
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuperprotonic conductivity in crystalline and amorphous framework materials under anhydrous conditions
Nattapol Ma
 *
*
International Center for Young Scientists (ICYS), National Institute for Materials Science, 1-1 Namiki, Tsukuba, Ibaraki 305-0044, Japan. E-mail: ma.nattapol@nims.go.jp
First published on 6th May 2026
Abstract
Fast proton conduction under anhydrous conditions is pivotal for advancing intermediate-temperature (100–300 °C) hydrogen (H2) energy technologies, such as fuel cells and electrolysers. However, identifying suitable proton conductors for this temperature range remains challenging: the temperature is too high for water-mediated transport but too low for defect-driven conduction. Framework materials, including crystalline and glassy coordination polymers (CPs) or metal–organic frameworks (MOFs) and covalent organic frameworks (COFs), have emerged as promising candidates, offering tunable structures and unique pathways for anhydrous proton conduction, with some exhibiting “superprotonic-like” conductivity. This review covers the fundamental mechanisms and design principles governing proton transport, emphasising the roles of structural features, defects, dynamic disorder, and functional groups. Emerging materials, including CP/MOF glasses with isotropic structures and COFs with aligned one-dimensional nanoscale channels, are highlighted as promising candidates. Beyond intrinsic conductivity, we evaluate practical considerations essential for device integration, including mechanical processability, thin-film fabrication, long-term thermal and chemical stability, and stimuli-responsive conductivity. The comparative advantages and limitations of CPs/MOFs, their glass derivatives, and COFs are critically analysed. Finally, the review discusses key challenges and future directions toward realising stable, high-performance anhydrous proton conductors for practical hydrogen energy applications.
 Nattapol Ma | Nattapol Ma obtained his PhD in 2023 from Kyoto University, under the supervision of Prof. Satoshi Horike, and received the FWO postdoctoral fellowship for conducting research in the group of Prof. Rob Ameloot at KU Leuven. Since 2024, he has been an ICYS Research Fellow at the National Institute for Materials Science (NIMS), working with Dr Takashi Nakanishi and Dr Renzhi Ma. Since 2025, he has also been a JST PRESTO researcher in the “Development and Recycling of Materials” project supervised by Prof Susumu Kitagawa. His research focuses on developing amorphous coordination polymers and metal–organic frameworks with proton conductivity. |
1. Introduction
H2 production and utilisation via electrolysers and fuel cells operating at intermediate temperatures (100–300 °C) represents a promising pathway for replacing fossil fuels, as they offer optimised thermodynamic efficiency and enhanced electrode reaction kinetics (Fig. 1A and B). For example, electrolysers operated above 100 °C can directly utilise steam , which requires less energy for water splitting than liquid water
, which requires less energy for water splitting than liquid water 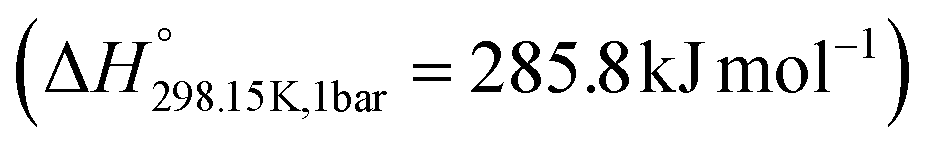 .1 Moreover, a portion of the energy required for the electrolysis process can be supplied as heat, which contributes an entropic term (TΔS) to the overall reaction. This heat can either be generated internally through cell losses or supplied externally, for example, from waste heat streams. As a result, the electrical energy input required, corresponding to the Gibbs free energy change (ΔG), is reduced according to ΔG = ΔH − TΔS. By providing part of the reaction enthalpy in the form of heat, the required cell voltage decreases and overall system efficiency increases, leading to greater hydrogen production per unit of electrical energy input. In addition, elevated operating temperatures accelerate electrode kinetics, reducing or potentially eliminating the need for precious metal catalysts such as Pt, IrO2, and RuO2,2 while also reducing cooling requirements. Collectively, these factors can significantly lower both capital and operating costs compared with conventional water electrolysers operating below 90 °C.
.1 Moreover, a portion of the energy required for the electrolysis process can be supplied as heat, which contributes an entropic term (TΔS) to the overall reaction. This heat can either be generated internally through cell losses or supplied externally, for example, from waste heat streams. As a result, the electrical energy input required, corresponding to the Gibbs free energy change (ΔG), is reduced according to ΔG = ΔH − TΔS. By providing part of the reaction enthalpy in the form of heat, the required cell voltage decreases and overall system efficiency increases, leading to greater hydrogen production per unit of electrical energy input. In addition, elevated operating temperatures accelerate electrode kinetics, reducing or potentially eliminating the need for precious metal catalysts such as Pt, IrO2, and RuO2,2 while also reducing cooling requirements. Collectively, these factors can significantly lower both capital and operating costs compared with conventional water electrolysers operating below 90 °C.
 | ||
| Fig. 1 (A) Schematic illustration of the green hydrogen cycle, including hydrogen production via electrolysis and utilisation in a fuel cell. (B) Standard thermodynamic functions of water electrolysis at 1 bar.6 (C) Examples of well-known proton conductors showing proton conductivity as a function of inverse temperature, highlighting the intermediate-temperature region (blue box).7 Coloured lines represent different classes of proton conductors. Solid acids (yellow): (NH4)3H(SO4)2,8 CsHSO4(1),9 CsHSO4(2),10 Rb3H(SeO4)2,11 β-Cs3(HSO4)2[H2−x(P1−xSx)O4],12 K3H(SO4)2,13 CsH2PO4,14 (CsH2PO4)0.85(H3PO4)0.15,14 (CsH2PO4)0.93(H3PO4)0.07,14 Rb5H7(PO4)4,15 and ZrH5(PO4)3.15 Perovskite (dark blue): BaCe0.9Y0.1O2.95−y/2(OH)y,16 La0.99Ca0.01NbO3.995−y/2(OH)y,17 La26(BO3)8O27−y/2(OH)y,18 BaScO2(OH),19 BaZr0.4Sc0.6O2.7−y/2(OH)y,20 BaSc0.67O(OH)2,21 BaSc0.8Mo0.2O2.8−y/2(OH)y,22 BaSc0.75Mo0.25O2.875−y/2(OH)y,22 BaSn0.3Sc0.7O3−δ,23 and BaSc0.8Mo0.1W0.1O3−δ at P(H2O) = 0.021 atm and dry conditions.24 Polymers (black): acid-doped polybenzimidazole25 and Nafion at 90% RH.26 Liquid acids (red): H3PO4.27 Brackets indicate the corresponding references. | ||
A major challenge in implementing electrolysers or fuel cells within this intermediate-temperature range is the absence of solid-state proton (H+) conductors that simultaneously exhibit sufficiently high proton conductivity (σ > 10 mS cm−1, Fig. 1C), operate without external humidification, and maintain thin membrane formability along with long-term chemical and mechanical stability. Proton conduction below 100 °C under humidified conditions, as in polymer electrolyte membrane (PEM) electrolysers, relies mainly on the hopping of protons between water molecules within the pore networks of the separator membrane.3,4 Sulfonated fluoropolymers (Nafion) are considered the benchmark, with proton conductivities up to 1 S cm−1 at high relative humidity (RH).5 However, Nafion is easily dehydrated above 80 °C or at low RH, resulting in a severe decline in conductivity. Moreover, its low softening point prevents the application of Nafion above 100 °C due to deformation. Most other materials are also limited to operating temperatures below 100 °C, as they rely on weakly adsorbed water molecules for proton conduction. Extending the use of solid electrolytes designed for high-temperature electrolysers and fuel cells into the intermediate-temperature range is not feasible either. Solid oxides, particularly perovskite-type materials, conduct ions through mobile defects; however, their diffusion decreases significantly at temperatures below 300 °C, especially under anhydrous conditions.23,24,28 The absence of suitable solid-state proton conductors in the intermediate-temperature ‘gap’ was highlighted in 1999 (Fig. 1C).7 More than two decades later, the practical application of proton conductors in this intermediate-temperature regime remains challenging, especially when factors such as processability and long-term stability must be considered alongside proton conductivity.
In some compounds, solid-state protonic or ionic conductivities become exceptionally high, approaching or even exceeding those of the molten state without the need for external humidification. This typically occurs alongside solid-to-solid phase transitions from a ‘low conductivity’ to a ‘high conductivity’ state. For instance, the solid acid CsHSO4 undergoes a phase transition from a static monoclinic structure (phase II, space group: P21/c) to a more dynamic tetragonal structure (superprotonic phase I, space group: I41/amd) at temperatures above 141 °C.29 This transition results in a dramatic increase in proton conductivity to nearly 10−2 S cm−1, which is more than three orders of magnitude higher than that of the monoclinic phase, even below its melting point (Tm = 206 °C). Moreover, a considerably low activation energy of ca. 0.1 eV was observed. CsHSO4 is a well-known example of an ionic solid that exhibits ‘liquid-like’ proton or ion conductivities, commonly referred to as ‘superprotonic’ or ‘superionic’ conduction, depending on the type of mobile ion involved (Fig. 2).30–32 However, the risk of dissolution in the presence of water condensation, particularly during start-up and shut-down processes, along with the narrow operational temperature window of the superprotonic phase, can significantly limit the practical applicability of solid acids.31
 | ||
| Fig. 2 Comparison of the “superprotonic” transition between low and high conductivity states, involving the rotational disorder of protic species observed in CsHSO4 solid acid and [Zn(HPO4)(H2PO4)2](ImH2)2 coordination polymer. Crystal structures and conductivity data were obtained from ref. 40–42. | ||
In recent years, the search for improved proton conductors to bridge the ‘gap’ in the intermediate-temperature range has continued and expanded beyond traditional ionic solid acids and acid-doped polymers to include a broad class of framework materials, such as coordination polymers (CPs), metal–organic frameworks (MOFs), covalent organic frameworks (COFs), and others, which offer structural tunability and diverse chemical functionalities.33–35 These materials facilitate the rational design of proton conduction pathways through well-defined architectures, functionalised channels, dynamic hydrogen-bond networks, and confined acid–base interactions. Such features contribute to enhanced anhydrous proton conductivity and improved thermal stability. Notably, members of this material class have exhibited high proton conductivity over a broad temperature range (25–190 °C).34 Furthermore, the recent discovery of crystal-to-glass transformation has opened new avenues, enabling not only improved processability but also homogeneous incorporation of guest molecules and the retention of liquid-like dynamics within a solid matrix.36–38
Before delving into the details, it is essential to clarify the term “superprotonic conductivity” as used in this review and define its scope. Here, materials are considered to exhibit superprotonic-like conductivity not necessarily based on their high proton conductivity (typically >10 mS cm−1), but rather if they meet either of the following criteria: (1) a low activation energy for proton transport (<0.4 eV) or (2) the presence of a phase transition from a low-conductivity to a high-conductivity state, behaviours characteristic of superprotonic acid salts (Fig. 2).39 A low activation energy ensures sustained conductivity even at temperatures well below the melting point (if applicable), while the occurrence of a phase transition in certain framework materials mirrors the superprotonic phase transition behaviour observed in solid acids, where rapid proton migration is facilitated by rotational disorder of protic moieties.29 Another important term frequently used in this review is “anhydrous conditions”. This term generally refers to an environment that is essentially free of water. In practice, it typically describes measurement conditions conducted under a dry inert atmosphere, often in a glovebox, where the water content is maintained at the parts-per-million (ppm) level.
At the time of writing this review, several reviews have addressed the broader topic of proton conductivity in framework materials. Examples include comprehensive discussions of proton conductivity in crystalline CPs and MOFs,34,43–46 design strategies for crystalline CPs and MOFs in the wider context of ion conduction,33,47 recent advances in MOF-based proton-exchange membranes,48 intermediate-temperature proton conduction in crystalline porous frameworks,49 and anhydrous proton conduction in crystalline systems encompassing MOFs, COFs, hydrogen-bonded organic frameworks (HOFs), and polyoxometalates (POMs).50
This review explores the fundamental concepts, design principles, and recent advances in proton-conductive framework materials, including CPs, MOFs, and COFs, under anhydrous conditions, with particular emphasis on systems exhibiting superprotonic-like conductivity. Instead of focusing purely on performance-based metrics, this review highlights distinctive features exclusive to these framework materials, covering direct structure–property relations, host–guest interactions, melt processability, and more. The discussion extends further to highlight the emerging class of CP/MOF glasses, which present new opportunities for enhancing proton transport and processability by leveraging structural disorder and liquid-like dynamics within the solid state.
2. Conducting mechanisms and design principles
2.1 Proton transport mechanisms
Achieving fast ion conductivity (σ) requires maximising the product of the number of charge carriers (ni), their charge (zi), and their mobility (µi), as described by the relation σ = Σni·zi·µi. A classical example that embodies the essential features of fast ion conductors is AgI.32,51 Upon heating above 148.9 °C, AgI undergoes a phase transition from β-AgI (wurtzite) or γ-AgI (sphalerite) phases to the body-centred cubic α-AgI phase, accompanied by an abrupt, four-order-of-magnitude increase in conductivity, reaching ca. 2 S cm−1.39,52 At this temperature, the Ag+ sublattice becomes highly disordered and exhibits “liquid-like” behaviour, often described as a melting of the cation sublattice. This interpretation is supported by entropy gains of +14.5 J mol−1 K−1 for the phase transition and +11.3 J mol−1 K−1 for melting of AgI at 555.9 °C, values comparable to the entropy of fusion for typical ionic solids like NaCl (ca. 24 J mol−1 K−1). However, structural studies suggest that this dynamic behaviour is better described by rapid, site-to-site hopping of Ag+ ions within a well-defined lattice. AgI thus exemplifies the key criteria for high ionic conductivity: (1) a high concentration of mobile ions, (2) a favourable site network with low-energy migration pathways formed by the arrangement of oppositely charged ions, (3) highly polarisable anions that facilitate ion movement, (4) minimal trapping of mobile ions, (5) low mobile ion charge, and (6) a small ionic radius that lowers the activation energy for migration.39Proton conduction operates through mechanisms distinct from those seen in other ionic conductors, largely due to the unique bonding characteristics of protons. In materials designed for pure proton conduction, protons commonly form a hydrogen bond composed of a short bond with one oxygen atom (ca. 0.95 Å) and a weaker hydrogen bond with a neighbouring oxygen. This creates a double potential well. When the O⋯O separation reaches around 2.4 Å, the potential well flattens into a single minimum, allowing the proton to be equally bonded between both oxygens.53,54 Although protons cannot migrate freely like other cations due to the relatively strong O–H bond, they can still be transported via a low-energy mechanism in which proton migration is coupled to the motion of the bonded oxygen, either locally, through the Grotthuss mechanism, or over longer distances via diffusion motion of the host molecules (vehicle mechanism).55–58 In most cases, proton conductivity follows Arrhenius behaviour, where the product of proton conductivity and temperature (σT) increases linearly with the inverse temperature (1/T). The activation energy (Ea) can be determined from the Arrhenius equation: σT = σ0exp(−Ea/kT), where σ0 and k represent the pre-exponential factor and Boltzmann constant, respectively.34
In contrast, temperature-dependent proton conductivity in glasses and polymeric materials often deviates from Arrhenius behaviour, particularly above their glass transition temperature. Although these materials appear macroscopically solid due to their relatively high viscosity, proton transport occurs through cooperative migration that is coupled with local structural dynamics and facilitated by structural relaxation. As a result, their conductivity typically follows the Vogel–Fulcher–Tammann (VFT) law: σ = σ0exp(−B/(T − T0)),59–61 where σ0, B, and T0 represent the pre-exponent factor, the activation factor, and the ideal transition temperature, respectively. In addition, more advanced models consider system viscosity and electrical relaxation time to reveal decoupling effects or highlight strong correlations between electrical and mechanical relaxation processes.62–64
At this point, it is important to note that understanding the underlying mechanism of proton conductivity cannot be achieved solely through conductivity measurements and model fitting.39 A more comprehensive insight requires complementary techniques, such as neutron scattering or nuclear magnetic resonance (NMR), which can provide detailed information on proton positions, migration pathways, and the cooperative role of structural dynamics in the material of interest.65–67 These advanced characterisation methods are essential for unveiling the complex interplay between structural relaxation, hydrogen bonding networks, and proton mobility, thus benefiting the rational design of next-generation proton-conductive materials.
2.2 Overview of CPs, MOFs, COFs, and hybrid frameworks
Coordination polymers (CPs) and metal–organic frameworks (MOFs) are materials constructed through the self-assembly of repeating coordination units that extend indefinitely in one, two, or three dimensions.68–72 They are composed of metal cations or clusters serving as nodes, interconnected by linkers through coordination bonds (Fig. 3A). The field of CPs/MOFs was established following the discovery of the permanent porosity and robust framework structures in these framework materials, such as [Co2(4,4′-bipyridyl)3(NO3)4] and [Zn(1,4-benzenedicarboxylate)].68,69 The stability and predictability of coordination bonds allow for precise three-dimensional atomic arrangement, while the use of organic linkers enables the intentional incorporation of functional side groups, giving rise to truly “designer” materials. Since then, over 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 crystalline CPs/MOFs have been reported, exhibiting diverse structural motifs and a wide range of applications, including gas sorption/separation, catalysis, magnetism, optics, sensing, and more.34,70–82
000 crystalline CPs/MOFs have been reported, exhibiting diverse structural motifs and a wide range of applications, including gas sorption/separation, catalysis, magnetism, optics, sensing, and more.34,70–82
 | ||
| Fig. 3 Examples of crystal structures and their precursors for (A) coordination polymers (CPs) or metal–organic frameworks (MOFs) and (B) covalent organic frameworks (COFs). (C) Schematic representation of approaches for obtaining different states of coordination polymers (CPs) and metal–organic frameworks (MOFs). Crystalline CPs/MOFs are synthesised by combining metal ions and ligand precursors. Liquid and glassy states have been recently discovered and are generally prepared by melting and vitrification, respectively. | ||
The discovery of crystal–liquid–glass transformations in CPs/MOFs in 2015 significantly boosted interest in their amorphous counterparts (Fig. 3C).36,83,84 The concept of CP/MOF glasses presents new opportunities for materials design by integrating two important features into a single material system.38,85 The first is the potential for systematic structural design, inherited from their crystalline counterparts; second is the broad compositional flexibility characteristic of amorphous and glassy materials. Additionally, their softer mechanical properties and reversible phase transitions improve processability and suggest compatibility with current industrial processes. The approaches for achieving glass formation are no longer limited to melt-quenching but have expanded to include mechanical vitrification86 and direct synthesis routes.87
Apart from frameworks constructed through metal–ligand coordination, covalent organic frameworks (COFs) are composed entirely of light elements connected via strong covalent bonds in a periodic fashion, typically forming well-ordered one-dimensional channels (Fig. 3B).88–90 These features endow COFs with low mass density as well as high thermal and chemical stability. Furthermore, the pore shape, size, and chemical environment can be precisely tailored using predesigned molecular building blocks and controlled self-assembly processes.
2.3 Background of proton conductivity in framework materials
The earliest example of proton conductive CPs dates to 1979 and describes a family of two-dimensional catena-µ-N,N′-disubstituted dithiooxamido copper(II) complexes featuring dimeric Cu(II) centres with square-planar coordination geometries, investigated under humidified conditions.91 In the study, protonic conduction was proved through the spectroscopic detection of hydrogen evolution during direct current electrolysis, complemented by the observation of an isotope effect under H2O and D2O vapours. Subsequent investigations of these copper CPs containing dithiooxamide-derived ligands, using alternating-current measurements, reported proton conductivities up to 1.2 × 10−6 S cm−1 at ca. 27 °C and 93% relative humidity.92–96 The compound also exhibits proton-coupled redox behaviour. The proton conductivity in these materials was attributed to the formation of H3O+ species generated through the reaction between water molecules and acid–base polymers at elevated relative humidity.93 Since then, the development of proton-conductive CPs/MOFs has primarily focused on optimising interactions between pore-lining functional groups and charge-carrying guest molecules and has mostly involved humidified conditions.34,43,44,97 To date, many proton-conductive CPs/MOFs operating under humidified conditions have surpassed conductivities of 10−1 S cm−1, many of which incorporated sulfonate groups.98–100In 2009, early demonstrations of proton conductivity under anhydrous conditions in porous CPs/MOFs employed non-volatile azole-based guests confined within the pores to facilitate long-range proton migration.101,102 These systems will be discussed in detail in later sections. Briefly, incorporation of imidazole guest molecules into the one-dimensional channels of Al-based MOFs suppresses guest aggregation while maintaining molecular dynamics (Fig. 4A), resulting in a relatively high proton conductivity of 2.2 × 10−5 S cm−1 at 120 °C (Fig. 5).101 Another example involves the incorporation of 1,2,4-triazole guests into Na-based MOFs featuring one-dimensional pores lined with sulfonate groups (Fig. 4B). The compound with optimised guest loading exhibits a proton conductivity of 5 × 10−4 S cm−1 at 150 °C (Fig. 5). In addition to azole incorporation, other proton-conducting guests, including non-volatile acids103,104 and ionic liquids,105–109 have been explored as potential proton carriers to achieve proton conductivity under anhydrous conditions.
 | ||
| Fig. 4 (A–H) Crystal structures of representative CPs/MOFs discussed in Section 3.1 (guest-assisted conduction) and representative guest molecules used to promote conductivity. | ||
 | ||
| Fig. 5 Temperature-dependent proton conductivity of CPs/MOFs exhibiting proton conduction via incorporated guest molecules described in Section 3.1 of this review. Coloured lines represent different types of guests used. Azole-based guests (dark blue): Al(OH)(1,4-ndc) with imidazole101 and histamine,117 Na3(2,4,6-trihydroxy-1,3,5-benzenetrisulfonate) with 1,2,4-triazole (β-PCMOF2(Tz)0.3),102 UiO-67 with imidazole.118 Quinone guest (blue): Zn3(tz)2(bdc)2 with hydroquinone (Htz = 1H-1,2,3-triazole, H2bdc = benzene-1,4-dicarboxylic acid).119 Acid guests (green): Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 (MIL-101) with H2SO4 and H3PO4 (0.1–20% RH),103 CsHSO4.104 Ionic salts/liquids (beige): MIL-101 with imidazolium triflate,106 SA-EIMS,108 and HTFSA-EMIS.107 Zn(2-methylimidazolate)2 (ZIF-8) with EMI-TFSA.105 Mg6(2,5-dioxidiobenzene-1,4-dicarboxylate)3 (CPO-27) with formamidinium thiocyanate (SCNFA).120 (H3O)[(UO2)4(2-pmb)3(H2O)3]·0.5H2O with LiBr.121 Brackets indicate the corresponding references. | ||
Beyond the guest-assisted strategies, anhydrous proton conductivity has also been achieved in CPs/MOFs in which the proton-conducting species, such as phosphates and azoles, are intrinsically embedded within the coordination networks.42,110 Some of these systems undergo crystal-to-glass transformation. For example, a mechanically vitrified two-dimensional coordination polymer, Cd(H2PO4)2(1,2,4-triazole)2 (Fig. 8B), exhibits an increase in anhydrous proton conductivity of more than two orders of magnitude, from 8 × 10−7 S cm−1 at 150 °C in the crystalline state to 1.0 × 10−4 S cm−1 at 125 °C in the glassy state (Fig. 9).86 This enhancement was attributed to faster proton dynamics, driven by the increased acidity and isotropy of the H2PO4−.
In the case of COFs, an early demonstration of proton conductivity was reported in a system incorporating H3PO4 into a COF constructed from triformylphloroglucinol and 4,4′-azodianiline, leveraging the aligned nanochannels and the intrinsic structural stability of the material (Fig. 11A).111 While the parent COF exhibits negligible conductivities, the H3PO4-loaded COF shows proton conductivity of 6.7 × 10−5 S cm−1 under anhydrous conditions (ca. 67 °C) and 9.9 × 10−4 S cm−1 at 98% relative humidity (ca. 59 °C) (Fig. 12). Beyond acid-loaded systems, only a few COFs exhibit intrinsic proton conductivity under guest-free conditions, which typically requires the incorporation of well-aligned, framework-embedded sulfonic acid groups.112,113
2.4 Role of functional groups, defects, and structural features
Strategies for modulating anhydrous proton conductivity in these frameworks can be categorised based on the source of charge carriers. In cases where conductivity originates from protic guests, three main factors must be considered: (1) the type of guest molecule, (2) the pore dimensions, and (3) the surface functionality of the pores. This design concept is broadly applicable to both porous crystalline CPs/MOFs and COFs. (1) The selection of protic guest molecules is relatively straightforward, and in many cases, the overall performance closely follows the intrinsic conductivity of the guest in its liquid state. However, one important consideration is the chemical stability of the host framework toward the selected guest. (2) Pore dimensions influence conductivity primarily through pore size and pore volume. In general, larger pore sizes tend to lower the activation energy for proton migration, and frameworks with greater pore volume often exhibit higher proton conductivity due to the increased contribution of the conductive guest. A representative example is provided by a systematic study demonstrating how variations in pore size and pore volume of COF hosts affect the conductivity of H3PO4, as discussed later in this review (Section 5.2).114 (3) Functional groups on the pore surface contribute to proton conductivity in two ways: first, by serving as anchoring sites that prevent leaching of acidic guests; and second, by providing additional hydrogen-bonding sites that facilitate more efficient proton transport. However, the density and positioning of these functional groups must be carefully optimised to avoid obstructing guest connectivity within the pore channels.In cases where proton conductivity originates from the framework itself, the strategies for enhancement differ fundamentally from those relying on mobile guest species. These frameworks are typically dense and incorporate protic groups as integral components of the structure. Achieving high proton conductivity under anhydrous conditions often mirrors the design principles found in superprotonic solid acids, focusing on two main factors: (1) the connectivity of protic species and (2) the dynamics of the protic components.115 For example, the activation energy for proton migration is known to be significantly reduced when the distance between a proton donor and an adjacent oxygen acceptor falls within ∼2.4 Å. In contrast, this energy barrier increases to approximately 1 eV when the distance extends to ∼3 Å.53,116 Local molecular dynamics, such as rotational motions of protic groups, play a crucial role in reducing the effective proton-hopping distance and facilitating closer interactions with neighbouring acceptor sites. Furthermore, the incorporation of defects and structural disorder tends to enhance local molecular mobility by reducing intermolecular constraints, while simultaneously introducing loosely bound moieties that further facilitate proton transport.
3. Proton conduction in crystalline CPs/MOFs
3.1 Guest-assisted conduction
This approach relies on encapsulating non-volatile protic organic molecules or inorganic acids, typically through post-synthetic guest incorporation, within the porous networks of inert MOFs. In such systems, the host framework itself lacks proximate protic moieties capable of supporting long-range proton transport, making the incorporated guests the primary contributors to conductivity. One of the early examples of anhydrous proton conduction in MOFs is the incorporation of protic organic guest molecules into one-dimensional channels (1 nm in diameter) of Al(OH)(1,4-ndc) (1,4-ndc = 1,4-naphthalenedicarboxylate) (Fig. 4A).101 The resulting materials are thermally stable up to 130 °C. The hydrophobic and flat pore surface allows imidazole guest molecules to rotate freely within the aligned channels, resulting in anhydrous proton conductivities of 2.2 × 10−5 S cm−1 at 120 °C and 5.5 × 10−8 S cm−1 at 30 °C, with an activation energy of 0.6 eV (Fig. 5). The values here are based on pressed powder samples. Importantly, this strategy relies on a precise dimensional match between the host framework and guest molecules. When Al(OH)(1,4-benzenedicarboxylate) is used as the host framework (Fig. 3A), the proton conductivity drops to 1 × 10−7 S cm−1 at 120 °C, with an activation energy of 0.9 eV, despite its ability to accommodate larger amounts of imidazole. The oversized pore spaces and unfavourable hydrophilic–hydrophobic host–guest interactions must promote imidazole aggregation, confirmed by 2H solid-state NMR, which restricts guest mobility and, in turn, lowers the efficiency of proton transport.Replacing imidazole with histamine (Fig. 4A), which contains an additional amine group that can serve as extra proton-hopping sites, further enhances the proton conductivity of pelletised powder to 3.0 × 10−5 S cm−1 at room temperature and up to 1.7 × 10−3 S cm−1 at 150 °C (Fig. 5).117 The activation energy for proton transport is reduced to 0.25 eV. Notably, the bulk conductivity of histamine alone is significantly lower, measured at 5.4 × 10−11 S cm−1 at room temperature and 9.4 × 10−6 S cm−1 near its melting point, with a much higher activation energy of 2.3 eV. Thermogravimetric analysis under N2 atmosphere indicates that the sample remains stable with no distinct weight loss up to 170 °C. In both examples, the dynamic azole guest molecules are likely oriented in conformations that promote efficient proton exchange within the confined pore spaces, resulting in a shallower and more uniform potential energy landscape.101,117 This favourable alignment significantly reduces the activation energy for proton migration compared to that of their respective bulk systems.
Another early example involves the incorporation of 1,2,4-triazole guests within the pores of Na3(2,4,6-trihydroxy-1,3,5-benzenetrisulfonate) (Fig. 4B).102 The framework features one-dimensional pores lined with sulfonate groups, resembling the aggregated sulfonic acid channels found in Nafion. In its dehydrated form above 70 °C, the MOF exhibits proton conductivities of less than 10−8 S cm−1. Upon introducing 1,2,4-triazole guests during synthesis, the anhydrous proton conductivity at 150 °C increases to 2 × 10−4, 5 × 10−4, and 4 × 10−4 S cm−1 for (Na3(2,4,6-trihydroxy-1,3,5-benzenetrisulfonate)-(1,2,4-triazole)x) with x = 0.3, 0.45, and 0.6, respectively (Fig. 5). For the sample with x = 0.3, a single activation energy of 0.51 eV was calculated over the entire measurement range (23–150 °C). In contrast, the other two samples exhibit a change in activation energy, indicating a transition in the conduction mechanism.
For the x = 0.45 sample, the activation energies of 1.8 and 0.34 eV were determined below and above 90 °C. A similar trend was observed for x = 0.6, with activation energies of 1.87 and 0.56 eV below and above 80 °C, respectively. This behaviour resembles the reduction in activation energy typically observed in the superprotonic phase of inorganic acids. The MOF remains thermally stable up to at least 300 °C. However, the incorporated 1,2,4-triazole begins to undergo sublimation at approximately 250 °C. To prove the concept, a membrane-electrode assembly (MEA) was prepared using the sample with x = 0.45. The MEA was prepared by pressing the powdered sample (x = 0.45) between porous Pt/C electrodes in a die, followed by attachment of Pt wires to both electrodes. On the anode side, the MEA was connected via a gas-tight seal to a hollow ceramic tube supplying H2 gas, while the cathode was exposed to ambient air. The MEA exhibited an open-circuit voltage (OCV) of 1.18 V, which remained stable for 72 h at 100 °C. At higher temperatures, the OCV decreased to 0.93 V (120 °C) and 0.77 V (140 °C) due to fuel crossover.
Beyond the aforementioned systems, the incorporation of azole guests, especially imidazole, to promote anhydrous proton conductivity has been widely explored in various porous MOFs. Examples include Zr6O4(OH)4(4,4′-biphenyldicarboxylate)6 (UiO-67), derivatives of Al(OH)(1,4-benzenedicarboxylate), Al(OH)(4,4′-sulfonyldibenzoate), Ti(2,4,6-tris(3,4-dioxophenyl)-1,3,5-triazine), Ti2O3(C4O4), Zr6O4(OH)4(C4O4)6, and more.118,122–125 However, only one example using imidazole as the guest and UiO-67 (Fig. 4C) as the host has demonstrated a superprotonic-like transition to a low activation energy state (0.36 eV) near 90 °C, achieving a proton conductivity of 1.52 × 10−3 S cm−1 at 130 °C (Fig. 5).118 Further heating above 140 °C leads to the release of imidazole molecules. Upon exposure to water for 1 hour, the sample degrades and loses its conductivity.
Organic hydroxyl compounds such as hydroquinone (pKa ≈ 9.9), cyclohexanol (pKa ≈ 17), and butanol (pKa ≈ 18) have also been used as guest molecules to promote proton conductivity in a flexible MOF Zn3(tz)2(bdc)2 (Htz = 1H-1,2,3-triazole, H2bdc = benzene-1,4-dicarboxylic acid) (Fig. 4D).119 Guest inclusion was achieved by synthesising the MOF in the presence of these molecules in the reaction solution. Unlike previous examples, the exact positions of the guest molecules in this system were determined using single-crystal X-ray diffraction. These hydroxyl compounds were selected over the more commonly used azole family due to their lower pKa values, which potentially enhance proton dissociation and lead to lower activation energies for proton conduction within the MOF framework. Among the three hydroxyl guests, hydroquinone exhibits the most promising anhydrous conductivity, reaching 2.65 × 10−4 S cm−1 at 125 °C along with a relatively low activation energy of 0.18 eV (Fig. 5). This enables a wide operating temperature window, with a conductivity of 1.49 × 10−5 S cm−1 maintained at −30 °C. This wide operating temperature window may relate to the high boiling point (285 °C) and melting point (172.4 °C) of hydroquinone itself.
Incorporation of non-volatile acids such as H2SO4, H3PO4, or the solid acid CsHSO4 into the mesoporous framework Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 (MIL-101, Fig. 4E) represents another strategy for imparting anhydrous proton conductivity to an otherwise non-conductive host (conductivity lower than 10−10 S cm−1 at 140 °C).103,104 The hybrid samples were prepared by mixing the acid-tolerant MIL-101 and aqueous solutions of H2SO4 or H3PO4, followed by removal of excess liquid and drying at elevated temperatures.103 The resulting materials contain ca. 4.47H2SO4 and 3.69H3PO4 molecules per formula unit of MIL-101. The samples were pressed into pellets at 100–300 MPa. Although the conductivity measurements were not performed under strictly anhydrous conditions, the samples exhibit relatively high proton conductivities of 1 × 10−2, and 3 × 10−3 S cm−1 at 150 °C and 0.13% relative humidity for H2SO4- and H3PO4-loaded MIL-101, respectively (Fig. 5). For the H2SO4-loaded MIL-101, the decrease in conductivity observed above 80 °C is attributed to the progressive removal of H2O molecules. The corresponding activation energies were determined to be 0.42 eV for the H2SO4 composite and 0.25 eV for the H3PO4 composite. In the case of CsHSO4, the hybrid samples were prepared by mixing MIL-101 with varying amounts of CsHSO4, followed by heating to a temperature near the melting point of CsHSO4 to promote melt infiltration into the pores of MIL-101 ((1 − x)CsHSO4-xMIL-101).104 The superprotonic phase transition of CsHSO4 remains observable in the hybrid materials, although the onset temperature decreases slightly by approximately 14 °C. In the superprotonic phase, the proton conductivity of the hybrid material remains nearly comparable to that of pure CsHSO4 at low MIL-101 loadings (x ≤ 0.02) but decreases with increasing MIL-101 content due to the conductor–insulator effect. Additionally, MIL-101 composites incorporating toluenesulfonic and triflic acids have been prepared, exhibiting promising conductivities as high as 0.08 S cm−1 at 60 °C and 15% relative humidity.126 However, the authors did not perform further measurements under anhydrous conditions.
In addition to acids, incorporating proton-conductive ionic liquids or salts into MOFs presents a promising strategy to combine the high liquid-like anhydrous proton conductivity of these materials with the solid-state stability of MOFs.105–109 One of the earliest examples demonstrating ionic conductivity in a MOF via ionic liquid incorporation involves embedding EMI-TFSA (1-ethyl-3-methylimidazolium bis(trifluoromethyl-sulfonyl)amide) into ZIF-8 (Zn(MeIM)2, H(MeIM) = 2-methylimidazole) (Fig. 4F).105,127 When used alone, EMI-TFSA exhibits a sharp decline in ionic conductivity below its melting point (ca. −16 °C). However, when confined within the pores of ZIF-8, EMI-TFSA can remain in a liquid-like state even below its bulk melting point, preserving its molecular mobility and sustaining ionic conductivity at sub-zero temperatures. Introducing imidazolium triflate or benzimidazolium triflate salts into MIL-101 enables high anhydrous proton conductivities.106 The MIL-101 host was mechanically mixed with the respective salts, followed by pelletisation under a uniaxial pressure of 200 MPa and subsequent annealing at 230 °C for 30 minutes to promote melt infiltration. A proton conductivity as high as 9 × 10−2 S cm−1 was achieved at 230 °C (Fig. 5). Although this temperature exceeds the melting points of the salts, the MOF host matrix helps preserve the solid-like nature of the hybrid material while enabling the molten salts to deliver liquid-like proton transport behaviour.
Similarly, incorporating binary ionic liquids based on Brønsted acid–base buffers, such as HTFSA–EIMS, SA–EIMS, MSA–EIMS, and PTSA–EIMS into MIL-101 (Fig. 4E) enables anhydrous proton conductivity to be achieved in the solid state.107,108 The abbreviations are defined as follows: HTFSA = (N,N-bis(trifluoromethanesulfonyl)amide) (pKa ≈ 2), SA = sulfuric acid (pKa ≈ −2.9), MSA = methanesulfonic acid (pKa ≈ −2.6), PTSA = p-toluenesulfonic acid (pKa ≈ −2.8), EIMS = 1-(1-ethyl-3-imidazolium)propane-3-sulfonate (pKa ≈ 6.8). Protonated and unprotonated species in these systems serve as proton-donating and proton-accepting sites, contributing to long-range proton transport within pore networks of MOFs. The anhydrous proton conductivities of pelletised MIL-101 containing SA–EIMS, MSA–EIMS, and PTSA–EIMS at 150 °C are 1.89 × 10−3, 1.02 × 10−4, and 2.78 × 10−4 S cm−1, respectively, and follow the same trend across the entire temperature range measured (Fig. 5).108 Although the acidic components exhibit comparable acid strengths (pKa ≈ −2.6 to −2.9) and the ionic liquids show similar pore occupancy (density ≈ 1.42–1.32 g cm−3; molecular weight 160–197 g mol−1), the notable differences in their van der Waals volumes (Vvdw: SA = 62.23 Å3, MSA = 70.74 Å3, PTSA = 143.36 Å3) suggest that smaller anionic species may facilitate more efficient proton conduction pathways within the system. This trend is also reflected in the activation energies, which increased from 0.26 eV for SA–EIMS to 0.30 eV for MSA–EIMS and 0.42 eV for PTSA–EIMS. These values are notably lower than those of the pristine ionic liquids, for which activation energies of 0.33, 0.43, and 0.51 eV were measured for SA–EIMS, MSA–EIMS, and PTSA–EIMS, respectively. Interestingly, this behaviour contrasts with that observed in MIL-101 incorporating HTFSA–EIMS, where the activation energy trend does not follow the same pattern.107 This discrepancy may be attributed to poor compatibility between HTFSA–EIMS and the MOF framework, which likely induces ion clustering rather than uniform dispersion throughout the pore network.
Introducing protic salts with coordinating anions into MOFs featuring open metal sites is another strategy to achieve anhydrous proton conductivities.120 Solvent-free grinding of Mg6(2,5-dioxidiobenzene-1,4-dicarboxylate)3 or Ni6(2,5-dioxidiobenzene-1,4-dicarboxylate)3 (also known as CPO-27 or MOF-74) (Fig. 4G) with formamidinium thiocyanate (NCSFA) or methylammonium thiocyanate (NCSMA) enables dense pore filling, reaching up to a 1:1 metal-to-thiocyanate ratio. All pelletised modified samples exhibit anhydrous proton conductivity, with the highest values observed for NCSFA-loaded materials: 7.8 × 10−4 S cm−1 for Mg-based and 2.6 × 10−4 S cm−1 for Ni-based MOFs at 140 °C (Fig. 5). In contrast, samples incorporating NCSMA showed conductivities approximately one to two orders of magnitude lower. Density functional theory (DFT) calculations reveal that the key difference between NCSFA and NCSMA guests lies in the hydrogen-bonding networks formed within the [001] channels. Despite similar donor–acceptor distances, the additional amino group in formamidinium facilitates a more extended hydrogen-bonding network, thereby enhancing proton mobility. This difference is also reflected in the significantly lower activation energies for proton migration: 0.23 eV and 0.39 eV for the Mg- and Ni-based MOFs with NCSFA, respectively, compared to 0.54 eV for the corresponding MOFs with NCSMA.
Protonation of amphiprotic amine guests in the chalcogenide framework (C2N2H10)(C2N2H9)2Cu8Sn3S12, achieved by soaking the original material in 0.2 mol per L HCl for 8 days, offers another strategy to induce anhydrous proton conductivity in framework materials.128 During the process, C2N2H9+ monocations accept additional protons to form C2N2H102+ dications, and Cl− ions assemble into a clathrate-like lattice within the framework cavities. The proton conductivity of the protonated sample, measured under a dry N2 atmosphere, reaches 1.14 × 10−5 S cm−1 at 25 °C and increases sharply to 3.62 × 10−2 S cm−1 at 169 °C. A corresponding decrease in activation energy from 0.34 to 0.20 eV is also observed around this temperature, resembling the characteristic transition seen in superprotonic conductors.
Thus far, the examples discussed above have focused on sustaining proton conductivity under anhydrous conditions by incorporating non-volatile molecules as substitutes for water, thereby preserving the hydrogen-bonding network essential for proton transport. An alternative and equally compelling strategy, however, is to enhance the thermal stability of water itself, allowing it to remain within the framework even above its normal boiling point.121 This can be achieved by encapsulating LiBr within a negatively charged uranyl phosphonate MOF (Fig. 4H), (H3O)[(UO2)4(2-pmb)3(H2O)3]·0.5H2O (2-pmbH3 = 2-(phosphonomethyl)benzoic acid). α-LiBr·H2O is known to be thermally stable up to 156 °C without releasing water molecules, and its confinement within a negatively charged framework may further suppress undesirable Li+ migration through coulombic interaction. Thermogravimetric analysis confirms the retention of two lattice and three coordination water molecules even after heating the sample to 110 °C. The pelletised LiBr-containing MOF exhibits promising proton conductivities of 1.28 × 10−2 S cm−1 at 30 °C and 1.55 × 10−1 S cm−1 at 110 °C, with an activation energy of 0.36 eV under dry N2 at equilibrium (Fig. 5). Further heating above 140 °C leads to a steep decline in proton conductivity, attributed to the loss of water molecules.
3.2 Guest-free conduction
This section highlights examples where anhydrous proton transport occurs in the absence of external protic guest molecules. In these systems, proton-conducting species are inherently embedded within the framework or intrinsically integrated into the original crystal structure, resulting in guest-free proton conductivity. An early example of superprotonic-like behaviour was observed in a one-dimensional zinc-phosphate-azole coordination polymer, [Zn(HPO4)(H2PO4)2]-(ImH2)2 (ImH2 = imidazolium) (Fig. 6A).42 The anionic chain consists of tetrahedrally coordinated Zn2+ ions linked by two types of orthophosphate groups (HPO42− and H2PO4−), with imidazolium cations (ImH2+) acting as counterions. These cations form extensive hydrogen-bonding networks with the phosphate groups, facilitating efficient proton transport. Upon heating, no weight loss was observed below 200 °C, and the PXRD patterns remained unchanged between 25 and 140 °C, indicating that the crystal structure remains unchanged within this temperature range. Under anhydrous conditions, the proton conductivity at 25 °C of the pressed pellet was measured at 3.3 × 10−8 S cm−1. As the temperature increased to around 55 °C, the conductivity rose non-linearly, eventually reaching an equilibrium value of 2.6 × 10−4 S cm−1 at 130 °C with an activation energy of 0.47 eV (Fig. 7). The differential scanning calorimetry (DSC) profile also revealed an endothermic peak at 70 °C, accompanied by an entropy change of 6.6 J mol−1 K−1, allowing the crystal to be classified as a plastic crystal. | ||
| Fig. 6 (A–H) Crystal structures of representative CPs/MOFs discussed in Section 3.2 (guest-free conduction). | ||
 | ||
| Fig. 7 Temperature-dependent proton conductivity of CPs/MOFs exhibiting proton conduction in the absence of externally added guest species (Section 3.2). Coloured lines represent different structure types. Metal-phosphate-azole CPs (dark blue): [Zn(HPO4)(H2PO4)2](ImH2)2,42 [Zn3(H2PO4)6](Hbim),129 [Zn3(H2PO4)6](HBTA),130 Zn(HTr)2(H2PO4)2,110 and Zn(HTr)2(H2PO4)2 with H3PO4.131 Ionic framework with aligned protonic species (green): [(Me2NH2)3(SO4)]2[Zn2(ox)3],132 [In(C8H6O4)2][((CH3)2NH2)(DMF)],133 (Me2NH2)[Eu(C9H9O7P)] (C9H13O7P = 5-(phosphonomethyl)isophthalic acid),134 and (NH4)3[Zr(H2/3PO4)3].135 Framework retaining structural water (beige): [Eu2(CO3)(ox)2(H2O)2]·4H2O.136 Brackets indicate the corresponding references. | ||
Since the crystal structure remains intact throughout the temperature range used for conductivity measurements, as confirmed by PXRD, the observed enhancement in conductivity is attributed to increased local dynamics of the imidazolium cations. To further investigate the conductivity transition, structural changes were directly examined by comparing single-crystal structures at −30 and 75 °C. At the higher temperature, one of the two ImH2+ cations becomes significantly disordered within the plane of the five-membered ring (anisotropic), in contrast to its well-ordered configuration at −30 °C (Fig. 2). Meanwhile, the unit cell volume remains largely unchanged, and the thermal parameters of the zinc-phosphate framework increase only marginally compared to those of the ImH2+ cation. The enhanced mobility of the ImH2+ above the threshold temperature was further confirmed by solid-state 2H and 31P NMR spectroscopy. This pronounced increase in the dynamic behaviour of the ImH2+ cation at elevated temperatures is reminiscent of superprotonic transitions in solid acids, where rapid reorientation of oxyanion species around their central atoms plays a crucial role in facilitating proton transport.39
The same transition has also been observed in another pair of zinc-phosphate-azole coordination polymers: [Zn3(H2PO4)6(H2O)3](Hbim) and [Zn3(H2PO4)6(H2O)3](HBTA), where Hbim and HBTA refer to 1,3-benzimidazole and 1,2,3-benzotriazole, respectively (Fig. 6B and 8C).129,130 Both compounds feature one-dimensional neutral Zn-phosphate chains stacked in the bc plane and arranged via hydrogen-bonding interactions. Hbim or HBTA molecules are located between these chains, each surrounded by six chains and alternately stacked along the a-axis, engaging in π–π interactions. The compounds contain two types of phosphates: µ2-phosphates, which bridge two Zn ions via a single oxygen, and µ3-phosphates, which bridge three Zn ions via two oxygens. Among them, only the rotatable µ2-phosphates can support the Grotthuss mechanism, as it requires freely reorientable hopping sites. The µ2- and µ3-phosphates alternate along the chain and are isolated from each other. Additionally, the rotatable Hbim or HBTA moieties can also facilitate proton transfer via the Grotthuss mechanism. Heating the as-synthesised compounds to around 60 °C releases three water molecules and induces a structural transformation from the triclinic (P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) to their dehydrated forms (monoclinic, P21/c), which were subsequently used for anhydrous conductivity measurements.
) to their dehydrated forms (monoclinic, P21/c), which were subsequently used for anhydrous conductivity measurements.
 | ||
| Fig. 8 (A–C) Crystal structures of representative glass-forming CPs/MOFs, (D) RMC-simulated structure of a proton-conductive CP glass obtained via direct synthesis, (E–H) additives or dopants for modifying CP/MOF glasses, and (I and J) structures of structurally modified MOFs in which glass formation is induced by methylsulfonic acid guests, as discussed in Section 4 on proton conduction in CP/MOF glasses. | ||
Dehydrated [Zn3(H2PO4)6](Hbim) conducts protons under anhydrous conditions, exhibiting a conductivity of 1.2 × 10−7 S cm−1 at 30 °C, which rapidly increases to 1.5 × 10−5 S cm−1 at 60 °C and 1.3 × 10−3 S cm−1 at 120 °C (Fig. 7, measured using a pressed pellet).129 Interestingly, these values are significantly higher than those of the hydrated form, which shows conductivities of 1.4 × 10−7 S cm−1 at 30 °C and 6.1 × 10−7 S cm−1 at 60 °C. This suggests that the structural transformation upon dehydration, in this case, promotes more continuous hydrogen bonding networks than the presence of water molecules alone. The dehydrated form exhibits a change in slope in the Arrhenius plot, suggesting a superprotonic-like transition around 70 °C. The activation energy above this temperature is 0.5 eV, significantly lower than the 1.5 eV observed below the transition point. To investigate the origin of this transition, the mobilities of the phosphate groups and Hbim moieties were examined using two sets of variable-temperature static 2H solid-state NMR analyses: one using [Zn3(H2PO4)6](Hbim) with deuterated phosphates, and the other with deuterated Hbim. The results suggest that below 70 °C, proton migration is primarily facilitated by the phosphate groups, which exhibit increasing mobility with temperature. In contrast, Hbim shows similarly low mobility at both 25 and 50 °C, despite the rise in proton conductivity. This indicates that, below 70 °C, protons predominantly hop along the one-dimensional Zn-phosphate chains. However, above 70 °C, Hbim begins to exhibit an isotropic centre peak, which becomes markedly sharper at 100 °C, accompanied by a corresponding increase in the phosphate centre peak intensity. These observations suggest that Hbim undergoes isotropic rotation above 70 °C, and that both Hbim and the phosphate groups become highly mobile at 100 °C. The liquid-like behaviour of Hbim confined within the solid matrix of one-dimensional Zn-phosphate chains is characteristic of plastic crystals and is responsible for the observed superprotonic-like transition. A similar behaviour is observed in the HBTA counterpart.130 Dehydrated [Zn3(H2PO4)6](HBTA) exhibits proton conductivities of 3.3 × 10−7 S cm−1 at 30 °C and 9.0 × 10−5 S cm−1 at 60 °C, with an activation energy of 1.22 eV (Fig. 7). Above 60 °C, the mobility of HBTA increases, reducing the activation energy to 0.53 eV and enabling a conductivity of 1.2 × 10−3 S cm−1 at 100 °C.
Achieving liquid-like motion of proton-conducting species in a solid CP is observed upon incorporation of free H3PO4 into the defect sites of zinc-phosphate-azole CP, [Zn(H2PO4)2(1,2,4-triazole)2] (Fig. 6C).110,131 The pristine compound consists of octahedral Zn2+ centres coordinated by monocoordinated orthophosphates and bridging 1,2,4-triazole ligands. In this case, proton transport occurs via in-plane hopping between rotatable orthophosphate groups, resulting in a conductivity of 1.2 × 10−4 S cm−1 at 150 °C with an activation energy of 0.6 eV (Fig. 7).110 Replacing monocoordinated H2PO4− with mobile, uncoordinated H3PO4 at defect sites, induced through a modified synthesis procedure, significantly enhanced proton conductivity under anhydrous conditions. The conductivity values reach 2 × 10−5 S cm−1 at 30 °C and 4.6 × 10−3 S cm−1 at 150 °C (Fig. 7).131 The activation energy for proton conductivity also decreases to 0.53 eV. Notably, this improvement occurs without increasing the overall concentration of orthophosphate groups in the structure but rather by enhancing the local mobility of proton carriers. H2/O2 fuel cell was prepared using the sample as a proton conductor. The CP, in powder form, was pressed between two platinum-loaded carbon electrodes (Pt loading of 2 mg cm−1) under a pressure of 80 kN for 2 minutes to form the MEA. The assembly was subsequently attached to a PTFE gasket using an epoxy resin sealant to prevent gas leakage and crossover. The cell achieved a maximum OCV of 0.88 V, which was sustained for at least 1 h, and delivered a maximum power density of 2.8 mW cm−2.
Another strategy to achieve high proton conductivity under anhydrous conditions involves designing anionic frameworks that incorporate uncoordinated cationic protic guests. For example, a relatively low activation energy for proton transport has been demonstrated in three-dimensional [(Me2NH2)3(SO4)]2[Zn2(ox)3] (ox = oxalate), which is composed of an anionic framework, [Zn2(ox)3]2−, and a cationic supramolecular network of [(Me2NH2)3SO4]+ (Fig. 6D).132 The compound was synthesised by reacting oxalic acid with ZnSO4·7H2O in dimethylformamide (DMF). The asymmetric unit contains Zn2+ ions with 1/6 occupancy, coordinated by oxalate ligands with 1/4 occupancy, forming tris-chelated [Zn2(ox)3]2− subunits. The resulting three-dimensional channels are filled with an extended electrostatic and hydrogen-bonding network composed of dimethylammonium cations, generated via solvent decomposition, and disordered sulfate anions. Proton conductivity (pressed pellet) measured under a nitrogen atmosphere reached 7 × 10−5 S cm−1 at 30 °C and 1 × 10−4 S cm−1 at 150 °C, corresponding to a relatively low activation energy of 0.13 eV within this temperature range (Fig. 7). Pairing In3+ with an isophthalate ligand yields another cationic MOF, [In(C8H6O4)2][((CH3)2NH2)(DMF)] (Fig. 6E).133 The compound adopts a two-dimensional extended structure that stacks into a three-dimensional arrangement. In this case, the spaces are filled with [(CH3)2NH2]+ cations and DMF molecules. The material exhibits proton conductivity of 2.6 × 10−5 S cm−1 at 25 °C and 2.7 × 10−5 S cm−1 at 77 °C under anhydrous conditions, with an estimated activation energy of 0.08 eV (Fig. 7). Above 90 °C, the conductivity is lost due to the removal of DMF molecules.
In another example, proton conductivity above 100 °C without additional humidification is achieved through the retention of coordinated water molecules in [Eu2(CO3)(ox)2(H2O)2]·4H2O (Fig. 6F).136 The polycrystalline sample (pressed pellet) exhibits a conductivity of 8.08 × 10−4 S cm−1 at 100 °C and 2.08 × 10−3 S cm−1 at 150 °C, with an activation energy of 0.28 eV in the temperature range of 100–150 °C (Fig. 7). This value is lower than the 0.49 eV estimated from measurements conducted between 25 and 90 °C. Mechanistic studies reveal that hydrogen-bonded arrays between coordinated water molecules and oxalate ligands along the crystallographic a-axis are primarily responsible for proton transport. As the temperature increases, the rotation of coordinated water molecules and the vibration of their O–H bonds facilitate proton hopping from the water ligands to adjacent oxalate groups within these hydrogen-bonded arrays. This dynamic process surprisingly offsets the expected decrease in conductivity due to the loss of crystallised (non-coordinated) water.
Combining layered anionic frameworks [Eu(C9H9O7P)]− (C9H13O7P = 5-(phosphonomethyl)isophthalic acid) with non-volatile protic countercations Me2NH2+ yields two-dimensional MOFs exhibiting single-crystal anhydrous proton conductivity reaching 1.25 × 10−3 S cm−1 at 150 °C along the c-axis (Fig. 6G and 7).134 The activation energy is measured to be 0.21 eV. Along this direction, the countercations are embedded between the layers, forming one-dimensional arrays stabilised by strong N–H⋯O hydrogen bonds with the phosphonate groups. No conductivity is observed along the other two directions, confirming that proton transport occurs exclusively through these acid–base arrays. Moreover, a compacted pellet from microcrystals exhibits significantly lower proton conductivity (10−8 to 10−7 S cm−1) under identical conditions. However, the conductivity increases to 3.76 × 10−3 S cm−1 at 100 °C and 98% relative humidity, as water molecules bridge and maintain the connectivity of the hydrogen-bonding network. These results highlight the critical role of continuous hydrogen-bonded pathways, either through cation–phosphonate interactions or hydration-assisted bridges, in facilitating efficient proton conduction in this material.
Another design features highly crystalline one-dimensional zirconium phosphate anionic chains and well-ordered NH4+ counterions, (NH4)3[Zr(H2/3PO4)3] (Fig. 6H).135 A key feature of this compound is that the charge-balancing NH4+ ions are located in between partially protonated phosphate oxo atoms, forming continuous hydrogen-bonding networks that facilitate one-dimensional proton transport. The crystal structure consists of stacked one-dimensional anionic zirconium phosphate chains aligned along the c-axis, with NH4+ ions occupying the spaces between the chains. Neutron powder diffraction and DFT calculations indicate that the charge-balancing protons on the phosphate groups are fully disordered and evenly distributed between adjacent phosphates. The compound is thermally stable up to 200 °C and gradually converts to ZrP2O7 above this temperature. Interestingly, it remains structurally stable in water across a pH range of 2–12, as well as in methanol or ethanol over a month-long period, and even in boiling water for two weeks. Under strictly anhydrous conditions, proton conductivity of the pelletised sample was measured at 1.69 × 10−5 S cm−1 at 90 °C and 1.45 × 10−3 S cm−1 at 180 °C, with an activation energy of 0.26 eV over the temperature range (Fig. 7). DFT calculations and molecular dynamics simulations suggest a mechanism involving thermal activation of NH4+ as a proton carrier and proton migration from one phosphate pair to another via the NH4+ medium. Additionally, a proof-of-concept H2/O2 fuel cell was assembled. A dense membrane with a thickness of ca. 0.3 mm was prepared by a hot press technique with 10 wt% polyvinylidene fluoride (PVDF). The membrane was positioned between a PtRu/C anode and a Pt/C cathode, and the electromotive force was measured over the temperature range of 100–180 °C. An OCV potential of 0.72 V was obtained. The lowered cell potential compared to the theoretical value of 1.16 V is likely due to the fuel crossover. Overall, the cell delivered a maximum power density of 12 mW cm−2 at 180 °C and stable operation was sustained for at least 15 h. In addition, a direct methanol fuel cell (DMFC) was evaluated using the fabricated membrane. The DMFC showed a maximum power density of 0.13 mW cm−2 at 180 °C, with an OCV of 0.44 V.
4. Proton conduction in CP/MOF glasses
4.1 Crystal-to-glass transformations
With several examples now discussed, we can begin to form a clearer picture of the conditions that may promote liquid-like proton conductivity under anhydrous conditions. Among the favourable characteristics, at least two can be realised by transforming CPs/MOFs into a glassy state. First, the material should exhibit high structural dynamics, particularly in the moieties involved in proton transport. Second, isotropic conductivity is preferred, especially given the persistent challenge of fabricating large single crystals for device applications. In addition, the glassy state enhances macroscopic processability, offering practical advantages for real-world applications.Before turning to systems that exhibit behaviour potentially classifiable as superprotonic conductivity, it is instructive to examine two early examples demonstrating how structural disorder can enhance proton conductivity. Prior to the discovery of CP/MOF glasses in 2015,36,83,84 a notable case of order-to-disorder transformation affecting proton conduction was reported in [ImH2][Cu(H2PO4)2Cl]·H2O (ImH2 = protonated imidazole) (Fig. 8A).137 The crystal structure features one-dimensional chains composed of two crystallographically distinct octahedral Cu2+ centres and H2PO4− ions, along with a single Cl− ion. ImH2 and water molecules are aligned in the interstitial spaces between the anionic chains along the b-axis. The as-synthesised compound exhibits anhydrous conductivity, measured at 1 × 10−11 S cm−1 at 25 °C and 4 × 10−11 S cm−1 at 70 °C. Upon heating to 100–120 °C, the structural water molecules (6 wt%) and a portion of the Cl− ions (released as HCl gas) are removed, inducing a transition to a highly disordered state. This order-to-disorder transformation leads to a significant enhancement in conductivity, increasing to 2 × 10−7 S cm−1 at 25 °C and reaching a maximum of 2 × 10−2 S cm−1 at 130 °C, with an activation energy of 1.1 eV (Fig. 9). At 70 °C, the conductivity is already 106 times higher than that of the as-synthesised form. 2H NMR of deuterated samples suggests that the order-to-disorder transformation disrupts some of the hydrogen bonds that restrict the mobility of ImH2. As a result, the mobility of the ImH2 proton carrier is significantly enhanced in the disordered state, substantially improving the overall proton conductivity.
 | ||
| Fig. 9 Temperature-dependent proton conductivity of CP/MOF glasses discussed in Section 4 (proton conduction in CP/MOF glasses). Coloured lines represent different groups of samples. The grey line represents the amorphous counterpart of [ImH2][Cu(H2PO4)2Cl]·H2O.137 The beige lines represent the crystalline and glassy (-g) forms of Cd(H2PO4)2(1,2,4-triazole)2.138 The green lines represent [Zn3(H2PO4)6](HBTA) derivatives: ZnPBTA (dehydrated), ZnPBTA-g (glass),130 and the modified glasses ZnPBTA-m1, ZnPBTA-m2, and ZnPBTA-m3.139 The brown lines represent Zn-based glasses from the direct synthesis method: (dema)0.35[Zn(H2.22PO4)3] (Zn-1), (dema)0.33[Zn0.875(H2.31PO4)3] (Zn-2), and (dema)0.45[Zn0.75(H2.35PO4)3] (Zn-3).87,140 The yellow line represents (dema)0.9[Al(H2O)1.8(H2PO4)3.9(H3PO4)1.1] (Al-CP) glass from the direct synthesis method.141 The dark blue lines represent [Zn(HPO4)(H2PO4)2](ImH2)2 derivatives: ZnPIm (crystalline),42 ZnPIm-g (glass),37 [CsHSO4]0.5[ZnPIm]0.5-g (binary glass),37 Zr(HPO4)2 modified glasses, ZnPIm0.8ZrP0.2-g and ZnPIm0.5ZrP0.5-g,142 and Rubpy-ZnPIm-g doped glass under irradiation and in the dark state (dashed lines).143 The purple lines represent melt-quenched glasses from methylsulfonic acid (MSA) incorporating Zr6O4(OH)4(4,4′-biphenyldicarboxylate-MIMS)5.4 (MIMS = (1-methyl-3-imidazolio)propane-3-sulfonate) (UiO-67-MIMS-MSA)144 and Zr6O4(OH)4(1,4-benzenedicarboxylate-APS)6 (APS = amino-1-propanesulfonate) (UiO-66-APS-MSA).145 Brackets indicate the corresponding references. | ||
Another early example of enhanced anhydrous proton conductivity following a crystal-to-glass transformation was demonstrated in the two-dimensional layered compound Cd(H2PO4)2(1,2,4-triazole)2 (Fig. 8B).138 The structure is nonporous and is isostructural with Zn(H2PO4)2(1,2,4-triazole)2, as discussed in the previous section. Mechanical vitrification induces a crystal-to-glass transformation that increases the anhydrous proton conductivity by more than two orders of magnitude, reaching 1 × 10−4 S cm−1 at 125 °C (Fig. 9). This improvement is attributed to increased acidity and isotropy of the H2PO4− groups in the glassy state, which collectively promote greater overall proton mobility. Moreover, a millimetre-sized transparent glass monolith was prepared by hot pressing at 4 GPa and 70 °C for two hours.
4.2 Proton conductivity in glasses and supercooled liquids
The crystal-to-glass transformation not only improves overall proton conductivity by enhancing molecular dynamics but also broadens the temperature range over which the high-conductivity state is maintained. This behaviour also improves processability and provides more immersive electrode-conductor interfaces. One example is the CP [Zn3(H2PO4)6(H2O)3](HBTA) (Fig. 8C), previously discussed in Section 3.2, which melts at a relatively low melting point of 114 °C, and forms a glass upon subsequent cooling.130 This glass system exhibits relatively high anhydrous proton conductivity, measured at 6.5 × 10−3 S cm−1 at 110 °C and 8 × 10−3 S cm−1 at 120 °C (Fig. 9). Compared to its crystalline counterparts, the glassy state shows significantly lower activation energies for proton transport. The values calculated were 0.59 eV in the lower conductivity regime and 0.39 eV in the higher regime, in contrast to 1.22 eV and 0.53 eV for the crystalline form. As a result, the glassy state offers substantial advantages at lower temperatures, with approximately 124-fold and 500-fold higher proton conductivities at 30 °C and 50 °C, respectively. Changes in local dynamics upon the crystal-to-glass transformation were further investigated using variable-temperature 1H magic-angle spinning (MAS) NMR. The results revealed a significant increase in the dynamics of both phosphate and HBTA moieties, even at lower temperatures, promoted by the disordered structure. As mentioned, one key advantage of reversible solid-to-liquid phase transformations lies in device fabrication. Processing the device above the melting point enables the formation of grain-boundary-free structures and promotes well-integrated electrode–electrolyte interfaces (Fig. 10A). In this study, the material was used as a solid electrolyte for proton batteries, delivering a discharge capacity of 55.4 mAh g−1 at 10 mA g−1 under anhydrous conditions. | ||
| Fig. 10 (A) Cross-sectional SEM images (150× magnification) of the electrode–solid-state electrolyte interface (left) and electrode (right). Scale bar = 150 µm. Adapted under terms of the CC-BY licence.130 Copyright 2021, N. Ma et al., published by The Royal Society of Chemistry. (B) Walden plots of glass samples. The green lines represent ZnPBTA-g, ZnPBTA-g-m1, ZnPBTA-g-m2, and ZnPBTA-g-m3.139 The brown lines represent Zn-based glasses from the direct synthesis method: (dema)0.35[Zn(H2.22PO4)3] (Zn-1), (dema)0.33[Zn0.875(H2.31PO4)3] (Zn-2), and (dema)0.45[Zn0.75(H2.35PO4)3] (Zn-3).87,140 The dark blue lines represent [Zn(HPO4)(H2PO4)2](ImH2)2 glass (ZnPIm-g) and Zr(HPO4)2-modified glass ZnPIm0.8ZrP0.2-g.142 Brackets indicate the corresponding references. | ||
Another advantage of glassy states compared to crystals is the absence of stoichiometric constraints. This flexibility allows the properties of CP/MOF glasses to be modulated by introducing additional components.85 For example, the anhydrous proton conductivity and viscosity of the original CP [Zn3(H2PO4)6(H2O)3](HBTA) can be tuned by varying the amount of H3PO4 introduced during synthesis (Fig. 8C and E). Samples with different H3PO4 equivalents, 6.0 (stoichiometric ratio), 7.75 (ZnPBTA-g-m1), 9.5 (ZnPBTA-g-m2), and 11.25 (ZnPBTA-g-m3) mol equivalents, were prepared to investigate these effects.139 In this context, excess H3PO4 acts as a network modifier, disrupting the extended coordination network during melting and inducing fragmentation by replacing bridging phosphate groups with monodentate species. This effect is clearly evidenced by the shift of viscosity and glass transition temperature toward lower temperatures with increasing modifier content. The viscosity of these modified glasses is low enough that ZnPBTA-g-m1, ZnPBTA-g-m2, and ZnPBTA-g-m3 exhibit flow behaviour comparable to molten soda-lime glass at the working temperature (η = 103 Pa s) at temperatures as low as 39, 26, and 10 °C, respectively. The maximum proton conductivities achieved were 1.38 × 10−2 S cm−1 at 120 °C (ZnPBTA-g-m1), 1.72 × 10−2 S cm−1 at 100 °C (ZnPBTA-g-m2), and 2.7 × 10−2 S cm−1 at 80 °C (ZnPBTA-g-m3), all of which exceed the 8.0 mS cm−1 measured for the original glass at 120 °C (Fig. 9).
Although the conductivity values presented here appear promising, an important question may arise: should these be classified as decoupled ion transport, or are they simply the result of proton conduction via the long-range migration of charge species in a liquid state (vehicle mechanism)? This distinction can be especially ambiguous in glassy systems, where the boundary between a solid and a viscous liquid is often unclear. However, it is possible to differentiate between superionic, pure ionic, and subionic conductivities by examining the relationship between viscosity and equivalent conductivity using a Walden plot (Fig. 10B).146,147 Equivalent conductivities that exceed the predictions of Walden's rule indicate a decoupling of charge carrier motion from the structural relaxation processes that govern viscosity. This behaviour suggests that proton transport occurs primarily via the Grotthuss mechanism. The weak correlation between conductivity and viscosity allows for tuning the viscosity without significantly compromising proton conductivity. Along the ideal Walden line, molar conductivity is directly proportional to the fluidity of the medium, indicating that ion migration is fully coupled with the structural dynamics of the system (fluid-diluted solutions). In the case of ZnPBTA-g, ZnPBTA-g-m1, ZnPBTA-g-m2, and ZnPBTA-g-m3, all samples are positioned in the region above the ideal Walden line. As the content of the network modifier increases, the conductivity trend gradually approaches the ideal Walden line. The shift is attributed to reduced viscosity and an increased contribution from the vehicle mechanism, facilitated by free phosphoric acid and the formation of smaller coordination network fragments with higher H3PO4 content. However, even with the highest modifier content, ZnPBTA-g-m3 remains above phosphoric acid on the Walden plot. This suggests that ZnPBTA-g-m3 still relies heavily on the Grotthuss mechanism for proton conductivity, similar to phosphoric acid, which exhibits approximately 97% Grotthuss contribution.148
Glasses can be prepared not only by melt-quenching but also via direct synthesis using protic ionic liquids as starting materials (Fig. 8D).87,140,141 In such cases, the protic ionic liquid (dema)(H2PO4) (dema = diethylmethylammonium) functions both as the reaction medium and a source of bridging phosphate ligands. The size and connectivity of the resulting coordination networks can be tuned by adjusting the concentration of metal ions, such as Zn2+ or Al3+, during synthesis. For Zn2+-based systems, the highest metal content reported corresponds to a Zn/H3PO4 ratio of 1:3 (Zn-1), with the composition (dema)0.35[Zn(H2.22PO4)3]. The Zn/H3PO4 ratio of 0.875:3 (Zn-2) is formulated as (dema)0.33[Zn0.875(H2.31PO4)3], while the sample with the lowest metal content contains a Zn/H3PO4 ratio of 0.75:3 (Zn-3), formulated as (dema)0.45[Zn0.75(H2.35PO4)3].87,140 The (dema)0.35[Zn(H2.22PO4)3] exhibits an anhydrous proton conductivity of 1.3 × 10−2 S cm−1 at 120 °C, which is approximately twice that of the pristine ionic liquid at the same temperature (6.5 × 10−3 S cm−1) (Fig. 9). A higher proton transport number of 0.94, compared to 0.49 for the ionic liquid, is also observed, indicating that the coordination network effectively suppresses the movement of anionic species. Reverse Monte Carlo (RMC) structural modelling, based on the experimental X-ray structural factor S(Q) and extended X-ray absorption fine structure (EXAFS) analysis, reveals that the size of the coordination network is directly influenced by the Zn/H3PO4 ratio. As the Zn2+ content decreases, the network becomes increasingly fragmented, resulting in significantly lower viscosity. A slight increase in proton conductivity is also observed, reaching 2.1 × 10−2 S cm−1 at 120 °C in (dema)0.45[Zn0.75(H2.35PO4)3]. The larger degree of fragmentation also results in a lower proton transport number due to the reduced availability of Zn2+ to restrict counterion migration. The proton transport numbers are 0.88 and 0.83 for (dema)0.33[Zn0.875(H2.31PO4)3] and (dema)0.45[Zn0.75(H2.35PO4)3], respectively. The Walden plot shows that samples with larger coordination networks deviate more strongly from the ideal line, shifting toward the superionic region (Fig. 10B). The plot also suggests that the effect of viscosity on conductivity is negligible across different Zn/H3PO4 ratios, as all samples share a similar conductivity mechanism. In addition to conductivity values, the MEAs of all samples were evaluated in H2/O2 fuel cells. The electrolyte membranes were prepared by impregnating glass samples into polytetrafluoroethylene (PTFE) membranes, which were then sandwiched between two gas catalyst-coated gas diffusion electrodes. The resulting MEAs exhibit relatively high H2/O2 fuel cell OCVs of 0.95–0.98 V at 120 °C and maximum power densities ranging from 74 to 150 mW cm−2, depending on the glass composition.
Pairing (dema)(H2PO4) with Al3+ yields a water-stable, proton-conductive CP glass, formulated as (dema)0.9[Al(H2O)1.8(H2PO4)3.9(H3PO4)1.1].141 The enhanced hydrolytic stability in this system is attributed to the octahedral coordination environment of Al3+, which maintains a coordination number of six. This geometry is more resistant to hydration compared to the four- to five-coordinate Zn2+ systems discussed earlier, due to its being more sterically constrained and having a significantly slower ligand–water exchange rate. Under anhydrous conditions, the material exhibits a proton conductivity of 2.2 × 10−2 S cm−1 at 120 °C (Fig. 9), slightly higher than that of (dema)0.45[Zn0.75(H2.35PO4)3] under the same conditions. The MEA using the sample-impregnated PTFE membrane delivers maximum power densities of 120 and 299 mW cm−2 with 20 vol% and 100 vol% O2 gas, respectively, and maintains an OCV of 0.93 V for over 20 h without degradation under anhydrous H2/O2 fuel cell testing.
Incorporating a solid acid, such as CsHSO4, into the glassy state of [Zn(HPO4)(H2PO4)2](ImH2)2 (original CP is denoted as ZnPIm, Fig. 6A) has been demonstrated to be an effective strategy for preserving the high-conductivity state of CsHSO4 below its transition temperature (Fig. 8F).37 Physical mixing of crystalline ZnPIm (Tm = 154 °C) with CsHSO4 (Tm = 206 °C) results in a composition-dependent decrease in the mixture's melting temperature, exhibiting eutectic behaviour. The onset of melting occurs at temperatures as low as 85.8 °C at the eutectic composition, [CsHSO4]0.75[ZnPIm]0.25. Upon cooling, the melt vitrifies into a glass, with a miscibility limit observed at a 1:1 mol ratio ([CsHSO4]0.5[ZnPIm]0.5-g). Adding CsHSO4 beyond this ratio leads to partial crystallisation of Cs3(HSO4)2(H2PO4) phase. [CsHSO4]0.5[ZnPIm]0.5-g exhibits a proton conductivity of 6.1 × 10−6 S cm−1 at 40 °C, which increases to 1.7 × 10−3 S cm−1 at 120 °C (Fig. 9). The temperature-dependent conductivity follows a VFT trend. Notably, the conductivity continues to increase steadily with temperature, reaching 6.3 × 10−3 S cm−1 at 180 °C, without showing a distinct jump near 141 °C. This temperature is known as the superprotonic phase transition temperature of CsHSO4. The absence of this transition indicates that the high-conductivity state is maintained throughout the entire 40–180 °C range. Moreover, the proton conductivity of the resulting mixed glass is not a simple average of the parent compounds. For instance, [CsHSO4]0.5[ZnPIm]0.5-g exhibits a conductivity of 3.8 × 10−4 S cm−1 at 90 °C, over an order of magnitude higher than that of the original ZnPIm glass and more than 5000 times greater than that of crystalline CsHSO4 at the same temperature. The enhanced conductivity observed in these systems originates from structural changes induced by the role of CsHSO4 as a network modifier for ZnPIm. Specifically, partial reorientation of bridging phosphate ligands into a monodentate configuration is likely caused by oxyanion exchange with HSO4−. This exchange disrupts the original one-dimensional chain structure of ZnPIm and helps maintain overall dynamics even at lower temperatures.
Up to this point, enhancements in proton conductivity in structurally modified CP/MOF glasses have been accompanied by decreases in viscosity. However, the introduction of a small amount of amorphous Zr(HPO4)2 (referred to as ZrP) into ZnPIm led to simultaneous increases in both proton conductivity and viscosity (Fig. 6A and 8G).142 The incorporation of ZrP resulted in a continuous increase in glass transition temperature and temperature-dependent viscosity, analogous to the effect of a secondary network former in conventional oxide glasses. Under anhydrous conditions, the sample containing a 0.2 mole fraction of ZrP (denoted as ZnPIm0.8ZrP0.2-g) exhibited enhanced proton conductivity, reaching 5.1 × 10−4 S cm−1 at 110 °C and 2.6 × 10−3 S cm−1 at 150 °C (Fig. 9). The value at 110 °C is nearly fivefold higher than that of the pristine ZnPIm glass. However, when the mole fraction of ZrP increased to 0.5, clear phase segregation was observed, leading to a significant decrease in proton conductivity due to the formation of low-conductivity Zr-rich domains.
Incorporating methylsulfonic acid (MSA) in an amount equivalent to the total pore volume of desolvated Zr6O4(OH)4(4,4′-biphenyldicarboxylate-MIMS)5.4 (MIMS = (1-methyl-3-imidazolio)propane-3-sulfonate) induces crystal melting above 120 °C in a material that is otherwise non-melting (Fig. 8I).144,149 The compound is isostructural with UiO-67 (Fig. 4C) but features zwitterionic groups anchored directly onto the linkers, with MSA occupying the pores. The resulting glass exhibits a proton conductivity of 1.96 × 10−3 S cm−1 at 80 °C (Fig. 9). A sharp increase in conductivity, associated with the glass transition point, is observed above this temperature, reaching 1.65 × 10−2 S cm−1 at 110 °C. However, these values remain lower than those of the crystalline counterpart, likely due to disrupted pore connectivity caused by structural disorder. Such behaviour is commonly observed in systems where proton conduction relies heavily on guest molecules occupying well-defined pore channels.
Another example of guest-molecule-induced melting behaviour in structurally modified MOFs was observed in MSA-incorporated Zr6O4(OH)4(1,4-benzenedicarboxylate-APS)6 (APS = amino-1-propanesulfonate) (Fig. 8J). This material is structurally analogous to Zr6O4(OH)4(1,4-benzenedicarboxylate)6 (UiO-66).145 Similar to the previous example, incorporating MSA guests results in a melting temperature of 153 °C, followed by a glass transition temperature of 143 °C in the subsequent heating scan (10 °C min−1). The crystalline sample exhibits anhydrous conductivity of 5.89 × 10−6 S cm−1 and 1.99 × 10−2 S cm−1 at −60 and 90 °C, respectively. The melt-quenched glass shows comparable conductivity values of 1.47 × 10−6 S cm−1 and 2.02 × 10−2 S cm−1 at −60 and 90 °C, respectively (Fig. 9). Importantly, the glassy state enables fabrication of a symmetric electric double-layer capacitor via a melt-infiltration strategy. Although the crystalline sample exhibits higher proton conductivity, the improved interfacial contact in the melt-infiltrated device results in approximately a twofold increase in specific capacitance, from 106.67 mF per cm2 (crystalline) to 213.87 mF per cm2 (glassy) at 25 °C.
A transition to a state with significantly lower activation energy for proton transport upon photoexcitation can be achieved by incorporating a small amount of light-responsive guests into a transparent, proton-conductive CP glass.143 In this example, 0.1 mol% of a tris(bipyrazine)-ruthenium(II) complex (Rubpz) was homogeneously mixed with crystalline [Zn(HPO4)(H2PO4)2](ImH2)2, followed by melt-quenching to produce the doped glass, Rubpz-ZnPIm-g (Fig. 6A and 8H). Both the undoped and Rubpz-doped glasses exhibit similar features in pair distribution function analysis, indicating that the incorporation of Rubpz does not disrupt the glass structure. However, spectroscopic measurements suggest partial proton transfer from phosphate groups to Rubpz even immediately after melt quenching.
Unlike most photoswitchable framework materials, where the mechanism typically relies on photoisomerisation of framework components to modulate the connectivity of guest proton carriers (usually water), the switching behaviour in this system is governed by direct proton transfer between the phosphate groups of the CP glass and the Rubpz dopant.143 This process is initiated by a shift in the pKa of Rubpz upon entering the metal-to-ligand charge-transfer (MLCT) excited state, which generates proton-deficient sites that facilitate enhanced proton transport. Upon light irradiation, the anhydrous proton conductivity increases by nearly two orders of magnitude, from 6.6 × 10−7 to 1.2 × 10−4 S cm−1 at 30 °C, in less than 5 minutes under the measurement conditions (Fig. 9). Additionally, the activation energy for proton migration decreases from 0.76 eV in the dark to 0.30 eV under photoexcitation.
It is important to note that photo-induced modulation of proton conductivity is not limited to the glassy counterparts of CPs/MOFs.150–153 However, in most reported examples, proton transport relies primarily on external humidification or saturated vapour of protic molecules. For example, optical switching between high- and low-conductivity states was observed in Cs1.1Fe0.95[Mo(CN)5(NO)]·4H2O.151 The material exhibits a high conductivity value of 4.4 × 10−3 S cm−1 at 45 °C at 100% relative humidity, with an activation energy of 0.15 eV. Upon irradiation with 532 nm light, photoisomerisation of the Mo0–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O moiety disrupts the hydrogen-bonding network responsible for proton transport, resulting in a decrease in conductivity from 1.3 × 10−3 to 6.3 × 10−5 S cm−1 at 30 °C.
O moiety disrupts the hydrogen-bonding network responsible for proton transport, resulting in a decrease in conductivity from 1.3 × 10−3 to 6.3 × 10−5 S cm−1 at 30 °C.
5. Proton conduction in covalent organic frameworks (COFs)
Most examples of COFs exhibiting proton conduction involve two-dimensional frameworks with one-dimensional nanochannels that accommodate proton carriers. Due to the high degree of pore alignment and the small channel sizes, guest molecules are prevented from aggregating and are likely to maintain liquid-like dynamics. This results in a low activation energy for proton migration. Strategies to enhance proton conductivity in COFs typically include (1) selecting appropriate guest molecules, (2) tuning pore sizes, and (3) modifying pore surface functionalities.5.1 Guest selection
As briefly mentioned in Section 2.3, an early example of proton conductivity in COF involves the incorporation of phosphoric acid into the one-dimensional channels of a structurally robust COF.111 The azole-functionalised COF, Tp-Azo (Fig. 11A), was synthesised via a Schiff base reaction between triformylphloroglucinol (Tp) and 4,4′-azodianiline (Azo). This framework demonstrates high chemical stability, maintaining its structure even after prolonged exposure to boiling water and concentrated acid (9 N HCl). Upon treatment with 1.5 M H3PO4, approximately 5.4 wt% of acid was incorporated into the pores, inducing protonation of the –N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moieties. As a result, the pelletised sample exhibits a proton conductivity of 6.7 × 10−5 S cm−1 at 67 °C under anhydrous conditions.
N– moieties. As a result, the pelletised sample exhibits a proton conductivity of 6.7 × 10−5 S cm−1 at 67 °C under anhydrous conditions.
 | ||
| Fig. 11 (A–E) Structures of COFs (and crystal structures for D and E) and incorporated guest molecules used to achieve anhydrous proton conductivity, as discussed in Section 5.1 (guest selection). | ||
N-heterocyclic compounds have also been employed as proton carriers in mesoporous COFs.154 In this case, the target framework, TPB–DMTP, was synthesised via the condensation of 1,3,5-tri(4-aminophenyl)benzene (TPB) and 2,5-dimethoxyterephthalaldehyde (DMTP) (Fig. 11B), resulting in a hexagonally aligned structure with densely packed one-dimensional mesopores. The framework exhibits a pore size of 3.26 nm and a pore volume of 1.34 cm3 g−1, along with Brunauer–Emmett–Teller (BET) and Langmuir surface areas of 2072 and 3257 m2 g−1, respectively. In terms of stability, the sample retains its crystallinity after treatment with various organic solvents, boiling water, strong acids (12 M HCl at 25 °C), and strong bases (14 M NaOH at 25 °C). Additionally, the material remains thermally stable up to 400 °C under nitrogen and 340 °C under oxygen, as indicated by thermogravimetric analysis. To enable proton conductivity, TPB–DMTP was subsequently treated with triazole and imidazole at 120 °C for seven days, yielding triazole- and imidazole-loaded frameworks. The resulting composites contained 180 wt% triazole (relative to a theoretical maximum of 186 wt%) and 155 wt% imidazole (relative to 164 wt% theoretical). The pelletised triazole-loaded TPB–DMTP exhibited proton conductivities of 7.0 × 10−4 and 1.1 × 10−3 S cm−1 at 100 and 130 °C, respectively (Fig. 12). These values were slightly lower than those of the imidazole-loaded counterpart, which showed conductivities of 1.79 × 10−3 S cm−1 and 4.37 × 10−3 S cm−1 at the same temperatures. Moreover, when the imidazole content was reduced to 82 wt%, the conductivity dropped significantly to 2.03 × 10−4 S cm−1, highlighting the critical role of guest packing within pores. The activation energies for triazole- and imidazole-loaded COFs were 0.21 and 0.38 eV, respectively. The lower conductivity in the triazole system was attributed to a relatively lower proton concentration, likely due to the presence of two lone-pair nitrogen atoms in the triazole ring, which reduce the availability of mobile protons. Although the framework remains stable at higher temperatures, thermal release of the incorporated guests from the loaded TPB–DMTP was observed to begin at 210 and 220 °C for the triazole- and imidazole-containing systems, respectively.
 | ||
| Fig. 12 Temperature-dependent proton conductivity of COFs discussed in Sections 5.1 (guest selection) and 5.2 (pore size modification). Coloured lines represent different groups of samples. The grey lines represent COFs with azole guests: TPB–DMTP with imidazole and triazole guests,154 and Td-PPI and Td-PNDI with imidazole guests.155 The dark blue lines represent TPB–DMeTP COFs with different H3PO4 loadings.156 The orange lines represent systems in which host–guest interactions are resolved using single-crystal X-ray diffraction: USTB-5o and USTB-5r COFs with H3PO4 (ref. 157) and azole-functionalised COF-300.158 The green lines correspond to a series of hexagonal-pore COFs with systematically varied pore size and volume, each incorporating phosphoric acid guests.114 The beige lines represent a series of COFs with different pore shapes, pore sizes, and pore volumes, each containing phosphoric acid guests.159 Brackets indicate the corresponding references. | ||
Substituting N-heterocyclic guests with phosphoric acid in a slightly modified COF, TPB–DMeTP (DMeTP = 2,5-dimethylterephthalaldehyde), yields a COF with significantly enhanced proton conductivity (Fig. 11C).156 TPB–DMeTP features six inward-facing nitrogen sites from CN linkages per 3.36 nm hexagonal channel, all aligned along the z direction. These nitrogen sites are expected to stabilise and confine H3PO4 molecules through hydrogen bonding interactions. Following synthesis, H3PO4 was introduced into the COF channels via vacuum impregnation, resulting in a loading of 266.6 wt%, very close to the theoretical maximum of 269.6 wt% based on pore volume. Although the pristine framework is insulating, the pelletised H3PO4-impregnated TPB–DMeTP exhibited outstanding anhydrous proton conductivity, reaching 4.43 × 10−2 S cm−1 at 100 °C and 1.91 × 10−1 S cm−1 at 160 °C (Fig. 12). This exceeds the conductivity of molten H3PO4 by almost twofold, and the performance is stable upon continuous run over 20 hours. The activation energy for proton migration was determined to be 0.34 eV. When the H3PO4 loading was reduced to 75% and 50%, the conductivity at 160 °C dropped to 3.05 × 10−2 and 9.04 × 10−3 S cm−1, respectively. The activation energy also increased to 0.40 and 0.50 eV. The relatively small decrease in proton conductivity is likely attributed to the aligned nitrogen sites, which help maintain a continuous H3PO4 network even at lower loading levels.
Although many examples of COFs (including those discussed later in Sections 5.2 and 5.3) exhibit promising conductivity, most discussions are based on the properties of polycrystalline samples. This limitation arises from the difficulty of growing single crystals of COFs. Recently, a clearer picture of how protic guests, in this case H3PO4, interact with the framework has been demonstrated in a flexible COF using single-crystal X-ray diffraction.157 A flexible COF with an interpenetrated qtz topology was synthesised through the polymerisation of tetra(phenyl)bimesityl-based tetraaldehyde and tetraamine building blocks to form USTB-5 (Fig. 11D). The material undergoes a single-crystal-to-single-crystal transformation upon reduction or oxidation of its imine linkage to amine (USTB-5r) or amide (USTB-5o), respectively. These transformations produce materials with improved stability towards strong acid (12 M HCl or 14 M H3PO4). H3PO4-impregnated USTB-5r and USTB-5o were prepared by immersing activated samples into H3PO4–tetrahydrofuran solution, followed by solvent removal under vacuum. USTB-5r and USTB-5o, with pore volumes of 1.06 and 1.26 cm3 g−1, contain 194 wt% and 231 wt% H3PO4, respectively. H3PO4-loaded USTB-5r (and USTB-5o) exhibits anhydrous proton conductivity of 5.3 × 10−2 S cm−1 (and 6.3 × 10−2 S cm−1) at 140 °C, which decreases to 3.5 × 10−2 S cm−1 (and 3.8 × 10−2 S cm−1) at 100 °C (Fig. 12). Two distinct ranges of activation energies were observed. Between 140 and 100 °C, activation energies of 0.17 and 0.20 eV were obtained for USTB-5r and USTB-5o, respectively, before increasing to 0.47 and 0.37 eV between 80 and 0 °C. To clarify the interaction between the acid guests and the COF framework, the crystal structure of H3PO4-exchanged USTB-5o was examined (Fig. 11D). The analysis revealed that a disordered H3PO4 molecule, among other disordered guests, interacts with amide oxygen atoms of the host framework, confirming the role of the amide groups as binding sites. Moreover, anisotropic proton conductivity was observed from single-crystal impedance measurements, showing values of 1.3 × 10−2 S cm−1 along the c axis (pore direction) and 5.1 × 10−4 S cm−1 along the ab plane. The work also demonstrates the applicability of COFs as solid electrolytes for H2/O2 fuel cells. USTB-5r and USTB-5o were mixed with polyvinylidene fluoride (15 wt% PVDF of H3PO4-loaded COFs), followed by hot pressing to form a membrane. The membrane was then sandwiched with Pt/C-coated gas diffusion electrodes. The dry H2/O2 fuel cell based on H3PO4-loaded USTB-5r and USTB-5o exhibited an OCV between 60–140 °C of 0.776–0.812 and 0.872–0.882 V, respectively. The maximum power density and current density of the H2/O2 fuel cells based on H3PO4-loaded USTB-5r and USTB-5o were 78 mW cm−2 and 307 mA cm−2, and 30 mW cm−2 and 108 mA cm−2, respectively, at 140 °C. The PXRD patterns of the composite membranes after testing remained comparable to the pristine samples, confirming the stability of these COFs.
A direct observation of proton-conducting pathways and mechanisms in COFs has been studied using single crystals of archetypal imine-linked COF-300, synthesised from tetrakis(4-aminophenyl)methane (TAM) and benzene-1,4-dicarboxaldehyde (BDA).158 1,2,3-Triazole, 1,2,4-triazole, and pyrazole were introduced into the single-crystalline COF through solvent-free melt-phase post-synthetic modification. This approach avoids solvent occlusion by operating above the melting point of azoles without requiring the use of a solvent. Single-crystal X-ray diffraction analysis revealed the conversion of imine bonds (CN, 1.245 (7) Å) to amine bonds (C–N, 1.415 (5) Å), along with the ligation of azoles to the COF skeleton (C–N, 1.487 (5)) (Fig. 11E). These changes were also supported by FTIR and solid-state 13C NMR. All azole-modified samples retained a structure similar to that of pristine COF-300, with the primary differences being the conversion of imine to amine bonds and a change in the dimension of the diamondoid units to accommodate the azole molecules. In the representative COF-300-1,2,3-triazole system, triazole molecules are positioned within the channels of COF-300 in helical arrangement. Additional free azole molecules are organised and stabilised through π–π stacking interactions (3.53 Å) with adjacent coordinated triazole groups, forming a static one-dimensional helical hydrogen-bond network. Robust intermolecular hydrogen bond (N5–H5⋯N7, 2.50 (2) Å) are observed between neighbouring free triazole molecules. PLATON calculations indicate that the triazole molecules fully occupy the pores, leaving no residual solvent-accessible voids. When imidazole, 2-methyl-imidazole, 2-ethylimidazole, benzimidazole, and benzotriazole were introduced as guests, they formed composites in which the azole guests were physically adsorbed through van der Waals interactions and assembled into one-dimensional columnar arrays with packing density depending on molecular size. Single-crystal proton conductivities of COF-300 with 1,2,3-triazole, 1,2,4-triazole, and pyrazole were studied under anhydrous conditions using interdigital electrodes. The one-dimensional channels intersect the measurement axis at 45°, and the contact between the electrodes and the crystal was improved by using the ionic liquid 1-butyl-3-methylimidazolium bis(trifluoromethylsulfonyl)-imide. It should be noted that anisotropic conductivity along different crystallographic axes cannot be evaluated using this technique. At 170 °C, the proton conductivities of COF-300 containing 1,2,3-triazole, 1,2,4-triazole, and pyrazole were 4.23 × 10−3 S cm−1, 4.15 × 10−3 S cm−1, and 4.35 × 10−3 S cm−1, respectively (Fig. 12). These values decreased to 2.73 × 10−3 S cm−1, 2.27 × 10−3 S cm−1, and 2.78 × 10−3 S cm−1, respectively, at 110 °C. The corresponding activation energies were calculated to be 0.153, 0.186, and 0.155 eV, respectively. In contrast, when azole guests that form composites with COF-300 (without bond formation) were used, conductivity values at least two times lower were observed, along with higher activation energies (0.228–0.385 eV). This behaviour arises from the absence of a rigid and well-defined hydrogen-bonding network. The weak intermolecular hydrogen bonds between physically adsorbed guests are susceptible to thermal disruption.
5.2 Pore size modification
Variations in pore size influence both the packing density and dynamics of proton carriers, thereby affecting proton hopping efficiency. Imidazole has also been employed as a proton carrier in two tetrahedral mesoporous polyimides with micro- and mesoporous structures, enabling proton conductivity over a wide temperature range.155 Both frameworks were synthesised via the condensation of tetrahedral tetrakis(4-aminophenyl)-methane (TAPM) with rigid dianhydride linkers, perylene dianhydride for Td-PPI and naphthalene dianhydride for Td-PNDI. The reaction yields an amorphous solid with permanent porosity. Td-PNDI exhibits a type-I N2 adsorption isotherm at 77 K, with a BET surface area of 465 m2 g−1 and major pore sizes centred around 0.8 and 1.2 nm, as determined by nonlocal density functional theory (NLDFT). In contrast, Td-PPI displays mesoporous characteristics, with a BET surface area of 325 m2 g−1 and pore sizes ranging from 3 to 13 nm. Imidazole-loaded samples were prepared through a vapour infiltration process at 120 °C for 36 hours, resulting in the incorporation of 4.9 and 6.9 imidazole molecules per TAPM unit for Td-PNDI and Td-PPI, respectively. However, the imidazole loading density in Td-PPI (0.94 g cm−3) was lower than that of Td-PNDI (1.15 g cm−3). Although the host frameworks remain stable up to 500 °C under a nitrogen atmosphere, the release of guest molecules begins at 130 and 120 °C for the imidazole-loaded Td-PNDI and Td-PPI systems, respectively. Imidazole-loaded Td-PPI exhibited higher anhydrous proton conductivity across the entire measurement range from −40 to 90 °C. At −40 °C, the proton conductivities were 4.58 × 10−7 and 2.23 × 10−6 S cm−1 for imidazole-loaded Td-PNDI and Td-PPI, respectively. These values increased linearly with temperature, reaching 9.04 × 10−5 and 3.49 × 10−4 S cm−1 at 90 °C. The corresponding activation energies were 0.33 eV for Td-PNDI and 0.30 eV for Td-PPI (Fig. 12). Overall, the lower loading density potentially leads to a higher mobility of imidazole guests in the larger mesopores of Td-PPI, resembling the behaviour observed in previously described imidazole-loaded Al(OH)(1,4-ndc) MOFs.101A direct comparison of the influence of pore size and pore volume on anhydrous proton conductivity in COFs was conducted using a series of phosphoric acid-loaded hexagonal frameworks.114 Five COFs, including ACOF-1, TTA–TFB, TTA–TFPB, TPB–DMeTP, and TPB–TMBPDA, were investigated, with pore sizes of 0.9, 1.6, 2.2, 3.3, and 3.8 nm, respectively (Fig. 13). The building blocks included C3-symmetric units such as 1,3,5-tri(4-aminophenyl)benzene (TPB), 4,4′,4″-(1,3,5-triazine-2,4,6-triyl)trianiline (TTA), and 1,3,5-triformylbenzene (TFB) as the knot component. C2-symmetric 2,2′,5,5′-tetramethoxy-[1,1′-biphenyl]-4,4′-dicarbaldehyde (TMBPDA), 2,5-dimethylterephthalaldehyde (DMeTP), and hydrazine were used as linkers, along with C3-symmetric 5′-(4-formylphenyl)-[1,1′:3′,1″-terphenyl]-4,4″-dicarbaldehyde (TFPB) and 1,3,5-triformyl-benzene (TFB). Azine linkages (–CN–NC–) were employed in ACOF-1, in contrast to the imine linkages (–CN–) used in the other COFs. All COFs possess six single-file nitrogen chains per pore, serving as anchoring sites to stabilise H3PO4 molecules. In addition to differences in pore size, the BET surface areas of ACOF-1, TTA–TFB, TTA–TFPB, TPB–DMeTP, and TPB–TMBPDA were measured to be 933, 1366, 1700, 2890, and 1657 m2 g−1, respectively, with corresponding pore volumes of 0.41, 0.61, 0.93, 1.60, and 1.31 cm3 g−1. H3PO4 was introduced into each COF using a solution-based vacuum impregnation method, with loading amounts determined based on pore volume and the density of H3PO4. Regarding anhydrous proton conductivity, the results revealed a clear trend: increasing pore size correlates with a decrease in activation energy for proton migration. The activation energies were calculated to be 0.20, 0.27, 0.46, 0.57, and 0.75 eV for COF with pore sizes of 3.8, 3.3, 2.2, 1.6, and 0.9 nm, respectively. Interestingly, the highest anhydrous proton conductivity was observed in H3PO4-loaded TPB–DMeTP, reaching 3.06 × 10−1 S cm−1 at 160 °C, nearly three times higher than that of neat H3PO4. The conductivity, on the other hand, increased progressively across the series: from ACOF-1 (6.82 × 10−5 S cm−1 at 160 °C) to TTA–TFB, TTA–TFPB, TPB–TMBPDA, and peaked at TPB–DMeTP (Fig. 12). This trend correlates with increasing pore volume. The enhanced performance is attributed to accelerated proton mobility, where acid concentration-normalised conductivity was found to increase exponentially with pore volume.
 | ||
| Fig. 13 Structures of selected COFs featuring hexagonal pores (top row) and different pore geometries (bottom row), illustrating variations in pore size, pore volume, and activation energy for proton migration. Anhydrous proton conductivity in these materials is enabled by the incorporation of H3PO4. The materials are arranged from left to right according to decreasing proton conductivity under anhydrous conditions. In general, larger pore sizes facilitate more efficient proton migration, reflected by lower activation energies, while larger pore volumes contribute to higher overall proton conductivity. | ||
In addition to examining how pore size and pore volume affect anhydrous proton conductivity in COFs with hexagonal pores, the influence of pore shape was also investigated (Fig. 13).159 A series of H3PO4-loaded COFs with trigonal, tetragonal, hexagonal, and Kagome topologies, featuring a broad pore size range from 1.1 to 6.0 nm, were examined. The series involves five COFs with distinct pore characteristics (BET surface area, pore size, and pore volume): two trigonal COFs, HFPTP–PDA (510 m2 g−1, 1.1/1.4 nm, 0.32 cm3 g−1) and HFPTP–BPDA (843 m2 g−1, 1.2/1.6 nm, 0.48 cm3 g−1); one tetragonal Py-2,2′-BpyPh COF (1710 m2 g−1, 2.5 nm, 0.78 cm3 g−1); one hexagonal TPB–DMTP COF (2900 m2 g−1, 3.3 nm, 1.34 cm3 g−1); and one Kagome Py–TMQPDA (1500 m2 g−1, 2.5/6.0 nm, 1.22 cm3 g−1). The building blocks used in the synthesis included 2,3,6,7,10,11-hexakis(4-formylphenyl)triphenylene (HFPTP), benzene-1,4-diamine (PDA), 1,1′-biphenyl-4,4′-diamine (BPDA), 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl) tetraaniline (PyTTA), [2,2′-bipyridine]-5,5′-dicarbaldehyde (2,2′-BPyDCA), 1,3,5-tri(4-aminophenyl)-benzene (TPB), 2,5-dimethoxyterephthal-aldehyde (DMTP), 2,2‴,5,5‴-tetramethoxy-[1,1′:4′,1″:4″,1‴-quaterphenyl]-4,4‴-dicarbaldehyde (TMQPDA). Similar to the previously discussed hexagonal series, pore volume played a critical role in enhancing proton conductivity. Among the COFs, hexagonal TPB–DMTP exhibited the highest anhydrous proton conductivity, reaching 5.02 × 10−2 S cm−1 at 160 °C (Fig. 12). Moreover, the activation energy for proton migration followed a trend that correlated with pore size, increasing in the following order: Py–TMQPDA (0.24 eV), TPB–DMTP (0.30 eV), Py–2,2′-BpyPh (0.45 eV), HFPTP–BPDA (0.67 eV), and HFPTP–PDA (0.78 eV). These results confirm the direct influence of pore dimensions on anhydrous proton conductivity in COFs: larger pore sizes facilitate more efficient proton migration (reflected in lower activation energies), while larger pore volumes contribute to higher overall conductivity.
5.3 Pore surface functionality
Modifying pore characteristics is another important strategy for controlling proton mobility within COFs. One approach involves the periodic alignment of sulfonic acid groups along the two-dimensional layers, which promotes proton hopping within the hexagonally arranged one-dimensional channels.113 This was demonstrated through the synthesis of TpPa-SO3H, formed by condensing 1,3,5-triformylphloroglucinol (Tp) with 2,5-diaminobenzenesulfonic acid (Pa-SO3H) (Fig. 14A). The resulting COF exhibits intrinsic, guest-free proton conductivity without the need for additional guest molecules. Under anhydrous conditions, pressed-pellet measurements show conductivities of 1.2 × 10−5 S cm−1 at 25 °C and 1.7 × 10−5 S cm−1 at 120 °C (Fig. 15). Two additional analogues were synthesised in this study: TpPa-Py and TpPa-(SO3H-Py). The former was obtained by replacing Pa-SO3H with 2,5-diaminopyridine (Pa-Py), while the latter was synthesised using an equimolar mixture of Pa-SO3H and Pa-Py. Neither framework exhibited guest-free proton conductivity under the same conditions, likely due to the absence of protogenic groups in TpPa-Py and proton immobilisation caused by pyridinic nitrogen in TpPa-(SO3H-Py). To overcome this limitation, phytic acid was introduced into the pores of all COFs. Phytic acid was chosen over phosphoric acid for two main reasons: (1) its lower volatility, which extends applicability to higher temperatures, and (2) its molecular size (∼11 Å), which is comparable to the COF pore size (14.5 Å), potentially preventing leaching from the framework. However, leaching of acid guests from the COFs upon contact with water was found to occur, with the process completed between 10 and 60 minutes. Phytic acid-loaded TpPa-SO3H exhibited only a slight improvement in proton conductivity, reaching 7.5 × 10−5 S cm−1 at 120 °C. This was not the case for TpPa-Py and TpPa-(SO3H-Py), in which 3.0 × 10−4 and 5.0 × 10−4 S cm−1 at 120 °C were achieved, respectively. The activation energies for phytic acid-loaded TpPa-Py and TpPa-(SO3H-Py) were determined to be 0.10 and 0.16 eV, respectively, indicating the effect of host–guest interactions. | ||
| Fig. 14 (A–G) Structures of selected COFs discussed in Section 5.3 (pore surface functionality). Functional groups are highlighted in blue. | ||
 | ||
| Fig. 15 Temperature-dependent proton conductivity of COFs discussed in Section 5.3 (pore surface functionality). Coloured lines represent different groups of samples. Purple lines represent COFs showing guest-free proton conductivity.112,113 Blue lines represent COFs in which the proton conductivity of guests is influenced by thiophene rings160 and pyridinic nitrogen.161 Dark red lines represent systems where proton dissociation energy of guests is influenced by functional groups on the COF framework.162,163 Black lines represent COFs containing different –CF3 contents in concave dodecagonal nanopores.164 Yellow lines represent COF systems where crystallinity and proton conductivity are influenced by triazine rings.165 Green lines represent COFs containing extended fluorocarbon alkyl chains.166–168 Orange lines represent COFs containing oxygen-containing chains.169 Brackets indicate the corresponding references. | ||
Another example of a COF featuring sulfonic acid groups anchored on the pore surface exhibited a guest-free proton conductivity of 1.52 × 10−4 S cm−1 at 120 °C in pellet form (Fig. 15).112 In this study, a fully sulfonated COF, TpBD-(SO3H)2 (Fig. 14A), was synthesised via the condensation of 2,4,6-triformylphloroglucinol (Tp) and 4,4′-diaminobiphenyl-3,3′-disulphonic acid [BD-(SO3H)2]. To enhance its proton conductivity, a protic ionic liquid, 1-methyl-3-(3-sulfopropyl)imidazolium hydrogensulfate ([PSMIm]-[HSO4]), was introduced into the pores at a loading of 25.7 wt%. This incorporation led to a significant increase in conductivity, reaching 2.21 × 10−3 S cm−1 at 120 °C. In addition to improving conductivity under anhydrous conditions, the ionic liquid also enhanced proton transport efficiency by introducing additional proton-hopping sites, as reflected by the reduced activation energy of 0.30 eV compared to 0.37 eV for the pristine COF.
The influence of thiophene rings in COFs on hydrogen bonding interactions between imine groups and imidazole guests has been investigated by comparing the proton conductivities of three isoreticular COFs: Py-BT, Py-TT, and Py-BD COFs (Fig. 14B).160 These COFs were synthesised using 1,3,6,8-tetrakis(4-aminophenyl)pyrene as the knot and 2,2′-bithiophene-5,5′-dicarboxaldehyde, thieno[3,2-b]thiophene-2,5-dicarboxaldehyde, and 4,4′-biphenyldicarboxaldehyde as the respective linkers. The calculated pore widths of Py-BT, Py-TT, and Py-BD were 2.5, 2.3, and 2.6 nm, respectively, whereas only the calculated pore volume of Py-BT was reported (1.02 cm3 g−1). Imidazole was subsequently introduced via an evaporation method, resulting in guest loadings of 49.4, 51.5, and 50.6 wt% for Py-BT, Py-TT, and Py-BD, respectively. The imidazole-loaded samples were then pressed into pellets for proton conductivity measurements. The resulting anhydrous proton conductivities at 130 °C were 2.92 × 10−3, 3.08 × 10−3, and 8.20 × 10−4 S cm−1, with activation energy values of 0.31, 0.31, and 0.32 eV, respectively (Fig. 15). Despite the nearly identical pore volumes among the samples, the presence of thiophene rings enhances the hydrogen-bond donating ability of the imine linkers. This facilitates stronger hydrogen bonding interactions with imidazole, resulting in an overall improvement in proton conductivity.
The density of pyridinic nitrogen within one-dimensional channels plays a critical role in enhancing overall proton conductivity of H3PO4 guests by promoting the formation and disruption of Ntriazine⋯H+⋯H2PO4− hydrogen-bonding pairs.161 In this study, two H3PO4-loaded triazine-based COFs were synthesised using terephthalamidine dihydrochloride as the knot and either terephthalaldehyde or 4,4′-biphenyldicarboxaldehyde as the linker, yielding frameworks with high and low pyridinic nitrogen densities, respectively (Fig. 14C). The pelletised COF with higher nitrogen (Tz-Pa) density exhibited a proton conductivity of 1.6 × 10−1 S cm−1 at 150 °C, while the COF with lower nitrogen density (Tz-BD) showed a conductivity nearly three times lower at 5.1 × 10−2 S cm−1 under the same conditions (Fig. 15). Both samples displayed comparable activation energies of 0.25 and 0.26 eV, respectively, indicating that the improved conductivity is primarily attributed to closer hydrogen-bonding sites provided by pyridinic nitrogen. Tz-Pa exhibits negligible performance decay after 18 months of storage in air.
The proton dissociation energy of guest proton carriers such as H3PO4 and 1,2,4-triazole within COF pores can be modulated by the chemical nature of functional groups decorating the pore surface.162 This effect is evaluated by monitoring changes in the dielectric properties of the incorporated proton carriers, where a noticeable increase in the dielectric constant indicates the formation of ionic species within the channels. To investigate this, COFs with different pore surface functionalities, including neutral, polar, Lewis basic, and positively charged, were synthesised, represented by TP–TAP, TP–PD, TP–DAAQ, and TP–EB, respectively. These COFs were constructed using the trigonal 2,4,6-triformylphloroglucinol (TP) aldehyde as the common knot and 2,4,6-triaminopyrimidine (TAP), p-phenylenediamine (PD), 2,6-diaminoanthraquinone (DAAQ), and ethidium bromide (EB) as linkers. The resulting BET surface areas, pore sizes, and pore volumes are as follows: 75.0 m2 g−1, 0.67 nm, 0.20 cm3 g−1 for TP–TAP; 519.0 m2 g−1, 1.20 nm, 0.40 cm3 g−1 for TP–PD; 1163.4 m2 g−1, 2.25 nm, 0.51 cm3 g−1 for TP–DAAQ; and 670.3 m2 g−1, 1.66 nm, 0.35 cm3 g−1 for TP–EB. The proton dissociation energies were further evaluated using DFT calculations. In the free state, H3PO4 and triazole require 3.86 and 4.07 eV to dissociate one proton. These values decrease upon incorporation into COFs in the following trend (H3PO4/triazole): TP–TAP (3.68 eV/3.83 eV), TP–PD (3.17 eV/3.66 eV), TP–DAAQ (2.89 eV/3.93 eV), and TP–EB (2.38 eV/2.85 eV). Notably, the lowest dissociation energies observed in TP–EB are likely due to strong coulombic interactions between the cationic framework and the anionic H2PO4−. This trend is also reflected in the dielectric constants of the doped COFs, which increase with decreasing proton dissociation energy: TP–TAP (ε = 23), TP–PD (ε = 49), TP–DAAQ (ε = 76), and TP–EB (ε = 217). Correspondingly, the maximum proton conductivity of the H3PO4-loaded COF pellets (and activation energy) values measured at 140 °C are 2.65 × 10−3 S cm−1 (0.46 eV), 5.51 × 10−3 S cm−1 (0.24 eV), 9.20 × 10−3 S cm−1 (0.22 eV), and 6.63 × 10−3 S cm−1 (0.35 eV) for TP–TAP, TP–PD, TP–DAAQ, and TP–EB, respectively (Fig. 15). TP–EB reaches a maximum proton conductivity at 2.77 × 10−2 S cm−1 at 180 °C. It should be noted that a direct comparison of the effect of surface functionality on proton conductivity remains challenging in this example due to the non-negligible differences in pore size and pore volume.
Combining cationic and anionic units in zwitterionic COFs has been demonstrated as an effective strategy to promote the deprotonation of triazole or imidazole guest molecules.163 Three zwitterionic COFs were synthesised by pairing ethidium bromide (EB) with 2,5-diaminobenzenesulfonic acid (Pa-SO3H), 4,4′-diaminobiphenyl-2,2′-disulfonic acid (BDSA), or 4,4′-diamino-2,2′-stilbenedisulfonic acid (DASD), resulting in EB-Pa-SO3H, EB-BDSA (Fig. 14D), and EB-DASD, respectively. The NLDFT pore sizes of EB-Pa-SO3H, EB-BDSA, and EB-DASD are 1.5, 1.7, and 1.7 nm, respectively. Triazole and imidazole were introduced into the COFs via vapour-phase infiltration. According to DFT calculations, the proton dissociation energy of triazole (and imidazole) significantly decreases from 5 eV (4.8 eV) in their free molecular forms to 3.2 eV (4.1 eV) in EB-Pa-SO3H, 4.1 eV (4.0 eV) in EB-BDSA, and 2.8 eV (4.0 eV) in EB-DASD. Under anhydrous conditions, the proton conductivity (and corresponding activation energy) of triazole-loaded EB-Pa-SO3H, EB-BDSA, and EB-DASD pellets were measured to be 2.82 × 10−3 S cm−1 (0.25 eV), 6.91 × 10−3 S cm−1 (0.27 eV), and 2.88 × 10−3 S cm−1 (0.28 eV), respectively (Fig. 15). For imidazole-loaded samples, the conductivity values were 4.38 × 10−2 S cm−1 (0.21 eV) for EB-Pa-SO3H, 3.33 × 10−2 S cm−1 (0.24 eV) for EB-BDSA, and 2.43 × 10−2 S cm−1 (0.20 eV) at 140 °C for EB-DASD. Compared with the azole-guest COFs discussed earlier, the COFs in this series exhibit proton conductivities that are at least an order of magnitude higher. This work proposes that the presence of zwitterionic groups in the COFs promotes deprotonation of the azole guests, thereby enhancing overall proton transport.
Simultaneously engineering both the pore shape and pore surface enabled the design of three COFs with high anhydrous proton conductivity when loaded with H3PO4.164 The pore geometry was tailored into concave dodecagonal nanopores, and multiple functional groups, including –CN–, –OH, –NN–, and –CF3, were introduced to modulate host–guest interactions. Four building blocks were employed: Azo-mNH2, Azo-CF3 & NH2, 1,3,5-tris(p-formylphenyl) benzene (TFPB), and 1,3,5-tris(4-formyl-3-trifluoromethyl)-benzene (TFPB-CF3). These were combined to yield three isostructural COFs (pore widths of ca. 1.2–1.3 nm) with increasing –CF3 content and hydrophobicity: Azo-mNH2-TFPB, Azo-CF3 & NH2-TFPB, and Azo-CF3 & NH2-TFPB-CF3 (Fig. 14E). H3PO4 was incorporated via solution-assisted grinding, followed by drying, and the resulting materials were pressed into pellets. The resulting maximum anhydrous proton conductivities (and activation energies) at 160 °C were 1.12 × 10−3 S cm−1 (0.52 eV) for Azo-mNH2-TFPB, 1.24 × 10−2 S cm−1 (0.27 eV) for Azo-CF3 & NH2-TFPB, and 2.33 × 10−2 S cm−1 (0.29 eV) for Azo-CF3 & NH2-TFPB-CF3, following the increasing trend in –CF3 concentration (Fig. 15). Although the theoretical H3PO4 capacities were 167.7, 142.7, and 131.1 mg per 100 mg of Azo-mNH2-TFPB, Azo-CF3 & NH2-TFPB, and Azo-CF3 & NH2-TFPB-CF3, respectively, the presence of –CF3 groups improves the overall proton transport of H3PO4 guests.
Enhancing interlayer interactions by incorporating planar and rigid triazine units at the core of C3-symmetric monomers improves the structural stability of triazine-based COFs.165 This design minimises out-of-plane distortions and promotes self-complementary π–π stacking. The triazine-centric COF, TPT–TPT, synthesised from 2,4,6-tris(4-formylphenyl)-1,3,5-triazine and 2,4,6-tris(4-aminophenyl)-1,3,5-triazine, exhibits higher crystallinity, thermal stability, and 9–20 times greater proton conductivity (pellet) than non-triazine COFs (benzene-based, TPB–TPB) when loaded with 18 wt% H3PO4. The higher crystallinity also leads to a significant increase in pore volume, from 0.12 to 1.01 cm3 g−1, when comparing the non-triazine and triazine COFs, respectively (Fig. 15). Under full H3PO4 loading, TPT–TPT achieves a maximum anhydrous proton conductivity of 1.27 × 10−2 S cm−1 at 160 °C, with an activation energy of 0.17 eV.
In addition to pore surface functionalisation, incorporating functional groups onto extended alkyl chains within the pores has also proven effective. In particular, hydrazone-linked COFs functionalised with perfluoroalkyl chains exhibited superhydrophobicity.168 This study evaluated the influence of perfluoroalkyl chain length on acid stability, hydrophobicity, and anhydrous proton conductivity of H3PO4-loaded COFs. Three COFs, designated as COF-Fx (x = 6, 8, or 10), were synthesised by pairing 1,3,5-triformylbenzene with fluorocarbon-based hydrazide monomers of varying chain lengths, where x corresponds to the number of carbon atoms in the perfluoroalkyl chains (Fig. 14F). The contact angles are 144° for COF-F6, 142° for COF-F8, and 141° for COF-F10, all exceeding that of polytetrafluoroethylene (120°). Upon H3PO4 loading via a grinding process, the acid molecules were stabilised by hydrogen bonding interactions with NH groups (PO⋯H–N) within the one-dimensional channels, as well as O–H⋯NC and O–H⋯F–C interactions. Pelletised COF-F6, with 62 wt% H3PO4 loading, achieved an anhydrous proton conductivity of 4.2 × 10−2 S cm−1 at 140 °C with a low activation energy of 0.09 eV. At 42 wt% loading, COF-F8 and COF-F10 exhibited comparable but significantly lower conductivities than COF-F6 (1.65 × 10−2 S cm−1 at 140 °C), potentially due to partial pore blocking by the longer side chains (Fig. 15). The conductivity of COF-F6 loaded with 42 wt% H3PO4 remains stable for at least 40 hours at 140 °C. These fluorinated COFs demonstrated proton conductivities over four orders of magnitude higher than their non-fluorinated analogues (COF-42).
Another example demonstrates that attaching fluorocarbon chains to the pores of conjugated microporous polymers (CMPs) effectively enhances both framework stability and the anhydrous proton conductivity of H3PO4 guests.167 A series of fluorinated CMPs (CMP-F4, CMP-F6, and CMP-F8) were synthesised via Sonogashira cross-coupling between 1,3,5-tris(4-ethynylphenyl)benzene and 1,4-dibromobenzene-based perfluoroalkyl monomers, where Fx denotes the fluorocarbon chain length. Among these materials, CMP-F6 (Fig. 14F) in pellet form exhibited the highest anhydrous proton conductivity, reaching 4.39 × 10−3 S cm−1 at 120 °C (activation energy of 0.35 eV) with 60 wt% H3PO4 loading (Fig. 15). At a lower loading of 45 wt% (saturation point for CMP-F8), proton conductivity at 80 °C was 8.30 × 10−4 S cm−1 for CMP-F6, while CMP-F4 and CMP-F8 showed lower values of 3.27 × 10−4 and 1.27 × 10−4 S cm−1, respectively. These results highlight a trade-off between pore accessibility for guest connectivity and hydrogen bonding interactions between the fluorocarbon chains and H3PO4.
The influence of functional groups and side chains on the interaction between COF frameworks and protic ionic liquid (PIL) guests has been evaluated using three alkyl-functionalised, isostructural COFs.166 COF-C6 and COF-F6 were synthesised via the condensation of 1,3,5-triformylbenzene with alkyl-functionalised hydrazide monomers of varying chain lengths. COF-C6 is structurally analogous to COF-F6 but features –CH2– groups in place of –CF2– units. The ionic liquid (dema)HSO4 was introduced into the COFs by a grinding method, and the resulting materials were pressed into pellets for proton conductivity evaluation. At a PIL-to-COF weight ratio of 1.0, the anhydrous proton conductivity at 140 °C reached 2.84 × 10−3 S cm−1 (activation energy: 0.38 eV) for COF-F6, compared to 3.27 × 10−5 S cm−1 (0.59 eV) for COF-C6 (Fig. 15). Upon further PIL loading (1.5), the conductivity of COF-F6 increased to 1.33 × 10−2 S cm−1 at the same temperature. The sample maintains minimal decreases in conductivity values for at least 48 hours at 120 °C. As observed in previous systems with H3PO4 guests, the presence of perfluoroalkyl chains plays a critical role in stabilising hydrogen-bonding networks and facilitating proton transport.
In the final example, the influence of oxygen density, serving as a hydrogen-bonding acceptor, along the one-dimensional channels of COFs on anhydrous proton conductivity of H3PO4 guest was investigated.169 Two COFs, PyTTA-BMTP and PyTTA-DHTA, were synthesised by pairing 4,4′,4″,4‴-(pyrene-1,3,6,8-tetrayl)tetraalkane (PyTTA) with either 2,5-bis-(2-(2-methoxyethoxy)ethoxy)-1,4-phenylenedicarboxaldehyde (BMTP) or 2,5-dihydroxytere-phthalaldehyde (DHTA), respectively. PyTTA-BMTP contains 12 oxygen atoms per pore (Fig. 14G), while PyTTA-DHTA has 4. Despite its smaller pore volume (0.64 cm3 g−1 vs. 1.05 cm3 g−1), resulting from the longer side chains, pelletised PyTTA-BMTP exhibited a higher anhydrous proton conductivity of 2.6 × 10−2 S cm−1 at 140 °C, compared to PyTTA-DHTA (9.2 × 10−3 S cm−1) (Fig. 15). The higher conductivity observed in PyTTA-BMTP confirms the contribution of higher O density to proton transport, despite a much-reduced pore volume and higher activation energy (0.22 eV vs. 0.078 eV).
6. Processability, device fabrications, and comparison with other materials
6.1 Processability and device fabrications
Apart from focusing on achieving the highest proton conductivity and the lowest possible activation energy, other key parameters such as processability, mechanical strength, gas permeability, and the long-term stability of the fabricated membrane are often neglected. However, these parameters are particularly important when considering practical applications. Thin membranes with high mechanical strength and low gas permeability allow the membrane to be fabricated as thin as possible, thereby reducing ohmic losses across the membrane, improving overall cell performance, and increasing overall stack power density.170 However, thin-film formation often involves a trade-off with increased gas permeability, which leads to greater gas crossover and can negatively affect both safety and electrochemical performance.171In this review, most CP/MOF and COF examples evaluate the proton conductivity and, in some cases, the fuel cell performance, using samples in pellet form. Samples are typically pressed using a uniaxial hydraulic press, sometimes with heat (hot pressing), to improve the interfacial contact between particles. This method is widely used because it is simple, produces monolithic samples with well-defined dimensions, and allows performance evaluation without additives. The relative packing density, compared with the theoretical density estimated from the crystal structure, can be improved in some systems through sintering,172 a process long established in oxide materials.22,23
When pelletised samples are evaluated as electrolytes for devices such as H2/O2 fuel cells,102,110,135,157 the powder samples are usually pressed together with electrode components to ensure good contact.102,110 This technique has been used in other types of devices, such as solid-state fuel cells using solid acid electrolytes30 and solid-state batteries.173 In some cases, 10–15 wt% of polyvinylidene fluoride (PVDF) is added as a binder to increase the packing density and mechanical strength of proton conductors.157,174 Prominent challenges for the practical application of pelletised electrolytes in devices are the relatively large electrolyte thickness and fuel crossover.170 The latter becomes more significant at higher operating temperatures.102,135
In the case of CP/MOF glasses, the presence of liquid-like behaviour improves the processability of these materials. Samples can be heated to higher temperatures, usually above the working temperature, where the viscosity decreases to below 103 Pa s. This reduction in viscosity allows the material to be transferred into a mould to form monoliths with well-defined dimensions.37,130 The same approach can be used to fill conductivity cells with fixed dimensions for proton conductivity measurements. In this case, sample processing can be relatively straightforward. Established industrial processes can also be adopted for the processing of CP/MOF glasses due to their temperature-dependent viscosity. When compared with pelletised polycrystalline samples, melt-quenching improves the contact between the electrode and electrolyte because the melt can infiltrate the accessible spaces within the porous electrode.130,145 However, current examples of proton-conductive CP/MOF glasses usually feature relatively low melting and glass transition temperatures. This results in relatively poor mechanical properties at the operating temperature. Current examples of H2/O2 fuel cells using CP glass electrolytes require the impregnation of glass samples into polytetrafluoroethylene (PTFE) membranes, which are then sandwiched between two catalyst-coated gas diffusion electrodes. While this approach improves the mechanical strength of the membrane, it introduces additional ohmic loss due to the insulating nature of PTFE. In addition, the relatively low viscosity at operating temperatures poses a risk of gas crossover during high-temperature operation.
Apart from the aforementioned methods, other established approaches for forming self-standing COF films may represent promising alternatives. Although these methods have not yet been discussed for COFs exhibiting anhydrous proton conductivity, they may offer potential routes for membrane fabrication. For example, the synthesis of a COF at a liquid–liquid interface has been reported to produce self-standing crystalline COF films.175–177 This includes relatively thin (13 nm), centimetre-sized (2.5 cm2) crystalline COF films with a root mean square surface roughness of 0.5 nm over an analysis area of 25 µm2.177 Another method involves casting precursor suspensions, followed by heat treatment under controlled humidity. This enables the formation of COF membranes with lateral dimensions on the decimetre scale and thicknesses as low as the submicron level within 1 hour.178 Such advances could make the large-scale production of fuel cells and electrolysers based on these framework materials feasible.
6.2 Comparison of MOFs, COFs, and other proton conductors
When comparing the progress and performance of MOFs and COFs, a large similarity in the strategies employed can be observed. In cases where anhydrous proton conductivity relies primarily on protic guest molecules, several factors must be considered: (1) the framework must be sufficiently stable towards the guest molecules; (2) pore size and pore volume play important roles in accommodating the guests; and (3) the functionality of the pore surface. With these factors in mind, COFs possess several distinct advantages.As discussed in previous sections, COFs can be designed to withstand much harsher conditions than most CPs/MOFs, as they are primarily constructed from relatively strong covalent bonds. This includes resistance to strong acids, strong bases, and elevated temperatures. For example, the framework of TPB–DMeTP COF, which is among the top-performing systems when H3PO4 guests are introduced, retains its crystallinity and porosity under a wide range of conditions, including boiling water and aqueous 12 M HCl or 14 M NaOH for at least 7 days.156 It also exhibits oxidative stability in the Fenton test and remains stable up to 440 °C under nitrogen. This stability enables the use of a wider range of protic guest molecules. In addition, pore dimensions and surface functionality can be more readily tuned in COFs through the selection of diverse organic building blocks and control over the stacking of COF layers.114,159 However, one important consideration must be noted when discussing structure–property relationships in COFs. Apart from the few cases where single-crystal structures are available, most studies rely on powder X-ray diffraction refinement. In such cases, distinguishing between different stacking sequences, degrees of crystallinity, and random sheet orientations can be difficult, making the interpretation of proton transport over longer length scales challenging. Similar limitations have also been discussed for polycrystalline COF membranes used in molecular separations.179
The closest comparison in this category would be H3PO4-doped polybenzimidazole as it can be operated to ca. 200 °C. While these systems exhibit proton conductivity at levels approaching 0.2 S cm−1 at 160 °C,25,180 which is comparable to several COFs discussed in this review, the main challenge lies in a serious loss of mechanical strength due to the “plasticising effect” of the acid dopant.180 Moreover, another important issue is acid leaching during extended operation, which can not only lead to an increase in ohmic loss but also result in fuel crossover due to the porous nature of the host material. Although these issues have not yet been widely discussed for MOFs and COFs, further studies are required before these framework materials can be used in practical applications.
For systems that exhibit anhydrous proton conductivity without requiring external guests, CPs/MOFs appear to show more prominent advantages over COF systems. Proton conductivity under these conditions requires protic moieties to be sufficiently close to form a continuous hydrogen-bonding network. In COFs, the current examples mainly rely on well-aligned, framework-embedded sulfonic acid groups as proton-conducting pathways.112,113 However, further improvement in proton conductivity would require increasing the density of sulfonic acid groups within the framework, which can be challenging to achieve. In contrast, CPs/MOFs can enable dense packing of protic moieties through the formation of directional metal–ligand arrays, in which protic groups are directly coordinated and/or closely aligned, creating extended hydrogen-bonding connectivity.129,134,174 In such systems, the protic ligands are positioned sufficiently close to facilitate long-range proton migration. As a result, a larger variety of CP/MOF examples have been reported in this category. Some of these systems also exhibit crystal-to-liquid-to-glass transformations, which can be beneficial for device processing.130
When compared with other materials that exhibit proton conductivity without requiring external guest molecules, the reported proton conductivity of the best crystalline CPs/MOFs remains almost an order of magnitude lower than that of some solid acids in the temperature range of 100–300 °C (Fig. 1C and 7).8–15 This is particularly evident in light of the recent discovery of ZrH5(PO4)3, which exhibits high proton conductivity above 0.01 S cm−1 at temperatures as low as 50 °C without a drop in conductivity associated with a phase transition.15 The compound also shows phase stability under dry conditions over a wide temperature window (0–120 °C). However, one potential advantage of certain CPs/MOFs is their stability in the presence of water. Many solid acids can readily dissolve upon contact with water or transform into lower-conductivity phases. For example, ZrH5(PO4)3 converts into α-Zr(HPO4)2·H2O when exposed to humid environments at high temperatures.15 Such transformations may occur during unexpected flooding or condensation of water resulting from operational failure. In contrast, some CPs/MOFs have demonstrated high stability even under direct contact with water. For instance, (Me2NH2)[Eu(C9H9O7P)] is thermally stable up to 300 °C and retains its crystallinity even after soaking in water for six months or boiling in water for two weeks.134 Another example is (NH4)3[Zr(H2/3PO4)3], which is thermally stable up to 200 °C and retains its crystallinity after being soaked for more than one month in aqueous solutions with pH values ranging from 0 to 12, as well as ethanol and methanol, and after boiling in water for at least two weeks.135
7. Summary and perspective
The development of novel crystalline and amorphous framework materials for anhydrous proton conduction has witnessed remarkable progress in recent years (Table 1), driven by the growing interest in solid-state energy devices and the need for materials that can operate under long-standing challenging conditions. Advances in structural design and characterisation techniques now allow us to control the local structure of these materials and create new systems that exhibit promising proton conductivity approaching or even exceeding the practical requirement (10−2 S cm−1). At the same time, the systematic synthesis of isostructural series of materials has enabled systematic investigations into the influence of individual structural modifications on proton conductivity. This is particularly evident in COF materials, where pore dimensions, functional groups, and framework stability can be independently tuned through synthesis. In systems where proton conductivity is achieved through protic guest molecules, pore size, pore volume, and surface functionality play dominant roles in achieving high proton conductivity and low activation energy. Long-range hydrogen-bonding networks are formed between pore-confined guest molecules (and, in some cases, framework-bound functional groups), which exhibit liquid-like behaviour (and conductivity) while the material remains macroscopically in a solid state. In contrast, guest-free proton conductivity requires well-aligned protic moieties positioned sufficiently close to form continuous long-range hydrogen-bonding networks. Crystal-to-liquid-to-glass transformations in CPs/MOFs have also emerged as a promising strategy not only to enhance structural dynamics and proton conductivity but also to introduce improved processability, an advantage often lacking in their crystalline counterparts.| Material | Protonic species | Conductivity [S cm−1] | Temperature [°C] | Ea [eV] | Ref. |
|---|---|---|---|---|---|
| a These samples exhibit a transition to a lower activation energy state at elevated temperatures. The activation energies (Ea) listed in the table are presented in the format: lower Ea/higher Ea.b 0.13% RH.c The compound exhibits a superprotonic transition, but only the Ea value at the high-conductivity phase is reported.d The conductivity measurements were conducted without strictly controlled dry conditions.e Anhydrous conductivity is only observed along the c-axis.f The chemical formulas described in this section do not necessarily represent the final composition after the glass transformation.g The compound undergoes an order-to-disorder transformation, but no evidence of glass characteristics has been reported.h The activation energy was not calculated for these samples due to their Vogel–Fulcher–Tammann (VFT) behaviour, instead the B values obtained from VFT fitting are provided.i The activation energy of the sample under light irradiation/dark state. | |||||
| Crystalline CPs/MOFs with guest molecules | |||||
| Al(OH)(1,4-naphthalenedicarboxylate) with imidazole | Imidazole | 2.2 × 10−5 | 120 | 0.6 | 101 |
| Al(OH)(1,4-naphthalenedicarboxylate) with histamine | Histamine | 1.7 × 10−3 | 150 | 0.25 | 117 |
| Na3(2,4,6-trihydroxy-1,3,5-benzenetrisulfonate) with 1,2,4-triazole | 1,2,4-Triazole | 5 × 10−4 | 150 | 0.34/1.8a | 102 |
| Zr6O4(OH)4(4,4′-biphenyldicarboxylate)6 with imidazole | Imidazole | 1.5 × 10−3 | 130 | 0.36 | 118 |
| Zn3(triazolate)2(1,4-benzenedicarboxylate)2 with hydroquinone | Hydroquinone | 2.65 × 10−4 | 125 | 0.18 | 119 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with H2SO4 | H2SO4 | 1 × 10−2 | 150 | 0.42b | 103 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with H3PO4 | H3PO4 | 3 × 10−3 | 150 | 0.25b | 103 |
| Zn(2-methylimidazolate)2 with EMI-TFSA | EMI-TFSA | ∼10−4 | 68 | — | 105 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with (C3N2H5)CF3SO3 | (C3N2H5)-CF3SO3 | 9 × 10−2 | 230 | 0.18 | 106 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with SA-EIMS | SA-EIMS | 1.89 × 10−3 | 150 | 0.26 | 108 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with MSA-EIMS | MSA-EIMS | 1.02 × 10−4 | 150 | 0.30 | 108 |
| Cr3F(H2O)2O(1,4-benzenedicarboxylate)3 with PTSA-EIMS | PTSA-EIMS | 2.78 × 10−4 | 150 | 0.42 | 108 |
| Mg6(2,5-dioxidiobenzene-1,4-dicarboxylate)3 with NSCFA | NSCFA | 7.8 × 10−4 | 140 | 0.23 | 120 |
| Ni6(2,5-dioxidiobenzene-1,4-dicarboxylate)3 with NSCFA | NSCFA | 2.6 × 10−4 | 140 | 0.39 | 120 |
| (C2N2H10)(C2N2H9)2Cu8Sn3S12 with HCl | C2N2H102+ | 3.62 × 10−2 | 169 | 0.2/0.34a | 128 |
| [(H3O)[(UO2)4(2-pmb)3(H2O)3]·0.5H2O with LiBr | H2O | 1.55 × 10−1 | 110 | 0.36 | 121 |
|
|||||
| Crystalline CPs/MOFs with guest-free proton conductivity | |||||
| [Zn(HPO4)(H2PO4)2](ImH2)2 | HPO42−, H2PO4−, ImH2+ | 2.6 × 10−4 | 130 | 0.47c | 42 |
| [Zn3(H2PO4)6(H2O)3](HBim) | H2PO4−, Hbim | 1.3 × 10−3 | 120 | 0.5/1.5a | 129 |
| [Zn3(H2PO4)6(H2O)3](HBTA) | H2PO4−, HBTA | 1.2 × 10−3 | 100 | 0.57/1.22a | 130 |
| [Zn(H2PO4)2(1,2,4-triazole)2] with mobile H3PO4 | H2PO4−, H3PO4 | 4.6 × 10−3 | 150 | 0.53 | 131 |
| [(Me2NH2)3(SO4)]2[Zn2(ox)3] | Me2NH2+, SO43− | 1 × 10−4 | 150 | 0.13 | 132 |
| [In(C8H6O4)2][((CH3)2NH2)(DMF)] | Me2NH2+ | 2.7 × 10−5 | 77 | 0.08 | 133 |
| [Eu2(CO3)(ox)2(H2O)2]·4H2O | H2O | 2.08 × 10−3 | 150d | 0.28 | 136 |
| (Me2NH2)[Eu(C9H9O7P)] | Me2NH2+ | 1.25 × 10−3 | 150 | 0.21e | 134 |
| (NH4)3[Zr(H2/3PO4)3] | H2PO4−, H3PO4, NH4+ | 1.45 × 10−3 | 180 | 0.26 | 135 |
|
|||||
| Proton conductivity in CP/MOF glassesf | |||||
| [ImH2][Cu(H2PO4)2Cl]·H2Og | H2PO4−, ImH2 | 2 × 10−2 | 130 | 1.1 | 137 |
| [Zn3(H2PO4)6](HBTA) glass | H2PO4−, HBTA | 8.0 × 10−3 | 120 | 0.39/0.59a | 130 |
| Modified [Zn3(H2PO4)6](HBTA)·3.5(H3PO4) | H2PO4−, HBTA | 1.72 × 10−2 | 100 | 276 K−1h | 139 |
| (dema)0.35[Zn(H2.22PO4)3] | Dema, HnPO4 | 1.3 × 10−2 | 120 | 1400 K−1h | 87 |
| (dema)0.45[Zn0.75(H2.35PO4)3] | Dema, HnPO4 | 2.1 × 10−2 | 120 | 1640 K−1h | 140 |
| (dema)0.9[Al(H2O)1.8(H2PO4)3.9(H3PO4)1.1] | Dema, HnPO4 | 2.2 × 10−2 | 120 | 1256 K−1h | 141 |
| [CsHSO4]0.5[Zn(HPO4)(H2PO4)2](ImH2)2]0.5 glass | HPO42−, H2PO4−, ImH2+, HSO4− | 6.3 × 10−3 | 180 | 257 K−1h | 37 |
| [Zn(HPO4)(H2PO4)2](ImH2)2]0.8[Zr(H2PO4)]0.2-g | HPO42−, H2PO4−, ImH2+ | 2.6 × 10−3 | 150 | 1724.7 K−1h | 142 |
| Zr6O4(OH)4(4,4′-biphenyldicarboxylate-MIMS)5.4 glass with MSA | MIMS-MSA | 1.65 × 10−2 | 110 | n/a | 144 |
| Zr6O4(OH)4(1,4-benzenedicarboxylate-APS)6 glass with MSA | APS MSA | 2.02 × 10−2 | 90 | n/a | 145 |
| Rubpz doped [Zn(HPO4)(H2PO4)2](ImH2)2]0.5 glass | HPO42−, H2PO4−, ImH2+ | 1.2 × 10−4 | 30 | 0.30/0.76i | 143 |
|
|||||
| Proton conduction in covalent organic frameworks (COFs) | |||||
| H3PO4 in Tp-azo | H3PO4 | 6.7 × 10−5 | 67 | n/a | 111 |
| Triazole in TPB–DMTP | Triazole | 1.1 × 10−3 | 130 | 0.21 | 154 |
| Imidazole in TPB–DMTP | Imidazole | 4.37 × 10−3 | 130 | 0.38 | 154 |
| H3PO4 in TPB–DMeTP | H3PO4 | 1.91 × 10−1 | 160 | 0.34 | 156 |
| H3PO4 in USTB-5r | H3PO4 | 5.3 × 10−2 | 140 | 0.17 | 157 |
| H3PO4 in USTB-5o | H3PO4 | 6.3 × 10−2 | 140 | 0.20 | 157 |
| 1,2,3-Triazole functionalised COF-300 | 1,2,3-Triazole | 4.23 × 10−3 | 170 | 0.153 | 158 |
| 1,2,4-Triazole functionalised COF-300 | 1,2,4-Triazole | 4.15 × 10−3 | 170 | 0.168 | 158 |
| Pyrazole functionalised COF-300 | Pyrazole | 4.35 × 10−3 | 170 | 0.155 | 158 |
| Imidazole in Td-PPI | Imidazole | 3.49 × 10−4 | 90 | 0.30 | 155 |
| H3PO4 in TPB–DMeTP | H3PO4 | 3.06 × 10−1 | 160 | 0.27 | 114 |
| H3PO4 in TPB–DMTP | H3PO4 | 5.02 × 10−2 | 160 | 0.24 | 159 |
| Phytic acid in TpPa-(SO3H-Py) | Phytic acid, SO3H | 5.0 × 10−4 | 120 | 0.16 | 113 |
| [PSMIm][HSO4] in TpBD-(SO3H)2 | [PSMIm][HSO4], SO3H | 2.21 × 10−3 | 120 | 0.3 | 112 |
| Imidazole in Py-TT | Imidazole | 3.08 × 10−3 | 130 | 0.31 | 160 |
| H3PO4 in triazine COF | H3PO4 | 1.6 × 10−1 | 150 | 0.25 | 161 |
| H3PO4 in TP–EB | H3PO4 | 2.77 × 10−2 | 180 | 0.35 | 162 |
| Imidazole in EB-Pa-SO3H | Imidazole | 4.38 × 10−2 | 140 | 0.21 | 163 |
| H3PO4 in Azo-CF3&NH2-TFPB-CF3 | H3PO4 | 2.33 × 10−2 | 160 | 0.29 | 164 |
| H3PO4 in TPT–TPT | H3PO4 | 1.27 × 10−2 | 160 | 0.17 | 165 |
| H3PO4 in COF-F6 | H3PO4 | 4.2 × 10−2 | 140 | 0.09 | 168 |
| H3PO4 in CMP-F6 | H3PO4 | 4.39 × 10−3 | 120 | 0.35 | 167 |
| (dema)HSO4 in COF-F6 | Dema, HSO4− | 1.33 × 10−2 | 140 | 0.34 | 166 |
| H3PO4 in PyTTA-BMTP | H3PO4 | 2.6 × 10−2 | 140 | 0.22 | 169 |
Looking forward, the continued advancement of this field will likely depend on a deeper understanding of structure–property relationships, particularly within the emerging area of COFs and glassy CPs/MOFs, as well as the development of more sophisticated synthetic strategies. As discussed in this review, structure–property relationships in many examples are derived from single-crystal structures or from structures refined using powder X-ray diffraction data. However, the corresponding properties are usually measured using bulk pellets, which include contributions from particle size, packing density, grain boundaries, electrode–sample contact, and other extrinsic factors. These additional contributions can lead to misinterpretation of the intrinsic material properties. Increasing the number of systematic studies that measure proton conductivity in single crystals, or at least distinguish between bulk and grain-boundary contributions, would help clarify the underlying structure–property relationships.
Apart from achieving the highest possible proton conductivity and the lowest activation energy, other factors such as device performance, long-term stability under operating conditions, prolonged exposure to gases, membrane processability, mechanical strength, the potential for catalyst poisoning, and material management at the end of the life cycle are also important considerations that determine the practical applicability of these promising materials. Among the studies covered in this review, only device performance and long-term stability have been evaluated in a limited number of examples. Further assessments of these factors are therefore crucial for translating these well-performing materials into practical applications.
Fabrication of these promising materials into application-ready membranes is another important aspect that requires attention. Most examples presented in this review rely on uniaxial pressing or hot pressing, which are relatively simple methods for forming monolithic samples. However, translating such approaches into practical devices with larger dimensions remains challenging. Various methods have been developed to produce homogeneous membranes, particularly for crystalline COFs,175–178 but these approaches have rarely been applied to proton-conducting systems, especially when their performance is evaluated under anhydrous conditions.
Apart from pursuing ideal materials that perform well in all aspects through advanced materials design and synthesis, another strategy is to develop composite materials that combine the advantages of different components. For example, filler materials or rigid framework scaffolds could be introduced to improve the mechanical strength of CP/MOF glasses, which exhibit promising proton conductivity at elevated temperatures but tend to soften under operating conditions. In addition, a thin hydrophobic and selective layer could be incorporated into MOF- or COF-based systems in which proton conductivity relies on protic guest molecules. Such a layer could help mitigate guest leaching, thereby preventing fuel crossover and the associated decline in performance over time.
In addition to CPs, MOFs, and COFs discussed in this review, new opportunities may arise by challenging conventional assumptions. Framework materials constructed primarily through directional yet weaker interactions, such as hydrogen-organic frameworks (HOFs) and hydrogen-bonded organic crystals (HOCs), have recently emerged as promising contenders.181 Although it was long believed that combining structural stability with high anhydrous proton conductivity in such systems would be difficult, recent studies have demonstrated otherwise. For instance, an organic crystal stabilised by helical cooperativity between hydrogen-bonded lamellae and π–π stacked columns was shown to form interconnected proton pathways, achieving an anhydrous proton conductivity of 7.39 mS cm−1 at 200 °C with a low activation energy of 0.21 eV.182 The material maintained its stability even after exposure to aqueous solutions, strong acids, and boiling water, and it was further demonstrated to be applicable in a membrane–electrode assembly (MEA) for an H2/O2 fuel cell, delivering a maximum power density of 4.68 mW cm−2 with an OCV of 0.95 V at 150 °C.
Although many challenges remain before these materials can be considered practical for real-world applications, the unique ability to integrate tuneable chemical functionalities, with atomic-level precision, into dynamic framework structures offers a highly promising platform for the next generation of solid-state proton conductors. With continued advances in design strategies and synthetic methodologies, it is only a matter of time before novel framework materials emerge that successfully combine high anhydrous proton conductivity, long-term operational stability, processability, and structural versatility.
Author contributions
The manuscript was written by N. M.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by the ICYS research fellowship, the Japan Society of the Promotion of Science (JSPS) Grant-in-Aid for Research Activity Start-up (JP24K23109) and Grant-in-Aid for Early-Career Scientists (JP25K18055), the Sumitomo Foundation Basic Science grant number 2402150, the Iketani Science and Technology Foundation grant number 0371207-A, and the Japan Science and Technology Agency (JST) PRESTO grant (JPMJPR25MB).References
- M. W. Chase, NIST-JANAF Thermochemical Tables, American Institute of Physics, Washington, DC: New York, 4th edn, 1998 Search PubMed.
- S. Meyer, A. V. Nikiforov, I. M. Petrushina, K. Köhler, E. Christensen, J. O. Jensen and N. J. Bjerrum, Int. J. Hydrogen Energy, 2015, 40, 2905–2911 CrossRef CAS.
- K. D. Kreuer, J. Membr. Sci., 2001, 185, 29–39 CrossRef CAS.
- B. E. Logan and M. Elimelech, Nature, 2012, 488, 313–319 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4586 CrossRef CAS.
- J. O. Jensen, C. Chatzichristodoulou, E. Christensen, N. J. Bjerrum and Q. Li, in Electrochemical Methods for Hydrogen Production, ed. K. Scott, The Royal Society of Chemistry, 2019, pp. 253–285 Search PubMed.
- T. Norby, Solid State Ionics, 1999, 125, 1–11 CrossRef CAS.
- C. Ramasastry and K. S. Ramaiah, J. Mater. Sci., 1981, 16, 2011–2016 CrossRef CAS.
- A. I. Baranov, L. A. Shuvalov and N. M. Shchagina, JETP Lett., 1982, 36, 459–462 Search PubMed.
- T. Norby, M. Friesel and B. E. Mallander, Solid State Ionics, 1995, 77, 105–110 CrossRef CAS.
- A. Pawłowski, Cz. Pawlaczyk and B. Hilczer, Solid State Ionics, 1990, 44, 17–19 CrossRef.
- S. M. Haile, P. M. Calkins and D. Boysen, Solid State Ionics, 1997, 97, 145–151 CrossRef CAS.
- C. R. I. Chisholm and S. M. Haile, Solid State Ionics, 2001, 145, 179–184 CrossRef CAS.
- L. S. Wang, S. V. Patel, E. Truong, Y.-Y. Hu and S. M. Haile, Chem. Mater., 2022, 34, 1809–1820 CrossRef CAS.
- S. Fop, R. Vivani, S. Masci, M. Casciola and A. Donnadio, Angew. Chem., Int. Ed., 2023, 62, e202218421 CrossRef CAS PubMed.
- K. D. Kreuer, Annu. Rev. Mater. Res., 2003, 33, 333–359 CrossRef CAS.
- R. Haugsrud and T. Norby, Nat. Mater., 2006, 5, 193–196 CrossRef CAS.
- S. Noirault, S. Célérier, O. Joubert, M. T. Caldes and Y. Piffard, Adv. Mater., 2007, 19, 867–870 CrossRef CAS.
- R. B. Cervera, S. Miyoshi, Y. Oyama, Y. E. Elammari, T. Yagi and S. Yamaguchi, Chem. Mater., 2013, 25, 1483–1489 CrossRef CAS.
- J. Hyodo, K. Kitabayashi, K. Hoshino, Y. Okuyama and Y. Yamazaki, Adv. Energy Mater., 2020, 10, 2000213 CrossRef CAS.
- H. Kawamori, I. Oikawa and H. Takamura, Chem. Mater., 2021, 33, 5935–5942 CrossRef CAS.
- K. Saito and M. Yashima, Nat. Commun., 2023, 14, 7466 CrossRef PubMed.
- K. Tsujikawa, J. Hyodo, S. Fujii, K. Takahashi, Y. Tomita, N. Shi, Y. Murakami, S. Kasamatsu and Y. Yamazaki, Nat. Mater., 2025, 24, 1949–1956 CrossRef CAS PubMed.
- K. Umeda, K. Saito, T. Honda and M. Yashima, Angew. Chem., Int. Ed., 2026, 65, e21773 CrossRef CAS.
- J. S. Wainright, J.-T. Wang, D. Weng, R. F. Savinell and M. Litt, J. Electrochem. Soc., 1995, 142, L121 CrossRef CAS.
- M. Casciola, A. Donnadio and P. Sassi, J. Power Sources, 2013, 235, 129–134 CrossRef CAS.
- L. Vilčiauskas, C. C. de Araujo and K.-D. Kreuer, Solid State Ionics, 2012, 212, 6–9 CrossRef.
- E. Fabbri, D. Pergolesi and E. Traversa, Chem. Soc. Rev., 2010, 39, 4355–4369 RSC.
- A. V. Belushkin, C. J. Carlile and L. A. Shuvalov, J. Phys.: Condens. Matter, 1992, 4, 389–398 CrossRef CAS.
- S. M. Haile, D. A. Boysen, C. R. I. Chisholm and R. B. Merle, Nature, 2001, 410, 910–913 CrossRef CAS.
- D. A. Boysen, T. Uda, C. R. I. Chisholm and S. M. Haile, Science, 2004, 303, 68–70 CrossRef CAS PubMed.
- H. Stephen, Rep. Prog. Phys., 2004, 67, 1233 CrossRef.
- S. Horike, D. Umeyama and S. Kitagawa, Acc. Chem. Res., 2013, 46, 2376–2384 CrossRef CAS.
- D.-W. Lim and H. Kitagawa, Chem. Rev., 2020, 120, 8416–8467 CrossRef CAS PubMed.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS.
- D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2015, 137, 864–870 CrossRef CAS PubMed.
- N. Ma, N. Horike, L. Lombardo, S. Kosasang, K. Kageyama, C. Thanaphatkosol, K. Kongpatpanich, K. Otake and S. Horike, J. Am. Chem. Soc., 2022, 144, 18619–18628 CrossRef CAS.
- N. Ma, S. Kosasang, E. K. Berdichevsky, T. Nishiguchi and S. Horike, Chem. Sci., 2024, 15, 7474–7501 RSC.
- P. Colomban, Solid State Ionics, 2019, 334, 125–144 CrossRef CAS.
- Z. Jiráak, M. Dlouhá, S. Vratislav, A. M. Balagurov, A. I. Beskrovnyi, V. I. Gordelii, I. D. Datt and L. A. Shwalov, Phys. Status Solidi A, 1987, 100, K117–K122 CrossRef.
- T. Norby, M. Friesel and B. E. Mallander, Solid State Ionics, 1995, 77, 105–110 CrossRef CAS.
- S. Horike, D. Umeyama, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 7612–7615 CrossRef CAS PubMed.
- T. Yamada, K. Otsubo, R. Makiura and H. Kitagawa, Chem. Soc. Rev., 2013, 42, 6655–6669 RSC.
- P. Ramaswamy, N. E. Wong and G. K. H. Shimizu, Chem. Soc. Rev., 2014, 43, 5913–5932 RSC.
- Y. Ye, L. Gong, S. Xiang, Z. Zhang and B. Chen, Adv. Mater., 2020, 32, 1907090 CrossRef CAS.
- K. Otake and H. Kitagawa, Small, 2021, 17, 2006189 CrossRef CAS PubMed.
- W. Xue, C. D. Sewell, Q. Zhou and Z. Lin, Angew. Chem., Int. Ed., 2022, 61, e202206512 CrossRef CAS PubMed.
- Y.-R. Liu, Y.-Y. Chen, Q. Zhuang and G. Li, Coord. Chem. Rev., 2022, 471, 214740 CrossRef CAS.
- Y. Guo, J. Wei, Y. Ying, Y. Liu, W. Zhou and Q. Yu, Langmuir, 2023, 39, 11166–11187 CrossRef CAS PubMed.
- D. Mukherjee, A. Saha, S. Moni, D. Volkmer and M. C. Das, J. Am. Chem. Soc., 2025, 147, 5515–5553 CrossRef CAS PubMed.
- C. Tubandt and E. Lorenz, Z. Phys. Chem., 1914, 87U, 513–542 CrossRef.
- P. M. Woodward, P. Karen, J. S. O. Evans and T. Vogt, in Solid State Materials Chemistry, Cambridge University Press, Cambridge, 1st edn, 2021, ch. 13, pp. 529–578 Search PubMed.
- K.-D. Kreuer, Chem. Mater., 1996, 8, 610–641 CrossRef CAS.
- A. I. Baranov, Crystallogr. Rep., 2003, 48, 1012–1037 CrossRef CAS.
- N. Bjerrum, Science, 1952, 115, 385–390 CrossRef CAS PubMed.
- K.-D. Kreuer, A. Rabenau and W. Weppner, Angew. Chem., Int. Ed., 1982, 21, 208–209 CrossRef.
- N. Agmon, Chem. Phys. Lett., 1995, 244, 456–462 CrossRef CAS.
- B. Brzezinski and G. Zuńdel, Faraday Discuss., 1996, 103, 363–370 RSC.
- H. Vogel, J. Phys. Z., 1921, 22, 645–646 CAS.
- G. S. Fulcher, J. Am. Ceram. Soc., 1925, 8, 339–355 CrossRef CAS.
- G. Tammann and W. Hesse, Z. Anorg. Allg. Chem., 1926, 156, 245–257 CrossRef CAS.
- M. L. Williams, R. F. Landel and J. D. Ferry, J. Am. Chem. Soc., 1955, 77, 3701–3707 CrossRef CAS.
- C. A. Angell, Solid State Ionics, 1986, 18–19, 72–88 CrossRef CAS.
- C. A. Angell, Mater. Chem. Phys., 1989, 23, 143–169 CrossRef CAS.
- R. Sonntag, R. Melzer and K. S. Knight, Physica B, 1997, 234–236, 89–91 CrossRef CAS.
- I. Sosnowska, R. Przeniosło, W. Schäfer, W. Kockelmann, R. Hempelmann and K. Wysocki, J. Alloys Compd., 2001, 328, 226–230 CrossRef CAS.
- M. Catti and R. M. Ibberson, J. Phys. Chem. B, 2002, 106, 11916–11921 CrossRef CAS.
- M. Kondo, T. Yoshitomi, H. Matsuzaka, S. Kitagawa and K. Seki, Angew. Chem., Int. Ed., 1997, 36, 1725–1727 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. Horike, S. S. Nagarkar, T. Ogawa and S. Kitagawa, Angew. Chem., Int. Ed., 2020, 59, 2–15 CrossRef PubMed.
- A. Bétard and R. A. Fischer, Chem. Rev., 2012, 112, 1055–1083 CrossRef PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- I. Stassen, N. Burtch, A. Talin, P. Falcaro, M. Allendorf and R. Ameloot, Chem. Soc. Rev., 2017, 46, 3185–3241 RSC.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120, 8468–8535 CrossRef CAS PubMed.
- J. L. Mancuso, A. M. Mroz, K. N. Le and C. H. Hendon, Chem. Rev., 2020, 120, 8641–8715 CrossRef CAS PubMed.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130–8160 CrossRef CAS PubMed.
- Q. Qian, P. A. Asinger, M. J. Lee, G. Han, K. Mizrahi Rodriguez, S. Lin, F. M. Benedetti, A. X. Wu, W. S. Chi and Z. P. Smith, Chem. Rev., 2020, 120, 8161–8266 CrossRef CAS PubMed.
- A. M. Rice, C. R. Martin, V. A. Galitskiy, A. A. Berseneva, G. A. Leith and N. B. Shustova, Chem. Rev., 2020, 120, 8790–8813 CrossRef CAS PubMed.
- S. Rojas and P. Horcajada, Chem. Rev., 2020, 120, 8378–8415 CrossRef CAS PubMed.
- A. E. Thorarinsdottir and T. D. Harris, Chem. Rev., 2020, 120, 8716–8789 CrossRef CAS.
- T. D. Bennett, J.-C. Tan, Y. Yue, E. Baxter, C. Ducati, N. J. Terrill, H. H. M. Yeung, Z. Zhou, W. Chen, S. Henke, A. K. Cheetham and G. N. Greaves, Nat. Commun., 2015, 6, 8079 CrossRef CAS PubMed.
- Y. Hirai, T. Nakanishi, Y. Kitagawa, K. Fushimi, T. Seki, H. Ito, H. Fueno, K. Tanaka, T. Satoh and Y. Hasegawa, Inorg. Chem., 2015, 54, 4364–4370 CrossRef CAS PubMed.
- N. Ma and S. Horike, Chem. Rev., 2022, 122, 4163–4203 CrossRef CAS PubMed.
- W. Chen, S. Horike, D. Umeyama, N. Ogiwara, T. Itakura, C. Tassel, Y. Goto, H. Kageyama and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 5195–5200 CrossRef CAS PubMed.
- T. Ogawa, K. Takahashi, S. S. Nagarkar, K. Ohara, Y. Hong, Y. Nishiyama and S. Horike, Chem. Sci., 2020, 11, 5175–5181 RSC.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166–1170 CrossRef PubMed.
- X. Feng, X. Ding and D. Jiang, Chem. Soc. Rev., 2012, 41, 6010–6022 RSC.
- K. Geng, T. He, R. Liu, S. Dalapati, K. T. Tan, Z. Li, S. Tao, Y. Gong, Q. Jiang and D. Jiang, Chem. Rev., 2020, 120, 8814–8933 CrossRef CAS PubMed.
- S. Kanda, K. Yamashita and K. Ohkawa, Bull. Chem. Soc. Jpn., 1979, 52, 3296–3301 CrossRef CAS.
- M. Fujishima, S. Kanda, T. Mitani and H. Kitagawa, Synth. Met., 2001, 119, 485–486 CrossRef CAS.
- Y. Nagao, R. Ikeda, S. Kanda, Y. Kubozono and H. Kitagawa, Mol. Cryst. Liq. Cryst., 2002, 379, 89–94 CrossRef CAS.
- H. Kitagawa, Y. Nagao, M. Fujishima, R. Ikeda and S. Kanda, Inorg. Chem. Commun., 2003, 6, 346–348 CrossRef CAS.
- Y. Nagao, M. Fujishima, R. Ikeda, S. Kanda and H. Kitagawa, Synth. Met., 2003, 133–134, 431–432 CrossRef CAS.
- Y. Nagao, T. Kubo, K. Nakasuji, R. Ikeda, T. Kojima and H. Kitagawa, Synth. Met., 2005, 154, 89–92 CrossRef CAS.
- D.-W. Lim and H. Kitagawa, Chem. Soc. Rev., 2021, 50, 6349–6368 RSC.
- X.-M. Li, L.-Z. Dong, S.-L. Li, G. Xu, J. Liu, F.-M. Zhang, L.-S. Lu and Y.-Q. Lan, ACS Energy Lett., 2017, 2, 2313–2318 CrossRef CAS.
- F. Yang, G. Xu, Y. Dou, B. Wang, H. Zhang, H. Wu, W. Zhou, J.-R. Li and B. Chen, Nat. Energy, 2017, 2, 877–883 CrossRef CAS.
- S. Kim, B. Joarder, J. A. Hurd, J. Zhang, K. W. Dawson, B. S. Gelfand, N. E. Wong and G. K. H. Shimizu, J. Am. Chem. Soc., 2018, 140, 1077–1082 CrossRef CAS PubMed.
- S. Bureekaew, S. Horike, M. Higuchi, M. Mizuno, T. Kawamura, D. Tanaka, N. Yanai and S. Kitagawa, Nat. Mater., 2009, 8, 831–836 CrossRef CAS.
- J. A. Hurd, R. Vaidhyanathan, V. Thangadurai, C. I. Ratcliffe, I. L. Moudrakovski and G. K. H. Shimizu, Nat. Chem., 2009, 1, 705–710 CrossRef CAS PubMed.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, D. N. Dybtsev, E. S. Shutova and V. P. Fedin, J. Am. Chem. Soc., 2012, 134, 15640–15643 CrossRef CAS PubMed.
- V. G. Ponomareva, K. A. Kovalenko, A. P. Chupakhin, E. S. Shutova and V. P. Fedin, Solid State Ionics, 2012, 225, 420–423 CrossRef CAS.
- K. Fujie, K. Otsubo, R. Ikeda, T. Yamada and H. Kitagawa, Chem. Sci., 2015, 6, 4306–4310 RSC.
- V. G. Ponomareva, S. B. Aliev, E. S. Shutova, D. P. Pishchur, D. N. Dybtsev and V. P. Fedin, RSC Adv., 2017, 7, 403–407 RSC.
- X.-L. Sun, W.-H. Deng, H. Chen, H.-L. Han, J. M. Taylor, C.-Q. Wan and G. Xu, Chem.–Eur. J., 2017, 23, 1248–1252 CrossRef CAS PubMed.
- H. Chen, S.-Y. Han, R.-H. Liu, T.-F. Chen, K.-L. Bi, J.-B. Liang, Y.-H. Deng and C.-Q. Wan, J. Power Sources, 2018, 376, 168–176 CrossRef CAS.
- Y. Yoshida and H. Kitagawa, ACS Sustain. Chem. Eng., 2019, 7, 70–81 CrossRef CAS.
- D. Umeyama, S. Horike, M. Inukai, T. Itakura and S. Kitagawa, J. Am. Chem. Soc., 2012, 134, 12780–12785 CrossRef CAS PubMed.
- S. Chandra, T. Kundu, S. Kandambeth, R. BabaRao, Y. Marathe, S. M. Kunjir and R. Banerjee, J. Am. Chem. Soc., 2014, 136, 6570–6573 CrossRef CAS PubMed.
- Y. Guo, X. Zou, W. Li, Y. Hu, Z. Jin, Z. Sun, S. Gong, S. Guo and F. Yan, J. Mater. Chem. A, 2022, 10, 6499–6507 RSC.
- S. Chandra, T. Kundu, K. Dey, M. Addicoat, T. Heine and R. Banerjee, Chem. Mater., 2016, 28, 1489–1494 CrossRef CAS.
- S. Tao and D. Jiang, J. Am. Chem. Soc., 2024, 146, 18151–18160 CrossRef CAS PubMed.
- P. Žguns, K. Klyukin, L. S. Wang, G. Xiong, J. Li, S. M. Haile and B. Yildiz, Energy Environ. Sci., 2024, 17, 5730–5742 RSC.
- D. Borgis, G. Tarjus and H. Azzouz, J. Chem. Phys., 1992, 97, 1390–1400 CrossRef CAS.
- D. Umeyama, S. Horike, M. Inukai, Y. Hijikata and S. Kitagawa, Angew. Chem., Int. Ed., 2011, 50, 11706–11709 CrossRef CAS PubMed.
- S. Liu, Z. Yue and Y. Liu, Dalton Trans., 2015, 44, 12976–12980 RSC.
- Y. Ye, X. Wu, Z. Yao, L. Wu, Z. Cai, L. Wang, X. Ma, Q.-H. Chen, Z. Zhang and S. Xiang, J. Mater. Chem. A, 2016, 4, 4062–4070 RSC.
- M. Lupa, P. Kozyra and D. Matoga, ACS Appl. Energy Mater., 2023, 6, 9118–9123 CrossRef CAS.
- K. Zhang, G.-H. Wen, X.-J. Yang, D.-W. Lim, S.-S. Bao, M. Donoshita, L.-Q. Wu, H. Kitagawa and L.-M. Zheng, ACS Mater. Lett., 2021, 3, 744–751 CrossRef CAS.
- T. Homburg, C. Hartwig, H. Reinsch, M. Wark and N. Stock, Dalton Trans., 2016, 45, 15041–15047 RSC.
- J. Cao, W. Ma, K. Lyu, L. Zhuang, H. Cong and H. Deng, Chem. Sci., 2020, 11, 3978–3985 RSC.
- J.-X. Qu, Y.-M. Fu, X. Meng, Y.-O. He, C.-J. Li, H.-X. Sun, R.-G. Yang, H.-N. Wang and Z.-M. Su, Inorg. Chem., 2023, 62, 15992–15999 CrossRef CAS PubMed.
- J.-X. Qu, Y.-M. Fu, X. Meng, Y.-O. He, H.-X. Sun, R. Yang, H.-N. Wang and Z.-M. Su, Chem. Commun., 2023, 59, 1070–1073 RSC.
- D. N. Dybtsev, V. G. Ponomareva, S. B. Aliev, A. P. Chupakhin, M. R. Gallyamov, N. K. Moroz, B. A. Kolesov, K. A. Kovalenko, E. S. Shutova and V. P. Fedin, ACS Appl. Mater. Interfaces, 2014, 6, 5161–5167 CrossRef CAS.
- K. Fujie, T. Yamada, R. Ikeda and H. Kitagawa, Angew. Chem., Int. Ed., 2014, 53, 11302–11305 CrossRef CAS PubMed.
- H.-B. Luo, L.-T. Ren, W.-H. Ning, S.-X. Liu, J.-L. Liu and X.-M. Ren, Adv. Mater., 2016, 28, 1663–1667 CrossRef CAS PubMed.
- D. Umeyama, S. Horike, M. Inukai and S. Kitagawa, J. Am. Chem. Soc., 2013, 135, 11345–11350 CrossRef CAS PubMed.
- N. Ma, S. Kosasang, A. Yoshida and S. Horike, Chem. Sci., 2021, 12, 5818–5824 RSC.
- M. Inukai, S. Horike, T. Itakura, R. Shinozaki, N. Ogiwara, D. Umeyama, S. Nagarkar, Y. Nishiyama, M. Malon, A. Hayashi, T. Ohhara, R. Kiyanagi and S. Kitagawa, J. Am. Chem. Soc., 2016, 138, 8505–8511 CrossRef CAS PubMed.
- S. S. Nagarkar, S. M. Unni, A. Sharma, S. Kurungot and S. K. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 2638–2642 CrossRef CAS.
- T. Panda, T. Kundu and R. Banerjee, Chem. Commun., 2013, 49, 6197–6199 RSC.
- Y.-S. Wei, X.-P. Hu, Z. Han, X.-Y. Dong, S.-Q. Zang and T. C. W. Mak, J. Am. Chem. Soc., 2017, 139, 3505–3512 CrossRef CAS PubMed.
- D. Gui, X. Dai, Z. Tao, T. Zheng, X. Wang, M. A. Silver, J. Shu, L. Chen, Y. Wang, T. Zhang, J. Xie, L. Zou, Y. Xia, J. Zhang, J. Zhang, L. Zhao, J. Diwu, R. Zhou, Z. Chai and S. Wang, J. Am. Chem. Soc., 2018, 140, 6146–6155 CrossRef CAS PubMed.
- Q. Tang, Y. Liu, S. Liu, D. He, J. Miao, X. Wang, G. Yang, Z. Shi and Z. Zheng, J. Am. Chem. Soc., 2014, 136, 12444–12449 CrossRef CAS PubMed.
- S. Horike, W. Chen, T. Itakura, M. Inukai, D. Umeyama, H. Asakura and S. Kitagawa, Chem. Commun., 2014, 50, 10241–10243 RSC.
- W. Chen, S. Horike, D. Umeyama, N. Ogiwara, T. Itakura, C. Tassel, Y. Goto, H. Kageyama and S. Kitagawa, Angew. Chem., Int. Ed., 2016, 55, 5195–5200 CrossRef CAS.
- N. Ma, S. Kosasang, S. Horike and H. Yamada, Angew. Chem., Int. Ed., 2025, 64, e202504618 CrossRef CAS PubMed.
- T. Ogawa, K. Takahashi, T. Kurihara, S. S. Nagarkar, K. Ohara, Y. Nishiyama and S. Horike, Chem. Mater., 2022, 34, 5832–5841 CrossRef CAS.
- K. Takahashi, T. Ogawa, T. Itakura, K. Kami and S. Horike, ACS Appl. Energy Mater., 2024, 7, 11937–11945 CrossRef CAS.
- N. Ma, H. Ando, R. Ma and T. Nakanishi, Small, 2026, 22, e14459 CrossRef CAS PubMed.
- N. Ma, S. Impeng, S. Bureekaew, N. Morozumi, M. Haga and S. Horike, J. Am. Chem. Soc., 2023, 145, 9808–9814 CrossRef CAS PubMed.
- W.-L. Xue, G.-Q. Li, H. Chen, Y.-C. Han, L. Feng, L. Wang, X.-L. Gu, S.-Y. Hu, Y.-H. Deng, L. Tan, M. T. Dove, W. Li, J. Zhang, H. Dong, Z. Chen, W.-H. Deng, G. Xu, G. Wang and C.-Q. Wan, Nat. Commun., 2024, 15, 2040 CrossRef CAS PubMed.
- G.-Q. Li, H. Gu, F. Chen, W.-L. Xue, L.-M. Chen, Y.-H. Deng, W.-X. Song, H.-L. Dong, Q.-L. Chen, G. Wang, X.-Q. Kong and C.-Q. Wan, Chem. Mater., 2026, 38, 2346–2356 CrossRef CAS.
- W. Xu and C. A. Angell, Science, 2003, 302, 422–425 CrossRef CAS PubMed.
- C. Austen Angell, Y. Ansari and Z. Zhao, Faraday Discuss., 2012, 154, 9–27 RSC.
- L. Vilčiauskas, M. E. Tuckerman, G. Bester, S. J. Paddison and K.-D. Kreuer, Nat. Chem., 2012, 4, 461–466 CrossRef PubMed.
- W.-L. Xue, W.-H. Deng, H. Chen, R.-H. Liu, J. M. Taylor, Y. Li, L. Wang, Y.-H. Deng, W.-H. Li, Y.-Y. Wen, G.-E. Wang, C.-Q. Wan and G. Xu, Angew. Chem., Int. Ed., 2021, 60, 1290–1297 CrossRef CAS PubMed.
- K. Müller, J. Helfferich, F. Zhao, R. Verma, A. B. Kanj, V. Meded, D. Bléger, W. Wenzel and L. Heinke, Adv. Mater., 2018, 30, 1706551 CrossRef PubMed.
- S. Ohkoshi, K. Nakagawa, K. Imoto, H. Tokoro, Y. Shibata, K. Okamoto, Y. Miyamoto, M. Komine, M. Yoshikiyo and A. Namai, Nat. Chem., 2020, 12, 338–344 CrossRef CAS PubMed.
- A. B. Kanj, A. Chandresh, A. Gerwien, S. Grosjean, S. Bräse, Y. Wang, H. Dube and L. Heinke, Chem. Sci., 2020, 11, 1404–1410 RSC.
- H.-Q. Liang, Y. Guo, Y. Shi, X. Peng, B. Liang and B. Chen, Angew. Chem., Int. Ed., 2020, 59, 7732–7737 CrossRef CAS PubMed.
- H. Xu, S. Tao and D. Jiang, Nat. Mater., 2016, 15, 722–726 CrossRef CAS PubMed.
- Y. Ye, L. Zhang, Q. Peng, G.-E. Wang, Y. Shen, Z. Li, L. Wang, X. Ma, Q.-H. Chen, Z. Zhang and S. Xiang, J. Am. Chem. Soc., 2015, 137, 913–918 CrossRef CAS PubMed.
- S. Tao, L. Zhai, A. D. Dinga Wonanke, M. A. Addicoat, Q. Jiang and D. Jiang, Nat. Commun., 2020, 11, 1981 CrossRef CAS PubMed.
- B. Yu, R.-B. Lin, G. Xu, Z.-H. Fu, H. Wu, W. Zhou, S. Lu, Q.-W. Li, Y. Jin, J.-H. Li, Z. Zhang, H. Wang, Z. Yan, X. Liu, K. Wang, B. Chen and J. Jiang, Nat. Chem., 2024, 16, 114–121 CrossRef CAS PubMed.
- A. Yao, C. Zhu, J. Liu, H. Xu, K. Shao, C. Sun, C. Qin, X. Wang, H. Zang, Z. Su and D. Jiang, Nat. Commun., 2026, 17, 1115 CrossRef CAS PubMed.
- S. Tao and D. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202408296 CrossRef CAS PubMed.
- S. Li, Y. Liu, L. Li, C. Liu, J. Li, S. Ashraf, P. Li and B. Wang, ACS Appl. Mater. Interfaces, 2020, 12, 22910–22916 CrossRef CAS PubMed.
- M. Liu, W.-H. Deng, X. Wang, J. Liu, S. Jin, G. Xu and B. Tan, ChemSusChem, 2022, 15, e202201298 CrossRef CAS PubMed.
- S. Chen, Y. Wu, Y. Zhang, W. Zhang, Y. Fu, W. Huang, T. Yan and H. Ma, J. Mater. Chem. A, 2020, 8, 13702–13709 RSC.
- Y. Fu, Y. Wu, S. Chen, W. Zhang, Y. Zhang, T. Yan, B. Yang and H. Ma, ACS Nano, 2021, 15, 19743–19755 CrossRef CAS PubMed.
- L. Hao, S. Jia, X. Qiao, E. Lin, Y. Yang, Y. Chen, P. Cheng and Z. Zhang, Angew. Chem., Int. Ed., 2023, 62, e202217240 CrossRef CAS PubMed.
- G. Jiang, W. Zou, Z. Ou, L. Zhang, W. Zhang, X. Wang, H. Song, Z. Cui, Z. Liang and L. Du, Angew. Chem., Int. Ed., 2022, 61, e202208086 CrossRef CAS PubMed.
- X. Wu, Z. Liu, H. Guo, Y. Hong, B. Xu, K. Zhang, Y. Nishiyama, W. Jiang, S. Horike, S. Kitagawa and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 37172–37178 CrossRef CAS PubMed.
- X. Jiang, K. Zhang, Y. Huang, B. Xu, X. Xu, J. Zhang, Z. Liu, Y. Wang, Y. Pan, S. Bian, Q. Chen, X. Wu and G. Zhang, ACS Appl. Mater. Interfaces, 2021, 13, 15536–15541 CrossRef CAS PubMed.
- X. Wu, Y. Hong, B. Xu, Y. Nishiyama, W. Jiang, J. Zhu, G. Zhang, S. Kitagawa and S. Horike, J. Am. Chem. Soc., 2020, 142, 14357–14364 CrossRef CAS PubMed.
- S. Liu, M. Liu, X. Li, Q. Xu, Y. Sun and G. Zeng, J. Mater. Chem. A, 2023, 11, 13965–13970 RSC.
- T. Norby, Nature, 2001, 410, 877–878 CrossRef CAS PubMed.
- N. K. Moehring, A. B. Mansoor Basha, P. Chaturvedi, T. Knight, X. Fan, P. N. Pintauro, M. S. H. Boutilier, K. Karan and P. R. Kidambi, Nano Lett., 2025, 25, 1165–1176 CrossRef CAS PubMed.
- E. K. Berdichevsky, K. Kanamori and S. Horike, J. Mater. Chem. A, 2025, 13, 30349–30357 RSC.
- Y. Li, S. Song, H. Kim, K. Nomoto, H. Kim, X. Sun, S. Hori, K. Suzuki, N. Matsui, M. Hirayama, T. Mizoguchi, T. Saito, T. Kamiyama and R. Kanno, Science, 2023, 381, 50–53 CrossRef CAS PubMed.
- D. Gui, X. Dai, Z. Tao, T. Zheng, X. Wang, M. A. Silver, J. Shu, L. Chen, Y. Wang, T. Zhang, J. Xie, L. Zou, Y. Xia, J. Zhang, J. Zhang, L. Zhao, J. Diwu, R. Zhou, Z. Chai and S. Wang, J. Am. Chem. Soc., 2018, 140, 6146–6155 CrossRef CAS PubMed.
- K. Dey, M. Pal, K. C. Rout, S. Kunjattu H, A. Das, R. Mukherjee, U. K. Kharul and R. Banerjee, J. Am. Chem. Soc., 2017, 139, 13083–13091 CrossRef CAS PubMed.
- Z. Ou, B. Liang, Z. Liang, F. Tan, X. Dong, L. Gong, P. Zhao, H. Wang, Y. Zou, Y. Xia, X. Chen, W. Liu, H. Qi, U. Kaiser and Z. Zheng, J. Am. Chem. Soc., 2022, 144, 3233–3241 CrossRef CAS PubMed.
- Y. Yang, Y. Chen, F. Izquierdo-Ruiz, C. Schäfer, M. Rahm and K. Börjesson, Nat. Commun., 2023, 14, 220 CrossRef CAS PubMed.
- K. Eliasson, F. Jiang, M. Åhlén, M. Strømme and C. Xu, J. Am. Chem. Soc., 2025, 147, 29271–29281 CrossRef CAS PubMed.
- J. L. Fenton, D. W. Burke, D. Qian, M. Olvera de la Cruz and W. R. Dichtel, J. Am. Chem. Soc., 2021, 143, 1466–1473 CrossRef CAS PubMed.
- X. Li, H. Ma, P. Wang, Z. Liu, J. Peng, W. Hu, Z. Jiang, B. Liu and M. D. Guiver, Chem. Mater., 2020, 32, 1182–1191 CrossRef CAS.
- X. Song, Y. Wang, C. Wang, D. Wang, G. Zhuang, K. O. Kirlikovali, P. Li and O. K. Farha, J. Am. Chem. Soc., 2022, 144, 10663–10687 CrossRef CAS PubMed.
- C. Jia and F. Zhang, Adv. Mater., 2025, 37, e10756 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |