atropisomers with broader structural diversity.
Over the past decades, the transition-metal catalyzed50–53 asymmetric C–H activation has emerged as an atom-economical and versatile platform for accessing enantiopure molecules. However, forging C–O axially chiral compounds via this versatile approach has not been realized to date. More recently, the 3d-metal cobalt-catalyzed asymmetric C–H activation53–58 has attracted considerable attention since the independent establishment of cobalt/salicyloxazoline (Salox) catalytic system by the Shi group59 and our group.60 This 3d-metal platform has enabled facile access to diverse axially chiral architectures, including C–C, C–N, and N–N atropisomers.60–79 Inspired by these achievements, we hypothesized that the cobalt/Salox catalysis could offer a promising solution for synthesizing C–O atropisomers.
Herein, we report an unprecedented de novo synthetic strategy, delivering a new type of C–O axially chiral aryl-heterocyclic ethers that differ from the previously reported diaryl ether compounds (Fig. 1d). This protocol is successfully implemented through a cobalt-catalyzed atroposelective C–H activation/annulation of benzamides with rationally designed phenylethynyl ethers. The simultaneous construction of both the isoquinolinone heterocycle and the C–O chiral axis is faciliated in a single synthetic step. Notably, a traceless directing group approach is achieved using 2-(1-methylhydrazinyl)pyridine, which is cleaved in situ during the reaction. Meanwhile, a diverse array of C–O axially chiral compounds bearing multiple stereogenic elements are obtained with high stereoselectivity via the smoothly synchronous modulation of remote C–O and C–N axes. Moreover, high configurational stability is identified for the prepared C–O atropisomers through racemization studies. X-ray crystallography reveals a non-covalent π–π interaction between the isoquinolinone ring and the phenyl substituent of the phenylethynyl ether, likely contributing to both stereocontrol and the configurational stabilization of obtained products.
Results and discussion
Optimizing reaction conditions
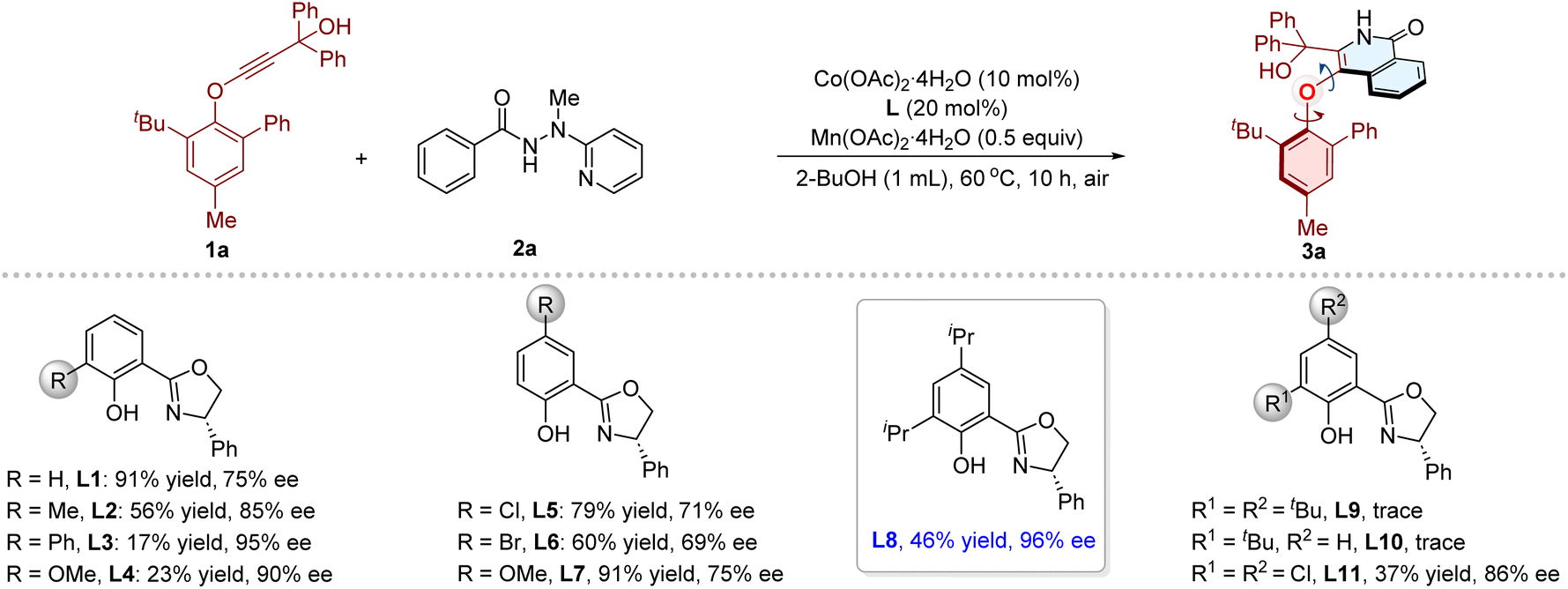
We began the investigations by subjecting phenylethynyl ether 1a and benzamide 2a preinstalled with 2-(1-methylhydrazinyl)pyridine74,80–83 as directing group to the cooperative Co/Salox catalytic system. Initially, various chiral Salox ligands were evaluated in combination with Co(OAc)2·4H2O as catalyst, Mn(OAc)2·4H2O as oxidant in 2-BuOH (0.1 M) at 60 °C for 10 h under air atmosphere (Table 1). Gratifyingly, the desired product 3a could be obtained in 91% yield with 75% ee in the presence of L1, and the directing group could be removed in situ during the transformation. Then the steric and electronic influence of substituents on the ligand were examined. The ortho-substituted L2, L3 and L4 afforded 3a with improved enantioselectivities (85%, 95%, and 90% ee, respectively), and the yields remained low (56%, 17%, and 23%, respectively). Introducing para-substituents (L5, L6, L7) had negligible effect on the stereoselectivity of this reaction. Notably, a superior result was obtained in the presence of L8, which features ortho- and para-isopropyl substituted phenol moiety, delivering 3a in 46% yield with 96% ee. The more sterically bulky L9 and L10 gave only trace amount of product. Subsequently, other reaction parameters, including cobalt salts, solvents, reaction temperature, and dosages of cobalt, ligand, and oxidants, were systematically optimized (Table 1 and S1–S9 in the SI). The best performance was attained under the conditions as shown in entry 15: Co(OAc)2·4H2O (10 mol%) as catalyst, L8 (20 mol%) as ligand, Mn(OAc)2·4H2O (0.5 equiv.) as co-oxidant, in 2-BuOH at 60 °C under air atmosphere, and the desired product 3a was obtained in 89% yield with 96% ee. Moreover, the use of other directing groups, such as 2-(1-ethylhydrazinyl)pyridine, 2-(1-propylhydrazinyl)pyridine, and 2-(1-benzylhydrazinyl)pyridine (Table S10), also afforded product 3a (81%, 96% ee; 77%, 96% ee; 18%, 96% ee, respectively).
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | [Co] | [Mn] (x equiv.) | Solvent | T (°C) | t (h) | Yield (%) | ee (%) |
| a Reaction conditions: 1a (0.12 mmol), 2a (0.1 mmol), cobalt catalyst (10 mol%), ligand (20 mol%), Mn(OAc)2·4H2O (x equiv.), solvent (1 mL), T/°C, t/h, air, isolated yields, the ee values were determined by HPLC analysis. | |||||||
| 1 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 60 | 10 | 46 | 96 |
| 2 | CoSO4·7H2O | 0.5 | 2-BuOH | 60 | 10 | Trace | — |
| 3 | CoBr2 | 0.5 | 2-BuOH | 60 | 10 | 18 | 95 |
| 4 | Co(acac)2 | 0.5 | 2-BuOH | 60 | 10 | Trace | — |
| 5 | Co(OAc)2·4H2O | 0.7 | 2-BuOH | 60 | 10 | 43 | 96 |
| 6 | Co(OAc)2·4H2O | 0.3 | 2-BuOH | 60 | 10 | 39 | 96 |
| 7 | Co(OAc)2·4H2O | 0.5 | t-BuOH | 60 | 10 | 45 | 96 |
| 8 | Co(OAc)2·4H2O | 0.5 | n-BuOH | 60 | 10 | 24 | 89 |
| 9 | Co(OAc)2·4H2O | 0.5 | n-PrOH | 60 | 10 | 17 | 93 |
| 10 | Co(OAc)2·4H2O | 0.5 | EtOH | 60 | 10 | 21 | 94 |
| 11 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 80 | 10 | 46 | 94 |
| 12 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 100 | 10 | 43 | 92 |
| 13 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 60 | 20 | 63 | 96 |
| 14 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 60 | 30 | 78 | 96 |
| 15 | Co(OAc)2·4H2O | 0.5 | 2-BuOH | 60 | 36 | 89 | 96 |
Reaction substrate scope
After establishing the optimal reaction conditions, we proceeded to evaluate the generality of this cobalt/Salox-catalyzed atroposelective synthesis of C–O axially chiral compounds. First, the substrate scope of benzamides was examined (Fig. 2). Substrates bearing diverse functionalities, including para-fluoro, chloro, bromo, iodo, methyl, tert-butyl, phenyl, methoxyl, trifluoromethoxy, and nitrile groups, reacted smoothly with phenylethynyl ether 1a, delivering the corresponding C–O axially chiral products (3b–3k) in good yields (69–95%) with excellent enantioselectivities (94–99% ee). The benzamides featuring para-sterically bulky anthracene or carbazole substituents were also able to give products 3l and 3m (54% yield, 65% ee, and 85% yield, 85% ee, respectively). Compound 3n was obtained as the sole product in 85% yield with 96% ee starting from meta-methyl substituted benzamide. The ortho-methyl substituted substrate reacted to deliver 3o in 35% yield with 95% ee. The substrates with extended aromatic systems, including either naphthalene or pyrene, were smoothly transformed into corresponding products 3p and 3q in 18% yield with 95% ee, and 71% yield with 94% ee, respectively. | ||
| Fig. 2 Scope of benzamides for the synthesis of C–O axially chiral aryl-isoquinolinol ethers. Reaction conditions: 1a (0.12 mmol), 2 (0.1 mmol), Co(OAc)2·4H2O (10 mol%), L8 (20 mol%), Mn(OAc)2·4H2O (0.5 equiv.), 2-BuOH (1 mL), 60 °C, 36 h, air, isolated yields, the ee values were determined by HPLC analysis. a48 h. | ||
Subsequently, the compatibility of phenylethynyl ethers was investigated with benzamide 2a under the optimized conditions (Fig. 3). The alkynes bearing divergent substituted aryl on the phenolic motif, including para-halogen, alkyl, and aryl substituted phenyl substituents, were well tolerated, furnishing the corresponding C–O axially chiral compounds (3r–3v) in 85–96% yield with 94–97% ee values. The strong electron-withdrawing CF3 group did not interfere with the reaction, giving 3w in 92% yield with 95% ee. The meta-alkyl substituted substrate also reacted well to afford 3x (80% yield, 96% ee). We then examined modifications on the gem-diaryl propargyl alcohol motif. The products (3y–3z, 3za–3zf) bearing para-halogen, electron-donating, electron-withdrawing groups and meta-chloro groups on the gem-diaryl motif were produced smoothly in 83–97% yields with 96–97% ee values. The absolute configuration of 3za was confirmed by X-ray crystallographic diffraction (Fig. 3, 3za, CCDC 2516693), and detailed analysis indicates a typical π–π interaction (typical π-stacking distances, face to face separations, 3.4–3.8 Å,84 find details in the SI, Fig. S8) between the isoquinolinone ring and the phenyl substituent of arylether moiety. This weak interaction might play a key role in the stereocontrol of the transformation, meanwhile potentially contribute to the configurational stability of C–O axially chiral products. The fluorene and thiophene derived alkynes were also suitable, affording 3zg and 3zh in 55% yield with 85% ee, and 81% yield with 80% ee, respectively. To evaluate steric effects of the tertiary propargyl alcohol motif, we replaced gem-diaryl group with less hindered alternatives. Using a gem-dimethyl, cyclopentyl- and cyclohexyl-substituted substrates furnished corresponding products 3zi–3zk in 73–95% yields with moderate enantioselectivities (77–81% ee values). Compound 3zl was obtained in 90% yield with only 27% ee. The low enantioselectivity likely arises from the loss of π–π interaction upon replacement of the phenyl group by a methyl group, which would reduce the rotational barrier. The alkynes containing unsymmetrical tertiary alcohol motif was also examined, furnishing products 3zm (ΔG‡ = 31.9 kcal mol−1, Table S19 and Fig. S4–S5) in 87% yield (8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr, 83% ee). Afterwards, we examined the scope of nonpropargyl substrates. Notably, the phenylethynyl ether bearing a TMS group in place of the tertiary alcohol motif provided both desired product 3zn (35%, 82% ee) and compound 3zn′ (21%, single diastereomer, 99% ee). The latter, with directing group still attached, possesses multiple chiral elements involving remote C–O and C–N axial chiralities.
:1 dr, 83% ee). Afterwards, we examined the scope of nonpropargyl substrates. Notably, the phenylethynyl ether bearing a TMS group in place of the tertiary alcohol motif provided both desired product 3zn (35%, 82% ee) and compound 3zn′ (21%, single diastereomer, 99% ee). The latter, with directing group still attached, possesses multiple chiral elements involving remote C–O and C–N axial chiralities.
 | ||
| Fig. 3 Scope of phenylethynyl ethers for the synthesis of C–O axially chiral aryl-isoquinolinol ethers. Reaction conditions: 1 (0.12 mmol), 2a (0.1 mmol), Co(OAc)2·4H2O (10 mol%), L8 (20 mol%), Mn(OAc)2·4H2O (0.5 equiv.), 2-BuOH (1 mL), 60 °C, 36 h, air, isolated yields, the ee values were determined by HPLC analysis. aThe dr value was determined by 1H-NMR. bCompound 3zn′ was obtained as single diastereomer, DG = 2-(methylamino)pyridine. | ||
In recent years, the enantioselective construction of molecules containing multiple axially chiral elements has garnered increasing attention from the synthetic community.9,61,69,72–77,85–99 However, the synthesis of such architectures featuring C–O axial chirality has not been achieved. Accordingly, we sought to explore the simultaneous installation of both C–O and C–N chiral axes under the aforementioned conditions.
Initially, the synthesis of compound 5a from phenylethynyl ether 1x and benzamide 4a bearing 8-aminoquinoline as directing group was set as a template to verify this concept. Gratifyingly, further optimization of this reaction (based on the previously established conditions, Table S11–S14 in SI) provided satisfactory results. Under the optimized conditions, the desired product 5a was obtained in 89% yield, 19:1 dr and 99% ee with Co(OAc)2·4H2O (10 mol%) as catalyst, L8 as ligand, Mn(OAc)2·4H2O/O2 as the oxidant, NaOPiv·H2O (1 equiv.) as the additive, in 2-BuOH (0.1 M) at 40 °C under air atmosphere. The absolute configuration of 5a was confirmed by X-ray crystallographic diffraction (Fig. 4, 5a, CCDC 2504636). The π–π interaction (find details in the SI, Fig. S9) between the isoquinolinone ring and the phenyl substiuent of phenolic moiety was also observed, which might be helpful for the stabilization of the configuration. Then the generality of this approach was scrutinized under the optimal conditions (Fig. 4). Benzamides substituted with para-halogen (including F, Cl, Br, I), or alkyl groups (Me, tBu) were all accommodated to give corresponding products (5b–5g) in 82–92% yield with 12:1–16:1 dr and 96–99% ee. Substrates with either electron-donating (OMe) or electron-withdrawing (OCF3, CN, CF3) groups at the para position were well tolerated, furnishing products 5h–5k in 90–96% yields with 13:1–17:1 dr and 99% ee. The meta-methyl-substituted benzamide provided 5l in 67% yield (16:1 dr, 99% ee). Compound 5m was obtained in 57% yield with 13:1 dr and 99% ee arising from 2-thiophenecarboxamide derivative. Afterwards, replacing the methyl with bulky isopropyl and tertiary butyl groups at 8-aminoquinoline furnished corresponding products 5n and 5o in good yields (84%, 93%, respectively) with 99% ee and moderate diastereoselectivity (9:1, 11:1 dr). Notably, the substrate with 5- bromo-substituted directing group afforded 5p in 74% yield with excellent diastereoselectivity (>20:1 dr, 99% ee). The compatibility of diverse phenylethynyl ethers was then evaluated. The alkynes bearing para-methyl, tert-butyl, phenyl, chloro, and CF3 substituted phenyl groups on the phenolic motif were well tolerated to give products (5q–5u) in 82–93% yield with >20:1 dr and 99% ee. The naphthyl-containing alkynes gave 5v in 90% yield (>20:1 dr, 96% ee). The meta-methyl substituted substrate afforded 5w in 97% yield (15:1 dr, 99% ee). The substrate with para-tBu in place of methyl at phenol moiety remained efficient, providing 5x in 92% yield (19:1 dr, 99% ee). Introducing a more bulky Ph2MeSi group instead of TMS smoothly afforded 5y in 97% yield (>20:1 dr and 99% ee). Replacing the phenyl with methyl afforded 5z in 87% yield with 6:1 dr and 99% ee. We also examined modifications of phenylethynyl ether by installing tertiary alcohol motif in place of TMS group. The compounds 5za–5zc were successfully obtained in good yields (71–91%) with excellent stereocontrol (single diastereomer, 95–99% ee). Finally, replacing the TMS group with a benzoyl group provided compound 5zd in 82% yield with 14:1 dr and 99% ee.
 | ||
| Fig. 4 Substrate cope for the synthesis of C–O/C–N diaxially chiral compounds. Reaction conditions: 1 (0.12 mmol), 4 (0.1 mmol), Co(OAc)2·4H2O (10 mol%), L8 (20 mol%), Mn(OAc)2·4H2O (0.5 equiv.), NaOPiv·H2O (1 equiv.), 2-BuOH (1 mL), 40 °C, 36 h, air, the dr values were determined by HPLC analysis. aThe dr value was determined by 1H-NMR. bLigand L9 was used instead of L8. | ||
Further transformations and synthetic applications
To demonstrate the synthetic utility of this protocol, gram-scale preparations of the representative C–O axially chiral compounds were carried out. Compound 3a was prepared in a reaction scale of 2.5 mmol, affording a good result without erosion on reaction efficacy and stereoselectivity (1.20 g, 83% yield, 96% ee) (Fig. 5a). Furthermore, a series of downstream transformations were performed on product 3a. The free N–H moiety of the isoquinolinone core could be readily functionalized: alkylations with methyl iodide, benzyl bromide, or propargyl bromide furnished products 6, 7, and 8 in good yields (86%, 85%, and 85%, respectively) without loss of enantiopurity. Additionally, treatment of 3a with PhNTf2 and Cs2CO3 in THF at 60 °C provided the C–O axially chiral aryl-isoquinoline ether compound 9 in 92% yield with 96% ee. This intermediate served as a versatile precursor for further diversifications. For instance, a Pd(PPh3)4-catalyzed Suzuki–Miyaura cross-coupling reaction with 4-tolylboronic acid delivered compound 10 in 88% yield with 96% ee. Subsequent removal of OTf group under Pd(OAc)2/dppf catalysis afforded compound 11 in 78% yield with 96% ee. Throughout all derivatizations, no erosion of enantiomeric purity was observed, underscoring the robustness of the chiral framework under diverse reaction conditions. The gram-scale synthesis of compound 5a maintained excellent stereoselectivity (19:1 dr, 99% ee), and only slight decreased yield (76%) was observed (Fig. 5b). Finally, desilylation of 5a with TBAF smoothly gave compound 12 in 99% yield with 3:1 dr and 99% ee. The results indicates that removal of TMS group decreases the rotational energy barrier of C–O axis, while with negligible effect on the stability of C–N axis. Furthermore, the experimental studies of racemization barriers (Fig. 5c) for compound 3a (recovery: 90%) afforded a ΔG‡ (C–O) value of 33.5 kcal mol−1, with a half-time (t1/2) of 6.0 × 103 years at 25 °C. The measurement of epimerization barriers for compound 5a (recovery: 91%, 100 °C; 89%, 120 °C) revealed a ΔG‡ (C–O) value of 33.4 kcal mol−1, and a ΔG‡ (C–N) value of 33.7 kcal mol−1. The half-times (t1/2) were determined to be 4.9 × 103 years for C–O axis and 8.8 × 103 years for C–N axis at 25 °C, respectively. These detailed studies underscore the high stability of prepared C–O axially chiral scaffolds.
 | ||
| Fig. 5 Investigations of synthetic utilities. (a) Gram-scale synthesis and divergent transformations of compound 3a. (b) Gram-scale synthesis and transformation of compound 5a. (c) Determination of racemization barriers for 3a and epimerization barriers for 5a. | ||
Afterwards, the ultraviolet-visible spectroscopy and fluorescence spectrum (Table S21–23) of representative C–O atropisomers were studied. The substitution patterns at either the benzamide or alkyne moieties had negligible effect on the maximum absorption wavelength in UV-vis of compounds 3 and 5. Among the C–O axially chiral products 3 and 5, introducing para-bromo (3d), iodo (3e), cyano (3k), and ortho-alkyl (3o) groups at the isoquinolinone moiety resulted in blue shift for the fluorescence spectrum, as well as compound 3zh with gem-dithiophene tertiary alcohol motif. Pronounced red shift was observed for compounds with para-phenyl (3h), carbazole (3m) group or extended aromatic fragments (3p, naphthalene, and 3q, pyrene, respectively). Compound 5a (446 nm) bearing diaxial architecture displayed a bathochromic shift compared with 3a (410 nm). Introducing para-tBu (5g), methoxy (5h), and cyano (5j) led to evident red shift. The longest maximum emission wavelength (540 nm) was obained with compound 5v bearing a naphthyl group at the phenol moiety. In comparison, compound 5zd with a benzoyl group instead of TMS exhibits the shortest maximum emission wavelength (414 nm). Notably, a dual emission was observed for compounds 3h, 3p, 3q, and 5w at 375 nm and 444 nm, 370 nm and 445 nm, 388 nm and 518 nm, 394 nm and 502 nm, respectively (Fig. S10).
Mechanistic studies
To gain insights into this transformation, several deuterium-labeling experiments were conducted (Fig. 6). The H/D exchange experiments (Fig. 6a) were carried out with 2a or [D5]-2a, as well as 4a or [D5]-4a in the absence of alkynes, detecting measurable H/D exchangement. The outcomes demonstrate that the C–H cleavage step is reversible. Meanwhile, we performed the H/D exchange experiments of 2a with 1a under standard conditions for 6 h, and no deuteration was observed. These findings are consistent with a reversible C–H activation, with the subsequent alkyne insertion being considerably faster. Subsequently, two sets of parallel (Fig. 6b) kinetic isotope effects (KIE) studies at low conversions gave kH/kD values of 1.40 and 1.69, respectively. The results suggest that C–H bond activation is likely not involved in the rate-determining step. Moreover, the non-linear effect (NLE) studies for the traceless directing group approach revealed a linear relationship between the ee values of chiral Salox ligand and the C–O axially chiral product 3a. However, a positive NLE outcomes was obtained for the synthesis of C–O/C–N diaxially chiral compound 5a, which indicates that multiple chiral ligands perhaps participate in the coordination to form the cobalt species in the catalytic cycle. | ||
| Fig. 6 Mechanistic studies. (a) H/D exchange experiments. (b) Parallel KIE experiments. (c) Non-linear effect studies. | ||
Based on the experimental results and literature reports,53–55 we proposed a plausible mechanism as shown in Fig. 7. In the presence of chiral ligand L8, the Co(II) is oxidized by O2/Mn(OAc)2·4H2O to form active Co(III) species Int-1. In the case with 7-methyl-8-aminoquinoline as the directing group, a positive NLE result indicates that two ligands might coordinates with Co to form complex [Co-L2]n species Int-1′.100 The Int-1 or Int-1′ undergoes coordination with benzamides 2a or 4a bearing different directing groups, and C–H activation to afford Int-2 and Int-2′, respectively. The following ligand exchange and migration insertion of Int-2 with alkyne 1a delivers a seven-membered alkenyl cobaltacycle intermediate Int-3. The subsequent reductive elimination provides a Co(I) species Int-4, which is likely to be stabilized by two non-covalent π–π interactions. The first interaction arises between the phenyl moiety of the oxazoline scaffold and the pyridyl unit of the directing group. The second one is established between the isoquinolinone ring and the phenyl substituent of the phenylethynyl ether. The latter is corroborated by X-ray crystallographic diffraction data for compounds 3za and 5a (Find details in Table S8–S9 in SI). Collectively, these two π–π stacking interactions are postulated to play a pivotal role in stabilizing the cobalt species and the axial chirality configuration of the products. The Int-4 is transformed to Co(III) Int-5 via an oxidative addition of Co(I) to N–N bond.80–83,101 Finally, protodemetallation and ligand exchange of Int-5 affords the desired C–O axially chiral aryl-isoquinolinone ether 3a, and simultaneously releases the Co(III) catalyst and L8. In the case of benzamide 4a bearing 7-methyl-8-aminoquinoline as the directing group, the corresponding intermediate Int-2′ undergoes migration insertion with alkyne 1x to give Int-6. After the subsequent reductive elimination, the final C–O/C–N diaxially chiral product 5a was produced. The regenerated Co(I) could be further oxidized to active Co(III) species.
 | ||
| Fig. 7 Proposed mechanism. | ||
Conclusions
In conclusion, we have developed an atroposelective construction of a new type of C–O axially chiral aryl-heterocyclic ethers through Co/Salox catalyzed asymmetric C–H activation/annulation of benzamides with phenylethynyl ethers. This strategy features a de novo synthetic approach, enabling simultaneous establishments of heteroaromatic ring and C–O chiral axis. A broad range of C–O axially chiral aryl-isoquinolinol ethers were efficiently synthesized with excellent stereoselectivity. Notably, the achievement of in situ removal of N,N-bidentate directing group secured the traceless directing approach. Moreover, an array of structurally diverse C–O axially chiral products bearing multiple chiral axes were afforded in good yields, accompanied by excellent enantio- and diastereoselectivity. High rotational energy barriers and configurational stability of the synthesized C–O atropisomers were identified through experimental studies. Furthermore, mechanistic investigations were conducted for gaining deep insights into this protocol. The synthetic utility was demonstrated through gram-scale synthesis and divergent transformations. This work provides a new synthetic paradigm for constructing C–O axially chiral architectures, and illuminates a promising perspective in the field of transition-metal-catalyzed C–H activation for the atroposelective construction of novel axial chirality.Author contributions
Y. D., D. Y., and J.-L. N. conceived the concept and prepared the manuscript. Y. Z., Y. X., and B. G. conducted the experiments and analyzed the data. Y. Z. and Y. X. contributed equally to this work. All the authors participated in the discussions of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2516693 (for 3za) and 2504636 (for 5a) contain the supplementary crystallographic data for this paper.102a,bThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, optimization studies, mechanistic studies, and characterization data of new compounds. See DOI: https://doi.org/10.1039/d6sc02299e.
Acknowledgements
This work was supported by the Natural Science Foundation of Henan Province (242300421033, 252300421178, 232301420007, 242301420059), and National Natural Science Foundation of China (22271260, 22401266).Notes and references
- J. Yang, Z.-Y. Xie, Y.-J. Ye, S.-B. Ye, Y.-B. Wang, W.-T. Wang, P.-C. Qian, R.-J. Song, Q. Sun, L.-W. Ye and L. Li, Sci. Adv., 2023, 9, eadk1704 CrossRef CAS PubMed.
- Y.-F. Jiang, S. Tong, J. Zhu and M.-X. Wang, Chem. Sci., 2024, 15, 12517–12522 RSC.
- Z.-Y. Zhang, T. Zhang, Y. Ouyang, P. Lu, J. X. Qiao and J.-Q. Yu, Chem. Sci., 2024, 15, 17092–17096 RSC.
- F. Wang, W. Xiang, Y. Xie, L. Huai, L. Zhang and X. Zhang, Sci. Adv., 2024, 10, eadq2768 CrossRef CAS PubMed.
- A. Chu, B. Zhu, X. Zhang, H. Zhu, J. Zhang and X. Liu, Sci. Adv., 2024, 10, eadr1628 CrossRef CAS PubMed.
- F.-R. Huang, P. Zhang, Q.-J. Yao and B.-F. Shi, CCS Chem., 2024, 6, 2783–2793 CrossRef CAS.
- Y. Zhang, Y.-Z. Gu, K. Lu, Y.-P. Wang, A.-F. Wang, S.-H. Wang, S.-Q. Wei, F.-M. Zhang, X.-M. Zhang, A.-J. Ma, J.-B. Peng and Y.-Q. Tu, CCS Chem., 2025, 7, 1067–1077 CrossRef CAS.
- M. Koner, N. Ballav, A. J. Varma, S. Ghosh, T. Mondal, R. Kuniyil and M. Baidya, Chem. Sci., 2025, 16, 19296–19303 RSC.
- Y. Ren, C. Lin, H. Zhang, Z. Liu, D. Wei, J. Feng and D. Du, Chem. Sci., 2025, 16, 7876–7883 RSC.
- Y.-F. Zhang, B. Wang, Z. Chen, J.-R. Liu, N.-Y. Yang, J.-M. Xiang, J. Liu, Q.-S. Gu, X. Hong and X.-Y. Liu, Science, 2025, 388, 283–291 CrossRef CAS PubMed.
- T. A. Schmidt, V. Hutskalova and C. Sparr, Nat. Rev. Chem., 2024, 8, 497–517 CrossRef PubMed.
- A. Naghim, J. Rodriguez, O. Chuzel, G. Chouraqui and D. Bonne, Angew. Chem., Int. Ed., 2024, 63, e202407767 CrossRef CAS PubMed.
- N. Kotwal and P. Chauhan, Chem. Commun., 2024, 60, 6837–6846 RSC.
- G.-J. Mei, W. L. Koay, C.-Y. Guan and Y. Lu, Chem, 2022, 8, 1855–1893 CAS.
- E. Kumarasamy, R. Raghunathan, M. P. Sibi and J. Sivaguru, Chem. Rev., 2015, 115, 11239–11300 CrossRef CAS PubMed.
- B. M. Duggan and D. J. Craik, J. Med. Chem., 1997, 40, 2259–2265 CrossRef CAS PubMed.
- K. C. Nicolaou, C. N. C. Boddy, S. Bräse and N. Winssinger, Angew. Chem., Int. Ed., 1999, 38, 2096–2152 CrossRef PubMed.
- L. Ciasullo, A. Casapullo, A. Cutignano, G. Bifulco, C. Debitus, J. Hooper, L. Gomez-Paloma and R. Riccio, J. Nat. Prod., 2002, 65, 407–410 CrossRef CAS PubMed.
- B. M. Crowley and D. L. Boger, J. Am. Chem. Soc., 2006, 128, 2885–2892 CrossRef CAS PubMed.
- D. G. Brown and H. J. Wobst, J. Med. Chem., 2021, 64, 2312–2338 CrossRef CAS PubMed.
- R. D. Kavthe, J. R. A. Kincaid and B. H. Lipshutz, ACS Sustainable Chem. Eng., 2022, 10, 16896–16902 CrossRef CAS PubMed.
- F. Zhao, H. Zhang, M. Xie, B. Meng, N. Liu, C. Dun, Y. Qin, S. Gao, E. De Clercq, C. Pannecouque, Y.-J. Tang, P. Zhan, X. Liu and D. Kang, J. Med. Chem., 2023, 66, 2102–2115 CrossRef CAS PubMed.
- P. Msaouel, K. Yu, Y. Yuan, J. Chen, X. Yan, M. Karki, F. Duan, R. A. Sheth, P. Rao, K. Sircar, A. Y. Shah, A. J. Zurita, G. Genovese, M. Li, C.-C. Yeh, M. Dang, G. Han, Y. Chu, M. Hallin, P. Olson, R. Yang, D. Slavin, H. Der-Torossian, C. D. Chin, N. M. Tannir, L. Wang and J. Gao, Nat. Commun., 2025, 16, 578 CrossRef CAS PubMed.
- Y. Sato, M. Abekura, T. Oriki, Y. Nagashima, H. Uekusa and K. Tanaka, Angew. Chem., Int. Ed., 2023, 62, e202304041 CrossRef CAS PubMed.
- G. M. Adams and A. S. Weller, Coord. Chem. Rev., 2018, 355, 150–172 CrossRef CAS.
- M. Mohankumar, M. Holler, E. Meichsner, J.-F. Nierengarten, F. Niess, J.-P. Sauvage, B. Delavaux-Nicot, E. Leoni, F. Monti, J. M. Malicka, M. Cocchi, E. Bandini and N. Armaroli, J. Am. Chem. Soc., 2018, 140, 2336–2347 CrossRef CAS PubMed.
- S. Cen, N. Huang, D. Lian, A. Shen, M.-X. Zhao and Z. Zhang, Nat. Commun., 2022, 13, 4735 CrossRef CAS PubMed.
- R. Wang, Y. Wu, J. Wang, H. Huang, Y. Wang, S. Xu and F. Zhao, J. Mol. Struct., 2022, 1257, 132642 CrossRef CAS.
- M. S. Betson, J. Clayden, C. P. Worrall and S. Peace, Angew. Chem., Int. Ed., 2006, 45, 5803–5807 CrossRef CAS PubMed.
- K. Fuji, T. Oka, T. Kawabata and T. Kinoshita, Tetrahedron Lett., 1998, 39, 1373–1376 CrossRef CAS.
- R. Farooqi, A. Mustafai, A. G. Woldegiorgis, X. Lin and P. Wang, ACS Catal., 2025, 15, 7891–7911 CrossRef.
- F. Hollmann, D. J. Opperman and C. E. Paul, Angew. Chem., Int. Ed., 2021, 60, 5644–5665 CrossRef CAS PubMed.
- J. Clayden, C. P. Worrall, W. J. Moran and M. Helliwell, Angew. Chem., Int. Ed., 2008, 47, 3234–3237 CrossRef CAS PubMed.
- B. Yuan, A. Page, C. P. Worrall, F. Escalettes, S. C. Willies, J. J. W. McDouall, N. J. Turner and J. Clayden, Angew. Chem., Int. Ed., 2010, 49, 7010–7013 CrossRef CAS PubMed.
- L. Dai, Y. Liu, Q. Xu, M. Wang, Q. Zhu, P. Yu, G. Zhong and X. Zeng, Angew. Chem., Int. Ed., 2023, 62, e202216534 CrossRef CAS PubMed.
- H. Bao, Y. Chen and X. Yang, Angew. Chem., Int. Ed., 2023, 62, e202300481 CrossRef CAS PubMed.
- J. Xu, W. Lin, H. Zheng and X. Li, ACS Catal., 2024, 14, 6667–6673 CrossRef CAS.
- S. Shee, S. Shree Ranganathappa, M. S. Gadhave, R. Gogoi and A. T. Biju, Angew. Chem., Int. Ed., 2023, 62, e202311709 CrossRef CAS PubMed.
- L. Li, W. Ti, T. Miao, J. Ma, A. Lin, Q. Chu and S. Gao, J. Org. Chem., 2024, 89, 4067–4073 CrossRef CAS PubMed.
- Y. Liu, L. Yuan, L. Dai, Q. Zhu, G. Zhong and X. Zeng, J. Org. Chem., 2024, 89, 7630–7643 CrossRef CAS PubMed.
- Y. Wu, X. Guan, H. Zhao, M. Li, T. Liang, J. Sun, G. Zheng and Q. Zhang, Chem. Sci., 2024, 15, 4564–4570 RSC.
- B.-A. Zhou, X.-N. Li, C.-L. Zhang, Z.-X. Wang and S. Ye, Angew. Chem., Int. Ed., 2024, 63, e202314228 CrossRef CAS PubMed.
- A. Naghim, P. Solai, M. Giorgi, J.-V. Naubron, S. Humbel, R. Gupta, J. Rodriguez, O. Chuzel, G. Chouraqui and D. Bonne, ChemistryEurope, 2026, 4, e70257 CrossRef CAS.
- L. Dai, X. Zhou, J. Guo, Q. Huang and Y. Lu, Chem. Sci., 2024, 15, 5993–6001 RSC.
- X. Han, L. Chen, Y. Yan, Y. Zhao, A. Lin, S. Gao and H. Yao, ACS Catal., 2024, 14, 3475–3481 CrossRef CAS.
- Y. Wang, R. Mi, S. Yu and X. Li, ACS Catal., 2024, 14, 4638–4647 CrossRef CAS.
- Y. Li, S. Liu, B. Yuan, N. Graw and L. Ackermann, Nat. Commun., 2026, 17, 743 CrossRef CAS PubMed.
- Z. Han, L. Wei, C. Nian, X. Hu, S. Liu, H. Huang and J. Sun, Sci. China Chem., 2026, 69, 1266–1272 CrossRef CAS.
- A. N. Dinh, R. R. Noorbehesht, S. T. Toenjes, A. C. Jackson, M. A. Saputra, S. M. Maddox and J. L. Gustafson, Synlett, 2018, 29, 2155–2160 CrossRef CAS PubMed.
- J. Wencel-Delord, A. Panossian, F. R. Leroux and F. Colobert, Chem. Soc. Rev., 2015, 44, 3418–3430 RSC.
- G. Liao, T. Zhou, Q.-J. Yao and B.-F. Shi, Chem. Commun., 2019, 55, 8514–8523 RSC.
- C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed.
- G. Liao and B.-F. Shi, Acc. Chem. Res., 2025, 58, 1562–1579 CrossRef CAS PubMed.
- Q.-J. Yao and B.-F. Shi, Acc. Chem. Res., 2025, 58, 971–990 CrossRef CAS PubMed.
- T. Yang, Y. Zhang, Y. Dou, D. Yang and J.-L. Niu, Sci. China Chem., 2026, 69, 659–679 CrossRef CAS.
- F.-R. Huang, M.-Y. Teng, H. Qiu, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2025, 64, e202506465 CrossRef CAS PubMed.
- S. Fukagawa, Y. Kato, R. Tanaka, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2019, 58, 1153–1157 CrossRef CAS PubMed.
- K. Ozols, S. Onodera, Ł. Woźniak and N. Cramer, Angew. Chem., Int. Ed., 2021, 60, 655–659 CrossRef CAS PubMed.
- Q.-J. Yao, J.-H. Chen, H. Song, F.-R. Huang and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202202892 CrossRef CAS PubMed.
- X.-J. Si, D. Yang, M.-C. Sun, D. Wei, M.-P. Song and J.-L. Niu, Nat. Synth., 2022, 1, 709–718 CrossRef CAS.
- Y. Zhang, C. Yang, X. Meng, M. Wu, D. Yang, M.-P. Song and J.-L. Niu, Org. Lett., 2025, 27, 6076–6081 CrossRef CAS PubMed.
- Y. Zhang, S.-L. Liu, T. Li, M. Xu, Q. Wang, D. Yang, M.-P. Song and J.-L. Niu, ACS Catal., 2024, 14, 1–9 CrossRef CAS.
- T. Li, Y. Zhang, C. Du, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2024, 15, 7673 CrossRef CAS PubMed.
- Y. Sun, T. Yang, Q. Wang, L. Shi, M.-P. Song and J.-L. Niu, Org. Lett., 2024, 26, 5063–5068 CrossRef CAS PubMed.
- T. Li, L. Shi, X. Zhao, J. Wang, X.-J. Si, D. Yang, M.-P. Song and J.-L. Niu, Org. Lett., 2023, 25, 5191–5196 CrossRef CAS PubMed.
- T. Li, L. Shi, X. Wang, C. Yang, D. Yang, M.-P. Song and J.-L. Niu, Nat. Commun., 2023, 14, 5271 CrossRef CAS PubMed.
- X.-J. Si, X. Zhao, J. Wang, X. Wang, Y. Zhang, D. Yang, M.-P. Song and J.-L. Niu, Chem. Sci., 2023, 14, 7291–7303 RSC.
- X. Wang, X.-J. Si, Y. Sun, Z. Wei, M. Xu, D. Yang, L. Shi, M.-P. Song and J.-L. Niu, Org. Lett., 2023, 25, 6240–6245 CrossRef CAS PubMed.
- P.-F. Qian, Y.-X. Wu, J.-H. Hu, J.-H. Chen, T. Zhou, Q.-J. Yao, Z.-H. Zhang, B.-J. Wang and B.-F. Shi, J. Am. Chem. Soc., 2025, 147, 10791–10802 CrossRef CAS PubMed.
- P.-F. Qian, G. Zhou, J.-H. Hu, B.-J. Wang, A.-L. Jiang, T. Zhou, W.-K. Yuan, Q.-J. Yao, J.-H. Chen, K.-X. Kong and B.-F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202412459 CrossRef CAS PubMed.
- Y.-J. Wu, Z.-K. Wang, Z.-S. Jia, J.-H. Chen, F.-R. Huang, B.-B. Zhan, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2023, 62, e202310004 CrossRef CAS PubMed.
- B.-J. Wang, G.-X. Xu, Z.-W. Huang, X. Wu, X. Hong, Q.-J. Yao and B.-F. Shi, Angew. Chem., Int. Ed., 2022, 61, e202208912 CrossRef CAS PubMed.
- X. Wu, Q. Tian, J. Ge, Y. Liu, Z. Li, J. Zhang and G. Cheng, Chin. J. Chem., 2025, 43, 2669–2676 CrossRef CAS.
- A. Das, S. Kumaran, P. Maity, J. R. Premkumar and B. Sundararaju, J. Am. Chem. Soc., 2025, 147, 26226–26237 CrossRef CAS PubMed.
- M. Tang and X. Yang, Chem Catal., 2023, 3, 100754 CAS.
- P. Boos, N. K. Pandit, S. Dana, T. von Münchow, A. Hashidoko, L. Haberstock, R. Herbst-Irmer, D. Stalke and L. Ackermann, ChemistryEurope, 2025, 3, e202500071 CrossRef.
- A. Luc, J. C. A. Oliveira, P. Boos, N. Jacob, L. Ackermann and J. Wencel-Delord, Chem Catal., 2023, 3, 100765 CAS.
- Y. Xu, Y. Lin, S. L. Homölle, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2024, 146, 24105–24113 CrossRef CAS PubMed.
- T. von Münchow, Y.-R. Liu, R. Parmar, S. E. Peters, S. Trienes and L. Ackermann, Angew. Chem., Int. Ed., 2024, 63, e202405423 CrossRef PubMed.
- S. Zhou, M. Wang, L. Wang, K. Chen, J. Wang, C. Song and J. Zhu, Org. Lett., 2016, 18, 5632–5635 CrossRef CAS PubMed.
- A. Dey and C. M. R. Volla, Org. Lett., 2020, 22, 7480–7485 CrossRef CAS PubMed.
- S. Zhai, S. Qiu, X. Chen, J. Wu, H. Zhao, C. Tao, Y. Li, B. Cheng, H. Wang and H. Zhai, Chem. Commun., 2018, 54, 98–101 RSC.
- R. Mei, N. Sauermann, J. C. A. Oliveira and L. Ackermann, J. Am. Chem. Soc., 2018, 140, 7913–7921 CrossRef CAS PubMed.
- B. Schramm, M. Gray and J. M. Herbert, J. Am. Chem. Soc., 2025, 147, 3243–3260 CrossRef CAS PubMed.
- H.-H. Zhang, T.-Z. Li, S.-J. Liu and F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202311053 CrossRef CAS PubMed.
- S.-C. Zhang, S. Liu, X. Wang, S.-J. Wang, H. Yang, L. Li, B. Yang, M. W. Wong, Y. Zhao and S. Lu, ACS Catal., 2023, 13, 2565–2575 CrossRef CAS.
- Y. Wang, X. Zhu, D. Pan, J. Jing, F. Wang, R. Mi, G. Huang and X. Li, Nat. Commun., 2023, 14, 4661 CrossRef CAS PubMed.
- S. Zhang, X. Wang, L.-L. Han, J. Li, Z. Liang, D. Wei and D. Du, Angew. Chem., Int. Ed., 2022, 61, e202212005 CrossRef CAS PubMed.
- S. Huang, H. Wen, Y. Tian, P. Wang, W. Qin and H. Yan, Angew. Chem., Int. Ed., 2021, 60, 21486–21493 CrossRef CAS PubMed.
- Q. Gao, C. Wu, S. Deng, L. Li, Z.-S. Liu, Y. Hua, J. Ye, C. Liu, H.-G. Cheng, H. Cong, Y. Jiao and Q. Zhou, J. Am. Chem. Soc., 2021, 143, 7253–7260 CrossRef CAS PubMed.
- T. A. Schmidt and C. Sparr, Acc. Chem. Res., 2021, 54, 2764–2774 CrossRef CAS PubMed.
- O. M. Beleh, E. Miller, F. D. Toste and S. J. Miller, J. Am. Chem. Soc., 2020, 142, 16461–16470 CrossRef CAS PubMed.
- X. Bao, J. Rodriguez and D. Bonne, Chem. Sci., 2020, 11, 403–408 RSC.
- X. Bao, J. Rodriguez and D. Bonne, Angew. Chem., Int. Ed., 2020, 59, 12623–12634 CrossRef CAS PubMed.
- W. Xia, Q.-J. An, S.-H. Xiang, S. Li, Y.-B. Wang and B. Tan, Angew. Chem., Int. Ed., 2020, 59, 6775–6779 CrossRef CAS PubMed.
- Y.-L. Hu, Z. Wang, H. Yang, J. Chen, Z.-B. Wu, Y. Lei and L. Zhou, Chem. Sci., 2019, 10, 6777–6784 RSC.
- Q. Dherbassy, J.-P. Djukic, J. Wencel-Delord and F. Colobert, Angew. Chem., Int. Ed., 2018, 57, 4668–4672 CrossRef CAS PubMed.
- Y. Yang, C. Yang, X. Zhang, Y. Tian, X. Cheng, T. Zeng and B. Li, ACS Catal., 2025, 15, 3442–3450 CrossRef CAS.
- N.-Y. Wang, S. Gao, Z.-D. Shu, B.-B. Cheng, C. Ma, Y.-C. Zhang and F. Shi, Sci. China Chem., 2025, 68, 3130–3137 CrossRef CAS.
- W. Zhang, S. Chen, Y. Zhang, S. Liu and X. Shen, Angew. Chem., Int. Ed., 2025, 64, e202422020 CrossRef CAS PubMed.
- A. Lerchen, S. Vásquez-Céspedes and F. Glorius, Angew. Chem., Int. Ed., 2016, 55, 3208–3211 CrossRef CAS PubMed.
- (a) CCDC 2516693: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2qgtnp; (b) CCDC 2504636: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2q28qt.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |