
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe characterisation and reactivity of a RhIII η1-σ-alkane complex and the role of a structurally responsive phosphine ligand in solid-state molecular organometallic chemistry
Joe C. Goodall *a,
M. Arif Sajjadb,
Samantha K. Furfaria,
Kristof M. Altusa,
Theo S. C. Hatcherb,
Graham J. Tizzardc,
Samuel J. Paged,
Mark R. Warrene,
Jason M. Lynam*a,
Stuart A. Macgregor*b and
Andrew S. Weller*a
*a,
M. Arif Sajjadb,
Samantha K. Furfaria,
Kristof M. Altusa,
Theo S. C. Hatcherb,
Graham J. Tizzardc,
Samuel J. Paged,
Mark R. Warrene,
Jason M. Lynam*a,
Stuart A. Macgregor*b and
Andrew S. Weller*a
aDepartment of Chemistry, University of York, York, YO10 5DD, UK
bEaStCHEM School of Chemistry, University of St Andrews, North Haugh, St Andrews, KY16 9ST, UK
cUK National Crystallography Service, School of Chemistry and Chemical Engineering, University of Southampton, Southampton, SO17 1BJ, UK
dDepartment of Chemistry, University of Durham, Durham, DH1 3LE, UK
eDiamond Light Source Ltd, Didcot OX110DE, UK
First published on 24th April 2026
Abstract
A combined experimental and computational study on the single-crystal to single-crystal addition of H2 to [Rh(κ2-dtbpb)(NBD)][BArF4] [dtbpb = tBu2P(CH2)4PtBu2, NBD = norbornadiene, ArF = 3,5-C6H3(CF3)2] to give the Rh(III) η1-σ-alkane complex [Rh(dtbpb’)(H)(η1-C7H12)][BArF4] (dtbpb’ = κ3-PCP-tBu2PCH2CHCH2CH2PtBu2) is reported, in which the supporting phosphine ligand has also undergone C–H activation of one of the methylene groups in the chelate backbone to form a trans-spanning κ3-pincer-type ligand. Characterisation by variable temperature single-crystal X-ray diffraction, solid-state NMR spectroscopy, periodic DFT, QTAIM, NBO and IGMH calculations support this assignment, and also that the norbornane (NBA) alkane ligand can access low energy conformational isomers in the solid-state. Dissolving in CD2Cl2 displaces the alkane and the corresponding solvent adduct is formed, [Rh(dtbpb’)(H)(κ1-ClCD2Cl)][BArF4]. DFT calculations, supported by experiment, indicate the C–H activation of the backbone occurs after full hydrogenation of the NBD to NBA, and not at a norbornene intermediate. Addition of propene to the crystalline σ-alkane complex displaces the NBA, and the phosphine ligand responds by reforming the κ2-motif, consistent with C–H activation being thermodyamically favoured when trans to a weakly coordinating σ-alkane. These reversible bond activations of the chelating ligand in response to changes in the co-ligands demonstrate that significant structural reorganisation is possible in the crystalline environment.
Introduction
The non-innocent role ligands can play in organometallic reaction mechanisms has become an important design principle that is used for the delivery of efficient, lower energy, and selective homogeneous catalytic processes. Central to such active ligand participation are the concepts of metal–ligand cooperativity (MLC) and structurally responsive ligands (SRL). MLC1–3 involves the ligand taking a role in bond activation processes, that often involve the shuttling of protons/hydrides in a catalytic manifold between the metal centre and ligand, e.g. in (de)hydrogenation reactions,4,5 Scheme 1a. SRLs show dynamic structural behavior;6–9 such as hemilability – in which a ligand can adapt to changes at the metal centre by offering a variety of accessible coordination modes, e.g. Scheme 1b.10 Large bite-angle κ2-chelating diphosphines, e.g. R2P(CH2)5PR2, have been shown to capture aspects of both MLC and SRL reactivity, undergoing (reversible) backbone C–H activation at the metal centre to form κ3-PCP-type carbometallated pincer ligands,11–13 that have the potential to facilitate bond activation processes such as H/D exchange, Scheme 1c.14–16 | ||
| Scheme 1 (a) Schematic metal–ligand co-operativity (R = alkyl, aryl).2–4 (b) Example of a hemilabile ligand.10 (c) Suggested mechanism for H/D exchange at a 14e intermediate using large bite-angle phosphines.15 | ||
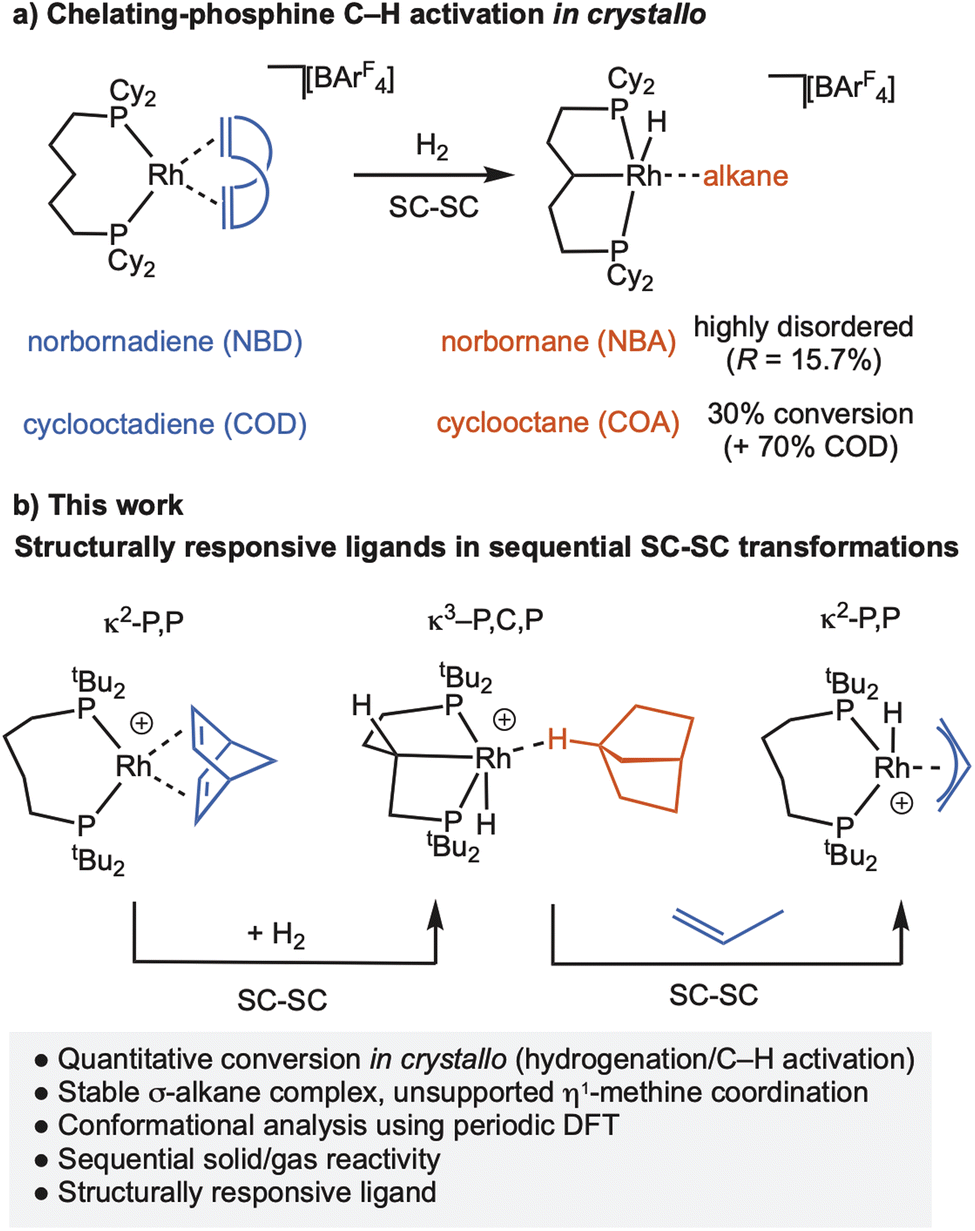
We have been developing the concept of solid-state molecular organometallic chemistry (SMOM17), where solid/gas reactivity in crystallo18,19 allows for the isolation and detailed characterisation of σ-alkane complexes by single-crystal to single-crystal (SC–SC) transformations.20,21 While σ-alkane complexes are generally unstable in solution, being characterised in situ at low temperature using photolysis22,23 or protonation24,25 routes; by optimal design of metal complex, solvent and reaction conditions, σ-alkane complexes can be recrystallised from solution.26–30 Using this SMOM approach we have also shown that a wide variety of fundamental organometallic mechanistic steps are supported in crystalline molecular reactivity. Examples include: (reversible) C–H activation of a bound alkane ligand,31–33 migratory insertion,34,35 ligand substitution,36–38 C–N bond formation,39 and isotope effects.32,33,40 We have also reported that [Rh(Cy2P(CH2)5PCy2)(alkene)][BArF4] (alkene = norbornandiene, NBD, or 1,5-cycloctadiene, COD; ArF = 3,5-(CF3)2C6H3), can undergo backbone C–H activation on addition of H2 to give a carbometallated κ3-PCP ligand complex with a σ-alkane ligand, Scheme 2a.41 However, the norbornane (NBA) is highly disordered precluding detailed characterisation, while the cyclooctane (COA) σ-alkane complex is only generated in ∼30% maximum conversion from the COD precursor in crystallo before onward decomposition occurs.
 | ||
| Scheme 2 (a) Prior SMOM work showing C–H activation of a P,P-ligand. (b) This work. [BArF4]− anions are not shown. | ||
In this contribution we show that by using the wide bite-angle, κ2-PP phosphine ligand, tBu2P(CH2)4PtBu2, the quantitative conversion to a stable rhodium η1-σ-alkane complex of NBA is achieved on hydrogenation of the NBD precursor. A rare example of a carbometallated κ3-PCP–ligand with a C4-backbone is concurrently formed through C–H activation of the phosphine backbone, Scheme 2b. We show that this is a structurally responsive ligand in crystallo, as addition of propene reforms the κ2-PP ligand; and provide evidence for the likely order of events in the mechanism of C–H activation. These reversible bond activations of the chelating ligand in response to changes in the co-ligands (NBD/NBA/propene) demonstrate that the molecular single-crystal environment can support significant structural reorganisation that is a characteristic of MLC and SRLs, so well-documented in the solution phase.
Results and discussion
Synthesis and characterisation of precursor [Rh(dtbpb)(NBD)][BArF4], 1[BArF4]
The precursor complex, [Rh(dtbpb)(NBD)][BArF4] 1[BArF4] [dtbpb = tBu2P(CH2)4PtBu2] was prepared by addition of dtbpb to [Rh(NBD)2][BArF4] in CH2Cl2 solution. Recrystallisation from CH2Cl2/hexane produced analytically pure deep-red crystals of 1[BArF4] in 80% yield (SI). The solid-state structure of 1[BArF4], as determined by single-crystal X-ray diffraction (P21/n, R1 = 3.15%), reveals a pseudo square-planar cation, Fig. 1a. The cation is located within an approximate octahedral cage of [BArF4]− anions (Fig. 1b and c) as we have noted for other [Rh(L2)(NBD)][BArF4] SMOM systems that undergo solid/gas reactivity.20,31,32,34,41 Some NBD alkene protons show close non-covalent42 C–H⋯F contacts with the CF3 groups of proximate anions, e.g. H1⋯F22 2.6148(15) Å; H2⋯F21 2.7007(15); H3⋯F13, 3.0951(17) Å. There is no crystallographically imposed symmetry in the cation, and this is reflected in the 31P{1H} SSNMR spectrum in which two environments are observed [δ 42.8 and 25.0; J(RhP) = 148 Hz], similar to those reported for [Rh(tBu2P(CH2)3PtBu2)(NBD)][BArF4].43 In the 13C{1H} SSNMR spectrum signals between δ 77.1 and 48.2 are assigned to the alkene groups of the NBD ligand. C2v symmetry is observed in the solution NMR spectra (SI). | ||
| Fig. 1 Solid-state structure of 1[BArF4] (a) structure of the cation, 1+. Displacement ellipsoids are shown at the 50% probability level, selected H-atoms shown, tBu groups shown in stick form. Selected bond lengths (Å) and angles (°): Rh–P1, 2.4412(5); Rh–P2, 2.4252(5); C1–C2, 1.390(3); C3–C4, 1.393(3); Rh⋯C18, 3.8103(19); P1–Rh–P2, 102.110(17). (b) Extended asymmetric unit showing the relationship between two proximal [BArF4]− anions and the metal centre. F⋯H contacts less than 3.1 Å shown (range 3.095–2.614 Å). (c) Arrangement of [BArF4]− anions around the 1+ cation. Cation atoms and anion surfaces shown at van der Waals radii. | ||
Synthesis of [Rh(dtbpb’)(H)(NBA)][BArF4], 2[BArF4]: ligand non-innocence
Addition of H2 (2 bar, 2 h) to single-crystalline 1[BArF4] results in a SC–SC transformation and a colour change from deep red to sandy yellow, Scheme 3, signalling the formation of form a new σ-alkane complex in which the supporting phosphine ligand has also undergone C–H activation (carbometallation) of one of the methylene groups in the chelate backbone to give [Rh(dtbpb’)(H)(η1-C7H12)][BArF4] 2[BArF4] (dtbpb’ = κ3-PCP-tBu2PCH2CHCH2CH2PtBu2). | ||
| Scheme 3 Synthesis of complex 2[BArF4]. Inset shows optical microscopy of the crystals pre- and post-addition of H2. | ||
On addition of H2, the NBD ligand in 1[BArF4] is hydrogenated to norbornane which remains coordinated to the Rh centre through a bridgehead methine C–H group, forming an η1-σ-(C–H)-NBA complex,25 Rh1⋯C5 = 3.009(5) Å. While in the initial structural refinements the NBA ligand was unrestrained, its associated displacement ellipsoids were large and some C–C bond lengths were long (∼1.7 Å), indicating unresolved disorder. These separate disordered components could not be satisfactorily modelled. For this reason, the C–C distances in the NBA were lightly restrained to lie within a range of 1.385–1.656 Å. Nevertheless, the conversion from NBD to NBA on hydrogenation is unambiguous based on the change in coordination mode from an η2η2-alkene ligand in 1[BArF4] to a η1-σ-alkane ligand in 2[BArF4]. While a η1-methine Rh⋯H–C interaction has been noted in a structurally characterised σ-alkane methylpentane44 complex, the Rh⋯H–C interaction in that case is also supported by being part of a chelating alkane ligand. An unsupported η1-Rh⋯H–C interaction has been observed in a cyclooctane complex (Scheme 2a), but here it arises from a methylene group.41 The unsupported η1-binding methine mode of the alkane in 2[BArF4] is new for a structurally characterised alkane complex, and thus adds to the rich variety observed for alkane σ-complex interactions in the solid-state42,45 (methyl, methylene, methine;26,27,44 η2η2 (ref. 20 and 40) through to η1 (ref. 41 and 44)), mirroring those measured in solution using low temperature in situ NMR methods.22,25,46
Hydrogenation of NBD is accompanied by one methylene group (C18) of the chelating phosphine undergoing C–H activation to form an unsymmetrical, trans spanning κ3-PCP pincer ligand: Rh–C18, 2.078(11) Å, P1–Rh–P2 = 154.86(4)°. The alkane ligand sits trans to this new bond [C18–Rh–C5 = 164.5(5)°]. This Rh–C bond in 2[BArF4] is disordered over two positions, with C18/C18A sitting above or below the RhP2 plane, modelled as a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :40 split, Fig. 3. The corresponding Rh–hydride and C18–H were not located. Their presence was confirmed by solution studies of the corresponding CH2Cl2 adduct, 3[BArF4], that shows the hydride sitting anti disposed relative to the C–H bond, vide infra. There are no close interactions (Rh⋯C < 3.5 Å) between the tBu groups and the vacant site trans to the hydride.
:40 split, Fig. 3. The corresponding Rh–hydride and C18–H were not located. Their presence was confirmed by solution studies of the corresponding CH2Cl2 adduct, 3[BArF4], that shows the hydride sitting anti disposed relative to the C–H bond, vide infra. There are no close interactions (Rh⋯C < 3.5 Å) between the tBu groups and the vacant site trans to the hydride.
C–H activation of a C5 phosphine backbone is relatively common in solution-based organometallics,11–13 but only one example involving a C4 phosphine is known,47 although related complexes have been reported.48 Intramolecular ligand activation in SC–SC in crystallo transformations have been reported, for example C–H32,33,49,50 and C–C activation.51,52
While the structural change on alkene hydrogenation and C–H activation of the phosphine is significant, the overall arrangement of [BArF4]− anions remain unchanged from 1[BArF4], with the similar C–H⋯F hydrogen bonding observed from the proximal anions, Fig. 2b. The structural plasticity of the [BArF4]− anions, likely facilitated by the –CF3 groups,37,53–55 allows for the arrangement of anions to breathe to accommodate the changes at the cation, with cross-cage B⋯B distances increasing slightly, alongside a 5% change in unit cell volume, Fig. 4.
 | ||
| Fig. 2 Solid-state structure of 2[BArF4] (a) Structure of the cation, 2+. Only one disordered component (C18/C18A disorder) shown. See main text and SI. Displacement ellipsoids are shown at the 30% probability level, selected H-atoms shown, tBu groups shown in stick form. The Rh–hydride was not located and is not shown. Selected bond lengths (Å) and angles (°): Rh–P1, 2.2968(9); Rh–P2, 2.3284(11); C1–C2, 1.516(7); C3–C4, 1.502(10)a; Rh1⋯C5, 3.009(5); Rh1–C18, 2.078(11); P1–Rh–P2, 154.86(4); C18–Rh–C5, 164.5(5) (b) extended asymmetric unit showing the relationship between two proximal [BArF4]− anions and the metal centre. F⋯H contacts less than 3.1 Å shown (range 2.880–2.696 Å). (c) Comparison of displacement ellipsoids (30% probability) of the structure of 2[BArF4] collected at 110 K and 298 K. Only Rh, P atoms and NBA group shown. Selected distances and angles (see (a)) Rh–C18: 110 K, 2.076(11); 298 K, 2.111(15) Å; 110 K, P1–Rh–P2, 154.86(4); 298 K, 155.27(9)°. [a NBA C–C distances lightly restrained to within a range of 1.385–1.656 Å. See SI.]. | ||
 | ||
| Fig. 3 Disorder model for the C4-backbone of 2[BArF4] at C18/C18A. | ||
 | ||
| Fig. 4 Comparison of anion cage metrics between 1[BArF4], and 2[BArF4]. Front [BArF4]− anion removed for clarity. Van Der Waals radii. Distances shown are B⋯B cross-cage distances. | ||
The formation of 2[BArF4] is supported by the analysis of bulk materials (∼50 mg sample size) using 31P{1H} and 13C{1H} SSNMR spectroscopy at 298 K, Fig. 5 (see SI for the 13C{1H} SSNMR spectrum). On hydrogenation the chemical shifts of the two 31P environments become more disperse, δ 100.6 and δ −8.7, consistent with the formation of 5 and 4 membered cyclometallated rings.47 The lower field signal at δ 100.6 is observed as a doublet of doublets [J(PP) = 280 Hz, J(RhP) = 115 Hz], while the higher field signal is observed as an apparent triplet [J(PP) ∼300 Hz] that is significantly broader [fwhm = 800 Hz]. These data are consistent with the generation of a Rh(III) centre with trans-spanning phosphines.56 The apparent triplet is interpreted as being formed of two overlapped doublets, in which the 103Rh–31P coupling is not resolved (i.e. it is small), assigned to each C18/C18A disordered component in 2[BArF4]. These are suggested to be coincident for the two components in the lower field signal. The 13C{1H} SSNMR spectrum shows an absence of signals in the alkene region (δ 110–50). When dissolved in CD2Cl2 solution only one set of signals are observed (vide infra).
 | ||
| Fig. 5 31P{1H} SSNMR spectra (162 MHz, 10 kHz spin rate, room temperature) of (a) 1[BArF4], and (b) 2[BArF4]. * Spinning-side bands. ‡ Unidentified product. Light blue highlighted areas indicate the isotropic chemical shifts, insets show these in more detail. | ||
The disorder of the bound NBA alkane ligand observed in 2[BArF4] was probed using variable temperature single-crystal X-ray diffraction studies (215 K and 298 K). These higher temperatures increase the size of the displacement ellipsoids, Fig. 2c, and the errors on the unrestrained C–C bond lengths and angles in the NBA, while C18/C18A of the phosphine backbone become undistinguishable. These data suggest there is a dynamic process occurring at the coordinated alkane, that results in a superposition of a variety of different conformers for the alkane ligand, that becomes more apparent on warming.
To probe this further, periodic-DFT calculations were employed in conjunction with conformational searching with CREST57 to model different η1-alkane binding modes in 2[BArF4] (SI). These confirmed the experimental structure bound through a bridgehead C–H bond (i.e. C5) to be lowest in energy while also locating alternative η1-isomers (Fig. 6a) bound through an endo-C–H bond (at +1.8 kcal mol−1), an exo-C–H bond (at +2.8 kcal mol−1), and a methylene bridge C–H bond (at +2.1 kcal mol−1). Additional rotamers of the experimental structure were also located at +0.4 and +6.7 kcal mol−1. Thus, a series of low energy alternative structures are readily accessible that, assuming low barriers for interconversion, are consistent with dynamic disorder of the NBA ligand at the Rh centre, as signalled by the large displacement ellipsoids evident in the experimentally-determined 298 K structure (Fig. 2c).
 | ||
| Fig. 6 (a) Relative energies of Rh⋯H–C interactions with the NBA ligand in 2[BArF4] (b) detail of the QTAIM molecular graph of the cation of 2[BArF4]. (c) IGMH plot for the 2+ cation (sign(λ2)ρ-coloured isosurfaces with δGinter = 0.001 au) (d) key donor–acceptor interactions for the Rh-NBA interaction as quantified via 2nd order perturbation NBO analyses. Back-donation from σRh–Hy and σRh–P2 contribute 2.4 kcal mol−1 and 1.7 kcal mol−1 respectively (see SI for orbital plots). | ||
The nature of the Rh⋯NBA interaction was assessed via QTAIM, NBO and Independent Gradient Model (Hirshfeld partitioning, IGMH) calculations. These used the experimental structure of 2[BArF4] with the H and F positions refined through a periodic-DFT optimisation. With this model Rh⋯H5 and H5–C5 distances of 1.99 Å and 1.12 Å are computed and a Rh⋯H5–C5 angle of 150°. The QTAIM molecular graph (Fig. 6b) exhibits a Rh⋯H5–C5 bond path with a bond critical point (BCP) electron density, ρ(r), of 0.042 au, consistent with previous examples of η1-C–H⋯Rh binding while somewhat lower than η2-C–H⋯Rh interactions.44 The C5–H5 BCP (ρ(r) = 0.252 au) indicates a weakening due to the σ-interaction with Rh; in comparison ρ(r) = 0.277 au at the BCP of the unperturbed C6–H6 bridgehead bond.
The IGMH plot (Fig. 6c) highlights the H5⋯Rh interaction as the strongest atom–atom interaction between the NBA and Rh fragments, with the localised blue disk characteristic of a η1-C–H⋯Rh interaction. Green isosurfaces between the NBA ligand and tBu substituents indicate additional van der Waals interactions29 aligning with weak C–HNBA⋯H–CtBu bond paths in the QTAIM study (ρ(r) = 0.003–0.009 au; omitted from Fig. 6b for clarity; see Fig. S29). A 2nd order perturbation analysis within the NBO framework (Fig. 6d) indicates σC5–H5 → σ*Rh–C18 donation is the strongest individual interaction at 12.3 kcal mol−1. σRh–C18 → σ*C5–H5 contributes most to back-donation (6.6 kcal mol−1), and once contributions from the σRh–hydride and σRh–P2 orbitals are included the total back-donation is similar in magnitude (Σ = 10.7 kcal mol−1) to σC5–H5 → Rh σ-donation. This is again consistent with previous η1-C–H⋯Rh motifs and contrasts with η2-C–H⋯Rh interactions where C–H → Rh σ-donation usually dominates.41,44
Synthesis of [Rh(dtbpb’)(H)(κ1-ClCD2Cl)][BArF4], 3[BArF4]
Vacuum transfer of CD2Cl2 onto single crystals of 2[BArF4], resulted in the quantitative formation of a complex in which NBA is no longer coordinated to the metal centre. Recrystallisation from CD2Cl2/heptane at 243 K afforded single crystals suitable for single-crystal X-ray diffraction, that allowed for the identification of the complex as [Rh(dtbpb’)(H)(κ1-ClCD2Cl)][BArF4], 3[BArF4], Fig. 7. The resulting solid-state structure is described first, followed by solution NMR spectroscopic data. | ||
| Fig. 7 (a) Synthesis of 3[BArF4] (b) solid-state structure of one of the crystallographically-independent cations of 3+. Displacement ellipsoids are shown that the 50% probability level, selected H-atoms shown, tBu groups shown in stick form. Selected bond distance (Å) and angles (°): Rh–P1, 2.3032(9); Rh–P2, 2.3366(8); Rh–C18, 2.044(4); Rh–Cl1, 2.5692(10); P1–Rh–P2, 154.50(4); C18–Rh–Cl1, 178.26(13). | ||
Complex 3[BArF4] crystallised with two crystallographically independent, but structurally very similar, cations in the unit cell. One of these was well defined, Fig. 7b, and the other refined best using a three-part disorder model for bound CD2Cl2. Only the bond metrics for the ordered component are discussed. These show a κ1-bound CD2Cl2 [Rh–Cl1, 2.5692(10) Å], that sits trans to the carbometallated phosphine backbone [Rh–C18, 2.044(4) Å]. The phosphine ligand remains trans spanning [P1–Rh–P2 = 154.50(4)°]. The Rh–H and C18–H protons were located in the difference map, and sit anti to one another. The structure of 3[BArF4] is similar to that reported for the analogue with the Cy2P(CH2)5PCy2 ligand;41 while CH2Cl2 (ref. 58–60) and related61 adducts of Rh are known more generally. The rather long Rh–Cl distance measured is consistent with its orientation trans to a strongly σ-donating alkyl ligand.60 The formation of solvent adducts, such as 3[BArF4], by displacement of weakly bound alkane ligands at low temperature is well-established.24,62 While the anti-arrangement of Rh–H and C18–H does not unambiguously define that the same orientation is present in 2[BArF4] it does offer strong support for such an orientation being present in this alkane complex.
Complex 3[BArF4] decomposes to a mixture of products at room temperature in CD2Cl2, and so NMR spectroscopic data were collected at 193 K. In the solution 31P{1H} NMR spectrum two environments are observed, at δ 98.8 [J(PP), 289 Hz; J(RhP) = 127 Hz] and at δ −16.5 [J(PP) = 289 Hz; J(RhP) = 90 Hz]. These data are similar to the SSNMR data for 2[BArF4], suggesting a closely related structure for the phosphine ligand. In the solution 1H NMR spectrum a hydride signal is observed at δ −28.43 as a doublet of doublets of doublets, that collapses to a doublet in the 1H{31P} NMR spectrum [J(PH) = 7, 12 Hz; J(RhH) 64 Hz]. The methine proton on C18 is identified at δ 1.67 by an unusually large coupling to 31P [J(PH) = 49 Hz, 1 H], that collapses in the 1H{31P} NMR spectrum. Vacuum transfer of the volatiles, dissolving in CD2Cl2 and interrogation by 1H NMR spectroscopy shows the formation of NBA, further supporting the assignment of 2[BArF4].
Identification of a probable intermediate: [Rh(dtbpb)(NBE)][BArF4], 4[BArF4]
Given the significant structural reorganisation that accompanies the in crystallo formation of the NBA complex, 2[BArF4], the order of events leading to this complex was of interest. In particular, when does C–H activation of the phosphine backbone occur? As the NBD ligand requires to be hydrogenated twice to form NBA, presumably via a norbornene (NBE) intermediate, the independent synthesis of this complex was attempted to see if C–H activation had occurred, Fig. 8a. | ||
| Fig. 8 (a) Synthesis of 4[BArF4] (b) solid-state structure of one of the crystallographically-independent cations of 4+. Displacement ellipsoids are shown that the 30% probability level, selected H-atoms shown, tBu groups shown in stick form. Selected bond distance (Å) and angles (°): Rh1–P1, 2.3222(12); Rh1–P2, 2.3678(11); Rh1–C1, 2.262(6); Rh1–C2, 2.225(9); Rh1⋯C7 2.366(9); P1–Rh1–P2, 102.03(5); P1–Rh1–C7, 155.29(16); P2–Rh1–mid(C1/C2) 154.02(4). | ||
[Rh(dtbpb)(NBE)][BArF4], 4[BArF4], was isolated as purple crystalline material, by the solution hydrogenation of excess NBD where H2 is the limiting reagent using 1[BArF4] as a catalyst, followed by low temperature recrystallisation (−30 °C). The molecular structure of 4[BArF4] (Fig. 8b) shows an NBE ligand coordinated to a Rh(I) centre by the exo-face of the alkene and an agostic Rh⋯H–C interaction, Rh⋯C7 = 2.366(9) Å. The phosphine backbone is not C–H activated, retaining the cis-κ2-P,P motif of the starting material. As the isolated yield of 4[BArF4] was poor (15%), characterisation by SSNMR or onward SC–SC reactivity was not attempted.63 Complex 4[BArF4] is unstable at room temperature in CD2Cl2, but low temperature solution NMR data for 4[BArF4] are fully consistent with both the solid-state structure, and previously-reported NBE complexes.31,64 Notably the agostic Rh⋯H–C group is observed at δ −5.67 as a broad signal in the 1H NMR spectrum; while in the 31P{1H} NMR spectrum two environments are observed at δ 90.3 [J(RhP) 220 Hz; J(PP) 19 Hz] and δ 30.0 [J(RhP) 142 Hz; J(PP) 19 Hz]. The arrangement of [BArF4]− anions around the cation is very similar to that observed for 1[BArF4], 2[BArF4] and 3[BArF4], with the unit cell volume only 1.6% larger than in the starting complex, 1[BArF4].
To probe the sequence of events that transform 1[BArF4] into 2[BArF4], C–H activation of the phosphine backbone was modelled via DFT calculations on the isolated cis-κ2-P,P cations [Rh(dtbpb)(L2)]+, where L2 = NBD (1+), NBE (4+) and NBA (2b+, a putative η2η2-NBA-bound precursor to C–H activation). The computed thermodynamics indicate backbone C–H activation is significantly endergonic at 1+ (ΔG = +15.3 kcal mol−1), becomes more accessible at 4+ (ΔG = +0.9 kcal mol−1), and is exergonic at 2b+ (ΔG = −6.8 kcal mol−1). The competition between backbone C–H activation and NBE hydrogenation was therefore computed at 4+ and showed the latter to proceed with a barrier of 14.8 kcal mol−1 to give 2b+ at −13.1 kcal mol−1. In comparison backbone activation in 4+ is kinetically less accessible (ΔG‡ = 18.9 kcal mol−1, see Fig. S49). This implies that 4[BArF4] is unlikely to have a significant lifetime in the presence of available H2 and will instead react to form 2[BArF4] via NBE hydrogenation, followed by backbone C–H activation. In the absence of H2, backbone C–H activation in 4+ is a kinetically accessible, if slightly endergonic (and hence reversible) process.
The computed reaction profile for backbone C–H activation at 2b+ (Fig. 9a) involves opening the P2–Rh–P3 angle, coupled with rearrangement of the C4-backbone to form agostic intermediate Int(2b+−2+) at −1.7 kcal mol−1. C–H activation then proceeds via TS(2b+−2+) at +12.2 kcal mol−1 to form the anti-isomer of 2+ at −19.9 kcal mol−1. Both Int(2b+−2+) and 2+ feature the NBA ligand bound primarily through an endo C–H bond. With this isolated cation model the bridgehead C–H-bound structure observed in the crystal structure is 1.6 kcal mol−1 higher in energy. C–H activation therefore has an overall barrier of +26.3 kcal mol−1 with the rate-limiting transition state, TS(2b+−2+)1, corresponding to P–Rh–P angle opening and backbone rearrangement (Fig. S49).
 | ||
| Fig. 9 (a) Computed reaction profile (free energies, kcal mol−1 relative to 4+ + H2) for backbone C–H activation in 2b+. Level of theory: PBE-D3(def2-tzvp)//PBE-D3(SDD(Rh, P, with d-orbital polarization on P); 6–31 g ** other atoms). (b) Computed structure of TS(2b+−2+)1 (selected distances in Å; tBu groups in wireframe). | ||
Scheme 4a shows the overall proposed order of events, which also support the work of Goldman and Krogh-Jespersen on oxidative addition of C–H bonds in d8-MXL2-type complexes.65 Their studies showed that stronger σ-donating ligands trans to the site of C–H activation disfavour oxidative addition. In the system here, C–H activation of the ligand backbone is thermodynamically preferred for σ-alkane intermediate 2b[BArF4], rather than the alkene complex 4[BArF4].
 | ||
| Scheme 4 (a) Proposed order of events in the in crystallo formation of 2[BArF4] from H2 addition to 1[BArF4]. (b) Formation of exo-endo- d4-NBA from addition of D2 to 1[BArF4]. | ||
Supporting evidence for the intermediate role of 4[BArF4] comes from addition of D2 to crystalline 1[BArF4] to ultimately form d4-2[BArF4], the heavy atoms of which were confirmed by SSNMR and single-crystal X-ray diffraction, Scheme 4b. Dissolving the resulting crystals in CD2Cl2, vacuum transfer of the volatiles, and analysis by 2H NMR spectroscopy and GC–MS shows the exclusive formation of exo–endo-d4-NBA.66,67 This is consistent with initial addition of D2 to the endo-face of NBD in 1[BArF4], a rearrangement to form 4[BArF4], and then addition of the second equivalent of D2 to the exo-face of NBE. We have previously noted both selective exo–endo21 and endo–endo31 NBD hydrogenation in crystallo, and suggest these differences come from variations in the primary and secondary microenvironment that influence the barrier to rearrangement of NBE.68 Addition of D2 to single crystals of 2[BArF4] resulted in no H/D exchange at the NBA (by GC/MS and 1H NMR spectroscopy), in contrast with [Rh(Cy2P(CH2)2PCy2)(η2η2-NBA)][BArF4] where selective exchange with the exo positions is observed.31,69
A structurally responsive ligand and reversible C–H activation: addition of propene to [Rh(dtbpb)(NBA)][BArF4]
The calculated differences in the thermodynamics of C–H activation of the phosphine backbone in 1+, 2b+ and 4+ suggested that modification of the ligand trans to the position of C–H activation might promote reductive elimination. We thus targeted the synthesis of complexes where the alkane ligand was replaced, in an in crystallo substitution. While addition of a variety of gases to single crystals of 2[BArF4] (CO, ethene, butene, 1,3-butadiene) did result in a reaction, there was also loss of crystallinity and the formation of intractable products. However, addition of propene to 2[BArF4] resulted in a clean SC–SC transformation over 24 h, to give a product of empirical formula [Rh(dtbpb)(C3H6)][BArF4], 5[BArF4], Scheme 5. While the crystals do not change colour, they become coated in an oily-solid, presumably liberated NBA, that could be removed by application of a dynamic vacuum (5 × 10−3 mbar) for 18 h. | ||
| Scheme 5 Synthesis of 5[BArF4]. | ||
As shown by a resulting single-crystal X-ray diffraction study at 110 K, Fig. 10a, substitution of NBA with propene reverses C–H activation of the phosphine bridge, reforming the κ2-cis-P,P ligand motif observed in the NBA and NBE adducts. There is a space group change to C2/c that results in a superposition of two cationic fragments related by a C2 axis, which sit in the ∼Oh cage of [BArF4]− anions, Fig. 10b. This complexity is exacerbated by a two-component twin model being needed for a reasonable structural model, R1 = 9.74%. While the resulting structural solution unambiguously showed formation the κ2-cis-P,P ligand and NBA had been lost, it did not allow for the discrimination between a propene or an allyl-hydride ligand, as the C–C distances were lightly restrained and crystallographically identical [1.45(5) and 1.46(3) Å].
 | ||
| Fig. 10 Solid-state structure of one of the disordered components 5[BArF4] (a) Structure of the cation, 5+. Displacement ellipsoids are shown at the 30% probability level, H-atoms not shown, tBu groups shown in stick form. The Rh–hydride was not located. Selected bond lengths (Å) and angles (°): Rh–P1, 2.363(5); Rh–P2, 2.327(5); C1–C2, 1.45(5); * C2–C3 1.46(3); * Rh–C1, 2.21(3); Rh–C2, 2.18(2); Rh–C3, 2.21(3); P1–Rh–P2, 105.39(15). * Restrained at 1.47(1) Å. (b) Arrangement of [BArF4]− anions around one of the cations. Van der Waals radii. (c) Variable temperature 31P{1H} SSNMR spectra (162 MHz, 10 kHz spin rate, −90 °C and +20 °C). | ||
Vacuum transfer of CD2Cl2 onto these single crystals resulted in a temperature sensitive solution of 5[BArF4]. NMR spectra recorded at 193 K showed a single environment in the 31P{1H} NMR spectrum at δ 65.3 [J(RhP) = 131 Hz]; at a chemical shift similar to 1[BArF4], but with a coupling constant to 103Rh more like 2[BArF4]. In the 1H NMR spectrum an upfield-shifted doublet of triplets (1 H) was observed at δ −32.55 [J(RhP) = 40 Hz; J(PH) 12 Hz], that collapses into a doublet on decoupling 31P. Three, mutually coupled, resonances are observed at δ 5.09 (1H), 4.82 (2H) and 2.36 (2H). Collectively these data identify the low temperature solution structure as being a Rh(III) allyl-hydride, [Rh(dtbpb)(H)(η3-C3H5)][BArF4]. Warming CD2Cl2 solutions of 5[BArF4] to room temperature resulted in decomposition to multiple products. In the solid-state the 31P{1H} SSNMR spectrum recorded at 183 K shows two signals at δ 59 and 73; while in the 13C{1H} SSNMR spectrum two broad environments are observed at δ 63 and 68 in the region associated with alkene or allyl ligands.56 Warming to +20 °C results in a small chemical shift change to δ 63 and 70 in the 31P{1H} SSNMR spectrum, Fig. 10c. However, the corresponding 13C{1H} SSNMR spectrum at +20 °C is now featureless in the region associated with alkene or allyl groups, δ 110 to 50. These data are all consistent with the chelating phosphine ligand adopting a κ2-motif throughout, as there is no-evidence for trans-P,P coupling, as observed for 2[BArF4], that is a reporter for a κ3-ligand binding mode.
These data are also consistent with a rapid, at room temperature, non-degenerate fluxional process occurring in the crystalline solid-state for the allyl ligand. Coupled with the solution-determined low temperature structure of an allyl hydride we propose an equilibrium is established in crystallo between Rh(III) allyl hydride and Rh(I) propene complexes, both of which have a κ2-cis-P,P ligand motif, that favours the former at low temperature. Fluxional behaviour between propene and allyl hydride tautomers has been noted before in crystallo for [Rh(Cy2P(CH2)2PCy2)(C3H5)][BArF4].34 In contrast with 5[BArF4], the Rh(I) propene tautomer for this complex is observed at low temperature, consistent with smaller bite angle phosphine ligands with less bulky groups favouring Rh(I) over Rh(III) oxidation states in such equilibria.70 Supporting this finely balanced equilibrium, the previously reported butenes complex [Rh(tBu2P(CH2)3PtBu2)(C4H8)][BArF4]43 exists as an equilibrium mixture of allyl hydride and alkene complexes at 183 K (CD2Cl2), in-line with the smaller bite angle associated with the C3-backbone backbone compared with the C4 backbone in 5[BArF4] [cf. NBD complexes P–Rh–P 96.75(2)° versus 102.11(2)° respectively].
The addition of propene to 2[BArF4] thus results in a change in the thermodynamics of C–H activation of the phosphine backbone, so that on swapping the very weak σ-donor NBA for propene/allyl hydride C–H activation is now disfavoured, and the structurally responsive phosphine ligand adopts a κ2-binding mode in a SC–SC transformation. This observation is also consistent with the initial formation of 2[BArF4], which occurs from C–H activation at the σ-alkane complex 2b[BArF4], rather than the NBE precursor, 4[BArF4].
Conclusions
Ligands that adapt their coordination mode in response to other changes around the metal centre play an important role in organometallic synthesis and catalysis. We demonstrate here that such adaptability is also possible in solid/gas in crystallo organometallic reactivity, in which a cis-chelating phosphine undergoes reversible C–H activation to form a trans-spanning pincer-type ligand in response to the identity of the ligand that sits opposite. Our combined experimental and DFT studies indicate that C–H activation (oxidative addition) occurs at the Rh(I) centre when this ligand is a weakly-binding alkane (NBA) rather than the more strongly binding alkene (NBD), and that when the alkane ligand is exchanged in crystallo for an alkene (propene) this C–H activation is reversed to reform the Rh(I) centre. As well as showcasing the power of the in crystallo approach for the synthesis, characterisation and stabilisation of σ-alkane complexes, it allows for the reversible adaptivity of the chelating phosphine ligand to be revealed, highlighting how versatile the crystalline solid-state environment can be. Given the wide-spread utilisation of cooperative and responsive ligands in solution homogeneous catalysis this suggests that similar non-innocence is possible in crystallo.Author contributions
The manuscript was written by contributions from all the Authors. J. C. G. led the experimental investigations, corresponding data analysis and writing the first draft. M. A. S. led the computational investigation. S. K. F. performed initial synthetic investigations. K. M. A. contributed to experimental work and data analysis. T. S. C. H. developed enabling code for the computational investigation. G. J. T. and M. R. W. contributed to data collection and analysis on the Diamond I19 beamline. S. J. P. performed solid-state NMR experiments. J. M. L. was a co-supervisor of J. C. G., alongside A. S. W. A. S. W. and S. A. M. were responsible for writing the final draft, and overall conceptualisation and supervision of the project.Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): the SI contains full experimental and characterisation data for the new complexes, including single crystal X-ray crystallographic collection and refinement details. Full details of computational studies, including xyz geometries. See DOI: https://doi.org/10.1039/d6sc02133f.CCDC 2472635–2472641 contain the supplementary crystallographic data for this paper.71a–g
Acknowledgements
The EPSRC (DTP award to JCG; EP/W015552/1, KA; EP/W015498/1, MAS); SCG Chemicals (SKF); ERC (PRECISION SMOM, Project Number 101198896, HORIZON-ERC, ERC-2024-ADG), Diamond Light Source (Beamline I19, proposal CY33007). JML is supported by a Royal Society Industry Fellowship (INF\R1\221057).Notes and references
- J. R. Khusnutdinova and D. Milstein, Metal–Ligand Cooperation, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 Search PubMed.
- L. Alig, M. Fritz and S. Schneider, First-Row Transition Metal (De)Hydrogenation Catalysis Based On Functional Pincer Ligands, Chem. Rev., 2019, 119, 2681–2751 Search PubMed.
- M. R. Elsby and R. T. Baker, Strategies and mechanisms of metal–ligand cooperativity in first-row transition metal complex catalysts, Chem. Soc. Rev., 2020, 49, 8933–8987 Search PubMed.
- L. V. A. Hale and N. K. Szymczak, Hydrogen Transfer Catalysis beyond the Primary Coordination Sphere, ACS Catal., 2018, 8, 6446–6461 Search PubMed.
- A. Rossin and M. Peruzzini, Ammonia–Borane and Amine–Borane Dehydrogenation Mediated by Complex Metal Hydrides, Chem. Rev., 2016, 116, 8848–8872 Search PubMed.
- J. M. Blacquiere, Structurally-Responsive Ligands for High-Performance Catalysts, ACS Catal., 2021, 11, 5416–5437 Search PubMed.
- G. M. Adams and A. S. Weller, POP-type ligands: Variable coordination and hemilabile behaviour, Coord. Chem. Rev., 2018, 355, 150–172 Search PubMed.
- Y. Guari, G. P. F. van Strijdonck, M. D. K. Boele, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Palladium-Catalyzed Amination of Aryl Bromides and Aryl Triflates Using Diphosphane Ligands: A Kinetic Study, Chem. – Eur. J., 2001, 7, 475–482 Search PubMed.
- S. Acosta-Calle and A. J. M. Miller, Tunable and Switchable Catalysis Enabled by Cation-Controlled Gating with Crown Ether Ligands, Acc. Chem. Res., 2023, 56, 971–981 Search PubMed.
- A. F. Orsino, M. Gutiérrez del Campo, M. Lutz and M.-E. Moret, Enhanced Catalytic Activity of Nickel Complexes of an Adaptive Diphosphine–Benzophenone Ligand in Alkyne Cyclotrimerization, ACS Catal., 2019, 9, 2458–2481 Search PubMed.
- H. D. Empsall, E. M. Hyde, R. Markham, W. S. McDonald, M. C. Norton, B. L. Shaw and B. Weeks, Synthesis and X-ray structure of an unusual iridium ylide or carbene complex, J. Chem. Soc., Chem. Commun., 1977, 589–590 Search PubMed.
- J. M. Brown, P. A. Chaloner, A. G. Kent, B. A. Murrer, P. N. Nicholson, D. Parker and P. J. Sidebottom, The mechanism of asymmetric homogeneous hydrogenation. Solvent complexes and dihydrides from rhodium diphosphine precursors, J. Organomet. Chem., 1981, 216, 263–276 Search PubMed.
- C. Crocker, R. J. Errington, W. S. McDonald, K. J. Odell, B. L. Shaw and R. J. Goodfellow, Rapid reversible fission of a C–H bond in a metal complex: X-ray crystal structure of [RhHCl(But2PCH2CH2CHCH2CH2PBut2)], J. Chem. Soc., Chem. Commun., 1979, 498–499 Search PubMed.
- A. Vigalok, Y. Ben-David and D. Milstein, Complexation of N2, H2, CO2, and Ethylene to a T-Shaped Rhodium(I) Core, Organometallics, 1996, 15, 1839–1844 Search PubMed.
- J. Zhou and J. F. Hartwig, Iridium-Catalyzed H/D Exchange at Vinyl Groups without Olefin Isomerization, Angew. Chem., Int. Ed., 2008, 47, 5783–5787 Search PubMed.
- D. Gelman and S. Musa, Coordination Versatility of sp3-Hybridized Pincer Ligands toward Ligand–Metal Cooperative Catalysis, ACS Catal., 2012, 2, 2456–2466 Search PubMed.
- K. M. Altus, S. K. Furfari, J. Goodall, M. R. Gyton, J. Heaton, C. Johnson, A. I. McKay, M. Sondrup Møller, S. D. Pike and A. S. Weller, Solid-state Molecular OrganoMetallic Chemistry (SMOM): A Users’ Guide to In Crystallo Single-Crystal to Single-Crystal Transformations using Solid/Gas Methods, Dalton Trans., 2026, 55, 6649–6666 Search PubMed.
- K. A. Reid and D. C. Powers, In crystallo organometallic chemistry, Chem. Commun., 2021, 57, 4993–5003 Search PubMed.
- A. Sur and D. C. Powers, Crystallo Photochemistry: Reimagining Synthetic Tractability with Transparent Single-Crystalline Flasks, ACS Cent. Sci., 2025, 11, 834–842 Search PubMed.
- S. D. Pike, F. M. Chadwick, N. H. Rees, M. P. Scott, A. S. Weller, T. Krämer and S. A. Macgregor, Solid-State Synthesis and Characterization of σ-Alkane Complexes, [Rh(L2)(η2,η2-C7H12)][BArF4] (L2 = Bidentate Chelating Phosphine), J. Am. Chem. Soc., 2015, 137, 820–833 Search PubMed.
- S. D. Pike, A. L. Thompson, A. G. Algarra, D. C. Apperley, S. A. Macgregor and A. S. Weller, Synthesis and Characterization of a Rhodium(I) σ-Alkane Complex in the Solid State, Science, 2012, 337, 1648–1651 Search PubMed.
- J. D. Watson, L. D. Field and G. E. Ball, Binding methane to a metal centre, Nat. Chem., 2022, 14, 801–804 Search PubMed.
- S. Geftakis and G. E. Ball, Direct Observation of a Transition Metal Alkane Complex, CpRe(CO)2(cyclopentane), Using NMR Spectroscopy, J. Am. Chem. Soc., 1998, 120, 9953–9954 Search PubMed.
- W. H. Bernskoetter, C. K. Schauer, K. I. Goldberg and M. Brookhart, Characterization of a Rhodium(I) σ-Methane Complex in Solution, Science, 2009, 326, 553–556 Search PubMed.
- P. J. Sempsrott, B. B. Trinh, C. F. Lovitt, N. E. Capra and G. S. Girolami, An osmium(II) methane complex: Elucidation of the methane coordination mode, Sci. Adv., 2023, 9, eadg8130 Search PubMed.
- M. Sellin, J. D. Watson, J. Fischer, G. E. Ball, L. D. Field and I. Krossing, Promoting Alkane Binding: Crystallization of a Cationic Manganese(I)-Pentane σ-Complex from Solution, Angew. Chem., Int. Ed., 2025, 64, e202507494 Search PubMed.
- J. C. Mullins, M. J. Evans, J. M. Parr, D. T. Nguyen, A. I. McKay, T. Rajeshkumar, R. O. Piltz, A. J. Edwards, L. Maron and C. Jones, Alkane Coordination by a Neutral, Lewis Acidic Magnesium Complex, J. Am. Chem. Soc., 2025, 147, 47584–47594 Search PubMed.
- N. R. Andreychuk and D. J. H. Emslie, Potassium–Alkane Interactions within a Rigid Hydrophobic Pocket, Angew. Chem. Int. Ed., 2013, 125, 1740–1743 Search PubMed.
- J. Jung, S. T. Löffler, J. Langmann, F. W. Heinemann, E. Bill, G. Bistoni, W. Scherer, M. Atanasov, K. Meyer and F. Neese, Dispersion Forces Drive the Formation of Uranium–Alkane Adducts, J. Am. Chem. Soc., 2020, 142, 1864–1870 Search PubMed.
- M. A. Bogachev, A. N. Selikhov, A. V. Cherkasov, R. R. Aysin, S. S. Bukalov and A. A. Trifonov, Intermolecular Lithium η2-Alkene and κ2-Alkane Complexes: Synthesis, Bonding, and Facile Interconversion, J. Am. Chem. Soc., 2025, 147, 34610–34619 Search PubMed.
- F. M. Chadwick, T. Krämer, T. Gutmann, N. H. Rees, A. L. Thompson, A. J. Edwards, G. Buntkowsky, S. A. Macgregor and A. S. Weller, Selective C–H Activation at a Molecular Rhodium Sigma-Alkane Complex by Solid/Gas Single-Crystal to Single-Crystal H/D Exchange, J. Am. Chem. Soc., 2016, 138, 13369–13378 Search PubMed.
- A. I. McKay, A. J. Bukvic, B. E. Tegner, A. L. Burnage, A. J. Martı́nez-Martı́nez, N. H. Rees, S. A. Macgregor and A. S. Weller, Room Temperature Acceptorless Alkane Dehydrogenation from Molecular σ-Alkane Complexes, J. Am. Chem. Soc., 2019, 141, 11700–11712 Search PubMed.
- M. R. Gyton, M. A. Sajjad, D. J. Storm, K. M. Altus, J. C. Goodall, C. L. Johnson, S. J. Page, A. J. Edwards, R. O. Piltz, S. B. Duckett, S. A. Macgregor and A. S. Weller, An Operationally Unsaturated Iridium-Pincer Complex That C–H Activates Methane and Ethane in the Crystalline Solid-State, J. Am. Chem. Soc., 2025, 147, 8706–8719 Search PubMed.
- F. M. Chadwick, A. I. McKay, A. J. Martinez-Martinez, N. H. Rees, T. Krämer, S. A. Macgregor and A. S. Weller, Solid-state molecular organometallic chemistry. Single-crystal to single-crystal reactivity and catalysis with light hydrocarbon substrates, Chem. Sci., 2017, 8, 6014–6029 Search PubMed.
- K. M. Altus, Y. Shi, P. Probst, J. H. Heaton, M. R. Gyton, L. Lari, M. R. Buchmeiser, P. W. Dyer and A. S. Weller, Room Temperature Ethene to Propene (ETP) Tandem Catalysis using Single Crystalline Solid-State Molecular Pre-Catalysts, Angew. Chem., Int. Ed., 2025, 64, e202419923 Search PubMed.
- C. L. Johnson, D. J. Storm, M. A. Sajjad, M. R. Gyton, S. B. Duckett, S. A. Macgregor, A. S. Weller, M. Navarro and J. Campos, A Gold(I)–Acetylene Complex Synthesised using Single-Crystal Reactivity, Angew. Chem., Int. Ed., 2024, 63, e202404264 Search PubMed.
- J. C. Goodall, M. A. Sajjad, E. A. Thompson, S. J. Page, A. M. Kerrigan, H. T. Jenkins, J. M. Lynam, S. A. Macgregor and A. S. Weller, In crystallo lattice adaptivity triggered by solid-gas reactions of cationic group 7 pincer complexes, Chem. Commun., 2023, 59, 10749–10752 Search PubMed.
- C. L. Johnson, K. M. Altus, M. Navarro, J. Campos, S. B. Duckett and A. S. Weller, Single-crystal to single-crystal synthesis of a gold(I)-ammonia complex and H/D exchange in crystallo with D2O, Dalton Trans., 2025, 54, 16673–16676 Search PubMed.
- K. M. Altus, M. A. Sajjad, M. R. Gyton, A. C. Whitwood, S. J. Page, S. A. Macgregor and A. S. Weller, Solid/Gas In Crystallo Reactivity of an Ir(I) Methylidene Complex, Organometallics, 2024, 43, 3137–3142 Search PubMed.
- L. R. Doyle, M. R. Galpin, S. K. Furfari, B. E. Tegner, A. J. Martínez-Martínez, A. C. Whitwood, S. A. Hicks, G. C. Lloyd-Jones, S. A. Macgregor and A. S. Weller, Inverse Isotope Effects in Single-Crystal to Single-Crystal Reactivity and the Isolation of a Rhodium Cyclooctane σ-Alkane Complex, Organometallics, 2022, 41, 284–292 Search PubMed.
- A. J. Martínez-Martínez, B. E. Tegner, A. I. McKay, A. J. Bukvic, N. H. Rees, G. J. Tizzard, S. J. Coles, M. R. Warren, S. A. Macgregor and A. S. Weller, Modulation of σ-Alkane Interactions in [Rh(L2)(alkane)]+ Solid-State Molecular Organometallic (SMOM) Systems by Variation of the Chelating Phosphine and Alkane: Access to η2,η2-σ-Alkane Rh(I), η1-σ-Alkane Rh(III) Complexes, and Alkane Encapsulation, J. Am. Chem. Soc., 2018, 140, 14958–14970 Search PubMed.
- M. A. Sajjad, S. A. Macgregor and A. S. Weller, A comparison of non-covalent interactions in the crystal structures of two σ-alkane complexes of Rh exhibiting contrasting stabilities in the solid state, Farad. Disc., 2023, 244, 222–240 Search PubMed.
- S. K. Furfari, B. E. Tegner, A. L. Burnage, L. R. Doyle, A. J. Bukvic, S. A. Macgregor and A. S. Weller, Selectivity of Rh⋯H−C Binding in a σ-Alkane Complex Controlled by the Secondary Microenvironment in the Solid State, Chem. – Eur. J., 2021, 27, 3177–3183 Search PubMed.
- A. J. Bukvic, A. L. Burnage, G. J. Tizzard, A. J. Martínez-Martínez, A. I. McKay, N. H. Rees, B. E. Tegner, T. Krämer, H. Fish, M. R. Warren, S. J. Coles, S. A. Macgregor and A. S. Weller, A Series of Crystallographically Characterized Linear and Branched σ-Alkane Complexes of Rhodium: From Propane to 3-Methylpentane, J. Am. Chem. Soc., 2021, 143, 5106–5120 Search PubMed.
- A. G. Algarra, A. L. Burnage, M. Iannuzzi, T. Krämer, S. A. Macgregor, R. E. M. Pirie, B. Tegner and A. S. Weller, Computational Studies of the Solid-State Molecular Organometallic (SMOM) Chemistry of Rh σ-Alkane Complexes, Struct. Bonding, 2020, 186, 183–228 Search PubMed.
- N. E. Capra, B. B. Trinh and G. S. Girolami, Weakly Bound but Strongly Interacting: The Structures, Stabilities, and Dynamics of Osmium(II) Ethane, Propane, and Butane Complexes, J. Am. Chem. Soc., 2025, 147, 7377–7390 Search PubMed.
- D. Amoroso, M. Haaf, G. P. A. Yap, R. West and D. E. Fogg, A Stable Silylene in a Reactive Environment: Synthesis, Reactivity, and Silicon Extrusion Chemistry of a Coordinatively Unsaturated Ruthenium Silylene Complex Containing Chloride and η3-P−C−P Ligands, Organometallics, 2002, 21, 534–540 Search PubMed.
- C. S. Durfy, J. A. Zurakowski, G. Jobin and M. W. Drover, An Investigation of Allyl-Substituted Bis(Diphosphine) Iron Complexes: Towards Precursors for Cooperative CO2 Activation, Chem. – Eur. J., 2024, 30, e202302721 Search PubMed.
- A. Das, C.-H. Wang, G. P. Van Trieste, III, C.-J. Sun, Y.-S. Chen, J. H. Reibenspies and D. C. Powers, In Crystallo Snapshots of Rh2-Catalyzed C–H Amination, J. Am. Chem. Soc., 2020, 142, 19862–19867 Search PubMed.
- Z.-J. Lv, K. A. Eisenlohr, R. Naumann, T. Reuter, H. Verplancke, S. Demeshko, R. Herbst-Irmer, K. Heinze, M. C. Holthausen and S. Schneider, Triplet carbenes with transition-metal substituents, Nat. Chem., 2024, 16, 1788–1793 Search PubMed.
- O. V. Ozerov, C. Guo, V. A. Papkov and B. M. Foxman, Facile Oxidative Addition of N−C and N−H Bonds to Monovalent Rhodium and Iridium, J. Am. Chem. Soc., 2004, 126, 4792–4793 Search PubMed.
- A. B. Chaplin, J. C. Green and A. S. Weller, C–C Activation in the Solid State in an Organometallic σ-Complex, J. Am. Chem. Soc., 2011, 133, 13162–13168 Search PubMed.
- I. J. Vitórica-Yrezábal, S. Libri, J. R. Loader, G. Mínguez Espallargas, M. Hippler, A. J. Fletcher, S. P. Thompson, J. E. Warren, D. Musumeci, M. D. Ward and L. Brammer, Coordination Polymer Flexibility Leads to Polymorphism and Enables a Crystalline Solid–Vapour Reaction: A Multi-technique Mechanistic Study, Chem. – Eur. J., 2015, 21, 8799–8811 Search PubMed.
- M. K. Panda, S. Ghosh, N. Yasuda, T. Moriwaki, G. D. Mukherjee, C. M. Reddy and P. Naumov, Spatially resolved analysis of short-range structure perturbations in a plastically bent molecular crystal, Nat. Chem., 2015, 7, 65–72 Search PubMed.
- I. J. Vitórica-Yrezábal, C. A. McAnally, M. P. Snelgrove, M. R. Warren, A. H. Hill, S. P. Thompson, M. Quinn, S. Mottley, S. Mottley, A. J. Fletcher and L. Brammer, Selective CO2 uptake mimics dissolution in highly fluorinated non-porous crystalline materials, Nat. Chem., 2025, 17, 1705–1711 Search PubMed.
- P. S. Pregosin, NMR in Organometallic Chemistry, Wiley-VCH, Weinheim, 2012 Search PubMed.
- P. Pracht, S. Grimme, C. Bannwarth, F. Bohle, S. Ehlert, G. Feldmann, J. Gorges, M. Müller, T. Neudecker, C. Plett, S. Spicher, P. Steinbach, P. A. Wesołowski and F. Zeller, CREST—A program for the exploration of low-energy molecular chemical space, J. Chem. Phys., 2024, 160, 114110 Search PubMed.
- B. K. Corkey, F. L. Taw, R. G. Bergman and M. Brookhart, Aromatic and aldehyde carbon–hydrogen bond activation at cationic Rh(III) centers. Evaluation of electronic substituent effects on aldehyde binding and C–H oxidative addition, Polyhedron, 2004, 23, 2943–2954 Search PubMed.
- G. M. Adams, F. M. Chadwick, S. D. Pike and A. S. Weller, A CH2Cl2 complex of a [Rh(pincer)]+ cation, Dalton Trans., 2015, 44, 6340–6342 Search PubMed.
- R. C. Knighton, J. Emerson-King, J. P. Rourke, C. A. Ohlin and A. B. Chaplin, Solution, Solid-State, and Computational Analysis of Agostic Interactions in a Coherent Set of Low-Coordinate Rhodium(III) and Iridium(III) Complexes, Chem. – Eur. J., 2018, 24, 4927–4938 Search PubMed.
- A. Longcake, M. R. Lees, M. S. Senn and A. B. Chaplin, Oxidative Addition of C–Cl Bonds to a Rh(PONOP) Pincer Complex, Organometallics, 2022, 41, 3557–3567 Search PubMed.
- M. D. Walter, P. S. White, C. K. Schauer and M. Brookhart, Stability and Dynamic Processes in 16VE Iridium(III) Ethyl Hydride and Rhodium(I) σ-Ethane Complexes: Experimental and Computational Studies, J. Am. Chem. Soc., 2013, 135, 15933–15947 Search PubMed.
- The in crystallo solid/gas addition of D2 to a precursor NBE complex closely related to 4[BArF4], gives exo-D2-NBA. See ref. 31.
- P. H. M. Budzelaar, N. N. P. Moonen, R. d. Gelder, J. M. M. Smits and A. W. Gal, Rhodium and Iridium β-Diiminate Complexes – Olefin Hydrogenation Step by Step, Eur. J. Inorg. Chem., 2000, 753–769 Search PubMed.
- D. Y. Wang, Y. Choliy, M. C. Haibach, J. F. Hartwig, K. Krogh-Jespersen and A. S. Goldman, Assessment of the Electronic Factors Determining the Thermodynamics of “Oxidative Addition” of C–H and N–H Bonds to Ir(I) Complexes, J. Am. Chem. Soc., 2016, 138, 149–163 Search PubMed.
- R. R. Schrock and J. A. Osborn, Catalytic hydrogenation using cationic rhodium complexes. II. The selective hydrogenation of alkynes to cis olefins, J. Am. Chem. Soc., 1976, 98, 2143–2147 Search PubMed.
- B. Nguyen and J. M. Brown, Stereoselectivity in the Rhodium-Catalysed Reductions of Non-Conjugated Dienes, Adv. Synth. Catal., 2009, 351, 1333–1343 Search PubMed.
- The observation of d4-NBA is also consistent with NBE hydrogenation preceding backbone C–H activation; an alternative pathway in which backbone C–H activation in 4+ occurred prior to D2 addition would lead to the formation of a Rh–D species (see Fig. S48/S49). Moreover, assuming facile formation of 2b+, such a process would be both kinetically (ΔG‡ = +31.8 kcal mol−1) and thermodynamically (ΔG = +13.8 kcal mol−1) disfavoured compared to backbone C–H activation directly at 2b+ (ΔG‡ = +26.3 kcal mol−1; ΔG = –6.8 kcal mol−1).
- T. Krämer, F. M. Chadwick, S. A. Macgregor and A. S. Weller, Solid-State Confinement Effects in Selective exo-H/D Exchange in the Rhodium σ-Norbornane Complex [(Cy2PCH2CH2PCy2)Rh(η2:η2-C7H12)][BArF4], Helv. Chim. Acta, 2023, 106, e202200154 Search PubMed.
- A. D. Wilson, A. J. M. Miller, D. L. DuBois, J. A. Labinger and J. E. Bercaw, Thermodynamic Studies of [H2Rh(diphosphine)2]+ and [HRh(diphosphine)2(CH3CN)]2+ Complexes in Acetonitrile, Inorg. Chem., 2010, 49, 3918–3926 Search PubMed.
- (a) CCDC 2472635: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzf2; (b) CCDC 2472636: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzg3; (c) CCDC 2472637: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzh4; (d) CCDC 2472638: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzj5; (e) CCDC 2472639: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzk6; (f) CCDC 2472640: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzl7; (g) CCDC 2472641: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nzzm8.
| This journal is © The Royal Society of Chemistry 2026 |