
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Regioselective [3 + 2] cycloaddition reactions of the phosphorus and arsenic analogues of the thiocyanate anion
Marc Baltrun a,
David Hanneberga,
Florian Hetta,
Michael Seidla,
Florian Weigend*bc and
Stephan Hohloch*a
a,
David Hanneberga,
Florian Hetta,
Michael Seidla,
Florian Weigend*bc and
Stephan Hohloch*a
aDepartment for General, Inorganic and Theoretical Chemistry, University of Innsbruck, Innrain 80 – 82, 6020 Innsbruck, Austria. E-mail: Stephan.Hohloch@uibk.ac.at
bInstitute for Quantum Materials and Technologies (IQMT), Karlsruhe Institute of Technology (KIT), Kaiserstr. 12, 76131 Karlsruhe, Germany. E-mail: Florian.Weigend@kit.edu
cFachbereich Chemie und Wissenschaftliches Zentrum für Materialwissenschaften (WZMW), Philipps-Universität Mar-burg, Hans-Meerwein-Straße 4, 35043 Marburg, Germany
First published on 24th April 2026
Abstract
Salt metathesis reactions of tris-amide zirconium iodide complex ([NRR′]3ZrI (1-I) (with NRR′ = 3,5-Xylyl-tert-butylamide, N(Xyl)(tBu)) and sodium 2-phosphaethynthiolate (Na(diox)3SCP) or 2-arsaethynthiolate (Na(diox)3SCAs) in THF result in a [3 + 2] cycloaddition of two SCE (E = P, As) units forming five membered 2-thio-1,3,4-thiadiphosphole and 2-thio-1,3,4-thiadiarsole heterocycles with an exocyclic sulfur atom, bridging two zirconium fragments (general formula (N(RR′)3Zr(κ-C,S-(SCE)2-Zr(NRR′)3 with E = P (2-PP) or As (3-AsAs)). The reactions are regioselective and only the P,P/As,As isomers are formed in THF and quantum chemical investigations suggest a concerted ring formation in line with a [3 + 2] cycloaddition reaction. Switching the solvent to toluene, salt metathesis with NaSCP results in the selective formation of the second possible [3 + 2] cycloaddition regioisomer 2-SP, with a 3-thio-1,2,4-thiadiphosphole bridge, while for NaSCAs both regioisomers with an As–As bond (3-AsAs) and an S–As bond (3-SAs; 3-thio-1,2,4-thiadiarsole bridge) are observed. Attempts to obtain the free diphosphole/diarsole rings using methyl triflate resulted in the cleavae of only one zirconium center, giving access to the triflate complex 1-OTf and the mono-metallated diphospholes/diarsoles 4-PP, 4-PS and 5-AsAs respectively.
Introduction
The chemistry of cyanates has unarguably led to the development of a plethora of milestones in chemical history, creating a large variety of textbook examples.1,2 For instance, cyanates were used to introduce the concepts of constitution isomerism by Liebig and Gay-Lussac on silver cyanate (AgOCN) vs. silver fulminate (AgONC),3 the theory of ambiphilic ligands using thiocyanates [SCN]−4 and Wöhler's connection between inorganic and organic chemistry by the synthesis of urea from ammonium cyanate.5 Since the early days of cyanate anions, chemists endeavored the exchange of either oxygen or nitrogen in the [OCN]− anion by heavier analogues of the chalcogen or pnictide series, which led among others to the development of the selenocyanate [SeCN]−6 and the tellurocyanate [TeCN]− anions.7 Turning to pnictogen atom exchange, one milestone was the synthesis of the phosphaethynolate anion [OCP]−. Its synthesis being already indicated in 1894 (but falsely identified as Na(CP)),8 Becker and co-workers were the first to describe its targeted synthesis in the form of [Li(DME)2][OCP].9 Shortly after, Westerhausen et al. synthesized the bis-[OCP]− salts of the alkaline earth metals Mg, Ca, Sr, and Ba with the general formula [M(DME)2](OCP)2] (M = Alkaline Earth Metal).10 However, these salts were not stable9,10 and it took another decade until Grützmacher11 and Goicoechea12 introduced the stable sodium and potassium salts, which led to a renaissance of heavy cyanate chemistry and a flourishing (phosphorus) chemistry was introduced around this molecule in the past decade.13–16 Soon after, Goicoechea and Hinz also introduced the arsenic congener [OCAs]−17 which has also been widely used in (coordination) chemistry.2 Finally, in 2018, Goicoechea, Tambornino and Hinz introduced a general method to also synthesize the heaviest cyanates known so far by introducing sulfur or selenium,18 resulting in the synthesis of the phosphaethynthiolate [SCP]−19 (also originally synthesized by Becker as its Li(DME)3 salt),20 the arsaethynthiolate [SCAs]−, the phosphaethynselenolate [SeCP]− and the arsaethynselenolate [SeCAs]− anions (Fig. 1, top). Contrasting the chemistry of their oxygen congeners ([OCP]− and [OCAs]−),2,14,15 almost no coordination chemistry of these anions has been described so far. In fact, except for one example from our group (Fig. 1, bottom, vide infra), only two other examples have been reported.19,21 The first example of [SCP]− coordination was mentioned by Goicoechea and co-workers,19 describing its (unselective) coordination towards tungsten, giving the coordination isomers A and A′, which cannot be separated. The second example was reported only recently by Braunschweig and co-workers,21 showing the stable κ1-S coordination of the [SCP]− anion towards a NHC/CAAC stabilized borane B. Notably, for the even heavier [SCAs]− anion, no examples on any reactivity or coordination has been reported yet. However, theoretical calculations have shown that these anions should exhibit prominent reactivity in a variety of cycloaddition reactions.22 | ||
| Fig. 1 Matrix of known [ChCPn]− (heavy) cyanate anions with Ch = chalcogen atom, O, S, Se, Te and Pn = Pnictogen atom, N, P, As) (top). Examples of phosphaethynthiolate complexes reported (bottom). | ||
We have recently synthesized the first example of a stable lanthanide complex of the [SCP]− anion (Complex C, Fig. 1),23 using a bis-PN chelated lanthanide center.24 Therein, the [SCP]− anion was neither found to adopt a κ1-S nor a κ1-P end-on coordination mode, but instead an unprecedented η3-side-on coordination mode was found.23 The complex was indefinitely stable and reacted with cyclic alkyl amino carbenes (CAACs) under the formation of a CAAC-stabilized heavy fulminate-type anion in complex D (Fig. 1), which stated the first defined reactivity of a heavier homologue of the thiocyanate anion. Inspired by these results we further aimed to explore the coordination motifs and chemistry of the phosphaethynthiolate anion ([SCP]−) as well as its arsenic analogue, the arsaethynthiolate anion ([SCAs]−) towards early transition metals, namely zirconium.18 Here, we report the unprecedented and regioselective [3 + 2] cycloaddition reaction between two heavy cyanate anions, giving access to thiadiphosphole and thiadiarsole bridging ligands, as well as the attempts to isolate their “free” form through cleavage from the zirconium centers using methyl triflate.
Results and discussion
To further examine the chemistry of the heavy thiocyanates we synthesized the tetra-coordinated zirconium amide complex (NRR′)3ZrI (1-I) (R = tBu, R′ = Xylyl), supported by the amide ligand 3,5-Xylyl-tert-butyl-amide.25 The complex can be synthesized in two steps, from the reaction of the lithium amide with ZrCl4 and subsequent exchange of the remaining chloride ligand by iodide using trimethylsilyl iodide (yield 60%, Scheme 1). Salt metathesis of the iodo complex 1-I with Na(diox)3SCP in toluene resulted in the initial formation of a new species showing a 31P NMR resonance at −30.6 ppm. Given the similarity of this value to complex C (−44.9 ppm),23 we attribute this resonance to the SCP complex 1-SCP (Scheme 1). Despite numerous attempts we have not been able to grow any crystals of this species, since it only exists “transiently” and reacts to a new compound 2-SP within several minutes, showing two 31P{1H} NMR resonances at intense low-field shifts of 305.2 and 276.7 ppm with a 2JPP coupling of 56 Hz (Fig. S32). In addition, the 1H NMR (Fig. S30) reveals two signal sets for the ligand, indicating the formation of a dimeric species in solution. This is further corroborated by the observation of two distinct 13C{1H} NMR resonances at 235.6 (doublet, 1JCP = 103.3 Hz) and 199.4 ppm (doublet of doublets, 1JCP = 86.1 Hz; 1JCP = 97.5 Hz; Fig. S31). Comparison of these NMR signals to the literature showed, that these are in the similar range of a 5-(trimethylsilyl)-3-((trimethylsilyl)thio)-1,2,4-thiadiphosphole E-PS (Scheme 2)26 originally synthesized by Appel and Moors starting from CS2, Li[P(SiMe3)2] and TMS-Cl. Furthermore, calculations on the 31P NMR shifts (exact two-component decoupling (X2C) including spin–orbit coupling,27 see SI for further information on calculations) of such a putative 3-thio-1,2,4-thiadiphosphole gave values of 300 and 293 ppm, and a 2JPP coupling constant of 55 Hz, which agrees well with the observed coupling constant of 56 Hz (vide supra).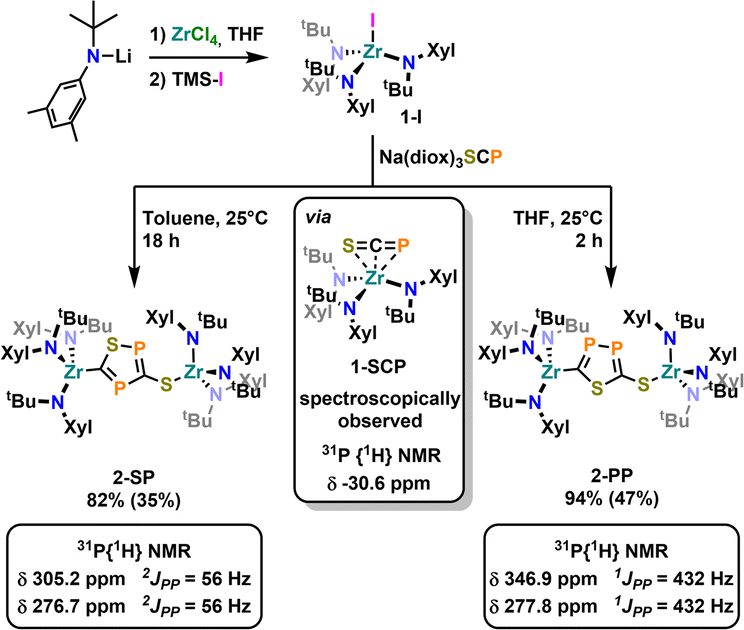 | ||
| Scheme 1 Synthesis of the iodide complex 1-I acting as a starting material for salt metathesis reaction in this manuscript, along with the regioselective cyclisation reactivity observed in its salt metathesis reaction with Na(diox)3SCP leading to the two heterocycles 2-SP and 2-PP. Yields in brackets indicate crystalline yields. | ||
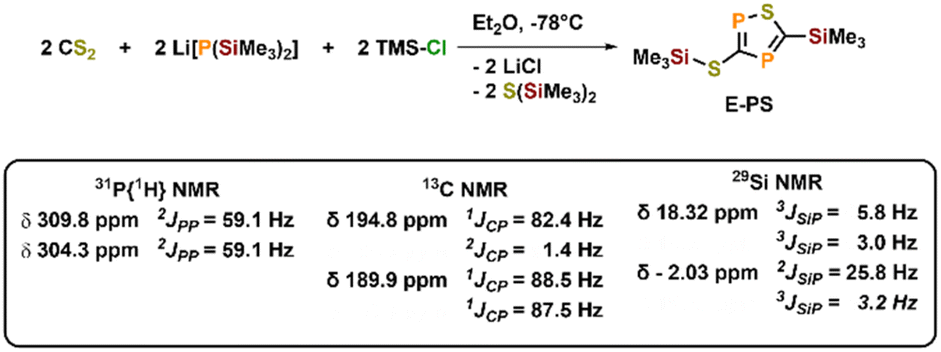 | ||
| Scheme 2 Synthesis of a silylated 3-thio-1,2,4-thiadiphosphole E-PS reported by Appel and Moors in 1986. | ||
The formation of the 3-thio-1,2,4-thiadiphosphole heterocycle was confirmed by X-ray diffraction analysis of single crystals of 2-SP grown by slow evaporation of a diethyl ether solution at room temperature (Fig. 2, bottom). The analysis revealed the presence of a five-membered heterocycle with an exo-cyclic sulfur atom, which most likely forms via an autogenic [3 + 2] cycloaddition reaction of a heavy cyanate. The cycle bridges two zirconium centers in an κ1-S and a κ1-C bound fashion with Zr1–S1 and Zr1–C2 distances of 2.554(1) and 2.297(8) Å respectively. The latter is in the range of typical Zr–Caryl/Zr–Calkyl distances,28 which is in line with a carbanionic donor atom. As indicated by 31P{1H} NMR spectroscopy, the two phosphorus atoms are separated by a carbon atom (C1), which results in the observed 2JPP coupling of 56 Hz (vide supra). The phosphor sulfur P1–S2 distance is 2.074(5) Å, which is in between a P–S single and double bond (P–S = 2.10–2.31 Å;29,30 P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) S = 1.95–2.03 Å,31 indicating electronic delocalization within the cycle. This is further supported by the calculation of localized32 molecular orbitals (LMOs, Fig. S90, for details of the calculation see SI): they reveal five σ-type orbitals representing the single bonds in the ring, and additionally three π-type orbitals. Two of them represent π-bonds mainly between one P and the neighbored C atom, but with a certain delocalization to the second P atom. The third one represents the free electron pair at S, but again somewhat delocalized to the neighbors. Overall, this suggests “close to double” P–C bonds and “more than single” for the other bonds in the ring. The C–S bond to the exocyclic sulfur atom was found to be 1.752(8) Å, which is representative for a C–S single bond,29,33
S = 1.95–2.03 Å,31 indicating electronic delocalization within the cycle. This is further supported by the calculation of localized32 molecular orbitals (LMOs, Fig. S90, for details of the calculation see SI): they reveal five σ-type orbitals representing the single bonds in the ring, and additionally three π-type orbitals. Two of them represent π-bonds mainly between one P and the neighbored C atom, but with a certain delocalization to the second P atom. The third one represents the free electron pair at S, but again somewhat delocalized to the neighbors. Overall, this suggests “close to double” P–C bonds and “more than single” for the other bonds in the ring. The C–S bond to the exocyclic sulfur atom was found to be 1.752(8) Å, which is representative for a C–S single bond,29,33
 | ||
| Fig. 2 Molecular structures of the two possible regioisomers 2-PP (top) and 2-SP (bottom) observed after the salt metathesis reaction between 1-I and Na(diox)3SCP in THF or toluene respectively. Hydrogen atoms are omitted for clarity, all ellipsoids are shown at a probability level of 50%. | ||
It should be noted at this point of the discussion, that Grützmacher34 and Goicoechea,35 have previously reported related cyclization reactions of the phosphaethynthiolate anion [OCP]−. However, contrasting the present case, neither of them led to thermally (or photo-) stable cyclic compounds and both examples decompose within days. Grützmacher reports the NHC-mediated coupling of two phosphaethynolate anions to obtain compound F (Fig. 3), which however can only be selectively obtained if the reaction is performed at very low-temperature (−78 °C) and decomposes within a few days.34 If the reaction is performed at room temperature, the zwitter ionic species G is observed as a main product. Turning to Goicoechea's examples,35 the group reported the synthesis of the bromoborane H and its thermal conversion (100 °C) in the presence of Na[OCP] to I-PP within 5 days, in which new P–P bond is formed (Fig. 3). However, I-PP was found to be highly light sensitive and decomposes under concomitant CO elimination to the new diphosphirene J. The authors spectroscopically see an intermediate in this transformation, which they computationally assign as the photo-isomerized product I-OP. Nevertheless, as noted, the authors were not able to structurally characterize and unambiguously validate this intermediate and its instability towards light preluded its isolation.
 | ||
| Fig. 3 Previously reported couplings of the phosphaethynolate anion [OCP]− mediate by NHCs or boranes (Ar = 2,6-diisopropylphenyl). | ||
Inspired by the fact that in related [OCP]− cyclization reactions a P–P heterocycle is postulated as a transitional product, we questioned if a selective synthesis of the second regioisomer of the autogenic [3 + 2] cycloaddition reaction with a 2-thio-1,3,4-thiadiphosphole bridge (2-PP) instead to the observed 3-thio-1,2,4-thiadiphosphole bridge in 2-SP is possible. A cyclisation to the putative regioisomer 2-PP would also be highly beneficial as the synthesis of related thia-1,2-diphospholes has so far not been accomplished selectively.36 Targeting the synthesis of this species 2-PP, we found that if the salt metathesis reaction between 1-I and Na(diox)3SCP is performed in THF instead of toluene, the reaction almost instantly gives access to a new product (Scheme 1). Notably, the 31P{1H} NMR shows the clean formation of a new product showing distinct low-field shifts at 346.8 and 276.8 ppm with a 1JPP coupling of 432 Hz (Fig. S25), which is strongly indicative of the successful formation of 2-PP. Quantum chemical calculations of a putative 2-thio-1,3,4-thiadi-phosphole proposed 31P NMR shifts of 361 and 280 ppm, with a 1JPP coupling constant of 422 Hz, well-fitting the observed values (vide supra; See SI for further information on NMR data calculations). Similar to the 1H NMR of 2-SP, the 1H NMR of the reaction in THF (Fig. S23) shows the presence of two signal sets for the ligand, further supporting the formation of a dimeric species in solution. Furthermore, the 13C NMR spectrum of 2-PP shows a doublet at 227.8 (1JCP = 117.2 Hz) and a doublet of doublets at 195.5 ppm (1JCP = 93.3 Hz; 2JCP = 6.7 Hz; Fig. S24). Ultimate proof for the formation of the regioisomer 2-PP was delivered by X-ray diffraction analysis on single crystals grown from slow evaporation of a diethyl ether solution at room temperature (Fig. 2, top). This revealed the formation of a new P–P bond showing a distance of 2.123(3) Å, which is in between a P–P single and double bond (P–P = 2.23–2.26 Å,37 PP = 2.03–2.05 Å38) also indicating an electronic delocalization in the heterocycle (compare P–P in cyclo-P5 ligands: 2.11 Å in (P5)FeCp,39 2.14–2.16 Å in [(P5)2Ti]2-40 and 2.12–2.13 Å substituted [cylco-C2RP3]− ligands41). Beyond this, the other bonding parameters are comparable to the regioisomer 2-SP and can be found in the SI (Tables S1 and S2). Notably all attempts to convert 2-SP into 2-PP (or vice versa) have failed and once formed, the two heterocycles are thermally stable up to at least 100 °C in solution over days.
With these results in hand, we wanted to investigate whether this reactivity translates to even heavier cyanates. Thus, we synthesized the arsaethynthiolate anion [SCAs]− in the form of Na(diox)3SCAs.18 To our delight, if the reaction between 1-I and Na(diox)3SCAs is performed in THF (Scheme 3) the formation of a clean single product is observed by 1H NMR spectroscopy (Fig. S37). The 13C{1H} NMR resonances were found at 241.3 and 206.1 ppm respectively (Fig. S38). X-ray diffraction analysis performed on single crystals grown from a concentrated hexane solution at room temperature, revealed solely the formation of the As–As isomer 3-AsAs showing an As–As distance of 2.3353(11) Å (Fig. 4). Similar to 2-PP, the As–As distance lies in between an As–As single and double bond (As–As = 2.42–2.48 Å;42 AsAs = 2.26–2.31 Å).43,44 Similarly, the C–As bonds C1–As1 (1.862(10) Å) and C2–As2 (1.839(10) Å) are in between a single and a double bond (C–As = 1.98–2.07 Å;43,45 CAs = 1.81–1.88 Å;46 arsabenzene derivatives: 1.83–1.95 Å;47 arsa-cyclopentadienyl ligands 1.89–1.90 Å).48 Calculated LMOs (Fig. S90) and their atomic contributions are very similar to that of the corresponding P compounds and thus confirm these findings. The Zr1–S1 and the Zr1–C1 distance are found to be 2.570(2) and 2.294(8) Å and are comparable to 2-SP and 2-PP. With this result in hand, the final question was, if we could also selectively form the regioisomer 3-SAs. Unfortunately, if the salt metathesis reaction between 1-I and Na(diox)3SCAs is performed in toluene (which yielded the selective formation of 2-SP) a mixture of two different products is observed, in a ratio of 53![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :47 (3-AsAs:3-SAs) by NMR spectroscopy (Scheme 3; Fig. S33 and S34). The major species could thereby be identified as 3-AsAs, wherefore we attribute the second species to the potential regioisomers 3-SAs. Carbon NMR analysis revealed the signals assigned for 3-AsAs (vide supra), and two additional singlets at 263.4 and 229.9 ppm, which we attribute to the 3-SAs regioisomer (Fig. S45). X-ray diffraction analysis of single crystals grown form diethyl ether further confirmed the presence of 3-AsAs in the crystal, along with the presence of the desired regioisomer 3-SAs (Fig. 4). Given the strong restraints, a discussion of the bond metrics in 3-SAs is not possible (see SI, Crystallographic Information for further information). Yet the model itself is reliable and the formation of the 3-thio-1,2,4-thiadiarsole heterocycle in 3-SAs can be unambiguously determined. To further elucidate the mechanism for the formation, we investigated the reaction from two separate κ1-S coordinated monomers to 2 and 3 for P and As by computational methods. Pathways for the ring formations were calculated without accounting for dispersion or environment effects at the economic dhf-SV(P)49/PBE50 level. For the identified stationary points additional single-point calculations were done with the larger dhf-TZVP basis49 and further employing the conductor-like screening model d-COSMO-RS51 employing the solvent-specific response function for THF at 25 °C (which accounts for the solvent's polarity) and/or employing the D3-BJ dispersion correction.52 Details on the computations may be found in the SI, as well as images of all stationary points (Fig. S91) and their energies for the different ways of calculation (Table S3), which are graphically shown in Fig. 5. Moreover, Cartesian coordinates are available in the file structures.txt, movies of the pathways in the files reaction_as-as.gif and reaction_as-s.gif.
:47 (3-AsAs:3-SAs) by NMR spectroscopy (Scheme 3; Fig. S33 and S34). The major species could thereby be identified as 3-AsAs, wherefore we attribute the second species to the potential regioisomers 3-SAs. Carbon NMR analysis revealed the signals assigned for 3-AsAs (vide supra), and two additional singlets at 263.4 and 229.9 ppm, which we attribute to the 3-SAs regioisomer (Fig. S45). X-ray diffraction analysis of single crystals grown form diethyl ether further confirmed the presence of 3-AsAs in the crystal, along with the presence of the desired regioisomer 3-SAs (Fig. 4). Given the strong restraints, a discussion of the bond metrics in 3-SAs is not possible (see SI, Crystallographic Information for further information). Yet the model itself is reliable and the formation of the 3-thio-1,2,4-thiadiarsole heterocycle in 3-SAs can be unambiguously determined. To further elucidate the mechanism for the formation, we investigated the reaction from two separate κ1-S coordinated monomers to 2 and 3 for P and As by computational methods. Pathways for the ring formations were calculated without accounting for dispersion or environment effects at the economic dhf-SV(P)49/PBE50 level. For the identified stationary points additional single-point calculations were done with the larger dhf-TZVP basis49 and further employing the conductor-like screening model d-COSMO-RS51 employing the solvent-specific response function for THF at 25 °C (which accounts for the solvent's polarity) and/or employing the D3-BJ dispersion correction.52 Details on the computations may be found in the SI, as well as images of all stationary points (Fig. S91) and their energies for the different ways of calculation (Table S3), which are graphically shown in Fig. 5. Moreover, Cartesian coordinates are available in the file structures.txt, movies of the pathways in the files reaction_as-as.gif and reaction_as-s.gif.
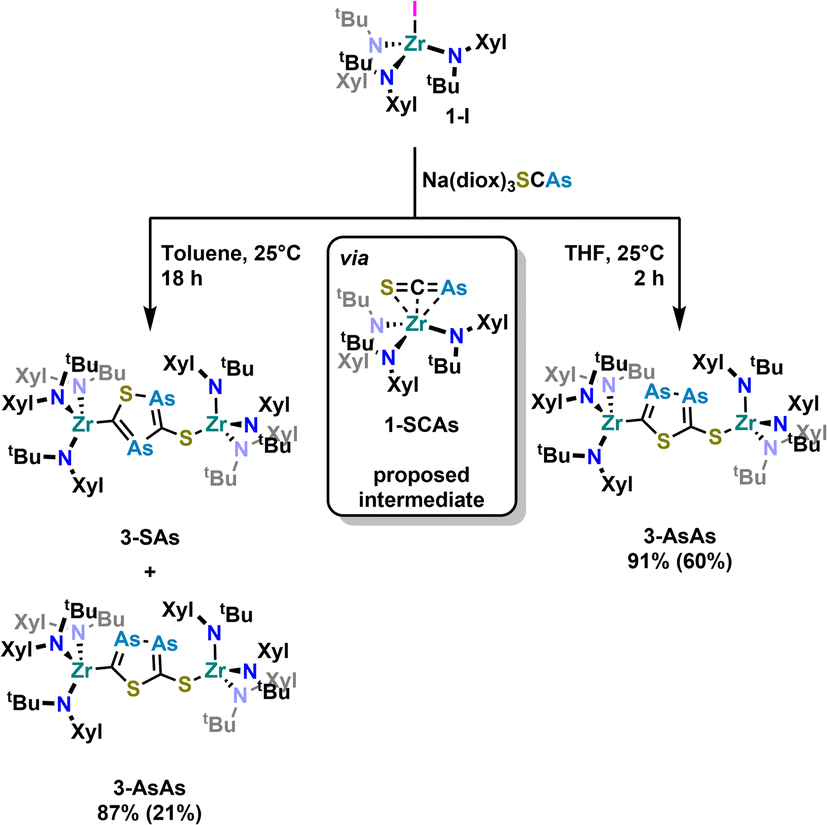 | ||
| Scheme 3 Salt metathesis reaction between 1-I and Na(diox)3SCAs and subsequent cyclisation of the [SCAs]− anion to form a new arsenic containing heterocycle. While the reaction is regioselective in THF leading to 3-AsAs, inseparable mixtures of both isomers 3-AsAs and 3-SAs (mixed yield: 87%, ratio 3-AsAs:3SAs = 53:47) are observed in toluene. Yields in brackets indicate crystalline yields. | ||
 | ||
| Fig. 4 Molecular structures of the two possible [3 + 2] cycloaddition regioisomers 3-AsAs (top) and 3-SAs (bottom) observed after the salt metathesis reaction between 1-I and Na(diox)3SCAs in THF or toluene, respectively. Please note that the structure of 3-SAs was obtained from a crystalline mixture of 3-AsAs and 3-SAs, in which the positions of the respective heterocycles superimpose each other (ration in crystals 60:40). Hydrogen atoms are omitted for clarity, all ellipsoids are shown at a probability level of 50%. | ||
 | ||
| Fig. 5 Energy profile for the reaction starting from two monomers with SCP/SCAs in κ1-S coordination via η3 coordination and ring closure to compounds 2-PP, 2-SP, 3-AsAs or 3-SAs at PBE level. Open symbols refer to gas phase calculations with the SV(P) basis, filled symbols to such with the TZVP basis with additional employment of the dCOSMO-RS model with the σ-profile for THF at 25 °C and the D3-BJ dispersion correction. The plotted structure is that of state VI, where the two monomers form a (weakly bound) dimer. | ||
As evident from Fig. 5, three transition states are passed, the first two (II, IV) for the rearrangement of each monomer from the κ1-S coordination to the η3 coordination, the third (VII) during the transformation of the two re-arranged monomers to the final compound. Without D3-BJ and dCOSMO-RS (open symbols in Fig. 5), the two different monomers are of almost the same energy and connected by low barriers (∼20 kJ mol−1).
The energy gain when approaching each other is small (energy difference between V and VI is ∼10 kJ mol−1) and the height of the third barrier is comparable to that of the first two. The energy gain of the reaction comes from the ring closure and amounts to 143 to 172 kJ mol−1 for the entire reaction, with the sequence As–As > As–S > S–P > P–P. Consideration of dispersion and environment produces rather subtle differences. First, the energies for the entire reaction are larger by ∼20 kJ mol−1, mainly due to the dispersion correction which increases the energy gain upon monomer approximation and leads to a slight preference of the η3 coordinating isomer over the κ1-S coordinating one. This means that rather state V than state I will be the starting point for the reactions, for which we get energy gains (energy difference between V and VIII) of 150 to 175 kJ mol−1, with the same sequence as before. This suggests that for thermodynamic reasons 3-AsAs is the (slightly) preferred product when using [SCAs]−, but 2-SP when using [SCP]−. The barrier heights are very similar to those obtained without employing D3-BJ and dCOSMO-RS. We may summarize that the three barriers for the ring formation in all cases can be easily surmounted under ambient conditions. Employing only D3-BJ but no dCOSMO-RS somewhat enhances the stabilization of states V to VIII but does not lead to qualitative changes; in passing we note that the simple COSMO model employed with a dielectric constant of 7.6 (THF) yields numbers between dCOSMO-RS and no COSMO, see Table S3. So, the calculations do not fully answer the question of the observed regioselectivity in different solvents. Given the fact that the calculations suggest 2-SP and 3-AsAs as the thermodynamic products of the reaction, we anticipate that regioisomer formation is largely controlled by kinetic effects which exponentially depend on the barrier height, and thus in a way that is too sensitive to be described with the comparably rough conductor-like screening model for solvation.
To set all these results into relation to their lighter homologues, we also investigated the salt metathesis reaction between 1-Cl or 1-I and Na(diox)3OCP or Na(diox)3OCAs. This revealed unexpected and severe differences between the [OCP]− and the [OCAs]− anions as well as to the thio-analogues [SCP]− and [SCAs]−. The reaction of Na(diox)3OCP with the halide complex 1-Cl gave clean access to the desired phosphaethynolate complex 1-OCP (Fig. 6). However, attempts to synthesize 1-OCAs via similar salt metathesis reactions resulted in the formation of wild mixtures (Fig. S22), from which so far no single product could be isolated. Investigations of the thermal stability of the complex 1-OCP further showed that even after prolonged time at 100 °C no degradation/further reaction is induced (Fig. S20 and S21), which contrasts Goicoechea's report on boron OCP compounds (vide supra, Fig. 3).35 These results furthermore clearly show that the reactivity of the lighter oxygen congeners [OCP]− and [OCAs]− is drastically different compared to their sulfur analogues [SCP]− and [SCAs]−. Formation of the defined complex 1-OCP is indicated by 1H NMR spectroscopy and its spectrum shows only one set of signals (Fig. S13) indicating the formation of a κ1-O coordinated product.13,53 Furthermore, the 31P phosphorus resonance was found at −323.6 ppm (Fig. S15) which is indicative of a κ1-O coordination.13,53 Unambiguous proof for the formation of the phosphaethynolate complex 1-OCP was finally given by X-ray diffraction analysis on a crystal grown from a concentrated pentane solution. The oxygen-zirconium distance was found at 2.0590(13) Å, and the Zr1–O1–C1 angle is 158.23(14)°. The C–P bond was found at 1.548(2) Å. All these parameters (NMR and structure) are in well agreement with previously reported κ1-O coordinated phosphaethynolate complexes of the early transition metals.13
 | ||
| Fig. 6 (Attempted) synthesis of the phospha- and arsaethynolate complexes 1-OCP and 1-OCAs and the molecular structure of 1-OCP. Hydrogen atoms have been omitted for clarity, ellipsoids are shown at a probability level of 50%. | ||
Finally, we attempted cleavage of the new formed heterocycles from the zirconium centers (Scheme 4). As an initial model reaction, we performed several test reactions using 2-PP as the substrate. While the addition of excess methyl iodide or TMS-Cl did not result in the formation of a new species, addition of approx. two equivalents of methyl triflate resulted in the formation of an equimolar mixture of two new species as observed by 1H NMR (Fig. S66). The 31P{1H} NMR of the crude mixture only shows the two resonances at 341.7 and 280.8 ppm (1JPP = 426 Hz, Fig. S67), proving that the diphosphole is still intact after the reaction. In addition, a singlet resonance in the crude 1H NMR at 2.45 ppm, integrating for three protons, clearly indicates the installation of a methyl group on the molecule. However, this also indicates that only one side of the molecule has been cleaved. As mentioned, along with the formation of the new diphosphole moiety, the crude 1H NMR also indicates the formation of a second species, which we initially assigned as the triflate complex 1-OTf. Indeed, 1-OTf can be independently synthesized from 1-Cl through the addition of AgOTf (Fig. S46–S52, Molecular structure: Fig. S63) and its 1H-NMR characteristics fits well with one of the newly observed products (Fig. S66). Upon fractional crystallization, we were able to obtain pure fractions of the 2nd reaction product 4-PP (Fig. S53–S59). 1H NMR spectroscopic investigations of the product (Fig. S53) clearly show that one zirconium tris-amide fragment is still present, indicating that complete cleavage of the diphosphole is not possible even in the presence of an overstoichiometric amount of MeOTf. To answer the question, if the C- or S- terminus of the diphosphole ligand was methylated, we grew single crystals suitable for X-ray diffraction analysis from hexane at −40 °C (Fig. 7). The structure clearly reveals cleavage of the S-terminus giving access to a new thioether carbon–sulfur bond S1–C3 (1.787(2) Å). The zirconium carbon distance Zr1–C2 was found at 2.3194(16) Å, which is marginally longer compared to the parent complex 2-PP (2.294(5) Å). In contrast the bond parameters of the diphosphole ring in 4-PP remain mostly unchanged and are comparable to the parent complex 2-PP (compare Tables S1 and S2).
 | ||
| Scheme 4 Attempted ring cleavage from the zirconium complexes using methyl triflate leading to the mono-methylated complexes 4-SP, 4-PP and 5-AsAs along with the “extrusion” of one equivalent of 1-OTf. | ||
 | ||
| Fig. 7 Molecular structure of 4-PP (left) and 5-AsAs (right). Hydrogen atoms have been omitted for clarity. Ellipsoids are shown at a probability level of 50%. | ||
With these results in hand, we became interested if (partial) ring cleavage was also possible in the other diphosphole/diarsole complexes 2-SP and 3-AsAs. Indeed, crude NMR investigations after exposing 2-SP and 3-AsAs to approx. 2 equivalents of MeOTf revealed similar results as already described for 2-PP, with the crude reaction mixtures showing the formation of equimolar mixtures of 1-OTf and new complexes, most likely 4-SP and 5-AsAs (Fig. S68 and S70 respectively). Furthermore, the phosphorus NMR of crude 4-SP also shows the retention of the diphosphole heterocycle displaying two doublets at 290.1 and 274.2 ppm (2JPP = 51 Hz, Fig. S69). The methyl group resonating at 2.46 ppm in the 1H NMR spectrum of 4-SP further indicates formation of a S-methylated heterocycle, similar to 4-PP (δ(CH3) = 2.45 ppm, vide supra). A similar resonance is found in the crude NMR of 5-AsAs (Fig. S60 and S70; 2.49 ppm) and we performed fractional crystallization of the reaction mixture to unambiguously proof S-methylation in 5-AsAs. X-ray quality crystals grown from concentrated hexane solution, clearly proof the formation of 5-AsAs (Fig. 7). A detailed discussion of the crystal/bonding parameters of the diarsole ring will be omitted here, since the differences between 5-AsAs/3-AsAs are only marginal (vide supra and Table S2). A detailed comparison of the bond parameters is presented in Tables S1 and S2 in the SI. Notably, TMS-OTf can also be applied as cleavage reagent, which gives access to the mono-silylated analogues of 4-PP and 4-SP as indicated by crude NMR analysis (see Fig. S71–S74), further indicating a surprising stability of the Zr–C bond in these complexes. However, it has to be stated, that the observed reactivity and the fact that only one zirconium atom can be cleaved off, somewhat hampers the general applicability of the new heterocycle in (coordination) chemistry. Future research in this field will be directed towards transmetallatable species, that allow a broader investigation of the spectroscopic, electrochemical and coordination properties of the new heterocycles.
Conclusions
In conclusion we present the first (autogenic) [3 + 2] cycloaddition reactions with heavy cyanate anions [SCP]− and [SCAs]−, mediated by zirconium. Regioselectivity of the observed cycloaddition reactions can be controlled by choice of solvent giving access to four (partly previously inaccessible) thiadiphosphole/thiadiarsole rings respectively. The straightforward and controllable access of all four heterocycles is notable, as simple synthetic routes to these entities have not yet been reported so far. All heterocycles described are thermally stable, with no signs of light-induced or thermal decomposition within several months.This report highlights the differences between the well-explored phospha- and arsaethynolate anions [OCP]− and [OCAs]− compared to their heavier sulfur analogues. The fact that neither the [SCP]− nor the [SCAs]− anion can undergo facile CS extrusion blocks the most prominent decomposition path of their lighter analogues, which often extrude CO. Although full cleavage of both metal centers, giving access to the “free” heterocycles was not feasible under the conditions investigated, and only one zirconium center could be cleaved, the general accessibility of these heterocycles gives ample opportunities in chemical synthesis and potential new applications of them in (in-)organic, electro or medicinal chemistry.
Author contributions
The project was designed by SH. Experimental work was performed by MB, DH and FH. X-Ray diffraction analysis was performed by MS and SH. Quantum chemical calculations were performed by FW. The manuscript was written by MB, MS, FW and SH. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2365786, 2365789–2365793, 2518191, 2526017, 2526018 and 2530439 contain the supplementary crystallographic data for this paper.54a–jAll data is available free of charge from our side if requested. Raw data is stored on the university servers and can be accessed via us if necessary. Supplementary information: contains all 1H, 13C, 31P and 2D NMR spectra, IR and UV-vis data as well as further information regarding X-ray crystallography and computational investigations. XYZ coordinates are provided as a serparate SI file. See DOI: https://doi.org/10.1039/d6sc01985d.
Acknowledgements
Roland Egger is kindly acknowledged for conducting elemental analyses. Dr Michael Bodensteiner is kindly acknowledged for providing access to the X-ray facility of the University of Regensburg. The University of Innsbruck is kindly acknowledged for funding of this work. Dedicated to the birthday of Prof. Dr John Arnold, whom SH would like to thank for his mentorship and support, which further encouraged him to pursue a career in academia.Notes and references
- N. N. Greenwood and A. Earnshaw in Chemistry of the Elements, 2nd edn, Elsevier Butterworth-Heinemann, Oxford, 1997 Search PubMed.
- S. Hohloch and F. Tambornino, Inorg. Chem., 2025, 64, 12900–12917 CrossRef CAS PubMed.
- (a) W. Beck, Eur. J. Inorg. Chem., 2003, 2003, 4275 Search PubMed; (b) S. Esteban, J. Chem. Educ., 2008, 85, 1201 CrossRef CAS; (c) J. Liebig and J. L. Gay-Lussac, Ann. Chem. Phys., 1824, 25, 285 Search PubMed.
- (a) C. F. Buchholz, Beiträge zur Erweiterung und Berichtigung der Chemie, Erfurt, 1799, vol. 1, pp. 81–92 Search PubMed; (b) H. Köhler Die Fabrikation von Cyanverbindungen Aus Tierischen Abfällen Und Produkten Der Trockenen Destillation, Die Industrie der Cyanverbindungen, Vieweg + Teubner Verlag, Wiesbaden, Germany, 1914, pp. 60–101 CrossRef; (c) S. Arlt, J. Harloff, A. Schulz, A. Stoffers and A. Villinger, Inorg. Chem., 2019, 58, 5305 CrossRef CAS PubMed; (d) J. Burmeister, Coord. Chem. Rev., 1990, 105, 77 CrossRef CAS; (e) J. L. Burmeister, Coord. Chem. Rev., 1968, 3, 225 CrossRef CAS; (f) C.-H. Hsieh, S. M. Brothers, J. H. Reibenspies, M. B. Hall, C. V. Popescu and M. Y. Darensbourg, Inorg. Chem., 2013, 52, 2119 CrossRef CAS PubMed.
- F. Wöhler, Ann. Chem. Phys., 1828, 88, 253 CrossRef.
- (a) J. J. Berzelius, Jahresber, 1822, 1, 49 Search PubMed; (b) A. Shlyaykher, M. Ehmann, A. J. Karttunen and F. Tambornino, Chem. Eur J., 2021, 27, 13552 CrossRef CAS PubMed; (c) J. L. Burmeister and L. E. Williams, Inorg. Chem., 1966, 5, 1113 Search PubMed.
- (a) N. N. Greenwood, R. Little and M. J. Sprague, J. Chem. Soc., 1964, 1292 RSC; (b) H. Günther, F. Weigend, X. Xie, W. Cao, X.-F. Gao, X.-B. Wang and F. Tambornino, Angew. Chem., 2025, e202507543 Search PubMed.
- W. S. Shober and F. W. Spanutius, Am. Chem. J., 1894, 16, 229–233 Search PubMed.
- G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72 CrossRef CAS.
- M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643–644, 189 CrossRef CAS.
- (a) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420 CrossRef CAS PubMed; (b) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 43, 831 RSC.
- A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064 CrossRef CAS PubMed.
- L. N. Grant and D. J. Mindiola, Chem. Eur J., 2019, 25, 16171 CrossRef CAS PubMed.
- J. M. Goicoechea and H. Grützmacher, Angew. Chem., Int. Ed., 2018, 57, 16968 CrossRef CAS PubMed.
- S. Basappa, R. Bhawar, D. H. Nagaraju and S. K. Bose, Dalton Trans., 2022, 51, 3778 RSC.
- C. J. Hoerger, F. W. Heinemann, E. Louyriac, L. Maron, H. Grützmacher and K. Meyer, Organometallics, 2017, 36, 4351 CrossRef CAS.
- (a) M. Bhunia, J. S. Mohar, C. Sandoval-Pauker, D. Fehn, E. S. Yang, M. Gau, J. Goicoechea, A. Ozarowski, J. Krzystek, J. Telser, K. Meyer and D. J. Mindiola, J. Am. Chem. Soc., 2024, 146, 3609 CrossRef CAS PubMed; (b) C. J. Hoerger, F. W. Heinemann, E. Louyriac, M. Rigo, L. Maron, H. Grützmacher, M. Driess and K. Meyer, Angew. Chem., 2019, 131, 1693 CrossRef; (c) M. Joost, W. J. Transue and C. C. Cummins, Chem. Commun., 2017, 53, 10731 RSC; (d) A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2016, 55, 8536 CrossRef CAS PubMed; (e) A. Hinz and J. M. Goicoechea, Angew. Chem., 2016, 128, 15741 CrossRef.
- F. Tambornino, A. Hinz, R. Köppe and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 8230 CrossRef CAS PubMed.
- A. R. Jupp, M. B. Geeson, J. E. McGrady and J. M. Goicoechea, Eur. J. Inorg. Chem., 2016, 2016, 639 CrossRef CAS PubMed.
- G. Becker and K. Hübler, Z. Anorg. Allg. Chem., 1994, 620, 405 CrossRef CAS.
- M. Jürgensen, T. Kunz, M. Arrowsmith, M. Dietz, S. Hagspiel and H. Braunschweig, Chem. Sci., 2025, 16, 8870–8877 RSC.
- (a) Á. Horváth and Z. Benkő, Inorg. Chem., 2022, 61, 7922 CrossRef PubMed; (b) Á. Horváth, B. D. Lőrincz and Z. Benkő, Chem. Eur J., 2023, 29, e202300611 CrossRef PubMed.
- F. A. Watt, L. Burkhardt, R. Schoch, S. Mitzinger, M. Bauer, F. Weigend, J. M. Goicoechea, F. Tambornino and S. Hohloch, Angew. Chem., Int. Ed., 2021, 60, 9534 CrossRef CAS PubMed.
- (a) F. A. Watt, A. Krishna, G. Golovanov, H. Ott, R. Schoch, C. Wölper, A. G. Neuba and S. Hohloch, Inorg. Chem., 2020, 59, 2719 CrossRef CAS PubMed; (b) F. A. Watt, K. N. McCabe, R. Schoch, L. Maron and S. Hohloch, Chem. Commun., 2020, 56, 15410 RSC; (c) B. Wittwer, F. Heim, K. Wurst and S. Hohloch, Chem. Commun., 2024, 60, 7299–7302 RSC; (d) B. Wittwer, K. N. McCabe, D. Leitner, M. Seidl, L. Maron and S. Hohloch, Inorg. Chem. Front., 2024, 11, 4158–4166 Search PubMed.
- (a) Y.-C. Tsai, F. H. Stephens, K. Meyer, A. Mendiratta, M. D. Gheorghiu and C. C. Cummins, Organometallics, 2003, 22, 2902 CrossRef CAS; (b) C. E. Laplaza and C. C. Cummins, Science, 1995, 268, 861 CrossRef CAS PubMed; (c) P. Vernaudon, H. G. Rajoharison and C. Roussel, Bull. Soc. Chim. Fr., 1987, 205 CAS.
- R. Appel and R. Moors, Angew. Chem., 1986, 98, 570 CrossRef CAS.
- (a) Y. J. Franzke and C. Holzer, J. Chem. Phys., 2023, 159 Search PubMed; (b) Y. J. Franzke and F. Weigend, J. Chem. Theory Comput., 2019, 15, 1028 Search PubMed.
- (a) S. El-Kurdi and K. Seppelt, Chem. Eur J., 2011, 17, 3956 CrossRef CAS PubMed; (b) M. R. Haneline and A. F. Heyduk, J. Am. Chem. Soc., 2006, 128, 8410 Search PubMed; (c) A. Kasani, S. Gambarotta and C. Bensimon, Can. J. Chem., 1997, 75, 1494 Search PubMed; (d) K. S. A. Motolko, J. S. Price, D. J. H. Emslie, H. A. Jenkins and J. F. Britten, Organometallics, 2017, 36, 3084 Search PubMed; (e) C. Romain, D. Specklin, K. Miqueu, J.-M. Sotiropoulos, C. Fliedel, S. Bellemin-Laponnaz and S. Dagorne, Organometallics, 2015, 34, 4854 CrossRef CAS; (f) I. A. Tonks, D. Tofan, E. C. Weintrob, T. Agapie and J. E. Bercaw, Organometallics, 2012, 31, 1965 Search PubMed; (g) H. Tsurugi, R. Ohnishi, H. Kaneko, T. K. Panda and K. Mashima, Organometallics, 2009, 28, 680 CrossRef CAS; (h) L. Turculet and T. D. Tilley, Organometallics, 2002, 21, 3961 Search PubMed.
- A. Linke, D. Decker, H.-J. Drexler and T. Beweries, Dalton Trans., 2022, 51, 10266 RSC.
- A. Schmidpeter, J. Gross, E. Schrenk and W. S. Sheldrick, Phosphorus Sulfur Silicon Relat. Elem., 1982, 14, 49 CrossRef CAS.
- (a) A. G. Jarvis, A. C. Whitwood and I. J. S. Fairlamb, Dalton Trans., 2011, 40, 3695 Search PubMed; (b) T. Schillmöller, R. Herbst-Irmer and D. Stalke, Adv. Opt. Mater., 2021, 9, 2001814 Search PubMed; (c) M. Arca, F. Demartin, F. A. Devillanova, A. Garau, F. Isaia, V. Lippolis and G. Verani, J. Chem. Soc., Dalton Trans., 1999, 3069 Search PubMed; (d) M. J. Alder, W. I. Cross, K. R. Flower and R. G. Pritchard, J. Chem. Soc., Dalton Trans., 1999, 2563 CAS.
- J. Pipek and P. G. Mezey, J. Chem. Phys., 1989, 90, 4916 CrossRef CAS.
- (a) S. Hohloch, L. Hettmanczyk and B. Sarkar, Eur. J. Inorg. Chem., 2014, 2014, 3164 CAS; (b) M. G. Sommer, D. Schweinfurth, F. Weisser, S. Hohloch and B. Sarkar, Organometallics, 2013, 32, 2069 CrossRef CAS.
- R. J. Gilliard, R. Suter, E. Schrader, Z. Benkő, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Chem. Commun., 2017, 53, 12325 Search PubMed.
- D. W. N. Wilson, A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 2188 CrossRef CAS PubMed.
- (a) Category 2, Hetarenes and Related Ring Systems, ed. R. C. Storr and T. L. Gilchrist, Georg Thieme Verlag, Stuttgart, 2004 Search PubMed; (b) Hetarenes and Related Ring Systems, ed. R. C. Storr and T. L. Gilchrist, Georg Thieme Verlag, Stuttgart, 2004, pp. 659–687 Search PubMed.
- (a) S. Kurz, H. Oesen, J. Sieler and E. Hey-hawkins, Phosphorus Sulfur Silicon Relat. Elem., 1996, 117, 189 Search PubMed; (b) D. Bevern, H. Görls, S. Krieck and M. Westerhausen, Z. Anorg. Allg. Chem., 2020, 646, 948 CrossRef CAS; (c) R. A. Barlett, H. V. R. Dias, K. M. Flynn, H. Hope, B. D. Murray, M. M. Olmstead and P. P. Power, J. Am. Chem. Soc., 1987, 109, 5693 CrossRef.
- (a) A. Schumann, F. Reiß, H. Jiao, J. Rabeah, J.-E. Siewert, I. Krummenacher, H. Braunschweig and C. Hering-Junghans, Chem. Sci., 2019, 10, 7859 RSC; (b) T. Sasamori, N. Takeda and N. Tokitoh, J. Phys. Org. Chem., 2003, 16, 450 CrossRef CAS; (c) M. Yoshifuji, I. Shima, N. Inamoto, K. Hirotsu and T. Higuchi, J. Am. Chem. Soc., 1981, 103, 4587 CrossRef CAS.
- C. Heinl, E. Peresypkina, G. Balázs, E. Mädl, A. V. Virovets and M. Scheer, Chem. Eur J., 2021, 27, 7542 CrossRef CAS PubMed.
- E. Urnius, W. W. Brennessel, C. J. Cramer, J. E. Ellis and P. v. R. Schleyer, Science, 2002, 295, 832 CrossRef PubMed.
- A. Petrov, L. Conrad, N. T. Coles, M. Weber, D. Andrae, A. Zagidullin, V. Miluykov and C. Müller, Chem. Eur J., 2022, 28, e202203056 CrossRef CAS PubMed.
- (a) A. J. Roering, J. J. Davidson, S. N. MacMillan, J. M. Tanski and R. Waterman, Dalton Trans., 2008, 4488 RSC; (b) H. Chen, M. M. Olmstead, D. C. Pestana and P. P. Power, Inorg. Chem., 1991, 30, 1783 CrossRef CAS; (c) K. Hedberg, E. W. Hughes and J. Waser, Acta Crystallogr., 1961, 14, 369 Search PubMed; (d) A. Schumann, J. Bresien, M. Fischer and C. Hering-Junghans, Chem. Commun., 2021, 57, 1014 RSC; (e) M. L. Tarlton, O. J. Fajen, S. P. Kelley, A. Kerridge, T. Malcomson, T. L. Morrison, M. P. Shores, X. Xhani and J. R. Walensky, Inorg. Chem., 2021, 60, 10614 CrossRef CAS PubMed; (f) M. L. Tarlton, X. Yu, R. J. Ward, S. P. Kelley, J. Autschbach and J. R. Walensky, Chem. Eur J., 2021, 27, 14396 CrossRef CAS PubMed.
- A. Petrov, T. Wellnitz, F. Hennersdorf, F. Reiß and C. Hering-Junghans, Chem. Eur J., 2024, 30, e202400575 Search PubMed.
- M. Bouslikhane, H. Gornitzka, J. Escudié and H. Ranaivonjatovo, J. Organomet. Chem., 2001, 619, 275 CAS.
- (a) A. Dashti-Mommertz and B. Neumüller, Z. Anorg. Allg. Chem., 1999, 625, 954 CAS; (b) T. Dunaj and C. von Hänisch, Chem. Eur J., 2022, 28, e202202932 CAS; (c) K. Mast, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1999, 625, 1475 CAS; (d) H. Schumann, S. Jurgis, E. Hahn, J. Pickardt, J. Blum and M. Eisen, Chem. Ber., 1985, 118, 2738 CAS.
- (a) D. Morales Salazar, E. Mijangos, S. Pullen, M. Gao and A. Orthaber, Chem. Commun., 2017, 53, 1120 CAS; (b) G. Ferguson and D. F. Rendle, J. Chem. Soc., Dalton Trans., 1975, 1284 CAS; (c) A. Decken, C. J. Carmalt, J. A. C. Clyburne and A. H. Cowley, Inorg. Chem., 1997, 36, 3741 CrossRef CAS PubMed.
- (a) C. Elschenbroich, J. Kroker, M. Nowotny, A. Behrendt, B. Metz and K. Harms, Organometallics, 1999, 18, 1495 CAS; (b) N. Avarvari, P. Le Floch, L. Ricard and F. Mathey, Organometallics, 1997, 16, 4089 CAS; (c) A. J. Ashe, X. Fang and J. W. Kampf, Organometallics, 2001, 20, 2109 CAS; (d) R. A. Aitken, A. J. Blake, I. Gosney, R. O. Gould, D. Lloyd and R. A. Ormiston, J. Chem. Soc., Perkin Trans. 1, 1998, 1801 CAS; (e) M. Olaru, D. Duvinage, Y. Naß, L. A. Malaspina, S. Mebs and J. Beckmann, Angew. Chem., Int. Ed., 2020, 59, 14414 CAS; (f) F. Sanz and J. J. Daly, J. Chem. Soc., Dalton Trans., 1973, 511 CAS; (g) J. Zechovský, V. Kremláček, M. Erben, M. Hejda, E. Rychagova, R. Jambor, A. Růžička, S. Ketkov and L. Dostál, Dalton Trans., 2022, 51, 15933 Search PubMed.
- (a) M. Kořenková, V. Kremláček, M. Hejda, J. Turek, R. Khudaverdyan, M. Erben, R. Jambor, A. Růžička and L. Dostál, Chem. Eur J., 2020, 26, 1144 Search PubMed; (b) M. Westerhausen, M. H. Digeser, C. Gückel, H. Nöth, J. Knizek and W. Ponikwar, Organometallics, 1999, 18, 2491 CAS; (c) A. J. Ashe, S. Mahmoud, C. Elschenbroich and M. Wünsch, Angew Chem. Int. Ed. Engl., 1987, 26, 229 CrossRef; (d) S. C. Sendlinger, B. S. Haggerty, A. L. Rbeingold and K. H. Theopold, Chem. Ber., 1991, 124, 2453 CrossRef CAS.
- F. Weigend and A. Baldes, J. Chem. Phys., 2010, 133, 174102 Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS PubMed.
- (a) A. Pausch, J. Chem. Theory Comput., 2024, 20, 3169 CrossRef CAS PubMed; (b) A. Klamt and M. Diedenhofen, J. Phys. Chem. A, 2015, 119, 5439 CrossRef CAS PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456 CrossRef CAS PubMed.
- C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379 RSC.
- (a) CCDC 2365786: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdspg; (b) CCDC 2365789: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdssk; (c) CCDC 2365790: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdstl; (d) CCDC 2365791: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdsvm; (e) CCDC 2365792: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdswn; (f) CCDC 2365793: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2kdsxp; (g) CCDC 2518191: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2qjczm; (h) CCDC 2526017: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2qsjfh; (i) CCDC 2526018: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2qsjgj; (j) CCDC 2530439: Experimental Crystal Structrue Determination, 2026, DOI:10.5517/ccdc.csd.cc2qy42x.
| This journal is © The Royal Society of Chemistry 2026 |