
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSubstrate-driven iridium photocatalysts enable diastereoselective dearomative polyoxygenation of hydroxyquinolines
Andrea Uroz a,
Laura Blancob,
Sergio Diaz-Tenderocde,
Claudia Febereroa,
Alba Collado*be,
Silvia Cabrera*be and
José Alemán*ae
a,
Laura Blancob,
Sergio Diaz-Tenderocde,
Claudia Febereroa,
Alba Collado*be,
Silvia Cabrera*be and
José Alemán*ae
aOrganic Chemistry Department, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: jose.aleman@uam.es
bInorganic Chemistry Department, Universidad Autónoma de Madrid, 28049 Madrid, Spain. E-mail: silvia.cabrera@uam.es; alba.collado@uam.es
cChemistry Department, Universidad Autónoma de Madrid, 28049 Madrid, Spain
dCondensed Matter Physics (IFIMAC), Universidad Autónoma de Madrid, 28049 Madrid, Spain
eInstitute for Advanced Research in Chemical Sciences (IAdChem), Universidad Autónoma de Madrid, 28049 Madrid, Spain
First published on 15th May 2026
Abstract
We report a novel strategy for the direct, diastereoselective dearomative polyoxygenation of 8-hydroxyquinolines, leveraging a substrate-coordinated Ir(III) complex as a self-activating photocatalyst under visible light and atmospheric oxygen. In this system, the hydroxyquinoline substrate coordinates to the Ir precatalyst generating an Ir–oxyquinolate complex. This species activates the substrate and acts as a photosensitizer, promoting the generation of singlet oxygen and enabling the formation of multiple C–O bonds in a single step with excellent regio- and stereocontrol. Mechanistic investigations, including ligand exchange experiments, control reactions, and DFT studies, reveal that the reaction proceeds via selective oxygen insertion into the coordinated quinoline, followed by water-assisted hydroxylation, yielding structurally complex polyoxygenated products. This substrate-driven approach eliminates the need for external photo-catalysts and demonstrates a unique strategy for the activation of heteroaromatic substrates, providing access to motifs of potential relevance in pharmaceuticals and functional materials. This work highlights how substrate–metal cooperation can be exploited to achieve transformations that are challenging or inaccessible with conventional photocatalytic systems.
Introduction
Oxidation reactions of organic compounds are key tools in the chemical industry for the production of a wide range of intermediates and products, from pharmaceuticals to fine chemicals.1,2 Maximizing efficiency and minimizing byproducts requires these oxidations to selectively target specific bonds or functional groups, making the process inherently challenging. Among oxidant reagents, molecular oxygen is considered ideal due to its low cost, high abundance, and minimal environmental impact, especially compared to oxidants like potassium permanganate and peracids, which generate large amounts of byproducts. In nature, selective oxyfunctionalization of organic molecules is remarkably achieved using molecular oxygen and specialized enzymes. For instance, dioxygenases are capable of directly activating O2 and catalyzing the oxidation of aromatic substrates by the incorporation of both oxygen atoms with incredible selectivity under mild conditions (Scheme 1A).3 However, natural products, pharmaceuticals, materials and biologically active compounds are usually heterocyclic complex molecules, containing more than one different oxygen and nitrogen functionalities.4 Typically, the installation of the corresponding oxygen-functional groups, particularly in heterocyclic chemistry, requires multistep synthetic sequences involving protection and deprotection strategies and the use of diverse oxidizing reagents, as precise control over the oxidation process is often challenging and commonly results in complex product mixtures (Scheme 1B).5 The most effective approach would be the oxidation of such heterocycles to form the desired Csp3–O bonds. It would be even more challenging to carry out the oxidation and subsequent dearomatization of these hetero-cycles in a single reaction step.6 Therefore, the development of novel, step-economical strategies for the direct dearomative oxyfunctionalization of heterocycles and aromatic starting materials would be highly desirable. | ||
| Scheme 1 Dearomative oxyfunctionalization of arenes and heteroarenes. | ||
Inspired by nature, scientists have developed synthetic catalysts that mimic the function of enzymes to developed oxidation processes using O2 as the oxidant.7–9 These catalysts are able to achieve the direct oxidation of organic compounds, as enzymatic processes, by direct C–H hydroxylation, olefin epoxidation, Baeyer–Villiger reaction, or heteroatom oxidation. As many oxygenase enzymes contain a metallic cofactor such as Fe, Cu, and Mn, a great number of the synthetic bioinspired catalysts developed are metallic complexes.10 The metal centre in these catalysts is key for the dioxygen activation and the formation of the reactive oxygen species as metal-oxo complexes, and radical oxygen species. Besides this strategy, the direct incorporation of oxygen to organic molecules (e.g. in sulfoxidation reactions, boronic acid oxidations or C–H oxygenations among others) can also be accomplished using photocatalysis.11–14 These transformations proceed via singlet oxygen activation (1O2) or the generation of radical oxygen species (O2˙−) and form only one C–O (or S–O) bond in the molecule. In many cases, these transformations are achieved using iridium and rhodium based photocatalysts (Scheme 1C). However, the polyoxygenation of N-containing heterocycles has not been reported so far despite the relevance of the resultant compounds. In this context, hydroxyquinolines are organic molecules with a wide variety of applications. They are well-studied drugs for Alzheimer’s, dementia and other diseases as well as antitumoral agents, along with exhibiting other biological activities.15–18 The C–H oxyfunctionalization of these quinoline derivatives is very challenging, as selectivity issues and the non-desired N-oxide formation are very likely to occur (Scheme 1B). Moreover, that transformation is of utmost importance for the pharmaceutical industry due to the necessity of preparation and identification of drug metabolites from oxidative pathways. Based on these precedents, we aim to develop the polyoxygenation of hydroxyquinolines under photocatalytic conditions using dioxygen as the oxidant to promote a dearomative oxidative reaction.
Due to the excellent metal-binding ability of hydroxyquino-lines, numerous oxyquinolinate–metal derivatives have been reported.19 These complexes have shown great biological activities or luminescence properties for organic light emitting diodes. More recently, our research group has developed very efficient Ir(III) and Pt(II) photocatalysts using substituted 8-oxyquinolinates (QO) as ligands.20–22 Regarding the [IrCp*Cl(QO)] complexes, we demonstrated that their photocatalytic activity can be also combined with traditional organometallic transformations thanks to their ability to undergo ligand substitution reactions.
Taking advantage of the diverse catalytic properties of [IrCp*Cl(QO)] mentioned above, we envision the development of a dearomatic photocatalytic oxyfunctionalization of hydroxyquinolines under a non-conventional photocatalytic approach (Scheme 1D). We sought to explore the in situ formation of the active photocatalyst by means of the coordination of the hydroxyquinoline molecule to an Ir precursor, added to the reaction mixture in catalytic amounts. Thus, the oxyquinolinate complex will act as the photocatalyst capable of activating molecular oxygen to generate singlet oxygen, eliminating the need for an additional external photocatalyst (Scheme 1D). Furthermore, coordination of the hydroxyquinoline to the iridium center activates the heterocycle toward a reaction with 1O2. In other words, the in situ-formed complex will function both as the photocatalyst and as the substrate to be oxidized, operating with a catalytic amount of iridium. Upon oxidation of the hydroxyquinoline ring, the resulting, more labile oxidized ligand can be displaced by coordination of a new hydroxyquinoline molecule. This process would enable a catalytic iridium cycle, effectively operating as a self-reacting photocatalyst system. We therefore propose that oxyfunctionalization of the quinoline derivative takes place while the substrate is coordinated to the metal center. This reactivity is anticipated to be unique to the catalytic properties of the [IrCp*Cl(QO)] family of complexes. Notably, such photocatalytic oxyfunctionalization is not feasible using [Ru(bpy)3]2+, fac-[Ir(ppy)3], or organic photocatalysts, as they are unable to interact directly with the hydroxyquinoline substrate (Scheme 1C).
Coordination-driven strategies in photocatalysis have emerged as a powerful means to access new reactivity, combining substrate activation with photoinduced processes. In particular, Meggers and co-workers pioneered chiral-at-metal photocatalysis, where substrate coordination to Ir(III) or Rh(III) complexes enables light-driven transformations.23 Complementary strategies based on non-covalent catalyst–substrate interactions have also been developed, as illustrated by the work of Yoon and co-workers, highlighting the role of preorganization in photochemical reactivity.24,25 Despite these advances, the direct oxidation of the coordinated ligand itself remains largely unexplored, particularly within polyoxygenation processes. In this work, we report for the first time a new reactivity of hydroxyquinolines based on their ability to coordinate to an Ir(III) center, resulting in a complex that both activates the hydroxyquinoline substrate and acts as an oxygen photosensitizer. Additionally, the study includes mechanistic experiments and theoretical investigations to elucidate the pathway of the dearomative oxyfunctionalization reaction.
Results and discussion
Catalytic oxidation
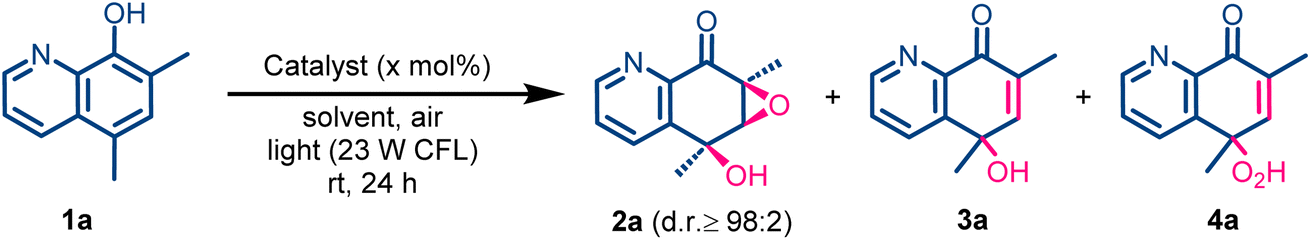
To evaluate our hypothesis, we tested the oxidation of 5,7-dimethyl-8-hydroxyquinoline (1a) in methanol using atmospheric oxygen and 2.5 mol% of an Ir(III) complex as a proof of concept. The reaction was irradiated with a 23 W commercial fluorescent bulb white light (CFL, see the SI for setup details) for 24 h (Scheme 2). The oxidation of 1a proceeded smoothly in the presence of the [IrCp*Cl2]2 dimer, giving 28% of the polyoxygenated product 2a (Table 1, entry 1). It is important to highlight that this catalytic oxidation protocol enables the simultaneous oxidation of four carbon atoms within the quinoline core in a single step, leading to the formation of a tertiary alcohol and a trisubstituted epoxide with complete diastereocontrol. Based on this excellent result, we next optimized the reaction conditions for the polyoxyfunctionalization of 1a (Table 1). First, to determine whether the observed reactivity is exclusive to [IrCp*Cl2]2, we tested other Ir(III) species and organic photosensitizers. IrCl3 showed no reactivity and the starting hydroxyquinoline 1a was recovered unaltered (Table 1, entry 2). Next, we tested the well-known singlet-oxygen photosensitizers [Ir(ppy)3] and Rose Bengal. Using these photocatalysts, a complete change in the outcome of the reaction was observed and dearomatized enone 3a or 4a were obtained as the only reaction products (Table 1, entries 3 and 4). This selectivity change highlights the need of a catalyst capable of both sensitizing oxygen and coordinating the hydroxyquinoline substrate for the reaction to proceed. Other light sources were also evaluated, but lower yields were obtained (see the SI). Afterwards, with [IrCp*Cl2]2 as the optimal catalyst, different solvents were evaluated (Table 1, entries 5–8). Thus, 2a was obtained in a similar yield (26%) in the reaction conducted in 1,2-dichloroethane (1,2-DCE); however the selectivity was improved compared with that of the reaction in methanol (Table 1, entry 8). During the solvent screening, we realized that the water content in the solvents has an influence on the reaction outcome. Thus, molecular sieves were added to the reaction mixture to remove water (Table 1, entry 9). Surprisingly, the oxidation was completely inhibited, suggesting that water might be important for the reaction mechanism and that rigorously dry conditions are detrimental to the transformation. On the other hand, when 1,2-DCE containing 0.2 ppm of water was employed, 2a was obtained in 71% yield as a single diastereomer, and in a very selective manner (Table 1, entry 10). Furthermore, these experiments seem to indicate the important role of water in the reaction mechanism as discussed below. | ||
| Scheme 2 Proof of concept of the dearomative oxyfunctionalization of hydroxyquinolines. | ||
|
||||
|---|---|---|---|---|
| Entry | Catalyst (x mol%) | Solvent | 2a/3a/4a ratio | 2a yieldb (%) |
| a Reaction conditions: a solution of 1a (0.05 mmol) and the corresponding catalyst (mol%) in 2.0 mL of solvent was irradiated using a 23 W CFL white light.b 1H NMR yield calculated using 1,3,5-trimethoxybenzene as the internal standard.c 99% 1H NMR yield of 4a.d 4 Å molecular sieves.e 0.2 ppm of H2O. | ||||
| 1 | [IrCp*Cl2]2 (2.5) | MeOH | 57![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:13 :30:13 |
28 |
| 2 | IrCl3 (5) | MeOH | — | 0 |
| 3 | [Ir(ppy)3] (5) | MeOH | 0:63:37 |
0 |
| 4 | Rose Bengal (5) | MeOH | 0:0:100 |
0c |
| 5 | [IrCp*Cl2]2 (2.5) | MeCN | 33:17:50 |
18 |
| 6 | [IrCp*Cl2]2 (2.5) | Toluene | 84:12:4 |
14 |
| 7 | [IrCp*Cl2]2 (2.5) | CH2Cl2 | 57:14:29 |
18 |
| 8 | [IrCp*Cl2]2 (2.5) | 1,2-DCE | 78:14:18 |
26 |
| 9 | [IrCp*Cl2]2 (2.5) | 1,2-DCEd | — | 0 |
| 10 | [IrCp*Cl2]2(2.5) | 1,2-DCEe | 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :8:0 :8:0 |
71 |
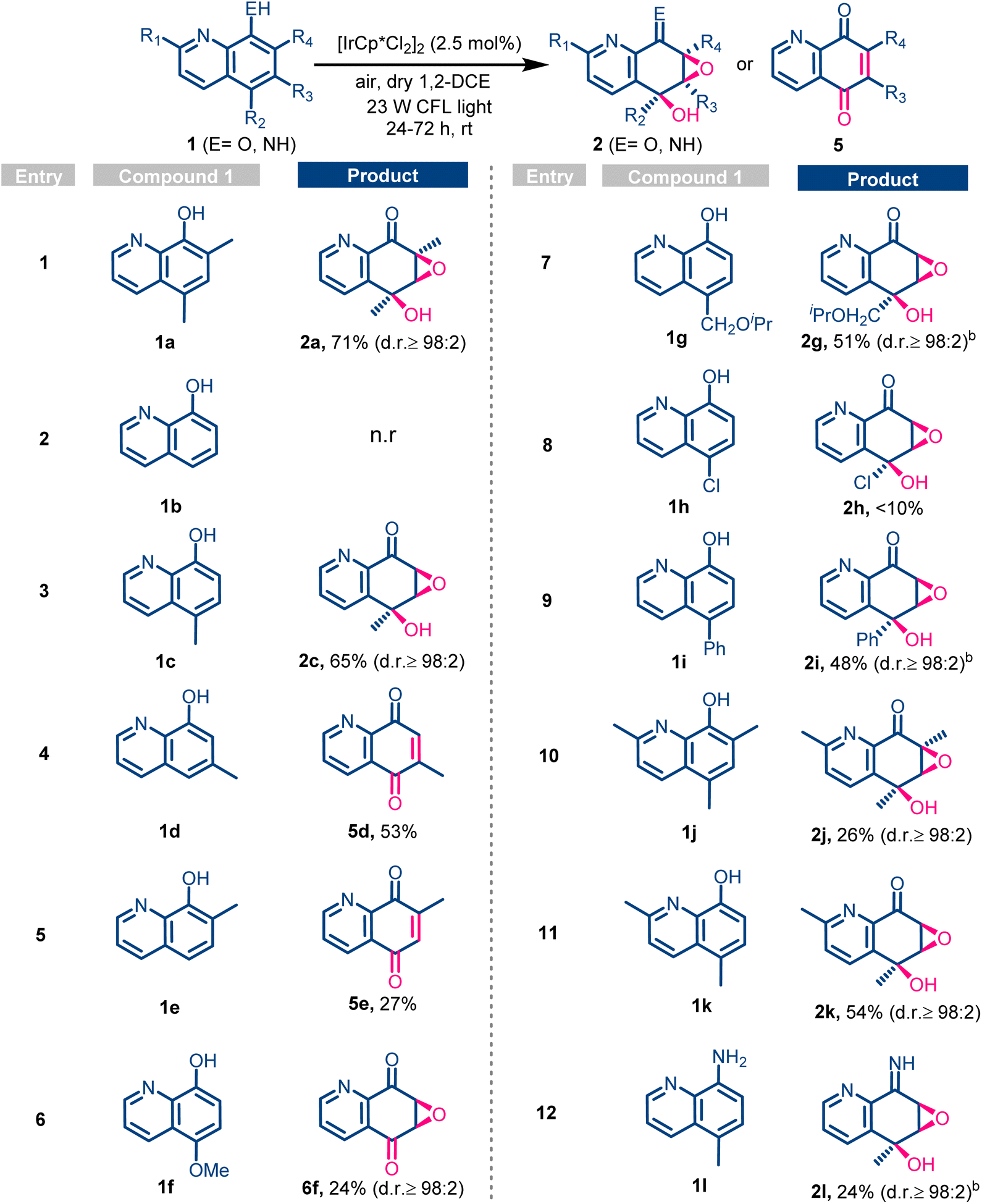
Having the optimal reaction conditions in hand, we next explored the applicability of the catalytic photooxidation protocol in other 8-hydroxyquinolines (Table 2). First, we found that the oxidation of the unsubstituted 8-hydroxyquinoline 1b was unsuccessful, and only the starting material (1b) was recovered after 24 h (Table 2, entry 2). This dramatic change in the reactivity can only be attributed to the substitution of the quinoline ring, which seems to be crucial for the photooxidation to proceed. Intrigued by this result, we decided to deeply study the substitution effect, including type and position, in the photooxidation transformation. As the model substrate 1a contained two methyl groups at the 5- and 7-position of the quinoline ring, we explored the photooxidation of monomethylated 8-hydroxyquinolines. Thus, the polyoxidation of 1c afforded compound 2c in 65% yield as a single diastereoisomer (Table 2, entry 3). Next, we evaluated the effect of the methyl substituent position, testing 6-methyl- and 7-methyl-8-hydroxyquinolines 1d and 1e, respectively (Table 2, entries 4 and 5). However, neither of these compounds gave the expected products 2d–e; instead quinone derivatives 5d and 5e were obtained in good yields. All the above experiments indicated that the polyoxidation of hydroxyquinolines to afford products 2 can only take place for 5-methylated derivatives. To analyse if other substituents different from methyl are also tolerated, compounds 1f–1i, bearing different substituents at position 5, were studied (Table 2, entries 6–9). 8-Hydroxyquinolines having methoxy (1f) and isopropoxymethyl (1g) groups were successfully oxidized, but two different products were obtained (Table 2, entries 6 and 7). While the photooxygenation of isopropyl ether derivative 1g afforded the expected compound 2g in 51% yield, the methoxy derivative 1f gave the epoxydiketone 6f. The formation of the latter (6f) could correspond to the evolution of the expected benzylic hemiketal of the oxygenated compound 2f into a ketone moiety. By contrast, the oxidation of the chlorinated compound 1h seemed to be difficult to accomplish, affording a complex reaction mixture from which 2h was identified in lower than 10% yield (Table 2, entry 8). Furthermore, the 5-phenyl analogue 1i led to the polyoxygenated derivatives 2i in moderate yield as single diastereoisomers (Table 2, entry 9), demonstrating the tolerance toward different substituents at the 5-position. Bulkier 2-methylated compounds 1j–k were also polyoxygenated to the corresponding products 2j–k under the photocatalytic conditions (Table 2, entries 10 and 11). Finally, the oxidation of the 8-aminoquinoline derivative 1l afforded the imine 2l in 24% yield (Table 2, entry 12).
| a Reaction conditions: a solution of the corresponding hydroxyquinoline 1 (0.05 mmol) and the [IrCp*Cl2]2 catalyst (2.5 mol%) in 2.0 mL of 1,2-DCE was irradiated using a 23 W CFL white light. 1H NMR yield was calculated using 1,3,5-trimethoxybenzene as the internal standard. n.r denotes no reaction.b Reaction carried out in the presence of K2CO3 (0.5 equiv.). |
|---|
 |
Mechanistic studies
Intrigued by the effect of the aryl substitution on the photooxidation reaction outcome and the different selectivities obtained for the well-stablish photocatalysts [Ir(ppy)3] and Rose Bengal, we decided to study the mechanism in detail, both from an experimental and a computational point of view. First, control experiments were performed (Table 3). The reaction did not proceed in the dark, under an argon atmosphere or without the [IrCp*Cl2]2 catalyst, indicating the key role of those variables in the reaction mechanism, i.e. light, oxygen and the Ir catalyst (Table 3, entries 2–4). As previously mentioned in the reaction condition optimization, the water content in the solvent affects the reaction outcome (see previous Table 1, entry 9 and 10 and Table 3, entry 5). Additionally, the photooxidation process could proceed via singlet oxygen oxidation, formed by an energy transfer process as will be detailed below. To confirm this, different experiments were conducted. First, the addition of 0.5 equivalents of DABCO, commonly used as a singlet oxygen (1O2) quencher,26,27 to the reaction mixture led to a decrease in the reaction yield (Table 3, entry 6). However, DABCO may also participate in single-electron transfer processes. To rule out the presence of reactive radicals or superoxide species, the reaction was performed in the presence of TEMPO, a well-known radical scavenger (Table 3, entry 7). The photooxidation of 1a was not inhibited upon addition of such a scavenger, confirming that radical or superoxide pathways are unlikely to be responsible for the observed reactivity. Furthermore, the reaction showed an increased yield when performed in deuterated solvent (Table 3, entries 8 and 9), which is consistent with the significant increase in the lifetime of 1O2 in such media.28 Finally, to detect the presence of 1O2, we performed the reaction in the presence of anthracene, as a chemical trapping probe (Table 3, entry 10).29 Notably, 10% of anthracene-endoperoxide was detected in the reaction media. All these experiments indicate that singlet oxygen is the reactive oxygen species involved in the transformation.
|
||
|---|---|---|
| Entry | Deviation from reaction conditions | 2a yieldb (%) |
| a Reaction conditions: a solution of 5,7-dimethylhydroxyquinoline (0.05 mmol) and a [IrCp*Cl2]2 catalyst (2.5 mol%) in 2.0 mL of dry 1,2-DCE was irradiating using a 23 W CFL white light for 24 h.b 1H NMR yield calculated using 1,3,5-trimethoxybenzene as the internal standard.c 10% of anthracene-endoperoxide is also formed. | ||
| 1 | — | 71 |
| 2 | Dark (no light) | 0 |
| 3 | Ar atmosphere | 0 |
| 4 | No catalyst | 0 |
| 5 | 4 Å molecular sieves | 0 |
| 6 | DABCO (0.5 equiv.) | 18 |
| 7 | TEMPO (0.5 equiv.) | 81 |
| 8 | CDCl3 as solvent | 55 |
| 9 | CHCl3 as solvent | 39 |
| 10 | Anthracene (0.5 equiv.) | 83c |

Oxyquinolinate–Ir(III) complexes are active photocatalysts in different transformations.20 According to this, we propose that the photocatalytic active species in the oxyfunctionalization reaction is the complex formed by the coordination of the corresponding deprotonated hydroxyquinoline to the Ir center. Based on this, we wanted to confirm whether the oxyfunctionalization of the heteroaromatic substrates takes place when the ligand is coordinated to the metal. Thus, we prepared and isolated the oxyquinolinate–Ir complexes Ir1–4 by reacting the dinuclear [IrCp*Cl2]2 complex with the corresponding 8-hydroxyquinoline in the presence of K2CO3 (see synthesis in the SI).20 Next, the 8-oxyquinolinate iridium complexes Ir1–4 were subjected to the photooxidation conditions, that is, white light irradiation in the presence of air and the reaction was monitored by 1H NMR spectroscopy (Scheme 3). After 24 h of irradiation, full conversion of Ir1–4 into new Ir complexes was observed. The corresponding oxyfunctionalized complexes, named Ir1–4Ox, were isolated in good yields (64–87%) and fully characterized. All the oxyfunctionalized Ir complexes, Ir1–4Ox, were unambiguously characterized by X-ray diffraction analysis. Single crystals were grown by slow diffusion of n-pentane into saturated dichloromethane solutions of the corresponding complexes. The structures obtained are shown in Fig. 1 and the most remarkable crystallographic data are collected in Table 4 (see full data in the SI). The four polyoxyfunctionalized Ir complexes preserved the expected three-legged piano-stool geometry. The length and angle data for Ir1-Ox, Ir2-Ox and Ir4-Ox were compared to that of the parent Ir1, Ir2, and Ir4 complexes.20,30
 | ||
| Scheme 3 Identification and isolation of the polyoxygenated quinolinate–Ir complex. | ||
 | ||
| Fig. 1 Structural views of complexes Ir1–4-Ox. Ellipsoids are shown at the 50% level and hydrogen atoms are omitted for clarity except those bonded to oxygen. The complexes crystallized as a racemic mixture. Only one enantiomer is shown. | ||
| Entry | Bond lengths | Ir1 | Ir1-Ox | Ir2 | Ir2-Ox | Ir3-Ox | Ir4 | Ir4-Ox |
|---|---|---|---|---|---|---|---|---|
| a Cp* distances and angles have been measured with respect to the ring centroid. | ||||||||
| 1 | Ir–N | 2.090(7) | 2.117(7) | 2.118(12) | 2.101(5) | 2.108(4) | 2.134(3) | 2.116(3) |
| 2 | Ir–O | 2.116(6) | 2.074(6) | 2.095(10) | 2.068(3) | 2.075(3) | 2.073(2) | 2.086(2) |
| 3 | Ir–Cl | 2.396(2) | 2.426(2) | 2.392(4) | 2.420(2) | 2.418(1) | 2.4046(10) | 2.403(9) |
| 4 | Ir–Cp* centroida | 1.772 | 1.771 | 1.774 | 1.765 | 1.776 | 1.778 | 1.776 |
| Entry | Angles | Ir1 | Ir1-Ox | Ir2 | Ir2-Ox | Ir3-Ox | Ir4 | Ir4-Ox |
|---|---|---|---|---|---|---|---|---|
| 5 | O–Ir–N | 78.3(3) | 77.45(15) | 78.6(4) | 77.43 (4) | 77.46(1) | 78.73(10) | 78.28(1) |
| 6 | O–Ir–Cl | 87.30(19) | 88.53(11) | 84.5(3) | 88.52(3) | 87.74(10) | 88.22(8) | 88.29(7) |
| 7 | N–Ir–Cl | 84.60(19) | 84.85(13) | 86.8(3) | 84.84(3) | 85.68(11) | 84.47(7) | 86.63(9) |
| 8 | Centroid Cp*–Ir–Cla | 128.33 | 126.79 | 126.35 | 127.94 | 128.21 | 128.34 | 128.06 |
| 9 | Centroid Cp*–Ir–Oa | 127.76 | 127.66 | 129.67 | 126.96 | 124.31 | 122.27 | 125.14 |
| 10 | Centroid Cp*–Ir–Na | 133.29 | 134.63 | 133.54 | 133.40 | 134.36 | 137.44 | 133.80 |
The Ir–N, Ir–O and Ir–Cl distances suffer from slight variations, but a clear trend was not observed. For instance, the Ir–O distances were found to be slightly shorter in complexes Ir1-Ox and Ir2-Ox than those of their precursors, while the Ir–O distance in Ir4-Ox was slightly longer than that of Ir4. Ir–N distances, however, increased in Ir1-Ox, and decreased in Ir2-Ox and Ir3-Ox, compared to that of their corresponding precursors. Overall, when compared amongst them, the lengths and angles of the oxyfunctionalized complexes were in the same range. It was surprising to find that the structure of the Ir1–4-Ox complexes contained three new oxygenated functional groups at the quinoline ligand, including a OH group at the 8-position. For the 5-methoxy derivative Ir3, its oxyfunctionalization afforded Ir3-Ox, bearing a ketone at the 5 position of the bicyclic ligand. This outcome aligns with the obtention of the diketone product 6f starting from the 5-methoxy-8-hydroxiquinoline substate 1f, as previously commented in the scope study of the catalytic process (Table 2). It is also important to note the high stereospecificity of the protocol, as the Ir complexes formed contain up to four chiral centres (three in the case of Ir3-Ox) in the bicyclic ligand, observing only one diastereoisomer by 1H NMR spectroscopy (Scheme 3). Moreover, the disposition of the new oxygenated groups is arranged as follows (Fig. 1 and Scheme 3): the hydroxyl group at the 5 position and the epoxide are in cis orientation whereas the hydroxyl located at the 8 position is trans with respect to both of them. According to the stereochemistry obtained in complexes Ir1–4Ox, the three oxygen-functional groups seem to come from two different reagents. The previous experiments help to partially understand the mechanism and corroborate that the coordination of the hydroxyquinoline derivative (1) to the Ir center happens prior to its oxyfunctionalization. Considering this, a plausible mechanism was proposed (Fig. 2). After formation of the quinolinate–Ir(III) complex (Ir1), the oxyfunctionalization of the ligand with singlet oxygen takes place. This singlet oxygen is generated via interaction between the excited state of the quinolinate–Ir(III) complex (Ir1*) and atmospheric oxygen. Indeed, we observed the quenching of the excited state of the quinolinate–Ir(III) complexes in the presence of oxygen (see the SI). Next, the oxyquinolinate ligand of the Ir1 complex will react with 1O2 to form the polyoxygenated complex Ir1-Ox. In addition, water is proposed to play a key role in such transformations, facilitating proton transfer processes and stabilizing reactive intermediates, ultimately enabling the formation of the observed polyoxygenated complexes. Absorption and emission measurements of both type of Ir complexes (oxygenated and non-oxygenated parent complexes) reveal that all parent Ir complexes exhibit stronger absorption in the visible region than the oxyfunctionalized analogues, while no emission was detected for the latter complexes (see the SI). These experiments are consistent with the proposed light-mediated steps.
 | ||
| Fig. 2 Simplified mechanism for the polyoxygenation of hydroxyquinolines based on experimental mechanistic studies. | ||
After the oxyfunctionalization of the complex (Fig. 2), a ligand exchange reaction will substitute the oxidized ligand in Ir1-Ox by the hydroxyquinoline derivative to recover the initial quinolinate–Ir complex (Ir1). Lastly, the 1,2-diol in the heterocycle (I) will be transformed into the final ketone derivative 2a, forming water as a byproduct. To demonstrate the ligand exchange between 1a and the Ir1-Ox complex, 1H NMR spectra were recorded to monitor the reaction (Fig. 3). First, the oxygenated complex Ir1-Ox (Fig. 3, spectrum A) was mixed with the dimethylated hydroxyquinoline 1a at room temperature in CD2Cl2. In the first spectrum, acquired just after mixing both reagents (Fig. 3, spectrum B), the full disappearance of the proton signals relative to complex Ir1-Ox was observed. In addition, the signals corresponding to Ir1 and compound 2a were identified, corroborating an instantaneous ligand exchange reaction and the formation of compound 2a. This evidence was further corroborated by DFT calculations, which afforded an equilibrium constant of 75.25 for the ligand exchange reaction (see the SI for more details). Next, the reaction mixture containing Ir1 and compound 2a was irradiated under visible light in the presence of air (Fig. 3, spectrum C). After 24 h of irradiation, complex Ir1 was fully converted into Ir1-Ox, showing that the oxyfunctionalization occurs upon coordination of the hydroxyquinoline to the Ir center and the reaction of that Ir complex with singlet oxygen.
 | ||
| Fig. 3 1H NMR spectra of the ligand exchange experiment. | ||
To gain deeper insight into the elemental steps and the diasteroselectivity of the reaction, we have carried out simulations with quantum chemistry starting from the simplified mechanism for the oxyfunctionalization of 8-hydroxyquinolines (see Fig. 2 and 4). The calculations were performed using density functional theory (DFT), specifically with the M06L functional.31 The electron density was expressed using the 6-31G(d,p) basis set for C, O, H, N, and Cl atoms.32,33 The Ir atom was described with an effective core potential (ECP) in combination with the corresponding LanL2DZ basis set.34–37 Solvent effects were considered through a Polarizable Continuum Model (PCM) of the solvation model based on density (SMD) type.38 All calculations were carried out with the Gaussian16 program.39
 | ||
| Fig. 4 Potential energy surface showing minima and transition states of the proposed mechanism. (a) The upper row shows calculations including one water molecule and (b) lower row, four water molecules. Relative energies ΔE are given in kcal mol−1 and referred to the ground state complex with triplet oxygen 3O2. ΔE values marked with an asterisk * have been estimated from relaxed scan simulations. | ||
A mechanistic proposal is presented in Fig. 4. In the top row of the figure, we consider the quinolinate–Ir complex with one oxygen molecule and one water molecule, both weakly bonded. The first step consists of the photoexcitation of oxygen 3O2 into 1O2, resulting in a minimum on the singlet potential energy surface; this structure will be taken as the energy reference and therefore has ΔG = 0.0 kcal mol−1. We have studied two cycloaddition reactions of insertion of singlet oxygen into the quinolinic ring: (i) the [4 + 2] cycloaddition leads to an almost degenerate structure. The very low energy barrier of ∼3 kcal mol−1, both in the direct and inverse reactions, leads us to conclude that an equilibrium of O2 insertion/expulsion is established (left-top, Fig. 4). (ii) The [2 + 2] cycloaddition produces a structure almost 4 kcal mol−1 more stable (right-top, Fig. 4). The barrier of this reaction can be easily overcome because the first absorption band in the photoexcitation takes place at higher energies. This band appears approximately at 25 kcal mol−1 above the reference minimum with singlet spin multiplicity.20 We rule out the equilibrium in this reaction because the newly formed structure after the [2 + 2] addition is protected with a barrier of ∼17 kcal mol−1. In addition, this structure evolves very rapidly through the cleavage of the O–O bond with a very low barrier of ∼3 kcal mol−1 (structure A shows the associated electron density redistribution schematically). As a result, an epoxide and a carboxyl on the quinoline ring are formed (see structure B). We have considered two pathways for this step, one with the water molecule behind the quinoline plane and another where the water molecule is located near O2. In both cases, the barrier is of similar height, indicating that the presence of water does not assist in breaking the O–O bond. However, water does help in stabilizing the new structure by establishing a hydrogen bond with the carboxyl group formed. Note that the new structure is at a relative energy of −30 kcal mol−1 with respect to the singlet multiplicity complex, and is even below the starting point with triplet multiplicity. That is, once the activation barrier for [2 + 2] addition is overcome, the complex evolves rapidly, stabilizing at an energy jump of more than 30 kcal mol−1. The role of water in the proposed mechanism is highlighted in Fig. 4B, where it is essential for the transformation from structure B to Ir1-Ox.
The following steps were studied assuming the presence of some water molecules around the complex. The relative energies are also referred to the complex with the weakly bound O2 molecule with singlet spin multiplicity. Protonation of the alkoxide anion by a nearby water molecule occurs in the first step. The hydroxyl generated is stabilized by hydrogen bonds with the other water molecules. Through a Grotthuss mechanism (proton jumping), the hydroxyl group is transferred and finally inserted on the opposite face of the quinoline ring. A cooperative process with very low energy barriers ensures the effectiveness of these last steps of the mechanism. It is worth noting that the final polyoxygenated complex Ir1-Ox is more than 50 kcal mol−1 below the initial complex with singlet spin multiplicity, formed in an additional stabilization step. The mechanistic proposal, in which the [2 + 2] cycloaddition and hydroxylation occur on opposite faces of the quinoline plane, account for the formation of the experimentally observed diastereomer.
Conclusions
In this work, we have developed a novel photocatalytic strategy for the direct dearomative polyoxygenation of 8-hydroxyquinolines using molecular oxygen as the sole oxidant. This methodology enables the simultaneous installation of multiple oxygen functionalities within the heteroaromatic core in a single synthetic step, representing a significant advance over traditional multistep oxidative approaches. A key feature of this transformation is the dual role of the iridium precursor [IrCp*Cl2]2. Upon in situ coordination of the hydroxyquinoline substrate, the resulting oxyquinolinate–Ir(III) complex operates both as a photosensitizer, generating singlet oxygen, and as a substrate-activating platform. This self-reacting photocatalytic system eliminates the need for external photosensitizers. In contrast, commonly employed photocatalysts such as [Ir(ppy)3] or Rose Bengal, although capable of sensitizing oxygen, fail to promote the same transformation due to their inability to coordinate and activate the hydroxyquinoline substrate.The reaction proceeds with remarkable chemoselectivity and diastereoselectivity, affording a single diastereomer even in systems generating up to four stereogenic centers. Substrate scope studies reveal a strong dependence on substitution at the 5-position of the quinoline ring, highlighting the importance of electronic and steric effects in facilitating oxygen insertion and subsequent transformations.
Structural characterization of the oxygenated Ir complexes and ligand exchange experiments unequivocally demonstrate that oxyfunctionalization occurs while the substrate is coordinated to the metal center. Mechanistic investigations, supported by control experiments and DFT calculations, confirm the involvement of singlet oxygen as the reactive oxygen species. Computational studies are consistent with a [2 + 2] cycloaddition of 1O2 to the coordinated quinolinate ligand, followed by O–O bond cleavage and water-assisted proton transfers that ultimately furnish the observed polyoxygenated products. Notably, trace amounts of water play a crucial role in stabilizing intermediates and enabling proton shuttling processes, whereas complete water removal inhibits the reaction.
Author contributions
A. U. and L. B. carried out the optimization and explored the scope of the reaction. S. D. T. and C. F. conducted the DFT calculations. A. C., S. C. and J. A. conceived the project and prepared the manuscript, which was edited by all other authors.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2423907 (Ir2-Ox), 2466572 (Ir1-Ox), 2466580 (Ir4-Ox), 2466582 (Ir3-Ox) and 2466583 (Ir4) contain the supplementary crystallographic data for this paper.40a–eThe data that support the findings of this study are available in the supplementary information (SI) of this article. Supplementary information: experimental details, general procedures, optimization of reaction conditions, characterization of products, copies of NMR spectra of all products and computational details. See DOI: https://doi.org/10.1039/d6sc01835a.
Acknowledgements
This work was supported by MICINN projects (PID2024-155520NB-I00, PID2022-138470NB-I00, PID2021-122299NB-100, TED2021-129999B-C32, and TED2021-130470B-100), the “Comunidad de Madrid” for Laboratorios inteligentes para la ciencia del futuro: descubrimiento de materiales avanzados para fotosíntesis artificial (FOTOART5.0-CM) (Reference: TEC2024/TEC-308) and the proyectos sinérgicos I+D (Y2020/NMT-6469). A. C. is thankful for financial support from the Comunidad de Madrid for a “Proyecto I+D de la CAM para Jóvenes Doctores de la UAM” (SI3/PJI/2021-00520) and Agencia Estatal de Investigación for a “Consolidación Investigadora 2022” project (CN2022-135687). L. B. and A. U. thank Universidad Autónoma de Madrid and Ministerio de Ciencia, Innovación y Universidades, respectively, for their predoctoral fellowships. The authors acknowledge the generous allocation of computer time at the Centro de Computación Científica at the Universidad Autónoma de Madrid (CCC-UAM). The Laboratorio de DRX Monocristal and the Servicio Interdepartamental de Investigación (SIdI) are thanked for the single-crystal X-ray diffraction analyses.Notes and references
- S. Caron, R. W. Dugger, S. G. Ruggeri, J. A. Ragan and D. H. B. Ripin, Chem. Rev., 2006, 106, 2943–2989 CrossRef CAS PubMed.
- Handbook of Advanced Methods and Processes in Oxidation Catalysis: From Laboratory to Industry, ed. D. Duprez and F. Cavani, Imperial College Press, 2011 Search PubMed.
- D. R. Boyd, N. D. Sharma and C. C. R. Allen, Curr. Opin. Biotechnol., 2001, 12, 564–573 CrossRef CAS.
- E. Kabir and M. Uzzaman, Results Chem., 2022, 4, 100606 CrossRef CAS.
- E. Grandi, F. Feyza Özgen, S. Schmidt and G. J. Poelarends, Angew. Chem., Int. Ed., 2023, 62, e202309012 CrossRef CAS PubMed.
- N. Choukairi Afailal, M. Borrell, M. Cianfanelli and M. Costas, J. Am. Chem. Soc., 2024, 146, 240–249 CrossRef CAS PubMed.
- M. D. Nothling, Z. Xiao, A. Bhaskaran, M. T. Blyth, C. W. Bennett, M. L. Coote and L. A. Connal, ACS Catal., 2019, 9, 168–187 CrossRef CAS.
- M. M. Pereira, L. D. Dias and M. J. F. Calvete, ACS Catal., 2018, 8, 10784–10808 CrossRef CAS.
- L. Vicens, G. Olivo and M. Costas, ACS Catal., 2020, 10, 8611–8631 CrossRef CAS.
- L. Que and W. B. Tolman, Nature, 2008, 455, 333–340 CrossRef CAS PubMed.
- L. Luo, T. Zhang, M. Wang, R. Yun and X. Xiang, ChemSusChem, 2020, 13, 5173–5184 CrossRef CAS PubMed.
- Y. Zhang, W. Schilling and S. Das, ChemSusChem, 2019, 12, 2898–2910 CrossRef CAS PubMed.
- L. Péault, P. Nun, E. Le Grognec and V. Coeffard, Chem. Commun., 2019, 55, 7398–7401 RSC.
- L. Péault, A. Planchat, P. Nun, E. Le Grognec and V. Coeffard, J. Org. Chem., 2021, 86, 18192–18203 CrossRef PubMed.
- B. Y. Choi, B. G. Jang, J. H. Kim, J.-N. Seo, G. Wu, M. Sohn, T. N. Chung and S. W. Suh, Neurobiol. Dis., 2013, 54, 382–391 CrossRef CAS PubMed.
- C. M. Darby and C. F. Nathan, J. Antimicrob. Chemother., 2010, 65, 1424–1427 CrossRef CAS PubMed.
- H. R. Dholariya, K. S. Patel, J. C. Patel, A. K. Patel and K. D. Patel, Med. Chem. Res., 2013, 22, 5848–5860 CrossRef CAS.
- H. A. Saadeh, K. A. Sweidan and M. S. Mubarak, Molecules, 2020, 25, 4321 CrossRef CAS PubMed.
- A. Casado-Sánchez, C. Martín-Santos, J. M. Padrón, R. Mas-Ballesté, C. Navarro-Ranninger, J. Alemán and S. Cabrera, J. Inorg. Biochem., 2017, 174, 111–118 CrossRef.
- L. Blanco, A. Uroz, K. Gutiérrez, S. Cabrera, A. Collado and J. Alemán, ACS Catal., 2024, 14, 6413–6422 CrossRef CAS PubMed.
- A. Casado-Sánchez, R. Gómez-Ballesteros, F. Tato, F. J. Soriano, G. Pascual-Coca, S. Cabrera and J. Alemán, Chem. Commun., 2016, 52, 9137–9140 RSC.
- P. Domingo-Legarda, A. Casado-Sánchez, L. Marzo, J. Alemán and S. Cabrera, Inorg. Chem., 2020, 59, 13845–13857 CrossRef CAS PubMed.
- X. Huang and E. Meggers, Acc. Chem. Res., 2019, 52, 833–847 CrossRef CAS PubMed.
- K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186–17192 CrossRef CAS PubMed.
- S. J. Chapman, W. B. Swords, C. M. Le, I. A. Guzei, F. D. Toste and T. P. Yoon, J. Am. Chem. Soc., 2022, 144, 4206–4213 CrossRef CAS PubMed.
- Y. Nosaka and A. Y. Nosaka, Chem. Rev., 2017, 117, 11302–11336 CrossRef CAS PubMed.
- C. Ouannes and T. Wilson, J. Am. Chem. Soc., 1968, 90, 6527–6528 CrossRef CAS.
- J. Dad'ová, E. Svobodová, M. Sikorski, B. König and R. Cibulka, ChemCatChem, 2012, 4, 620–623 CrossRef.
- Y. You, Org. Biomol. Chem., 2018, 16, 4044–4060 RSC.
- T.-T. Thai, B. Therrien and G. Süss-Fink, Inorg. Chem. Commun., 2009, 12, 806–807 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101 CrossRef PubMed.
- P. C. Hariharan and J. A. Pople, Theor. Chim. Acta, 1973, 28, 213–222 CrossRef CAS.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys., 1985, 82, 284–298 CrossRef CAS.
- T. H. Dunning Jr and P. J. Hay, in Modern Theoretical Chemistry, ed. H. F. Schaefer III, Plenum Press, New York, 1977, vol. 3, pp. 1–28 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman; G. Scalmani; V. Barone; G. A. Petersson; H. Nakatsuji; X. Li; M. Caricato; A. V. Marenich; J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2019 Search PubMed.
- (a) CCDC 2423907: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mc8kv; (b) CCDC 2466572: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nsnvz; (c) CCDC 2466580: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nsp38; (d) CCDC 2466582: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nsp5b; (e) CCDC 2466583: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2nsp6c.
| This journal is © The Royal Society of Chemistry 2026 |