
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The mechanochemical activation of a pyrimidine dimer
Xiang
Gao
ab,
Gurudas
Chakraborty
 *b,
Felix Joel
Urhahne
c,
Marcus
Lantzius-Beninga
ab,
Regina
Lennarz
c,
Jilin
Fan
ab,
Kara
Stappert
a,
Jan
Meisner
c,
Robert
Göstl
bd and
Andreas
Herrmann
*ab
*b,
Felix Joel
Urhahne
c,
Marcus
Lantzius-Beninga
ab,
Regina
Lennarz
c,
Jilin
Fan
ab,
Kara
Stappert
a,
Jan
Meisner
c,
Robert
Göstl
bd and
Andreas
Herrmann
*ab
aInstitute of Technical and Macromolecular Chemistry, RWTH Aachen University, Worringerweg 2, 52074 Aachen, Germany. E-mail: herrmann@dwi.rwth-aachen.de
bDWI – Leibniz-Institute for Interactive Materials, Forckenbeckstraße 50, 52056 Aachen, Germany. E-mail: chakraborty@dwi.rwth-aachen.de
cInstitute for Physical Chemistry, Heinrich Heine University Düsseldorf, Universitätsstraße 1, 40225 Düsseldorf, Germany
dDepartment of Chemistry and Biology, University of Wuppertal, Gaußstraße 20, 42119 Wuppertal, Germany
First published on 17th April 2026
Abstract
The cyclobutane motif is a versatile force-reactive moiety enabling diverse mechanophores and their associated functions in polymer mechanochemistry. For example, the cyclobutane core has been applied in the context of stress-responsive polymers, self-healing materials, and self-sensing nanocomposites. However, leveraging the cyclobutane structure for the development of nucleobase- and nucleoside-derived mechanophores remains unexplored. Here, we introduce a pyrimidine dimer mechanophore based on a cyclobutane core (CPD), formed via photoinduced [2 + 2] cycloaddition of thymine under ultraviolet B (UVB) irradiation. Upon ultrasound exposure, the polymer-centered CPD undergoes formal cycloelimination. To better understand the individual mechanochemical contributions of the four possible CPD stereoisomers upon force input, we employ the CoGEF and FM-PES methods and compare the results. Experimental and computational methods suggest that the syn-diastereomers cleave preferentially compared to their anti-counterparts under mechanical force.
1 Introduction
Polymer mechanochemistry has rapidly advanced as a field focused on harnessing mechanical energy to drive specific chemical transformations.1,2 In this context, ultrasound serves as a key source of mechanical force, widely used in solution-phase polymer systems.3 Upon the collapse of cavitation bubbles, ultrasound generates elongational flow that overstretches segments of reasonably long polymer chains, eventually leading to bond scission.4,5 Building on this foundation, a wide variety of mechanochemically responsive motifs (mechanophores) have been developed6 and embedded in synthetic polymers to trigger distinct functions in response to mechanical force, including catalysis,7–9 initiating cross-linking,10,11 releasing small molecules,12–20 or visually reporting material damage.21–25These functions have also been uniquely realized using biomacromolecules, with demonstrations of ultrasound-induced drug activation by covalent and noncovalent bond cleavage within nucleic acid scaffolds, introducing a new research domain of sonopharmacology.26 This polyaptamer-based deactivation and ultrasound-triggered activation approach was also applied to activate thrombin, enabling it to catalyze the conversion of fibrinogen into fibrin.27 Additional advances include sonocontrolled optical and catalytic activity of genetically engineered proteins,28 mechanochemical activation of DNAzymes,29 and fracture detection in bio-glues using fluorescent-protein-based optical force probes.30
Within the specific context of sonopharmacology, ultrasound-responsive mechano-nanoswitches have also been developed,31 consisting of gold nanoparticles (AuNPs) functionalized with a defined number of DNA strands on their surfaces. These nanoparticles form dimers by Watson–Crick base pairing, enabling the incorporation of anticancer drugs into the double helix. Upon ultrasound application, the drugs are liberated, leading to killing of cancer cells in vitro. More importantly, the applicability of polymer mechanochemistry principles has recently been demonstrated in vivo.32 A universal polynucleotide framework was developed, enabling the binding and release of therapeutic oligonucleotides, both DNA and RNA, using biocompatible medical-imaging ultrasound as the mechanical trigger. However, hydrogen-bonding-based caging strategies are often susceptible to thermal denaturation and enzymatic degradation. To address these limitations, an alternative approach involves caging oligonucleotides by exploiting covalent linkages between adjacent nitrogenous bases along the same strand, such as through the formation of CPDs, thereby exploring the mechanochemical reactivity potential of nucleobase- and nucleoside-derived motifs.
CPDs are the primary DNA lesions formed upon exposure to UVB light. They result from a [2 + 2] cycloaddition between the C5–C6 double bonds of adjacent pyrimidine bases.33–35 Although such damage can be reversed through photorepair mechanisms, such as photolyase-mediated electron transfer that cleaves the four-membered ring,36,37 the application of mechanical force to induce CPD ring-opening remains unexplored. Investigating the mechanochemical response of CPDs under ultrasound could not only establish their function as mechanophores but also open new avenues for their use in biologically relevant systems.
Indeed, cyclobutane-core mechanophores are well-established in polymer mechanochemistry38–40 underpinning the plausibility of this hypothesis. Therefore, we employ synthetic polymers rather than long-chain DNA as carriers for mechanical force transmission. Our experimental design centers on synthesizing CPDs via UVB-induced [2 + 2] cycloaddition of thymine41 and incorporating them into linear poly(methyl acrylate) (PMA) chains of varying molar masses (Mn ∼33–104 kDa) using Cu(0)-mediated controlled radical polymerization (CRP, Scheme 1).42,43 The mechanochemical behavior of these polymers was then probed under ultrasound exposure. We applied established mechanochemical conditions in this work (20 kHz ultrasound and linear PMA polymers), paving the way for future work to exploit medically relevant ultrasound, such as focused ultrasound with a mechanical index below 1.9 – the FDA approval limit.44,45 Gel permeation chromatography (GPC) revealed distinct peaks corresponding to half the original molar mass, confirming ultrasound-induced chain scission. By varying the ultrasound intensity (3–14 W cm−2), we quantified the corresponding rate constants for ultrasound-induced polymer cleavage (Table 1). To determine the site of cleavage, nuclear magnetic resonance (NMR) spectroscopy was employed to detect characteristic olefinic proton resonances before and after ultrasonication, providing evidence of bond rupture at the CPD site. To complement these findings, constrained geometries simulate external force (CoGEF) and force-modified potential energy surface (FM-PES) computations were performed on the four CPD isomers (trans–syn, cis–syn, trans–anti, and cis–anti) to evaluate their ring-opening force profiles and associated energy requirements.46,47 Collectively, this study not only establishes CPDs as a new class of mechanophores through comprehensive experimental and computational analyses but also introduces a versatile model for investigating nucleobase- and nucleoside-derived mechanochemistry, possibly also applicable to other photodimers, including cytosine dimers.48
 | ||
| Scheme 1 The cycloaddition reaction under UVB light and Cu(0)-mediated CRP. | ||
2 Results and discussion
2.1 UV-induced [2 + 2] cycloaddition for CPD formation
Thymine generates a CPD structure under UVB light irradiation. It is noteworthy that this reaction strongly depends on the wavelength of UV light. In fact, reports indicate that under UVA or UVC irradiation, thymine produces 6–4 photoproducts.49,50 Moreover, the CPD structures formed under UV light are predominantly of the cis–syn configuration.51 Based on this, we irradiated thymidine with a 310 nm UVB source to obtain CPD products (Scheme 1). In the 1H NMR spectra, we observed the presence of four different structural isomers (Fig. 1). | ||
| Fig. 1 The 1H NMR spectrum of the 1-position H of thymidine in CPD. The cooled photoreaction was performed at 10 °C and the uncooled reaction was performed at room temperature. | ||
The chemical shift of the 1-position protons in the dimer experiences mutual coupling, and in different isomers, the chemical shifts also vary. Based on this, we were able to determine the content of the different structural isomers. The characteristic peaks for trans–syn appeared at ca. 6.20 ppm and 5.85 ppm, for trans–anti at ca. 6.20 ppm and 5.75 ppm, for cis–syn at ca. 5.85 ppm and 5.25 ppm, and for cis–anti at ca. 6.20 ppm and 6.00 ppm. Among the four isomers, we found that the cis–syn structure was the major product, albeit with a relatively low yield of only 46% (Fig. 1, top). In order to better mimic the natural formation of CPD, we attempted to cool the reaction mixture by immersing it in cold water, aiming to mitigate the potential impact of prolonged exposure to high temperatures caused by continuous light irradiation. As a result, we observed a significant increase in the content of the cis–syn isomer, reaching up to 78% (Fig. 1, bottom). While we still observed the formation of the other three isomers in low yield, our primary emphasis, however, was not to isolate all four species, but to directly investigate the effect of mechanical force on the natural photoproducts of the UVB-induced dimerization reaction, with a particular focus on monitoring CPD cleavage under ultrasound.
2.2 Synthesis and ultrasonication of CPD-containing polymers
After obtaining the CPD structure, we further synthesized initiator 2 and employed Cu(0)-mediated CRP to synthesize various linear polymers with different molar masses (Scheme 1). These included poly(methyl acrylate) PMA1 (Mn = 104 kDa, ĐM = 1.18), PMA2 (Mn = 71 kDa, ĐM = 1.27), PMA3 (Mn = 49 kDa, ĐM = 1.24), and PMA4 (Mn = 33 kDa, ĐM = 1.31) (Fig. S10–S13). The polymerization reaction was initiated on both sides of the CPD mechanophore under identical conditions, ensuring that the mechanophore structure was positioned in the center of the polymer. This approach accounts for the maximum mechanochemical reactivity in the subsequent ultrasonic experiments.4The ultrasonication experiments were conducted using a 20 kHz immersion probe sonicator, with the solution adequately cooled using ice water. Samples were taken every 10 min for GPC measurements to monitor the molar mass distribution after different durations of ultrasonication (Fig. 2 and 3). Aligning with the positioning of the mechanophore at the center of the polymer chain, we observed the formation of a new peak corresponding to half of the initial molar mass. Furthermore, we clearly observed that with decreasing ultrasound intensities, ranging from 14 W cm−2 to 3 W cm−2, there was a significant and expected reduction in the rate of polymer chain rupture (Fig. 2). Analogously, we observed the same trend for polymers with decreasing molar masses at constant intensity (14 W cm−2) (Fig. 3).
 | ||
| Fig. 2 GPC traces of PMA1 with an initial Mn of 104 kDa after ultrasonic treatment at different intensities. | ||
 | ||
| Fig. 3 The GPC traces of PMA1–4 with varying initial Mn after ultrasonic treatment at an intensity of 14 W cm−2. | ||
To quantify the influence of molar mass and ultrasound intensity on the mechanophore cleavage rates, we calculated the apparent scission rate constant for this mechanophore (the mixture of all isomers). We used a calculation method (eqn (1)) based on the approach of Malhotra, in which k′= k/M0, where M0 is the molar mass of the monomer unit, Mi is the initial number average molar mass of the polymer, Mt is the number average molar mass of the sonicated sample at time t, and k is the rate constant of polymer cleavage with initial molar mass Mi.52,53 Additionally, Moore and coworkers developed and efficiently applied this formula to calculate the rate constants for cyclobutane-based mechanophores.38
 | (1) |
The Mn data obtained from GPC measurements for products under different ultrasonic conditions can be fitted to the above-mentioned formula (Fig. 4a). The calculated rate constants k′ under different intensities and molar masses are presented in Tables 1 and 2. Furthermore, the cleavage rate constant k′ also demonstrates a linear relationship (Fig. 4b), providing strong evidence that ultrasound waves activate the cleavage process of CPD mechanophores.
 | ||
| Fig. 4 Experimentally determined rate constants of polymer cleavage as a function of initial polymer molar mass. (a) The fitting line of 1/Mt − 1/Mi against ultrasonication time, where the slope of the line represents the ultrasonic rate constant k′. (b) The fitting line of the rate constant k′ against the initial number average molar mass of the polymer Mi or ultrasonic intensity. | ||
| Ultrasound intensity IP/W cm−2 | Rate constant k/10−6 min−1 |
|---|---|
| 14 | 13.4 |
| 10 | 7.90 |
| 6 | 4.76 |
| 3 | 0.40 |
| Initial molar mass Mi/kDa | Rate constant k/10−6 min−1 |
|---|---|
| 104 | 13.4 |
| 71 | 9.12 |
| 49 | 3.78 |
| 33 | 1.63 |
However, the ultrasound-activated mechanophore cleavage competed with the random chain scission of the polymer backbone. While we have already demonstrated the cleavage of CPD mechanophores under ultrasound, it is crucial to further prove the exact cleavage positions. We employed NMR as a non-optical characterization method to detect the formation of double bond H signals, considering the non-fluorescent character of the designed mechanophore. Although observing a specific proton signal throughout the entire polymer can be challenging due to significant interference, there are no other signals in the vicinity of the double bond proton signals in the PMA, allowing us to detect the formation of double bonds after ultrasound exposure.
We conducted NMR measurements on the individual thymidine molecule and PMA1 polymers in DMSO-d6 solution before and after ultrasound irradiation. In the HH-COSY spectrum of the thymidine molecule (Fig. 5), the double bond proton signal (blue) is at 7.7 ppm, and the 1-position proton (red) signal is at 6.2 ppm. We also observed coupling relationships between the 1-position and 2-position proton signals. In the COSY spectrum of PMA1 after ultrasound exposure (Fig. 6), we could similarly determine the signal for the 1-position proton based on the coupling relationships. Due to the differences in chemical environments between the solvent and the polymer, there was a slight shift in the double bond proton signal. When comparing the NMR spectra before and after ultrasound exposure (Fig. 7), we found that there was no double bond proton signal before the CPD mechanophore was cleaved by ultrasound. After ultrasound irradiation, the double bond proton signal reappears. Based on this, we can confirm that the cleavage position of the mechanophore is primarily at the cyclobutane site, and the structure after cleavage restores the double bond structure of thymidine.
 | ||
| Fig. 5 The HH-COSY spectrum of thymine molecules, with a magnified section showing the correlation of the double bond H (blue) and the 1-position H (red) individually. | ||
 | ||
| Fig. 6 The HH-COSY spectrum of PMA1 after ultrasound exposure, with a magnified section showing the correlation of the double bond H (blue) and the 1-position H (red) individually. The intermediate appearance of the double peak corresponds to the residual BHT in the polymer. | ||
 | ||
| Fig. 7 The 1H NMR spectrum of PMA1 before and after ultrasonication, showing the reappearance of the double bond H signal after 30 min of ultrasonication. BHT was used as the internal standard. | ||
2.3 Experimental and computational investigations of the isolated isomers
Hereafter, we investigated whether different isomeric structures of CPD exhibit significant differences in their response to ultrasound. The mechanical cleavage of syn-configured cyclobutane-type mechanophores generally involves two main processes: single bond cleavage, leading to the formation of a diradical intermediate, followed by rapid diradical-mediated ring opening and formation of a diene (Scheme 2a).3 Upon the first C–C bond breaking, the second C–C bond is strongly activated. The responsiveness to force depends on the linker and on the cis/trans configuration. The mechanical cleavage of anti-configured cyclobutane is expected to require higher forces, because the two bonds to be broken are not aligned parallel to the tensile axis and, thus, the force is transduced less directly (Scheme 2b).3 We isolated the cis–syn, cis–anti (−), cis–anti (+), and trans–anti isomers of CPD by preparative high-performance liquid chromatography (HPLC) (the trans–syn isomer is only formed in traces) and functionalized the cis–syn and trans–anti isomers with initiators for CRP. The resulting PMA polymers with comparable Mn (cis–syn: 51.0 kDa, trans–anti: 50.2 kDa, Fig. S12) were subjected to ultrasonication at low intensity to resolve any differences in reactivity of the isomers (0–30 min, 20% amplitude, 3 W cm−2) and the scission rates were determined as demonstrated above (vide supra). The analysis of the ultrasound-induced bond scission by GPC showed activation of the cis–syn PMA, while the scission rate of the trans–anti PMA was the same as for the control PMA. These findings indicate a reactivity difference of the individual isomers (Fig. S20–S22). We further investigated the mechanochemical behaviour of the isomers by computational methods for a complementary comprehension of the responsiveness of these isomers to the applied force.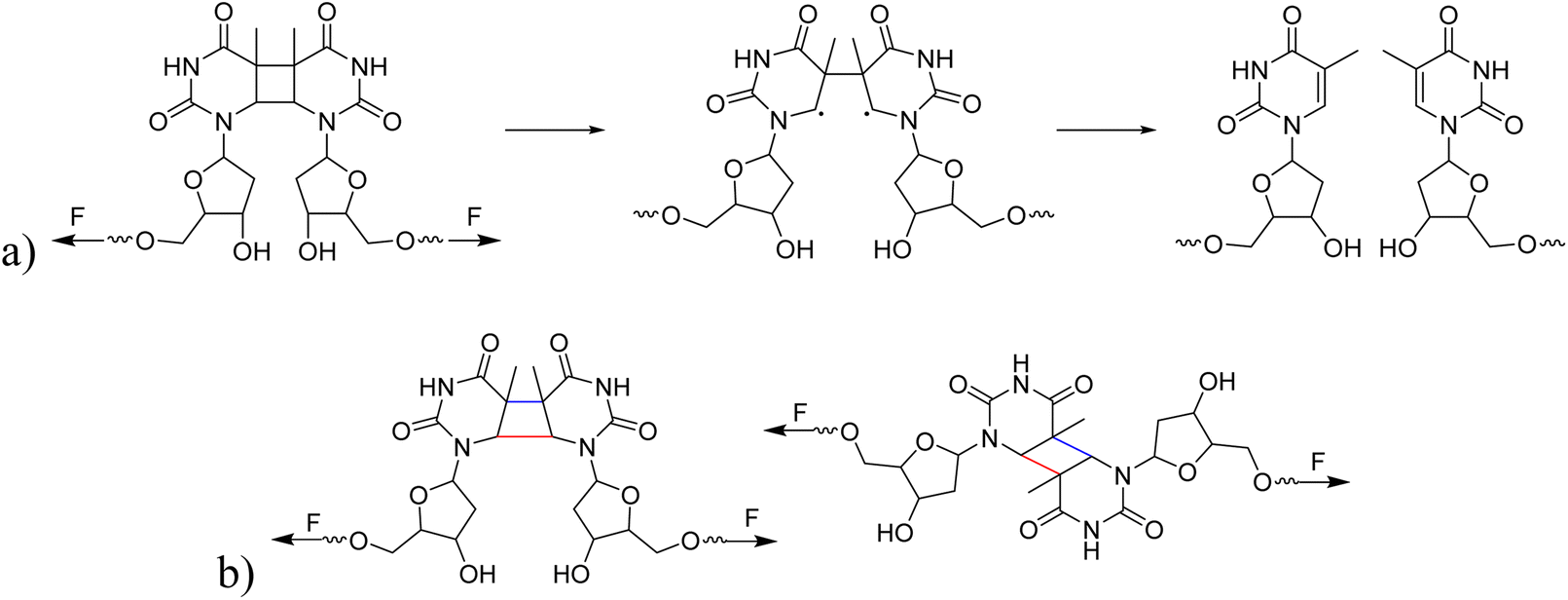 | ||
| Scheme 2 (a) The process of CPD mechanophore cleavage induced by mechanical force. (b) The potential for different cleavage outcomes of syn- and anti-conformations of CPD mechanophores under mechanical force. The bonds marked in red indicate the bond at which the first C–C scission occurs, while the bonds marked in blue indicate the second C–C scission. | ||
We performed CoGEF46 and FM-PES54 calculations on the four isomeric models of CPD at the B3LYP-D3/6-31G* level55–61 of theory. Mechanical force was applied at the 5′ end of deoxyribose, propagating through the entire deoxyribose molecule onto the cyclobutane structure. While CoGEF is a straightforward and highly accessible method to compute the influence of force on molecular systems, it purely relies on optimized geometric structures of increased geometric displacement. Therefore, although dissociation energies and rupture forces Fmax are available, reaction pathways are not available, which means that reactive intermediates might be missed. Furthermore, the values of the rupture forces obtained are too large (compared to the experiment) because the method does not take thermal effects into account.62,63 Nevertheless, due to its low computational effort, it is well suited as a method to investigate the influence of mechanical force on a novel mechanophore. The FM-PES computations rely on a potential energy surface, which is distorted (force-modified), so that minimum structures, transition state structures, and reaction paths can be obtained for different forces under investigation. This significantly more expensive approach allows the computation of barrier heights depending on the applied force and, thus, enables mechanochemical and purely thermal reactions to be treated on equal footing.54,62
Our CoGEF results show significant differences for the four isomeric structures (Fig. 8): both syn-CPDs are predicted to open in a two-step fashion with similar values of 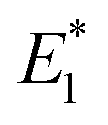 for the first C–C bond breaking (Fig. 8, upper panel). The cis–syn intermediate then requires less energy to break the second C–C bond, as the bond is already activated. However, the value of
for the first C–C bond breaking (Fig. 8, upper panel). The cis–syn intermediate then requires less energy to break the second C–C bond, as the bond is already activated. However, the value of  for the trans–syn intermediate is dubiously large. The reason for this is that the trans–syn intermediate twists, thereby reducing the force transduction to the second scissile bond. This large value of
for the trans–syn intermediate is dubiously large. The reason for this is that the trans–syn intermediate twists, thereby reducing the force transduction to the second scissile bond. This large value of  does not relate to a barrier that must be overcome for the second reaction step, but rather reflects the amount of energy introduced into the whole system during the stretching process. In the case of both anti-configured CPDs, one of the terminal CH3 groups breaks away at the end of the CoGEF simulations (Fig. 8, lower panel). This could, in principle, be regarded as a lack of mechanochemical activation, but the CoGEF method has been shown to fail to predict anti-configured cyclobutane mechanophores in the past.47
does not relate to a barrier that must be overcome for the second reaction step, but rather reflects the amount of energy introduced into the whole system during the stretching process. In the case of both anti-configured CPDs, one of the terminal CH3 groups breaks away at the end of the CoGEF simulations (Fig. 8, lower panel). This could, in principle, be regarded as a lack of mechanochemical activation, but the CoGEF method has been shown to fail to predict anti-configured cyclobutane mechanophores in the past.47
 | ||
| Fig. 8 CoGEF results for the four CPD mechanophore isomers investigated. For the syn-CPDs, the structures without displacement (I), just after the first C–C bond breaking (II), and after the second C–C bond breaking (III) are displayed. For the anti-CPDs, only the geometry after the dissociation of the CH3 group is displayed. The other black dots represent conformational changes induced by the mechanochemical stretching. | ||
To further elucidate the mechanochemical ring opening of the four isomeric CPDs, the transition state structures and reactant structures were localized with the FM-PES method (Fig. 9). It can be seen that the force transduction is stronger for the syn-CPDs compared to the anti-CPDs as the potential energy barriers decrease more strongly with increasing force for the syn-CPDs. We attribute this to the more direct force transduction to the first C–C bond of the syn-CPD, making them a good candidate for mechanochemical activation. For these syn-CPD motifs, the barrier of the second bond breaking is below 3.0 kcal mol−1 throughout, which will lead to an immediate, second C–C bond breaking once the barrier of the first C–C bond breaking is overcome. The difference in the  values computed with the CoGEF method for cis- and trans-isomers stems from twisting of the molecular structure, which leads to a larger Fmax, for the trans-isomer. The FM-PES computations indeed show existence of a minimum structure related to the twisted biradical intermediate up to 5.0 nN. However, with an associated barrier for the second C–C bond breaking of below 2.0 kcal mol−1 throughout, this biradical intermediate is not persistent, but will open to the products immediately once formed. In contrast, the diagonal configuration of the cyclobutane unit of the anti-CPDs leads to a smaller force transduction to the mechanophore. As a consequence, the barrier heights stay above 35 kcal mol−1 in the range of mechanochemical relevance (4 nN). Furthermore, only one single transition state structure involving breaking of both C–C bonds could be located for the anti-CPDs (Fig. 9, lower panel) and no biradical intermediate can be localized. We collected the most important parameters from CoGEF and FM-PES computations in Table 3. Altogether, both syn-CPDs can be regarded as functional mechanophores and, in agreement with the CoGEF computations, a lower mechanochemical reactivity can be expected for the anti-CPDs.
values computed with the CoGEF method for cis- and trans-isomers stems from twisting of the molecular structure, which leads to a larger Fmax, for the trans-isomer. The FM-PES computations indeed show existence of a minimum structure related to the twisted biradical intermediate up to 5.0 nN. However, with an associated barrier for the second C–C bond breaking of below 2.0 kcal mol−1 throughout, this biradical intermediate is not persistent, but will open to the products immediately once formed. In contrast, the diagonal configuration of the cyclobutane unit of the anti-CPDs leads to a smaller force transduction to the mechanophore. As a consequence, the barrier heights stay above 35 kcal mol−1 in the range of mechanochemical relevance (4 nN). Furthermore, only one single transition state structure involving breaking of both C–C bonds could be located for the anti-CPDs (Fig. 9, lower panel) and no biradical intermediate can be localized. We collected the most important parameters from CoGEF and FM-PES computations in Table 3. Altogether, both syn-CPDs can be regarded as functional mechanophores and, in agreement with the CoGEF computations, a lower mechanochemical reactivity can be expected for the anti-CPDs.
 | ||
| Fig. 9 Barrier heights of the first bond breaking of the four CPD mechanophore isomers investigated computed with the FM-PES method. The displayed geometries show the transition state structures with the corresponding C–C atom distances computed at a force of 2.0 nN. The potential activation energy of the syn-CPDs reaches 0 kcal mol−1, at Fmax = 2.9 nN (cis) and Fmax = 3.0 nN (trans), which is in line with CoGEF being able to detect mechanochemical activatability. For the anti-CPDs, there is only one transition state detectable along the total pathway from CPD to the diene products. This is reflected in the qualitatively different C–C distances of the transition state structures. The barrier heights for the second C–C bond breaking of the syn-CPDs can be found in the SI. | ||
 ,
,  (obtained from CoGEF computations), and the barriers at 2.0 nN are in kcal mol−1. The rupture forces Fmax were determined by the CoGEF method
(obtained from CoGEF computations), and the barriers at 2.0 nN are in kcal mol−1. The rupture forces Fmax were determined by the CoGEF method


3 Conclusions
In summary, we introduced CPDs as a novel class of mechanophores. Using complementary techniques including GPC, NMR, and computational simulations, we demonstrated that CPDs undergo cycloelimination under 20 kHz ultrasound irradiation, restoring the original thymidine structure in the sonication products. We quantified the rate constants of this mechanochemical scission across varying polymer molar masses and ultrasound intensities, further characterizing their force-responsive behavior. Ultrasound-induced bond scission of the different isomers suggested a reactivity difference between the cis–syn and the trans–anti isomers. Further, computational investigations using force application by CoGEF and FM-PES allowed inferring that syn-isomers are mechanochemically more active, while anti-isomers are less sensitive to force-induced cleavage. Together, these experimental and computational findings establish CPDs as nucleobase- and nucleoside-derived mechanophores, laying a foundation for their future application in biologically relevant systems and biohybrid materials.Author contributions
Conceptualization, X. G., G. C., J. M., R. G., and A. H.; methodology, X. G., J. M., R. G., and A. H.; investigation, X. G., F. J. U., M. L.-B., R. L., J. F., and K. S.; writing – original draft, X. G., G. C., F. J. U., M. L.-B., R. L., and J. M.; writing – review and editing, X. G., G. C., F. J. U., M. L.-B., R. L., J. M.; R. G., and A. H.; funding acquisition, J. M., A. H.; resources, J. M., A. H.; supervision, G. C., J. M., R. G., and A. H.Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6sc02165d.Acknowledgements
X. G. is grateful for support from the China Scholarship Council. Funding is acknowledged from the German Research Foundation for 503981124 (R. G.) and by the EU through an ERC Advanced Grant (SONOPHARMAGEN, No. 101142296, to A. H.). Moreover, this work was funded as part of the Leibniz ScienceCampus: ACTISONO, supported by the Leibniz Association (No. W89/2023, to A. H.). We acknowledge the support of computational infrastructure provided by the Centre for Information and Media Technology at Heinrich-Heine University Düsseldorf. The authors would like to thank Dr Michael Pohl and Rainer Haas for their help with preparative HPLC. J. M. is grateful for an allowance for material costs from the Fonds der Chemischen Industrie. M. L.-B. gratefully acknowledges support from a PhD scholarship from the German Scholarship Foundation. R. L. is grateful for support from the Jürgen Manchot Foundation.References
- R. T. O'Neill and R. Boulatov, Nat. Rev. Chem., 2021, 5, 148–167 CrossRef PubMed.
- Y. Chen, G. Mellot, D. van Luijk, C. Creton and R. P. Sijbesma, Chem. Soc. Rev., 2021, 50, 4100–4140 RSC.
- G. Cravotto, E. C. Gaudino and P. Cintas, Chem. Soc. Rev., 2013, 42, 7521–7534 RSC.
- R. T. O'Neill and R. Boulatov, Nat. Chem., 2023, 15, 1214–1223 CrossRef PubMed.
- M. E. McFadden, A. C. Overholts, S. K. Osler and M. J. Robb, ACS Macro Lett., 2023, 12, 440–445 CrossRef CAS PubMed.
- G. De Bo, Macromolecules, 2020, 53, 7615–7617 CrossRef CAS.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- P. Michael and W. H. Binder, Angew. Chem., Int. Ed., 2015, 54, 13918–13922 CrossRef CAS PubMed.
- D. Campagna and R. Göstl, Angew. Chem., Int. Ed., 2022, 61, e202207557 CrossRef CAS PubMed.
- A. L. B. Ramirez, Z. S. Kean, J. A. Orlicki, M. Champhekar, S. M. Elsakr, W. E. Krause and S. L. Craig, Nat. Chem., 2013, 5, 757–761 CrossRef CAS PubMed.
- H. Zhang, F. Gao, X. Cao, Y. Li, Y. Xu, W. Weng and R. Boulatov, Angew. Chem., Int. Ed., 2016, 55, 3040–3044 CrossRef CAS PubMed.
- C. E. Diesendruck, B. D. Steinberg, N. Sugai, M. N. Silberstein, N. R. Sottos, S. R. White, P. V. Braun and J. S. Moore, J. Am. Chem. Soc., 2012, 134, 12446–12449 CrossRef CAS PubMed.
- C. Nagamani, H. Liu and J. S. Moore, J. Am. Chem. Soc., 2016, 138, 2540–2543 CrossRef CAS PubMed.
- X. Hu, T. Zeng, C. C. Husic and M. J. Robb, J. Am. Chem. Soc., 2019, 141, 15018–15023 CrossRef CAS PubMed.
- Y. Lin, T. B. Kouznetsova and S. L. Craig, J. Am. Chem. Soc., 2020, 142, 99–103 CrossRef CAS PubMed.
- Z. Shi, J. Wu, Q. Song, R. Göstl and A. Herrmann, J. Am. Chem. Soc., 2020, 142, 14725–14732 CrossRef CAS PubMed.
- Z. Shi, Q. Song, R. Göstl and A. Herrmann, Chem. Sci., 2021, 12, 1668–1674 RSC.
- R. Küng, T. Pausch, D. Rasch, R. Göstl and B. M. Schmidt, Angew. Chem., Int. Ed., 2021, 60, 13626–13630 CrossRef PubMed.
- B. A. Versaw, T. Zeng, X. Hu and M. J. Robb, J. Am. Chem. Soc., 2021, 143, 21461–21473 CrossRef CAS PubMed.
- T. Zeng, L. A. Ordner, P. Liu and M. J. Robb, J. Am. Chem. Soc., 2024, 146, 95–100 CrossRef CAS PubMed.
- C. Löwe and C. Weder, Adv. Mater., 2002, 14, 1625–1629 CrossRef.
- C. R. Hickenboth, J. S. Moore, S. R. White, N. R. Sottos, J. Baudry and S. R. Wilson, Nature, 2007, 446, 423–427 CrossRef CAS PubMed.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- S. He, M. Stratigaki, S. P. Centeno, A. Dreuw and R. Göstl, Chem.–Eur. J., 2021, 27, 15889–15897 CrossRef CAS PubMed.
- B. V. Asya, S. Wang, E. Euchler, V. N. Khiêm and R. Göstl, Aggregate, 2025, 6, e70014 CrossRef CAS.
- S. Huo, P. Zhao, Z. Shi, M. Zou, X. Yang, E. Warszawik, M. Loznik, R. Göstl and A. Herrmann, Nat. Chem., 2021, 13, 131–139 CrossRef CAS PubMed.
- P. Zhao, S. Huo, J. Fan, J. Chen, F. Kiessling, A. J. Boersma, R. Göstl and A. Herrmann, Angew. Chem., Int. Ed., 2021, 60, 14707–14714 CrossRef CAS PubMed.
- Y. Zhou, S. Huo, M. Loznik, R. Göstl, A. J. Boersma and A. Herrmann, Angew. Chem., Int. Ed., 2021, 60, 1493–1497 CrossRef CAS PubMed.
- W. H. Rath, R. Göstl and A. Herrmann, Adv. Sci., 2024, 11, 2306236 CrossRef CAS PubMed.
- Y. Zhou, S. P. Centeno, K. Zhang, L. Zheng, R. Göstl and A. Herrmann, Adv. Mater., 2023, 35, 2210052 CrossRef CAS PubMed.
- S. Huo, Z. Liao, P. Zhao, Y. Zhou, R. Göstl and A. Herrmann, Adv. Sci., 2022, 9, 2104696 CrossRef CAS PubMed.
- A. Ishaqat, J. Hahmann, C. Lin, X. Zhang, C. He, W. H. Rath, P. Habib, S. E. M. Sahnoun, K. Rahimi, R. Vinokur, F. M. Mottaghy, R. Göstl, M. Bartneck and A. Herrmann, Adv. Mater., 2024, 36, 2403752 CrossRef CAS PubMed.
- T. Lindahl and R. D. Wood, Science, 1999, 286, 1897–1905 CrossRef CAS PubMed.
- J. L. Ravanat, T. Douki and J. Cadet, in Comprehensive Series in Photosciences, Elsevier, 2001, vol. 3, pp. 207–230 Search PubMed.
- R. P. Sinha and D. P. Häder, Photochem. Photobiol. Sci., 2002, 1, 225–236 CrossRef CAS PubMed.
- A. Mees, T. Klar, P. Gnau, U. Hennecke, A. P. Eker, T. Carell and L. O. Essen, Science, 2004, 306, 1789–1793 CrossRef CAS PubMed.
- D. B. Bucher, C. L. Kufner, A. Schlueter, T. Carell and W. Zinth, J. Am. Chem. Soc., 2016, 138, 186–190 CrossRef CAS PubMed.
- M. J. Kryger, A. M. Munaretto and J. S. Moore, J. Am. Chem. Soc., 2011, 133, 18992–18998 CrossRef CAS PubMed.
- Z. S. Kean, Z. Niu, G. B. Hewage, A. L. Rheingold and S. L. Craig, J. Am. Chem. Soc., 2013, 135, 13598–13604 CrossRef CAS PubMed.
- H. Zhang, X. Li, Y. Lin, F. Gao, Z. Tang, P. Su, W. Zhang, Y. Xu, W. Weng and R. Boulatov, Nat. Commun., 2017, 8, 1147 CrossRef PubMed.
- T. Suzuki, H. Ota, Y. Namba and T. Fujino, Chem. Pharm. Bull., 2019, 67, 130–134 CrossRef CAS PubMed.
- K. Matyjaszewski and J. Xia, Chem. Rev., 2001, 101, 2921–2990 CrossRef CAS PubMed.
- K. Matyjaszewski, Macromolecules, 2012, 45, 4015–4039 CrossRef CAS.
- J. Fan, R. Lennarz and K. Zhang, et al. , Nat. Commun., 2025, 16, 5380 CrossRef CAS PubMed.
- J. Fan, K. Zhang and M. Xuan, et al. , CCS Chem., 2024, 6, 1895–1907 CrossRef CAS.
- M. K. Beyer, J. Chem. Phys., 2000, 112, 7307–7312 CrossRef CAS.
- I. M. Klein, C. C. Husic, D. P. Kovács, N. J. Choquette and M. J. Robb, J. Am. Chem. Soc., 2020, 142, 16364–16381 CrossRef CAS PubMed.
- D. Roca-Sanjuán, G. Olaso-González and I. González-Ramírez, et al. , J. Am. Chem. Soc., 2008, 130, 10768–10779 CrossRef PubMed.
- P. H. Clingen, C. F. Arieti, L. Roza, T. Mori, O. Nikaido and M. H. L. Green, Cancer Res., 1995, 55, 2245–2248 CAS.
- Y.-H. You, P. E. Szabó and G. P. Pfeifer, Carcinogenesis, 2000, 21, 2113–2117 CrossRef CAS PubMed.
- J. Kim and D. Patel, Photochem. Photobiol., 1995, 62, 44–50 CrossRef CAS PubMed.
- S. L. Malhotra, J. Macromol. Sci., Part A:Pure Appl. Chem., 1982, 18, 1055–1085 CrossRef.
- S. L. Malhotra, J. Macromol. Sci., Part A:Pure Appl. Chem., 1986, 23, 729–748 CrossRef.
- M. T. Ong, J. Leiding, H. Tao, A. M. Virshup and T. J. Martínez, J. Am. Chem. Soc., 2009, 131, 6377–6379 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1992, 96, 2155–2160 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS PubMed.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- S. Aydonat, A. H. Hergesell, C. L. Seitzinger, R. Lennarz, G. Chang, C. Sievers, J. Meisner, I. Vollmer and R. Göstl, Polym. J., 2024, 56, 249–268 CrossRef CAS.
- T. Stauch and A. Dreuw, Chem. Rev., 2016, 116, 14137–14180 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |