
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceGoldilocks boronic esters: optimized properties through understanding hydrolysis kinetics
Reyner D. Vargas ,
Samantha S. Cox,
James O. Larkin,
Jingfei Dai,
Yuecheng Jiang,
Ivan J. Yuan,
Margaret A. Hankins and
Zachary T. Ball*
,
Samantha S. Cox,
James O. Larkin,
Jingfei Dai,
Yuecheng Jiang,
Ivan J. Yuan,
Margaret A. Hankins and
Zachary T. Ball*
Department of Chemistry, Rice University, Bioscience Research Collaborative, 6100 Main Street, Houston, Texas 77005, USA. E-mail: zb1@rice.edu
First published on 28th May 2026
Abstract
Boronic acids react reversibly with diols to form boronic ester derivatives, a process that can be used as a protecting group in multistep synthesis. Boronic ester formation is also an example of dynamic covalent chemistry that can be used for diverse applications, including polyol recognition, dynamic soft materials, and controlled drug release. Somewhat surprisingly, only limited quantitative structure–function relationships for boronic stability with different diols are available, with the consequence that it has been difficult to choose or design optimal reactivity, whether for multistep synthesis or for dynamic covalent applications. Here we report the first systematic study of a family of 1,2-diols that enable a series of boronic esters with tunable hydrolytic behavior and simple one-pot formation. Across a panel of aryl, heteroaryl, and bioactive boronic acids, we found that diol structure, electronics, and pH can be used together to modulate hydrolysis rates over a broad range while maintaining silica compatibility. This work establishes a practical dataset for rationally selecting boronic ester protecting groups to achieve predictable, mild, and temporally controlled access to free boronic acids in water under biocompatible conditions, and also provides a simple synthetic solution to some longstanding challenges in the preparation of complex boronic acids.
Introduction
Since the discovery of the Suzuki–Miyaura cross-coupling reaction,1 boronic acids and their derivatives have become versatile building blocks central to numerous C–C, C–N, C–O, and C–S bond-forming reactions.2–7 While synthetic applications are important, there are many other applications of boronic acids for which the ability to finely tune the kinetic and thermodynamic stability of boronic esters is essential. Their reversible binding with 1,2- and 1,3-diols enables broad use in polyol recognition,8–10 biosensing,11–13 and drug development (Fig. 1a).14–17 Boronic ester hydrolysis is a convenient and successful approach to controlled-release drug delivery.18,19 Molecule recognition and affinity purification depend on an understanding of relative affinity.20 Boronic acids are used as crucial components in the dynamic covalent assembly of biomaterials, where the dynamics and affinity of boronic ester structures affects macroscale properties.21,22 | ||
| Fig. 1 (a) Applications of boronic esters dependent upon tunable hydrolysis rates (b) novel boronic acid protecting groups (c) new stable boronic esters for easy boronic acid protection and tunable hydrolysis. | ||
For such a widely studied field, there is surprisingly little quantitative data about the kinetics of boronic ester stability, especially regarding structure–function relationships of varying diols, rather than boronic acids, under physiologically relevant conditions.23–26 At least in part, this limited knowledge stems from the fact that simple 1,2-diols and catechols form boronic esters that hydrolyze too quickly in water to be easily measured. One recent example is a systematic study established reactivity differences among pinacol boronic esters.27 Thermodynamic affinity measurements (Kd or Ka) have been reported by numerous groups,20,28–30 but even there published data is often qualitative or relative.9 Kinetic data is essential for designing optimal drug release profiles, for engineering properties of dynamic materials, and even for optimizing deprotection strategies in multistep synthesis.
The most widely used boronic ester is pinacol (pin). Widespread usage continues in spite of broad recognition that pin esters have poor properties. Pin esters hydrolyze within minutes in neutral water, are prone to decomposition, and are notoriously incompatible with silica chromatography, complicating isolation and storage.31–33 To overcome the limitations of pin esters, alternative structures such as N-methyliminodiacetic acid (MIDA),34 1,8-diaminonaphthalene (DAN),35 and potassium trifluoroborate salts (BF3K)36 have been developed to form the corresponding boronic esters (Fig. 1c). While each of these have benefits, there are also limitations. The recent development of Xpin,37 a photocleavable diol, highlights what has been a fundamental tradeoff: improving robustness and chemical orthogonality has come hand in hand with requiring harsh or nonselective hydrolysis conditions.38–40
It is worth comparing the situation around boronic esters with that of another metalloid ester common in organic chemistry: the silyl ether. Primarily due to its longstanding role as a hydroxyl protecting group, hundreds of different silyl ethers have been studied, and comparative stability studies are numerous. While TBS, TES, TIPS, and TBDPS ethers are well known, many other silyl ethers are also well studied, and significant data is at hand to optimize silyl ether stability for a specific synthetic challenge, allowing mild, chemoselective unmasking at desirable reaction rates.41 In contrast, there are few comparative studies of boronic ester stability. The MIDA, DAN, and trifluoroborate examples noted above are significant because they represent some of the relatively few efforts to engineer molecular structure of boronic esters to optimize chemical properties.
Our efforts in this area were inspired by the recent development of Epin, a more sterically demanding analog of pin. This simple and clever alteration enhances the stability of the corresponding boronic esters relative to traditional pin derivatives.33
We, however, began to see the limitations of the Epin esters in our laboratory. In several syntheses of polyfunctional targets, we were unable to hydrolyze the final Epin ester without resorting to harsh conditions that led to decomposition (vide infra). For context, Epin boronic esters are reportedly stable to K3PO4 in boiling toluene/water mixtures.33 Relatedly, we found that most Epin esters have extremely slow hydrolysis kinetics under physiological conditions, making them unsuitable for slow-release drug delivery applications.42
Against this background, we recognized a broader need for a more predictable and tunable boronic acid caging strategy. In this paper, we introduce two new diols with stability intermediate between pin and Epin (Fig. 1c). We report what is, to our knowledge, the first comprehensive study and dataset mapping hydrolysis behavior across a family of 1,2-diol boronic esters, establishing a practical framework for predictable optimization of pinacol ester stability based on local sterics. Furthermore, we demonstrate that the new diols, Gpin and 3pin, facilitate synthesis and hydrolysis/deprotection of challenging polyfunctional target molecules that proved impossible with pin or Epin.
Results and discussion
We synthesized two diols in a single step by addition of EtMgBr to an ester (Fig. 2a). We suggest the name “Gpin” for the diethyl analog, an abbreviation of “Goldilocks pinacol”, given its intermediate properties between the two extremes of pin and Epin. The triethyl analog we have called “3pin”, continuing the (rhyming) naming convention of Epin. | ||
| Fig. 2 Diols used for the formation of boronic esters in this study and their formation. (a) Commercially available diols. (b) Synthesis of the new diols designed for this study doing a simple Grignard addition. (c) Simple one-pot boronic ester formation using dry methanol as the solvent, HPLC traces of the reaction before Gpin addition, after mixing and rotovaping. | ||
Although boronic esters are traditionally formed in aprotic solvents under dry and/or reflux conditions,33,43 we frequently encountered poor solubility, especially for functionalized boronic acid precursors. Seeking a more universal and simple solution, we screened a range of solvents and found, unexpectedly, that anhydrous methanol thoroughly solubilized the boronic acid and enabled high-yield ester formation. Quantitative conversion was achieved simply by rotary evaporation of the reaction mixture, affirmed by TLC, LC and/or NMR (Fig. 2c). This simple protocol proved effective for the synthesis of all boronic esters, including with the sterically demanding Epin diol, as well as for boronic-acid-containing drugs and peptides (see Fig. 4).
Our primary means of assessing the stability of boronic esters was to examine hydrolysis kinetics in aqueous environments. This measurement is, obviously, germane to drug delivery and biomaterials applications. We were also mindful of our own synthetic challenges, and we envisioned that hydrolysis of boronic esters in neutral water would be a method mild enough for even sensitive substrates. Before extensive studies of hydrolysis kinetics, we also examined stability on silica gel, since silica instability is perhaps the biggest stability challenge in working with pin esters. As expected, pinacol esters (pin) frequently exhibited smearing and signs of decomposition on silica. We were gratified to find, however, that Gpin and 3pin boronic esters are stable on silica gel, providing a single focused spot and allowing isolation from a column in high yield, similar to that observed with Epin (Fig. 3a and b).33
 | ||
| Fig. 3 Boronic esters characterization. (a) HPLC traces of the formed boronic esters. (b) TLC analysis. (c) Measurement of pseudo-first-order rate constants (khydrol) and half-lives (t1/2). | ||
Moving on to aqueous hydrolysis, boronic esters were dissolved in pH 7.4 PBS buffer (10 mM) and monitored by HPLC over time. All esters displayed clean first-order kinetics, and pseudo-first-order rate constants (khydrol) and half-lives (t1/2) values were readily measured (Fig. 3c). As expected, most pin esters hydrolyzed within a few minutes and exhibited t1/2 values too small to be measured. At the other extreme, several of the Epin esters were effectively indefinitely stable in water, with t1/2 values of many days.
We examined how electronic and structural features influence hydrolysis under mild aqueous conditions (Fig. 4). As expected, all compounds followed the trend of increased hydrolytic stability (Epin > 3pin > Gpin, Fig. 4), consistent with our earlier observations. However, significant variations in pseudo-first-order rate constants were observed. Electronic effects have a significant effect on boronic ester stability. Electron-rich arylboronic esters were more stable than other compounds (4–6, Fig. 4b). Electron-poor analogs had a more modest impact, but did exhibit faster hydrolysis, at least for some examples (7–9, Fig. 4c). We also examined a set of boronic acids with ortho substituents that have been reported to offer improved diol affinity (Fig. 4d), including a Wulff-type boronic acid (13)44,45 and ortho-fluoro structures that we previously employed for diol recognition.8 Somewhat surprisingly, ortho-fluoro substitution had minimal impact of hydrolysis rates, while the Wulff-type aminomethyl-substitution gave increased hydrolysis rates (Fig. 5).
 | ||
| Fig. 4 Hydrolysis half-lives of boronic esters derived from structurally and electronically diverse boronic acids. Variation in measured khydrolysis values for all replicates was <10%. See SI for details. (a) Unsubstituted molecules. (b) Electron-donating groups. (c) Electron-withdrawing groups. (d) Mixed electronic substituents. (e) Heterocycles. (f) Bioactive molecules. (g) Peptides. a Ester insoluble in buffer. | ||
 | ||
| Fig. 5 Hammet plots measured for the hydrolysis of Gpin p-phenylboronic esters at two pH values. | ||
We expanded the scope to more complex boronic acid-containing structures, including heterocyclic compounds (14–16, Fig. 4e), drugs and other bioactive compounds (17–20, Fig. 4f), and even unprotected borylated peptides (21, 22, Fig. 4g). The Gpin and 3pin esters were successfully formed and the stability measured across this set. For a small number of cases (e.g. 14-Epin) solubility challenges prevented analysis, although the reduced hydrophobicity of new pinacols, Gpin and 3pin, improved aqueous solubility and allowed analysis, underscoring the practical value of having access to a suite of simple pinacol derivatives for the specific demands of individual applications. One important conclusion of this study is the wide differences in kinetic stability observed, indicating that the correct choice of boronic ester will be structure-dependent.
We next examined how pH influences boronic ester hydrolysis under biocompatible conditions46 (Table 1). While boronic ester hydrolysis is often thought to be a complex process, governed by competing equilibria,20 we found a consistent trend in the range of biologically relevant pH: hydrolysis occurs more slowly at lower pH. Changes in rate were generally modest, however.
|
||||
|---|---|---|---|---|
| Boronic ester | Hydrolysis half-life (h) | |||
| pH 6.8 | pH 7.4 | pH 8.0 | ||
| a Ester insoluble in buffer. | ||||
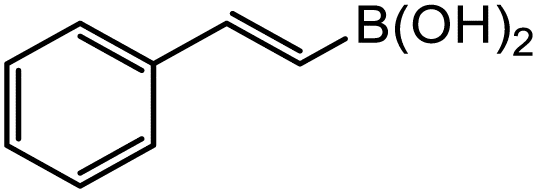 |
3-Gpin | 5.8 | 5.6 | 5.1 |
| 3-3pin | 18.1 | 15.3 | 7.2 | |
| 3-Epin | 22.3 | 20.2 | 9.6 | |
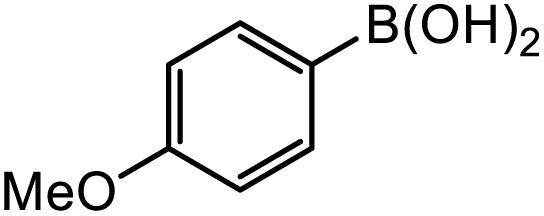 |
5-Gpin | 12.4 | 10.9 | 6.7 |
| 5-3pin | 69 | 61 | 49 | |
| 5-Epin | 112 | 99 | 61 | |
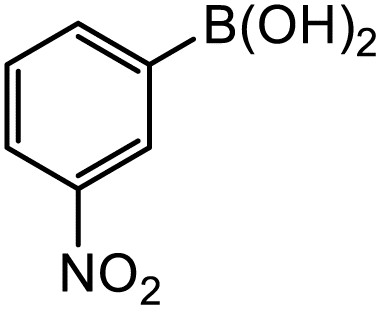 |
8-Gpin | 0.3 | 0.2 | <0.1 |
| 8-3pin | 2.9 | 1.2 | 1.1 | |
| 8-Epin | 31.5 | 24.2 | 13.8 | |
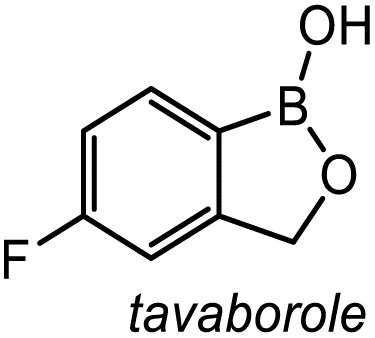 |
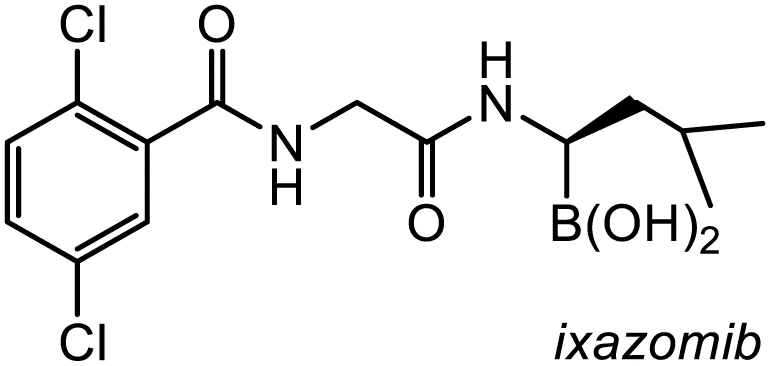
17-Gpin | 0.7 | <0.1 | <0.1 |
| 17-3pin | 2.8 | 2.3 | <0.1 | |
| 17-Epin | 40.0 | 13.6 | 4.7 | |
 |
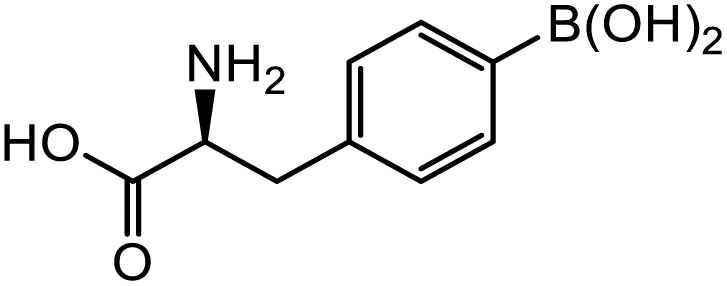
18-Gpin | 5.3 | 4.6 | 2.7 |
| 18-3pin | 47.8 | 18.2 | 15.7 | |
| 18-Epin | –a | –a | –a | |
 |
19-Gpin | 7.4 | 5.7 | 2.1 |
| 19-3pin | 29 | 22 | 15 | |
| 19-Epin | –a | –a | –a | |
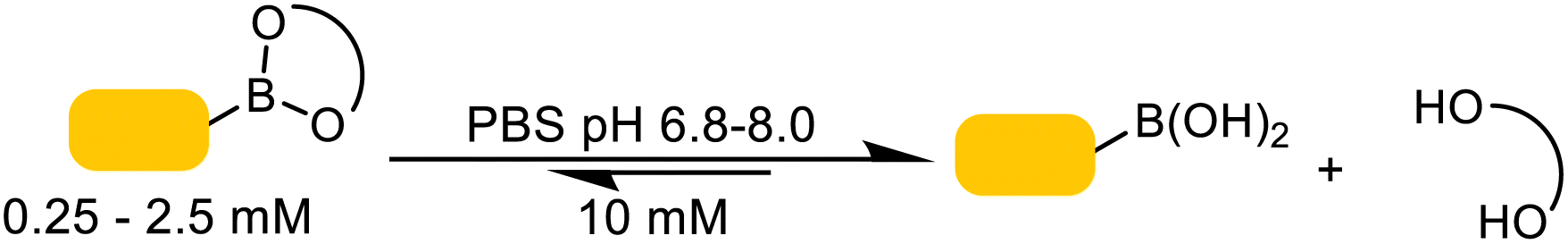
Hammett plots for a series of para-substituted Gpin phenylboronic esters were produced at pH 5.2 and pH 7.4. Both datasets yielded linear correlations with positive slopes, indicating that electron-withdrawing substituents accelerate hydrolysis.47 This behavior is consistent with a rate-determining nucleophilic attack at boron, during which negative character develops and is stabilized by electron-deficient aryl groups. More notably, the slope at pH 5.2 (ρ = 1.9) was nearly twice that at pH 7.4 (ρ = 1.0), revealing a stronger dependence on substituent electronics under acidic conditions.47 The data is consistent with our results (Table 1) that showed slower hydrolysis at lower pH levels.
The development of Gpin and 3pin solved two synthetic challenges within our lab (Fig. 6) that nicely illustrate the significant benefits of these diols for synthesis. In one project, we had a need to prepare pyridyl boronic acid derivative 24. Much to our surprise and frustration, both pin and Epin were incompatible with a simple, straightforward route (Fig. 6a). Pinacol esters were insufficiently stable: attempts to alkylate the hydroxy-pyridine 23-pin with alkyl halides led to complex mixtures, and we were unable to isolate the desired 24-pin. The improved stability of Epin nicely solves the issues related to intermediate instability, and we easily purified 24-Epin in reasonable yields. However, hydrolysis did not occur at mild temperatures, and elevated temperatures or other forcing conditions led to extensive decomposition. (For context, the Epin ester of a similar 3-pyridylboronic acid was reported to be stable to hydrolysis in the presence of K3PO4 in toluene/water up to 120 °C.33) In contrast, the corresponding 3pin ester was compatible with synthetic conditions and with silica chromatography of the intermediate (23-3pin), yet hydrolyzed readily in water at room temperature, affording the target alkyne-boronic acid in high yield (Fig. 6a).
 | ||
| Fig. 6 Boronic esters of Gpin and 3pin facilitate multistep synthesis and purification of organic and organometallic targets compounds. (a) Alkyne-boronic acid probe for copper-mediated chemistry. (b) A heteroleptic iridium-boronic acid complex. a Isolated yield. b Hydrolysis yield determined via HPLC. | ||
In a second, unrelated, project, we needed to prepare a boronic acid-iridium conjugate 25 for bioconjugation driven by dynamic covalent chemistry (Fig. 6b).8 Similar to our experience synthesizing 24 neither pin nor Epin esters were compatible with a designed route to boronic acid 25. Early in this effort, we found that pin esters were insufficiently stable under HATU coupling conditions and silica chromatography, preventing isolation of amide 12-pin. As anticipated, Epin again dramatically improved the stability of intermediates 12-Epin and 25-Epin, but we again observed extensive decomposition of the sensitive iridium complex under the elevated temperatures needed to hydrolyze the boronic ester, and we were unable to observe or isolate the desired boronic acid 25. The Gpin diol solved this issue: corresponding Gpin intermediates (12-Gpin and 25-Gpin) were again compatible with synthetic conditions and with silica chromatography, and again the target boronic acid 25 was easily formed upon hydrolysis of 25-Gpin in water at pH 7.4. Taken together, Fig. 6 illustrates that Gpin and 3pin offer significant synthetic advantages in synthesizing structurally diverse boronic acids.
The diboron reagent B2pin2 is a common precursor to organoboron compounds, and access to an analogous Gpin analog would provide improved synthetic options. We were thus pleased to find that the analogous B2Gpin2 is readily available and can be used in place of B2pin2 to directly synthesize more stable boronic esters. A simple synthesis from B2(OH)4 (see page S6 for details) yielded B2Gpin2, which—unlike B2pin2—is stable on silica gel (Fig. 7). Under standard borylation conditions, we obtained an arylboronic ester 5-Gpin in nearly quantitative yield (Fig. 7). We attribute this improvement to the enhanced stability under both reaction and purification conditions.
 | ||
| Fig. 7 A B2Gpin2 reagent enables borylation of halides with high stability. | ||
Conclusions
In summary, we present a comparative study of how steric demand affects boronic ester stability across a set of pinacol derivatives. This study adds to the limited kinetic data of boronic ester hydrolysis,23–25 supplementing existing thermodynamic measurements.20,28–30 The two new boronic esters, from Gpin and 3pin diols, form boronic esters that are stable to silica gel chromatography, yet are readily hydrolyzed in neutral aqueous solution. Across a structurally and electronically diverse panel of boronic acids, these esters display predictable trends in silica stability and aqueous hydrolysis, with Gpin and 3pin often striking a practical balance between robustness and facile hydrolysis. Boronic ester hydrolysis can be thus rationally tuned, enabling controlled access to free boronic acids under physiologically relevant conditions and providing a pathway to optimize synthetic schemes. The design of the Gpin diol avoids introducing a stereocenter, which may have practical advantages in complex-molecule synthesis. The wide range of hydrolysis half-lives observed indicate that, for controlled release and related applications, the optimal choice of diol structure is case-dependent.Author contributions
R. V., S. C. and Z. B. designed the experiments, R. V., S. C., J. L., J. D., Y. J., I. Y. and M. H. conducted the experiments. R. V., S. C., I. Y. and Z. B. analyzed the data. R. V. and Z. B. wrote the initial draft, which was reviewed and edited by all authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures; primary data used in preparation of Figures S1-S7 and Table S1; and characterization data for new compounds and peptides. See DOI: https://doi.org/10.1039/d6sc01941b.Acknowledgements
The authors acknowledge support from the Robert A. Welch Foundation Research Grant C-1680 (Z.T.B.) and the National Science Foundation under grant numbers CHE-1904865 and CHE-2203948.References
- N. . Miyaura and A. . Suzuki, Chem. Rev., 1995, 95, 2457–2483 CrossRef CAS.
- A. J. J. Lennox and G. C. Lloyd-Jones, Chem. Soc. Rev., 2014, 43, 412–443 RSC.
- J. Chen, J. Li and Z. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- V. A. Vil’, Y. A. Barsegyan, B. K. Chabuka, A. I. Ilovaisky, I. V. Alabugin and A. O. Terent’ev, ACS Catal., 2025, 15, 3636–3646 CrossRef.
- P. S. Devi, S. Saranya and G. Anilkumar, Catal. Sci. Technol., 2024, 14, 2320–2351 RSC.
- A. E. Mangubat-Medina, H. O. Trial, R. D. Vargas, M. T. Setegne, T. Bader, M. D. Distefano and Z. T. Ball, Org. Biomol. Chem., 2020, 18, 5110–5114 RSC.
- Y. Ding, Y. Jiang, N. Lorenzo Serrat, K. Zhong, Y. Lan and Z. T. Ball, J. Am. Chem. Soc., 2024, 10903 Search PubMed.
- R. D. Vargas, Y. Ding, H. O. Trial, R. Qian and Z. T. Ball, Chem. Commun., 2023, 59, 13030–13033 RSC.
- G. T. Williams, J. L. Kedge and J. S. Fossey, ACS Sens., 2021, 6, 1508–1528 CrossRef CAS PubMed.
- S. Kavoosi, K. Deprey, J. A. Kritzer and K. Islam, Chem. Commun., 2023, 59, 8692–8695 RSC.
- B. Akgun and D. G. Hall, Angew. Chem., Int. Ed., 2018, 57, 13028–13044 CrossRef CAS PubMed.
- D. Goldberg, I. Bentwich, Y. Haran and T. Getter, ACS Omega, 2025, 10, 10812–10825 CrossRef CAS PubMed.
- S. D. Bull, M. G. Davidson, J. M. H. Van Den Elsen, J. S. Fossey, A. T. A. Jenkins, Y.-B. Jiang, Y. Kubo, F. Marken, K. Sakurai, J. Zhao and T. D. James, Acc. Chem. Res., 2013, 46, 312–326 CrossRef CAS PubMed.
- Y.-M. Guo, Y. Cui, L. Yang and X.-Q. Wang, ACS Pharmacol. Transl. Sci., 2025, 8, 2384–2400 CrossRef CAS PubMed.
- J. P. M. António, R. Russo, C. P. Carvalho, P. M. S. D. Cal and P. M. P. Gois, Chem. Soc. Rev., 2019, 48, 3513–3536 RSC.
- B. C. Das, N. K. Nandwana, S. Das, V. Nandwana, M. A. Shareef, Y. Das, M. Saito, L. M. Weiss, F. Almaguel, N. S. Hosmane and T. Evans, Molecules, 2022, 27, 2615 Search PubMed.
- J. Plescia and N. Moitessier, Eur. J. Med. Chem., 2020, 195, 112270 Search PubMed.
- A. Stubelius, S. Lee and A. Almutairi, Acc. Chem. Res., 2019, 52, 3108–3119 Search PubMed.
- B. H. Pogostin, S. X. Wu, M. J. Swierczynski, C. Pennington, S.-Y. Li, D. Vohidova, E. H. Seeley, A. Agrawal, C. Tang, M. H. Yu, A. Dey, S. Hernandez, J. Cabler, O. Veiseh, E. L. Nuermberger, Z. T. Ball, J. D. Hartgerink and K. J. McHugh, Nat. Nanotechnol., 2025, 20, 1502–1513 CrossRef CAS PubMed.
- G. Springsteen and B. Wang, Tetrahedron, 2002, 58, 5291–5300 CrossRef CAS.
- Y. Dong, W. Wang, O. Veiseh, E. A. Appel, K. Xue, M. J. Webber, B. C. Tang, X.-W. Yang, G. C. Weir, R. Langer and D. G. Anderson, Langmuir, 2016, 32, 8743–8747 CrossRef CAS PubMed.
- N. Lagneau, L. Terriac, P. Tournier, J.-J. Helesbeux, G. Viault, D. Séraphin, B. Halgand, F. Loll, C. Garnier, C. Jonchère, M. Rivière, A. Tessier, J. Lebreton, Y. Maugars, J. Guicheux, C. Le Visage and V. Delplace, Biomater. Sci., 2023, 11, 2033–2045 Search PubMed.
- M. J. Swierczynski, Y. Ding and Z. T. Ball, Bioconjug. Chem., 2022, 33, 2307–2313 Search PubMed.
- J. A. Gonzalez, O. M. Ogba, G. F. Morehouse, N. Rosson, K. N. Houk, A. G. Leach, P. H.-Y. Cheong, M. D. Burke and G. C. Lloyd-Jones, Nat. Chem., 2016, 8, 1067–1075 CrossRef CAS PubMed.
- R. Bernardini, A. Oliva, A. Paganelli, E. Menta, M. Grugni, S. D. Munari and L. Goldoni, Chem. Lett., 2009, 38, 750–751 CrossRef CAS.
- M. Röttger, T. Domenech, R. Van Der Weegen, A. Breuillac, R. Nicolaÿ and L. Leibler, Science, 2017, 356, 62–65 Search PubMed.
- H. L. D. Hayes, R. Wei, M. Assante, K. J. Geogheghan, N. Jin, S. Tomasi, G. Noonan, A. G. Leach and G. C. Lloyd-Jones, J. Am. Chem. Soc., 2021, 143, 14814–14826 CrossRef CAS PubMed.
- W. L. A. Brooks, C. C. Deng and B. S. Sumerlin, ACS Omega, 2018, 3, 17863–17870 CrossRef CAS PubMed.
- A. M. Comiskey and E. V. Anslyn, J. Org. Chem., 2025, 90, 7161–7167 Search PubMed.
- Y. Suzuki, D. Kusuyama, T. Sugaya, S. Iwatsuki, M. Inamo, H. D. Takagi and K. Ishihara, J. Org. Chem., 2020, 85, 5255–5264 Search PubMed.
- T. Kégl and F. Ungváry, J. Organomet. Chem., 2007, 692, 1825–1833 CrossRef.
- D. H. Doherty, M. S. Rosendahl, D. J. Smith, J. M. Hughes, E. A. Chlipala and G. N. Cox, Bioconjug. Chem., 2005, 16, 1291–1298 CrossRef CAS PubMed.
- N. Oka, T. Yamada, H. Sajiki, S. Akai and T. Ikawa, Org. Lett., 2022, 24, 3510–3514 CrossRef CAS PubMed.
- D. M. Knapp, E. P. Gillis and M. D. Burke, J. Am. Chem. Soc., 2009, 131, 6961–6963 CrossRef CAS PubMed.
- Y. Mutoh, K. Yamamoto and S. Saito, ACS Catal., 2020, 10, 352–357 CrossRef CAS.
- S. R. Inglis, E. C. Y. Woon, A. L. Thompson and C. J. Schofield, J. Org. Chem., 2010, 75, 468–471 CrossRef CAS PubMed.
- J. J. Molloy and A. J. B. Watson, in ACS Symposium Series, ed. A. Coca, American Chemical Society, Washington, DC, 2016, vol. 1236, pp. 379–413 Search PubMed.
- C. He and X. Pan, Macromolecules, 2020, 53, 3700–3708 CrossRef CAS.
- G. A. Molander, L. N. Cavalcanti, B. Canturk, P.-S. Pan and L. E. Kennedy, J. Org. Chem., 2009, 74, 7364–7369 CrossRef CAS.
- A. K. L. Yuen and C. A. Hutton, Tetrahedron Lett., 2005, 46, 7899–7903 CrossRef CAS.
- T. W. Greene and P. G. M. Wuts, Protective Groups in Organic Synthesis, Wiley, 1st edn, 1999 Search PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 Search PubMed.
- D. M. Heard, M. C. Lessard and D. G. Hall, Angew. Chem., Int. Ed., 2025, 64, e202507571 Search PubMed.
- H. Li, Y. Liu, J. Liu and Z. Liu, Chem. Commun., 2011, 47, 8169 RSC.
- J. A. Ulloa, B. Boury, J. Aguilar, S. A. Sánchez, B. Ponce, A. Díaz-Barrera and B. F. Urbano, Eur. Polym. J., 2026, 250, 114678 CrossRef CAS.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 Search PubMed.
- C. D. Ritchie and W. F. Sager, in Progress in Physical Organic Chemistry, ed. S. G. Cohen, A. Streitwieser and R. W. Taft, Wiley, 1st edn, 1964, vol. 2, pp. 323–400 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |