
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aluminylation: a generalizable route towards low-valent aluminum under moderate conditions with controlled product nuclearity through precursor design
Paris C.
Reuel
 ,
Yogesh
Shandilya
,
M. Talha
Wattoo
and
Alison B.
Altman
*
,
Yogesh
Shandilya
,
M. Talha
Wattoo
and
Alison B.
Altman
*
Department of Chemistry, Texas A&M University, College Station, TX 77843, USA. E-mail: aaltman@tamu.edu
First published on 13th April 2026
Abstract
Recent advances in isolating and characterizing reactive aluminylenes and aluminyl anions have redefined the landscape of low-valent aluminum chemistry. Study of these molecules that span a wide range of structures and ligand environments have helped established new paradigms in main-group metal bonding and reactivity; however, their controlled access remains limited by aggregation, disproportionation, and oxidation of reactive Al(I) intermediates. Herein, we establish aluminylation as a generalizable redox-neutral synthetic route to low-valent aluminum complexes. By circumventing the use of external reductants through metathesis reactions between Al(I) precursors and alkali metal ligand salts, we can access novel heteroleptic clusters as well as known aluminyl complexes with improved yields or under more mild conditions. We demonstrate that the equilibrium of the Al(I) precursor between monomeric or oligomeric cyclopentadienyl aluminum species directly governs reaction outcomes and enables selective formation of products across multiple ligand classes.
Introduction
Low-valent aluminum complexes have emerged as a versatile class of main-group compounds capable of mediating transformations traditionally reserved for expensive, toxic transition metals or strong alkali metal reductants.1–7 As the field expands, chemical diversification is clarifying structure–function relationships that push the limits of aluminum-based redox chemistry. In particular, Al(I)-containing complexes, termed “aluminyls,” can span a range of electronic environments defined by the supporting ligand, from neutral aluminylenes8–18 to anionic aluminyl anions.12,19–30 (Fig. 1). This distinction is critical, as charge and coordination number strongly influence aggregation, redox behavior, and nucleophilicity of Al(I) centers.12,25,31 These aluminyl complexes exhibit rich redox behavior, tunable electronic properties, and the ability to engage in small-molecule activation and electron-transfer chemistry.1,3,4,7,32–37 Strong σ-donation and the polarizable aluminum center confer a unique balance of nucleophilicity and reducing ability, positioning low-valent aluminum complexes as promising next-generation reductants for selective transformations. | ||
| Fig. 1 Selection of low-valent aluminum complexes studied in this work, and their classification. Top left – anionic Al(I) complexes bearing two anionic ligands or a single dianionic ligand are defined as aluminyl anions. Top right – neutral Al(I) complexes bearing one anionic ligand are defined as aluminylenes. Bottom right – aluminylenes reacted through the cycloaddition of an aromatic solvent are defined as ‘masked’ aluminylenes. | ||
Despite this potential, access to low-valent aluminum species remains synthetically challenging as aluminum most commonly exists in the +3 oxidation state.38 Thus, synthetic routes to aluminyl complexes mostly rely on reductive salt elimination from Al(III) halide precursors, typically with harsh alkali metal reductants. However, the disparity between the extreme reduction potentials of these reagents and the intrinsic redox properties of the aluminum complexes often leads to poor yields.11,16,17,27,39–45 Low-valent aluminum is prone to forming metal clusters that bridge the size regimes between distinct molecules and nanoparticle precipitates as potential off-target products.46–57 These competing factors define a complex synthetic landscape where side reactions, ligand degradation, or formation of metallic aluminum compete with the desired chemistry, highlighting the need for milder, more controlled synthetic routes.
Aluminylation represents a promising alternative synthetic pathway, as first reported by Aldridge and coworkers.28–30,58,59 In this redox neutral reaction, an Al(I) center is transferred through a metathesis-type reaction, avoiding uncontrolled electron transfer, metal deposition, and heterogeneity inherent to reductive chemistry. This approach is enabled by scalable conversion of Cp*2AlH (Cp* = pentamethylcyclopentadienyl) to the tetramer [AlCp*]4,13 a known competent aluminylation reagent.30,58 This reaction occurs via reductive elimination of Cp*H, avoiding the use of external reductants at any stage. Importantly, the Cp* ligand can be readily exchanged with other Cp (Cp = cyclopentadienyl) derivatives to access new potential aluminylation reagents.59,60 In particular, the equilibrium between the tetrameric Al4 cluster and the monomeric Al(I) species is known to depend on the Cp ligand electronic and steric profile, directly influencing their solution state chemistry and providing a chemical handle that enables synthetic optimization.
Herein, we describe the divergent chemistry of different aluminylation reagents with a range of nitrogen and oxygen based substrates. The equilibrium between active monomeric and tetrameric aluminyl species in solution is found to provide a synthetic handle for accessing heteroleptic aluminum clusters. In comparison, by increasing the proportion of Al(I) monomers in solution, the yields of known reactions are improved, even under mild conditions. We demonstrate that Al(I) speciation, not only substrate identity, governs reaction outcomes to enable access to aluminyl species with selective nuclearities.
Results and discussion
Substrate-directed aluminylation pathways: stoichiometric vs. substoichiometric substitution
The equilibrium between tetrameric [AlCp*]4 and monomeric AlCp* is central to aluminylation reactivity. With only trace monomer concentrations at room temperature, elevated temperatures are required to shift this equilibrium.59,61 Such behavior rationalizes why heating is typically required in Cp*-based aluminylation chemistry, supporting monomeric AlCp* as the active aluminylation species rather than [AlCp*]4.11,29,30 In looking to expand this chemistry, we noted that bulkier Cp derivatives are known to give Al(I) complexes in which the tetramer–monomer equilibrium is shifted further to the monomer product. More generally, ligand bulk determines aluminyl nuclearity. Exceptionally bulky ligands can stabilize monomeric aluminyl species,9,26,59,62,63 whereas reduced steric demand typically promotes aggregation to aluminum dimers,41,43 trimers,64 or tetramers.13,56,59,65,66 These observations suggest that both the substrate ligand framework and the aluminylation reagent itself can be used as a powerful handle for tuning the outcome of aluminylation chemistry across ligand classes.To broaden aluminylation scope, we compared aluminylation products with substrates known to stabilize aluminyl molecules with different nuclearities across a range of steric profiles. We targeted both “masked” low-valent aluminum(I) amidinate dimers and asymmetric amide tetrameric clusters (where “masked” refers to cycloaddition of an aromatic solvent to an aluminylene fragment). In this context, amide and amidinate ligands offer a more flexible and tunable alternative to the commonly explored Cp frameworks. Such chemically diversifiable complexes provide a versatile platform for systematic studies of aluminylene reactivity,64 motivating the development of more direct synthetic routes away from reductive salt elimination.
Reaction of the lithium amidinate salt [(Li(NDipp)2C5H9)2·THF] (1; THF = tetrahydrofuran, Dipp = 2,6-diisopropylphenyl) with [AlCp*]4 in toluene at 60 °C for 36 h afforded the masked aluminylene dimer [(Al(NDipp)2C5H9)2C7H8]·C7H8 (2) in 43% isolated yield (Scheme 1) ,top, comparable to reported reductive preparations. In contrast, aluminylation of the less hindered amide system gave divergent behavior. Treatment of Na[N(SiMe3)(Dipp)]·3 Et2O with [AlCp*]4 (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) at 60 °C for 12 h afforded the substoichiometric heteroleptic cluster [Cp*AlAlN(SiMe3)(Dipp)]2·C6H14 (3) in 47% isolated yield (Scheme 1), middle. This product is a rare example of a heteroleptic aluminum cluster, and we do not observe evidence of either the fully substituted cluster, or other substoichiometric products, even by 27Al NMR spectroscopy. The 27Al resonances of 3 also do not change in relative intensity upon heating (see Fig. S6), consistent with the absence of a monomer–tetramer equilibrium. This suggests that AlCp* monomer is responsible for aluminyl transfer, but subsequent Al–Al aggregation of Al(I) monomers yields a heteroleptic cluster 3 that does not participate in a monomer–tetramer equilibrium.
:1 molar ratio) at 60 °C for 12 h afforded the substoichiometric heteroleptic cluster [Cp*AlAlN(SiMe3)(Dipp)]2·C6H14 (3) in 47% isolated yield (Scheme 1), middle. This product is a rare example of a heteroleptic aluminum cluster, and we do not observe evidence of either the fully substituted cluster, or other substoichiometric products, even by 27Al NMR spectroscopy. The 27Al resonances of 3 also do not change in relative intensity upon heating (see Fig. S6), consistent with the absence of a monomer–tetramer equilibrium. This suggests that AlCp* monomer is responsible for aluminyl transfer, but subsequent Al–Al aggregation of Al(I) monomers yields a heteroleptic cluster 3 that does not participate in a monomer–tetramer equilibrium.
 | ||
| Scheme 1 Aluminylation of various ligand scaffolds using [AlCp*]4 at elevated temperature to access the active monomer AlCp*. Top – aluminylation of amidinate ligand selects for stoichiometric substitution in the formation of [(Al(NDipp)2C5H9)2C7H8]·C7H8 (2). Middle – aluminylation of amide leads to substoichiometric substitution in the formation of heteroleptic cluster [Cp*AlAlN(SiMe3)(Dipp)]2·C6H14 (3). Bottom – aluminylation of dianionic bisamide leads to aluminyl anion appended to the [Al4Cp*3]+ cluster as [Al(N2Dipp2C3H6)(Al4Cp*3)]·0.5C6H6 (4). | ||
We next targeted aluminyl anions supported by dianionic ligands, which are typically prepared via a two-step reductive salt-elimination route that requires demanding purifications and often delivers lower yields. A one-step aluminylation reaction offers an appealing alternative, and so we aimed to access the previously reported aluminyl anion [Al(NDipp)2(CH2)3]− directly.26 Accordingly, we treated Li2[(NDipp)2(CH2)3]·2 Et2O with [AlCp*]4 (4:1 molar ratio) at 60 °C. The expected aluminylation product was not observed; instead, the reaction produced a blue cluster, [Al(N2Dipp2C3H6)(Al4Cp*3)]·0.5C6H6 (4), in <10% yield. Adjusting the ratio (Li2[(NDipp)2(CH2)3]·2Et2O:[AlCp*]4 = 4:5) increased the isolated yield to 23% (Scheme 1), bottom. Complex 4 can be described as [Al(NDipp)2(CH2)3]− associated with an [Al4Cp*3]+ fragment. We propose that the aluminyl anion Li[Al(NDipp)2(CH2)3] forms in situ and undergoes salt metathesis with [AlCp*]4 present in solution, although no intermediates were detected. Such a mechanism suggests that both monomeric AlCp* and tetrameric [AlCp*]4 can be reactive in the presence of sufficiently nucleophilic anions.
This potential mechanism is mirrored in the cluster connectivity, as this molecule presents, to the best of our knowledge, the first example of an Al cluster that does not conform to standard polyhedral skeletal electron-pair descriptions of these metal clusters. Instead, this molecule exhibits two chemically distinct aluminyl environments connected via a direct Al–Al bond. Compared with the heteroleptic tetramer 3, this substrate-dependent aluminylation behavior underscores the potential of clustered aluminylation reagents as a deliberate strategy for accessing heteroleptic, low-valent aluminyl architectures. These structures provide a foundation for targeting site-selective reactivity at differentiated aluminum centers, which will be the focus of future studies.
Crystallographic analysis of heteroleptic aluminum clusters
From a structural perspective, these new molecules also enable unusual comparisons to typical aluminum clusters with electron density delocalized isotropically over a homoleptic metal framework. In such cases, Al–Al bond distances provide a practical proxy for relative bond strength. For example, the average Al–Al separation in [AlCp*]4 is 2.769(7) Å,67 whereas [AlN(SiMe3)(Dipp)]4 exhibits a shorter average distance of 2.62(13) Å.11 This contraction is consistent with stronger Al–Al interactions in the amide system and rationalizes why monomer–tetramer equilibria are more pronounced for [AlCp*]4 than for [AlN(SiMe3)(Dipp)]4.In contrast, the heteroleptic clusters 3 and 4 are intrinsically less symmetric; therefore, electron density is expected to be polarized in the heteroleptic Al–Al bonds (Fig. 2). We note that in 3, its heteroleptic stoichiometry enforces three distinct classes of Al–Al interactions and distorts the framework away from ideal tetrahedral geometry of an Al4 core. The Cp*Al–AlCp* distances (2.785(3) Å) are close to those in [AlCp*]4 (2.769(7) Å), while the (Dipp)(SiMe3)NAl–AlN(SiMe3)(Dipp) distance (2.583(3) Å) tracks the shorter separations characteristic of the homoleptic amide cluster [AlN(SiMe3)(Dipp)]4 (2.62(13) Å). The mixed Cp*Al–AlN(SiMe3)(Dipp) contacts fall between these limits. Collectively, these metrics indicate that N(SiMe3)(Dipp) exerts a stronger influence on Al–Al bond contraction than Cp*, mirroring trends in the homoleptic analogues. The resulting asymmetric bonding environment affects both local bonding and global cluster shape, yielding a geometry more consistent with a nido-type arrangement than the idealized closo geometry expected from simple electron-counting arguments.68,69
 | ||
| Fig. 2 Solid-state molecular structures of the heteroleptic aluminum clusters [CpAlAlN(SiMe3)(Dipp)]2 (left; 3) and [Al(N2Dipp2C3H6)(Al4Cp*3)] (right; 4). Selected Al–Al bond lengths are shown to highlight ligand-dependent variations in Al–Al interactions and polarization within the clusters. Hydrogen atoms and outer sphere solvents are omitted for clarity. | ||
In comparison, complex 4 represents an even more significant departure away from idealized cluster geometry. The Al5 framework of Al(I) centers in 4 corresponds formally to an overall oxidation-state sum of +5. An even distribution of charge across the structure would treat the Al–Al4 bonding interaction as predominantly ionic. Alternatively, a covalent description of Al–Al bonding invokes an uneven oxidation-state distribution across the Al5 unit (e.g., R2Al(II)–Al(0)Al(I)3). Either formulation departs from the high symmetries exhibited by aluminum cluster precedents and points to significantly asymmetric and polarized Al–Al interactions.
X-ray diffraction studies on single crystals of 4 revealed the interfragment Al–Al4 linkage is 2.710(2) Å, longer than the sum of metallic radii for two Al atoms (2.50 Å) and other typical covalent metrics,70 yet shorter than an estimate for a purely ionic contact between [Al(N2Dipp2C3H6)]− and [Al4Cp*3]+ (2.82 Å; see SI for how the ionic-limit estimate is derived for these fragments). These comparisons place the interaction between covalent and ionic extremes, consistent with a mixed ionic–covalent contact. Together, these crystallographic results show how ligand identity and aluminylation pathway directly govern Al–Al bond character and cluster geometry, providing access to heteroleptic, low-valent aluminum frameworks not readily available from reductive synthesis pathways where redox chemistry competes with equilibria defined by all Al(I) species present in solution.
DFT, QTAIM, and ELF analysis of Al–Al bonding in 3 and 4
To further probe the Al–Al bonding in these clusters, we carried out density functional theory (DFT) calculations on truncated structural models (e.g., Cp* → Cp),71 with geometries constrained to match solid-state structures. Calculations employed the B3LYP functional with the def2-SVP basis set and the def2/J auxiliary basis.72–74 The model of 3 converged on a true minimum while the model of 4 converged on a low-energy saddle point (see SI for further details).For the model of 3, Kohn–Sham orbital analysis shows that the HOMO through HOMO−2 are dominated by the antisymmetric Al–Al bonding combinations characteristic of Al4 tetramers, while deeper orbitals (e.g., HOMO−23 and HOMO−31) correspond to fully symmetric Al4 bonding combinations. The low-lying virtual space mirrors this pattern: the LUMO through LUMO+2 predominantly comprise the antisymmetric antibonding combinations of the Al4 framework. Qualitatively, these frontier-orbital features are consistent with other Al(I) tetramer clusters; however, real-space analyses reveal key differences in bonding polarization within the heteroleptic core.
Quantum Theory of Atoms in Molecules (QTAIM)75,76 analysis (see SI, Section 3) differentiates the Al–Al interactions within 3 by quantifying electron density and its topology at the bond critical points (BCPs). The electron density (ρ(r)) at the Al–Al BCPs is low relative to typical covalent bonds (often ρ(r) > 0.1 e·Bohr−3, cf. ρ(r) = 0.0356–0.0465 e·Bohr−3), but the Laplacian is negative (∇2ρ(r) < 0), indicating charge concentration consistent with a covalent interaction (Table 1). At the same time, standard ionic descriptors (e.g., ΔG(r)/ρ(r) > 1; ΔG(r) = kinetic energy density and |ΔV(r)|/ΔG(r) < 1; ΔV(r) = potential energy density) do not support a purely ionic description. This intermediate regime is consistent with trends reported for other low-valent aluminum clusters.
| Bond lengtha, Exp./Calc. | Electron density ρ(r)b | Laplacian of electron density, ∇2ρ(r)b | Ellipticity, ε | |
|---|---|---|---|---|
| a Unit length Å. b Units for ρ(r) and ∇2ρ(r) in a.u. (e·Bohr−3, and e·Bohr−5 respectively). | ||||
| Structure 3 | ||||
| CpAl3-Al4Cp | 2.785(2)/2.754 | 0.036 | −0.010 | 1.387 |
| NAl1–Al2N | 2.583(3)/2.582 | 0.045 | −0.041 | 0.572 |
| CpAl1-Al4N | 2.650(2)/2.578 | 0.047 | −0.026 | 0.775 |
| CpAl2-Al4N | 2.651(3)/2.669 | 0.041 | −0.027 | 0.778 |
|
||||
| Structure 4 | ||||
| Al1–Al4 | 2.710(2)/2.710 | 0.048 | −0.034 | 0.030 |
| AlAl2–Al4Cp | 2.643(2)/2.600 | 0.046 | −0.033 | 0.630 |
| AlAl2–Al5Cp | 2.660(2)/2.632 | 0.044 | −0.037 | 0.537 |
| AlAl2–Al3Cp | 2.655(2)/2.601 | 0.046 | −0.034 | 0.637 |
| CpAl3-Al4Cp | 2.721(2)/2.671 | 0.039 | −0.025 | 1.268 |
| CpAl4-Al5Cp | 2.722(2)/2.717 | 0.037 | −0.016 | 0.850 |
| CpAl3-Al5Cp | 2.694(2)/2.668 | 0.040 | −0.025 | 0.844 |
Electron Localization Function (ELF)77,78 analysis further indicates both that the bonding density in 3 is displaced from the internuclear Al–Al axis and polarized along the bond. The ellipticity (ε) for each Al–Al bond is greater than 0.5, suggesting the electron density is biased toward the exterior of the cluster, whereas the BCP remains close to the internuclear line, near the boundary of the ELF-delineated bonding region (see Fig. S67–S72). Relative to homoleptic analogues, 3 also shows a systematic shift of the BCP position toward the amide-substituted vertices, concomitant with smaller ε values (0.572 vs. 1.387). This trend is consistent with stronger σ-donor ligands increasing σ-character in the Al–Al interactions via inductive polarization. Overall, these descriptors indicate that ligand σ-donation measurably strengthens the Al–Al bond, as reflected in lower ε values concomitant with larger values of ρ(r) and |∇2ρ(r)| at the BCP of the Al–Al bonds supported by amide ligands compared with those supported by Cp ligands, matching the observed bond distance metrics. These electronic effects install stability into the Al–Al bonds that define the Alnn+ framework, stabilizing higher-nuclearity clusters by preventing disproportionation to entropically favored lower nuclearity motifs. This phenomenon prevents the rearrangements of the cluster fragments present in 3 to other stoichiometries and indicates a path towards designing heteroleptic systems based on electronic structure requirements in addition to steric constraints.
For 4, we anticipated analogous polarization effects within the Al4 subunit, together with unique bonding at the interfragment Al–Al4 linkage. Accordingly, the HOMO is localized at the Al–Al4 and exhibits substantial lone-pair character localized at the formally anionic Al center, whereas lower-energy orbitals (notably HOMO−10 and HOMO−15) contain clear σ-bonding contributions associated with the Al–Al4 interaction. The LUMO and LUMO+1 are dominated by πx/πy-type bonding combinations between the Al and Al4 fragments (see Fig. S53 and S54), identifying the Al–Al4 linkage as a key frontier-orbital site for reactivity.
QTAIM analysis of 4 identifies polarized BCPs along the AlAl–AlCp contacts in the Al4 core toward the aluminyl anion containing vertex. This polarization, similar to that observed in 3, indicates that the aluminyl anion exerts a stronger σ-donating (and inductively polarizing) influence on the Al4 framework than the Cp ligand. Similarly, ε of the Al–Al contacts in Al4 core shows significant bias of electron density to the exterior of the Al4 subunit (ε = 0.537–1.268). In comparison, the BCP located along the interfragment Al–Al4 bond path exhibits a relatively low ρ(r) and a negative Laplacian, consistent with a weak shared interaction (Table 1). Unlike the Al–Al bonds within the Al4 core, where the ELF density is displaced from the internuclear axis, ELF analysis shows that the Al–Al4 interaction is nearly collinear, with the BCP positioned close to the internuclear line and a low value of ε = 0.030. The BCP lies at the boundary of the ELF-defined bonding region on the Al4 side, consistent with a strongly polarized interaction in which electron density is drawn toward the aluminyl fragment (Fig. 3). Analysis of the Al–Al4 disynaptic basin containing 2.033 electrons, shows this electron density partitioned unevenly between the aluminyl anion center (1.479 e) and the Al4 fragment (0.512 e). Together, the MO, QTAIM, and ELF results support describing this Al–Al4 linkage as a weakly covalent bond with pronounced polarization toward the aluminyl anion fragment. This homonuclear interaction is electronically heterogenous, with electron density being favorably localized on the aluminyl anion with only a small contribution of electron density being shared between the Al centers leading to the weak covalent metrics observed in the QTAIM. Other metal homonuclear M–M interactions do not commonly express this electronic heterogeneity, regardless of ligand differences,79 making this polarized interaction tractable for further studies.
 | ||
| Fig. 3 Top – ELF plot in the plane containing Al1, Al2, and the bond critical point (BCP) between them. Bottom – decomposition of the electron density at the BCP into contributions from the three most significant molecular orbitals (MOs). | ||
Suppressing cluster formation: leveraging monomeric AlCpiPr4 for milder aluminylation
This cluster chemistry also underscores an additional requirement for selective aluminylation: as the tetramer–monomer equilibrium is instrumental for the formation of heteroleptic clusters, stoichiometric products may be favored by shifting the equilibrium of the aluminylation reagent toward monomeric species. We identified AlCpiPr4, a known monomer in solution at room temperature,59 as a potential aluminylation candidate as opposed to the thermally accessed AlCp* monomer. Beyond this, at −40 °C AlCpiPr4 is monomeric, as determined by diagnostic shifts in the 27Al NMR spectra in our experiments. Thus, we predicted that AlCpiPr4 could perform aluminylation chemistry without the entropic penalty associated with tetramer dissociation, supporting mild reaction conditions and enabling direct comparisons of the temperature-dependent reactivity for AlCpiPr4versus [AlCp*]4 (Scheme 2).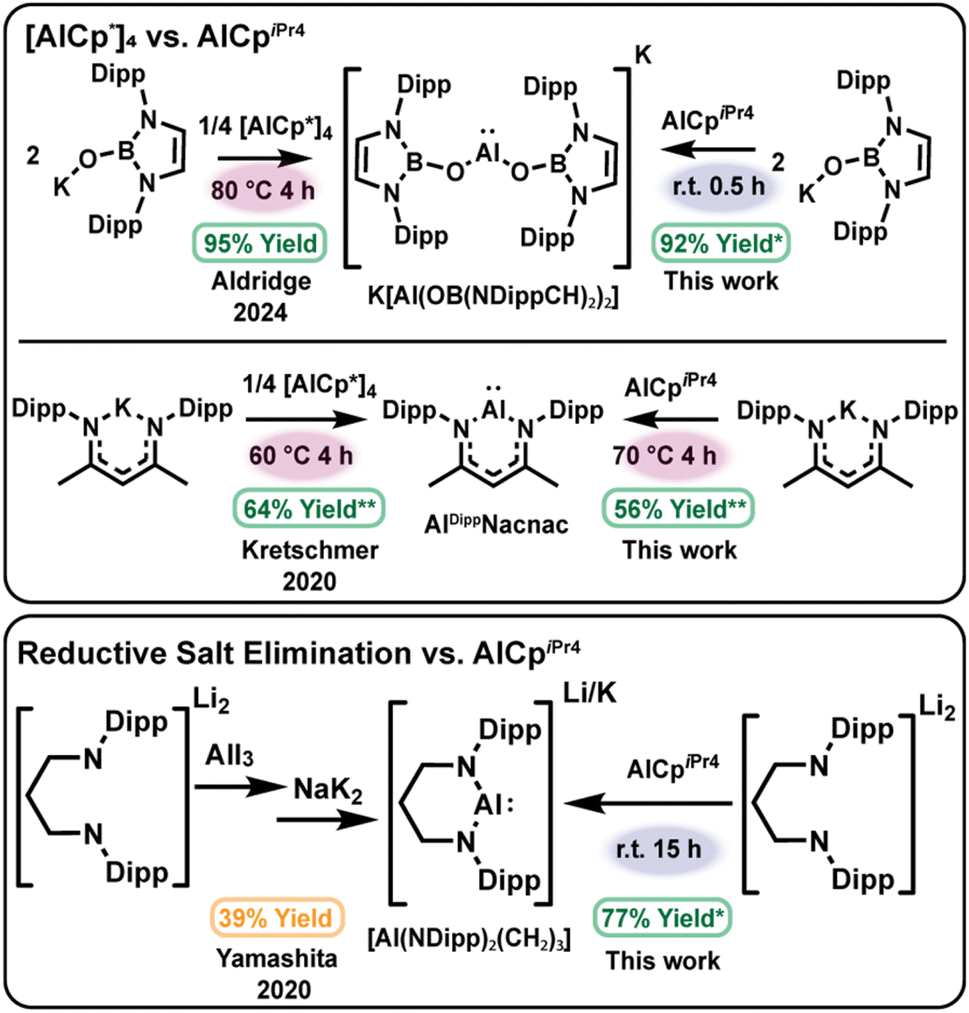 | ||
| Scheme 2 Comparison of aluminylation reagents [AlCp*]4 (top left) and AlCpiPr4 (top right) in the synthesis of aluminyl anion K[Al(OB(NDippCH)2)2] and aluminylene AlDippNacnac. Conditions highlight the thermal requirements that are necessary for reactivity with [AlCp*]4 while not necessarily required for AlCpiPr4. Use of mononuclear AlCpiPr4 in the aluminylation of dianionic salt Li2[(NDipp)2(CH2)3]·(Et2O)2 yields the aluminyl anion Li[Al(NDipp)2(CH2)3] analogous to the anion obtained through reductive elimination routes in one step higher yielding process (bottom). *In situ yield; **in situ yield/% consumed aluminylation reagent. | ||
A practical advantage of aluminylation over heterogeneous reductions is its compatibility with in situ NMR monitoring (see SI). Reactions were performed in sealed NMR tubes with an internal standard. Here, in situ yield is defined as the net increase in product concentration divided by the net decrease in the concentration of the limiting reagent. Where thermal decomposition complicates time-dependent yields (as similarly reported by Kretschmer and co-workers),58 values are normalized to the fraction of aluminylation reagent consumed at early reaction times to enable meaningful comparisons.
We tested two literature aluminylations with contrasting outcomes as a benchmark of this approach: clean formation of [Al(OB(NDippCH)2)2]−30 and the more side-reaction-prone formation of AlDippNacnac.58 Relative to AlCp* generated from [AlCp*]4 at 60–80 °C, AlCpiPr4 delivered K[Al(OB(NDippCH)2)2] rapidly at room temperature (92% in situ yield in 30 min; see SI). In comparison, no reaction with KDippNacnac was observed at room temperature, instead heating to 70 °C was required to form AlDippNacnac, indicating that temperature dependence can reflect substrate-specific activation barriers as well as precursor speciation (Scheme 2), top. Normalized early-time yields under heating were similar for AlCp* and AlCpiPr4 in the synthesis of AlDippNacnac (64% vs. 56%). Furthermore, reaction of the lithium amidinate salt with AlCpiPr4 in C6D6 at room temperature proceeded slowly (40 days) but ultimately furnished the masked dimer [(Al(NDipp)2C5H9)2C6D6]·C6H14 in an improved isolated yield of ∼60% compared to that of [AlCp*]4, demonstrating that aluminylation can be achieved at ambient temperature with a predominantly monomeric Cp reagent.
We also sought to compare the effect of a monomeric aluminylation reagent on the cluster formation described above. At room temperature, reaction of (THF)3Na[N(SiMe3)(Dipp)] with AlCpiPr4 over 36 h furnished the known tetramer [AlN(SiMe3)(Dipp)]4 in 66% crude yield.11 In a similar fashion, treatment of Li2[(NDipp)2(CH2)3]·(Et2O)2 with AlCpiPr4 (1:1) at room temperature for 15 h produced the aluminyl anion Li[Al(NDipp)2(CH2)3] in 77% in situ yield (Scheme 2), bottom, with comparable 1H NMR spectrum to the potassium analogue K[Al(NDipp)2(CH2)3] (Table S1).26 Together, these results demonstrate that aluminylation provides a tractable route to tune low-valent aluminum nuclearity through deliberate control of Al(I) reagent speciation and ligand environment.
Conclusions
This work establishes aluminylation as a general, redox-neutral strategy for accessing low-valent aluminum complexes across diverse ligand classes. However, the nuclearity and speciation of the Al(I) transfer reagent can govern product identity as strongly as substrate choice. In particular, [AlCp*]4 is not merely an Al(I) source: its monomer–tetramer equilibrium can divert reactivity toward heteroleptic Al–Al cluster motifs that are difficult to access using monomeric reagents or traditional reductive routes. Exploiting this dynamic behavior provides a platform for constructing structurally and electronically diverse low-valent aluminum clusters, expanding opportunities to tune aggregation, bonding polarity, and ultimately reactivity in aluminyl chemistry. In comparison, AlCpiPr4 functions as an effective aluminylation reagent under mild conditions, enabled by its persistent monomeric speciation at lower temperatures. Together, these results reveal a path toward enabling controlled Al(I) transfer and access to increasingly reactive low-valent aluminum species.Author contributions
P. C. R. was responsible for the majority of the investigation, including data curation, formal analysis and writing the original draft. Y. S. helped with the investigation of aluminyl anions and M. T. W. provided resources in the form of synthesized ligands. A. B. A was responsible for conceptualization, funding acquisition, project administration, supervision of other researchers as well as writing-review and editing responsibilities.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2531049–2531053 contain the supplementary crystallographic data for this paper.80a–eThe data supporting this article has been included as part of the supplementary information (SI). Supplementary information: full experimental procedures for synthesis of all compounds, characterization details and computational modeling results (1H, 13C, 27Al NMR spectra, crystallographic parameters, Fourier Transform Infrared spectra, UV-vis spectra, Molecular Orbital isosurfaces, Electron Localization Function plots, Tables of Quantum Theory of Atoms in Molecules Critical Points and Density Functional Theory calculated coordinates). See DOI: https://doi.org/10.1039/d6sc02187e.
Acknowledgements
P. C. R., Y. S. and M. T. W. acknowledge support from the Robert A. Welch Foundation (Grant A-2131-20230405) which also supported materials and supplies. A. B. A. acknowledges support from Texas A&M University start-up funds. The X-ray diffractometers and crystallographic computing systems in the X-ray Diffraction Laboratory at the Department of Chemistry, Texas A&M University used to perform the X-ray diffraction characterization were purchased with funds provided by the National Science Foundation (CHE-9807975, CHE-0079822 and CHE-0215838). The Bruker Venture, Quest and ECO diffractometers were purchased with funds provided by Texas A&M University Vice President of Research. Portions of this research were also conducted with the advanced computing resources provided by Texas A&M High Performance Research Computing and use of the TAMU/LBMS and Dr Yohannes Rezenom are acknowledged.Notes and references
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Synthesis, structure and reaction chemistry of a nucleophilic aluminyl anion, Nature, 2018, 557, 92–95 CrossRef CAS PubMed.
- G. Linti and H. Schnöckel, Low valent aluminum and gallium compounds — structural variety and coordination modes to transition metal fragments, Coord. Chem. Rev., 2000, 206–207, 285–319 CrossRef CAS.
- X. Zhang, Y. Mei and L. L. Liu, Free Aluminylenes: An Emerging Class of Compounds, Chem.–Eur. J., 2022, 28, e202202102 CrossRef CAS PubMed.
- D. Sorbelli, L. Belpassi and P. Belanzoni, Unraveling differences in aluminyl and carbene coordination chemistry: bonding in gold complexes and reactivity with carbon dioxide, Chem. Sci., 2022, 13, 4623–4634 RSC.
- C. Weetman and S. Inoue, The Road Travelled: After Main-Group Elements as Transition Metals, ChemCatChem, 2018, 10, 4213–4228 CrossRef CAS.
- P. P. Power, Main-group elements as transition metals, Nature, 2010, 463, 171–177 CrossRef CAS PubMed.
- X. Zhang and L. L. Liu, Aluminium redox catalysis enables cyclotrimerization of alkynes, Nature, 2026, 650, 353–360 CrossRef PubMed.
- X. Zhang and L. L. Liu, Modulating the Frontier Orbitals of an Aluminylene for Facile Dearomatization of Inert Arenes, Angew. Chem., Int. Ed., 2022, 61, e202116658 CrossRef CAS PubMed.
- X. Zhang and L. L. Liu, A Free Aluminylene with Diverse σ-Donating and Doubly σ/π-Accepting Ligand Features for Transition Metals, Angew. Chem., Int. Ed., 2021, 60, 27062–27069 CrossRef CAS PubMed.
- S. K. Mellerup, Y. Cui, F. Fantuzzi, P. Schmid, J. T. Goettel, G. Bélanger-Chabot, M. Arrowsmith, I. Krummenacher, Q. Ye, V. Engel, B. Engels and H. Braunschweig, Lewis-Base Stabilization of the Parent Al(I) Hydride under Ambient Conditions, J. Am. Chem. Soc., 2019, 141, 16954–16960 CrossRef CAS PubMed.
- M. Schiefer, N. D. Reddy, H. W. Roesky and D. Vidovic, Synthesis and Structural Characterization of an Exclusively N-Based Tetrameric Aluminum(I) Compound, Organometallics, 2003, 22, 3637–3638 CrossRef CAS.
- S. Grams, J. Mai, J. Langer and S. Harder, Synthesis, Structure, and Reactivity of a Superbulky Low-Valent β-Diketiminate Al(I) Complex, Organometallics, 2022, 41, 2862–2867 CrossRef CAS.
- C. Ganesamoorthy, S. Loerke, C. Gemel, P. Jerabek, M. Winter, G. Frenking and R. A. Fischer, Reductive elimination: a pathway to low-valent aluminium species, Chem. Commun., 2013, 49, 2858–2860 RSC.
- X. Chen, D. Yang, F. Cao and Z. Mo, Multielectron Reduction of Nitrosoarene via Aluminylene-Silylene Cooperation, J. Am. Chem. Soc., 2024, 146, 29278–29284 CrossRef CAS PubMed.
- J. M. L. Baeza, H. Xu, S. Takahashi, A. Baceiredo, R. S. R. Guerrero, D. Hashizume, N. Saffon-Merceron, V. Branchadell and T. Kato, Isolable Three-Coordinate Base-Stabilized Alumylene: A Precursor of Persistent Acceptor-Free Monomeric Aluminum Oxide, Angew. Chem., Int. Ed., 2025, 64, e202505181 CrossRef CAS PubMed.
- X. Li, X. Cheng, H. Song and C. Cui, Synthesis of HC[(CBut)(NAr)]2Al (Ar = 2,6-iPr2C6H3) and Its Reaction with Isocyanides, a Bulky Azide, and H2O, Organometallics, 2007, 26, 1039–1043 CrossRef CAS.
- C. Cui, H. W. Roesky, H. G. Schmidt, M. Noltemeyer, H. Hao and F. Cimpoesu, Synthesis and Structure of a Monomeric Aluminum(I) Compound [{HC(CMeNAr)2}Al] (Ar=2,6–iPr2C6H3): A Stable Aluminum Analogue of a Carbene, Angew. Chem., Int. Ed., 2000, 39, 4274–4276 CrossRef CAS PubMed.
- X. Zhang and L. L. Liu, Aluminylenes: Synthesis, Reactivity, and Catalysis, Acc. Chem. Res., 2026, 59, 726–739 CrossRef CAS PubMed.
- R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, A Stable Calcium Alumanyl, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed.
- A. Paparo, C. D. Smith and C. Jones, Diagonally Related s- and p-Block Metals Join Forces: Synthesis and Characterization of Complexes with Covalent Beryllium–Aluminum Bonds, Angew. Chem., Int. Ed., 2019, 58, 11459–11463 CrossRef CAS PubMed.
- S. Kurumada, R. Yamanashi, K. Sugita, K. Kubota, H. Ito, S. Ikemoto, C. Chen, T. Moriyama, S. Muratsugu, M. Tada, T. Koitaya, T. Ozaki and M. Yamashita, Mechanochemical Synthesis of Non-Solvated Dialkylalumanyl Anion and XPS Characterization of Al(I) and Al(II) Species, Chem.–Eur. J., 2024, 30, e202303073 CrossRef CAS PubMed.
- S. Kurumada, S. Takamori and M. Yamashita, An alkyl-substituted aluminium anion with strong basicity and nucleophilicity, Nat. Chem., 2019, 12, 36–39 CrossRef PubMed.
- K. Koshino and R. Kinjo, Construction of σ-Aromatic AlB2 Ring via Borane Coupling with a Dicoordinate Cyclic (Alkyl)(Amino)Aluminyl Anion, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS PubMed.
- R. A. Jackson, A. J. R. Matthews, P. Vasko, M. F. Mahon, J. Hicks and D. J. Liptrot, An acyclic aluminyl anion, Chem. Commun., 2023, 59, 5277–5280 RSC.
- S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Boosting Low-Valent Aluminum(I) Reactivity with a Potassium Reagent, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS PubMed.
- G. Feng, K. L. Chan, Z. Lin and M. Yamashita, Al–Sc Bonded Complexes: Synthesis, Structure, and Reaction with Benzene in the Presence of Alkyl Halide, J. Am. Chem. Soc., 2022, 144, 22662–22668 CrossRef CAS PubMed.
- D. Dhara, L. Endres, A. Roy, R. D. Dewhurst, R. Bertermann, F. Fantuzzi and H. Braunschweig, A Discrete Trialane with a Near-Linear Al3 Axis, J. Am. Chem. Soc., 2024, 146, 33536–33542 CrossRef CAS PubMed.
- F. Dankert, A. Kadriu and E. Hevia, A Hexametallic Copper Aluminylene Aluminyl, ChemRxiv, 2024 DOI:10.26434/chemrxiv-2024-r1wz8.
- F. Dankert and E. Hevia, Synthesis and Modular Reactivity of Low Valent Al/Zn Heterobimetallics Supported by Common Monodentate Amides, Chem.–Eur. J., 2024, 30, e202304336 CrossRef CAS PubMed.
- D. Sarkar, P. Vasko, A. F. Roper, A. E. Crumpton, M. M. D. Roy, L. P. Griffin, C. Bogle and S. Aldridge, Reversible [4+1] Cycloaddition of Arenes by a “Naked” Acyclic Aluminyl Compound, J. Am. Chem. Soc., 2024, 146, 11792–11800 CrossRef CAS PubMed.
- M. P. Coles and M. J. Evans, The emerging chemistry of the aluminyl anion, Chem. Commun., 2023, 59, 503–519 RSC.
- J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Reversible, Room-Temperature C—C Bond Activation of Benzene by an Isolable Metal Complex, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed.
- M. D. Anker and M. P. Coles, Aluminium-Mediated Carbon Dioxide Reduction by an Isolated Monoalumoxane Anion, Angew. Chem., Int. Ed., 2019, 58, 18261–18265 CrossRef CAS PubMed.
- R. J. Schwamm, M. S. Hill, H.-Y. Liu, M. F. Mahon, C. L. McMullin and N. A. Rajabi, Seven-Membered Cyclic Potassium Diamidoalumanyls, Chem.–Eur. J., 2021, 27, 14971–14980 CrossRef CAS PubMed.
- H. Zhu, J. Chai, H. Fan, H. W. Roesky, C. He, V. Jancik, H. G. Schmidt, M. Noltemeyer, W. A. Merrill and P. P. Power, A Stable Aluminacyclopropene LAl(η2-C2H2) and Its End-On Azide Insertion to an Aluminaazacyclobutene, Angew. Chem., Int. Ed., 2005, 44, 4989–5149 CrossRef.
- C. Cui, S. Köpke, R. Herbst-Irmer, H. W. Roesky, M. Noltemeyer, H.-G. Schmidt and B. Wrackmeyer, Facile Synthesis of Cyclopropene Analogues of Aluminum and an Aluminum Pinacolate, and the Reactivity of LAl[η2-C2(SiMe3)2] toward Unsaturated Molecules (L = HC[(CMe)(NAr)]2, Ar = 2,6-i-Pr2C6H3), J. Am. Chem. Soc., 2001, 123, 9091–9098 CrossRef CAS PubMed.
- N. J. Hardman, C. Cui, H. W. Roesky, W. H. Fink and P. P. Power, Stable, Monomeric Imides of Aluminum and Gallium: Synthesis and Characterization of [{HC(MeCDippN)2}MN-2,6-Trip2C6H3] (M=Al or Ga; Dipp=2,6-iPr2C6H3; Trip=2,4,6-iPr3C6H2), Angew. Chem., Int. Ed., 2001, 40, 1983–2181 CrossRef.
- K. Hobson, C. J. Carmalt and C. Bakewell, Recent advances in low oxidation state aluminium chemistry, Chem. Sci., 2020, 11, 6942–6956 RSC.
- X. Liu, S. Dong, J. Zhu and S. Inoue, Dialumene as a Dimeric or Monomeric Al Synthon for C–F Activation in Monofluorobenzene, J. Am. Chem. Soc., 2024, 146, 23591–23597 CrossRef CAS PubMed.
- A. Saddington, S. Dong, S. Yao, J. Zhu and M. Driess, Bis-Silylene-Supported Aluminium Atoms with Aluminylene and Alane Character, Angew. Chem., Int. Ed., 2024, 63, e202410790 CAS.
- D. Dhara, A. Jayaraman, M. Härterich, M. Arrowsmith, M. Jürgensen, M. Michel and H. Braunschweig, Trapping of a Transient Base-Stabilised Alumylene and Alumylene-Type Reactivity of a Self-Stabilising Dialumene towards Organic Azides, Chem.–Eur. J., 2023, 29, e202300483 CrossRef CAS PubMed.
- D. Dhara, A. Jayaraman, M. Härterich, R. D. Dewhurst and H. Braunschweig, Generation of a transient base-stabilised arylalumylene for the facile deconstruction of aromatic molecules, Chem. Sci., 2022, 13, 5631–5638 RSC.
- R. L. Falconer, K. M. Byrne, G. S. Nichol, T. Krämer and M. J. Cowley, Reversible Dissociation of a Dialumene, Angew. Chem., Int. Ed., 2021, 60, 24702–24708 CrossRef CAS PubMed.
- P. Bag, A. Porzelt, P. J. Altmann and S. Inoue, A Stable Neutral Compound with an Aluminum–Aluminum Double Bond, J. Am. Chem. Soc., 2017, 139, 14384–14387 CrossRef CAS PubMed.
- C. Weetman, A. Porzelt, P. Bag, F. Hanusch and S. Inoue, Dialumenes – aryl vs. silyl stabilisation for small molecule activation and catalysis, Chem. Sci., 2020, 11, 4817–4827 RSC.
- A. Purath, C. Dohmeier, A. Ecker, R. Köppe, H. Krautscheid, H. Schnöckel, R. Ahlrichs, C. Stoermer, J. Friedrich and P. Jutzi, Synthesis and Structure of a Neutral SiAl14 Cluster, J. Am. Chem. Soc., 2000, 122, 6955–6959 CrossRef CAS.
- A. Stasch, H. W. Roesky, D. Vidovic, J. Magull, H.-G. Schmidt and M. Noltemeyer, Synthesis of Carbaalane Halogen Derivatives, Inorg. Chem., 2004,(43), 3625–3630 CrossRef CAS PubMed.
- A. Stasch, M. Ferbinteanu, J. Prust, W. Zheng, F. Cimpoesu, H. W. Roesky, J. Magull, H.-G. Schmidt and M. Noltemeyer, Syntheses, Structures, and Surface Aromaticity of the New Carbaalane [(AlH)6(AlNMe3)2(CCH2R)6] (R = Ph, CH2SiMe3) and a Stepwise Functionalization of the Inner and Outer Sphere of the Cluster, J. Am. Chem. Soc., 2002, 124, 5441–5448 CrossRef CAS PubMed.
- H. Köhnlein, A. Purath, C. Klemp, E. Baum, I. Krossing, a. G. Stösser and H. Schnöckel, Synthesis and Characterization of an Al693- Cluster with 51 Naked Al Atoms: Analogies and Differences to the Previously Characterized Al772- Cluster, Inorg. Chem., 2001, 40, 4830–4838 CrossRef PubMed.
- M. Huber, A. Schnepf, C. E. Anson and H. Schnöckel, Si@Al56[N(2,6-iPr2C6H3)SiMe3]12: The Largest Neutral Metalloid Aluminum Cluster, a Molecular Model for a Silicon-Poor Aluminum–Silicon Alloy?, Angew. Chem., Int. Ed., 2008, 47, 8201–8206 CrossRef CAS PubMed.
- C. Klemp, R. Köppe, E. Weckert and H. Schnöckel, Al22Br20·12 THF: The First Polyhedral Aluminum Subhalide—A Step on the Path to a New Modification of Aluminum?, Angew. Chem., Int. Ed., 1999, 38, 1739–1743 CrossRef PubMed.
- K.-W. Klinkhammer, W. Uhl, J. Wagner and W. Hiller, K2[Al12iBu12], a Compound with Al12 Icosahedra, Angew. Chem., Int. Ed., 1991, 30, 179–180 CrossRef.
- H. Köhnlein, G. Stösser, E. Baum, E. Möllhausen, U. Huniar and H. Schnöckel, A Metalloid Al14 Cluster with the Structure of a “Nano-Wheel”, Angew. Chem., Int. Ed., 2000, 39, 799–801 CrossRef.
- J. Vollet, R. Burgert and H. Schnöckel, Al20X10 (X = Cl, Br): Snapshots of the Formation of Metalloid Clusters from Polyhedral AlnXm Molecules?, Angew. Chem., Int. Ed., 2005, 44, 6956–6960 CrossRef CAS PubMed.
- J. Vollet, J. R. Hartig and H. Schnöckel, Al50C120H180: A Pseudofullerene Shell of 60 Carbon Atoms and 60 Methyl Groups Protecting a Cluster Core of 50 Aluminum Atoms, Angew. Chem., Int. Ed., 2004, 43, 3186–3189 CrossRef CAS PubMed.
- P. Dabringhaus, J. Willrett and I. Krossing, Synthesis of a low-valent Al4+ cluster cation salt, Nat. Chem., 2022, 14, 1151–1157 CrossRef CAS PubMed.
- A. Purath, R. Köppe and H. Schnöckel, An Al12R8− cluster as an intermediate on the way from aluminium(I) compounds to aluminium metal, Chem. Commun., 1999, 1933–1934 RSC.
- O. Kysliak, H. Görls and R. Kretschmer, Salt metathesis as an alternative approach to access aluminium(I) and gallium(I) β-diketiminates, Dalton Trans., 2020, 49, 6377–6383 RSC.
- H. Sitzmann, M. F. Lappert, C. Dohmeier, C. Üffing and H. Schnöckel, Cyclopentadienylderivate von Aluminium(I), J. Organomet. Chem., 1998, 561, 203–208 CrossRef CAS.
- A. Hofmann, T. Tröster, T. Kupfer and H. Braunschweig, Monomeric Cp3tAl(I): synthesis, reactivity, and the concept of valence isomerism, Chem. Sci., 2019, 10, 3421–3428 RSC.
- J. Gauss, U. Schneider, R. Ahlrichs, C. Dohmeier and H. Schnoeckel, 27Al NMR spectroscopic investigation of aluminum(I) compounds: ab initio calculations and experiment, J. Am. Chem. Soc., 2002, 115, 2402–2408 CrossRef.
- G. M. Ballmann, M. J. Evans, T. X. Gentner, A. R. Kennedy, J. R. Fulton, M. P. Coles and R. E. Mulvey, Synthesis, Characterization, and Structural Analysis of AM[Al(NONDipp)(H)(SiH2Ph)] (AM = Li, Na, K, Rb, Cs) Compounds, Made Via Oxidative Addition of Phenylsilane to Alkali Metal Aluminyls, Inorg. Chem., 2022, 61, 19838–19846 CrossRef CAS PubMed.
- J. D. Queen, A. Lehmann, J. C. Fettinger, H. M. Tuononen and P. P. Power, The Monomeric Alanediyl :AlAriPr8 (AriPr8 = C6H-2,6-(C6H2-2,4,6-Pri3)2-3,5-Pri2): An Organoaluminum(I) Compound with a One-Coordinate Aluminum Atom, J. Am. Chem. Soc., 2020, 142, 20554–20559 CrossRef CAS PubMed.
- I. Squire, M. de Vere-Tucker, M. Tritto, L. Silva de Moraes, T. Krämer and C. Bakewell, A neutral cyclic aluminium (I) trimer, Nat. Commun., 2026, 17, 1732 CrossRef CAS PubMed.
- A. Purath, C. Dohmeier, A. Ecker, H. Schnöckel, K. Amelunxen, T. Passler and N. Wiberg, Synthesis and Crystal Structure of the Tetraaluminatetrahedrane Al4[Si(t-Bu)3]4, the Second Al4R4 Compound, Organometallics, 1998, 17, 1894–1896 CrossRef CAS.
- A. Purath and H. Schnöckel, Tetrakis[tris(trimethylsilyl)silylaluminium(I)] Al4[Si(SiMe3)3]4—eine siliziumreicheVerbindung mit zentralem tetraedrischem Al4-Kern, J. Organomet. Chem., 1999, 579, 373–375 CrossRef CAS.
- C. Dohmeier, C. Robl, M. Tacke and H. Schnöckel, The Tetrameric Aluminum(I) Compound [{Al(η5-C5Me5)}4], Angew. Chem., Int. Ed., 1991, 30, 564–565 CrossRef.
- K. Wade, The structural significance of the number of skeletal bonding electron-pairs in carboranes, the higher boranes and borane anions, and various transition-metal carbonyl cluster compounds, J. Chem. Soc. D: Chem. Commun., 1971, 792–793 RSC.
- D. M. P. Mingos, A General Theory for Cluster and Ring Compounds of the Main Group and Transition Elements, Nat. Phys. Sci., 1972, 236, 99–102 CrossRef CAS.
- P. Pyykkö, Additive Covalent Radii for Single-, Double-, and Triple-Bonded Molecules and Tetrahedrally Bonded Crystals: A Summary, J. Phys. Chem. A, 2014, 119, 2326–2337 CrossRef PubMed.
- F. Neese, Software Update: The ORCA Program System—Version 6.0, Wiley Interdiscip. Rev. Comput. Mol. Sci., 2025, 15, e70019 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Accurate Coulomb-fitting basis sets for H to Rn, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- T. Lu and F. Chen, Multiwfn: A multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- R. F. W. Bader, A quantum theory of molecular structure and its applications, Chem. Rev., 2002, 91, 893–928 CrossRef.
- L. Tian and C. Fei-Wu, Meaning and Functional Form of the Electron Localization Function, Acta Phys. Chim. Sin., 2011, 27, 2786–2792 Search PubMed.
- A. D. Becke and K. E. Edgecombe, A simple measure of electron localization in atomic and molecular systems, J. Chem. Phys., 1990, 92, 5397–5403 CrossRef CAS.
- R. J. Eisenhart, L. J. Clouston and C. C. Lu, Configuring Bonds between First-Row Transition Metals, Acc. Chem. Res., 2015, 48, 2885–2894 CrossRef CAS PubMed.
- (a) CCDC 2531049: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qyrr6; (b) CCDC 2531050: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qyrs7; (c) CCDC 2531051: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qyrt8; (d) CCDC 2531052: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qyrv9; (e) CCDC 2531053: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qyrwb.
| This journal is © The Royal Society of Chemistry 2026 |