DOI:
10.1039/D6SC01670G
(Review Article)
Chem. Sci., 2026, Advance Article
Advanced functional porous materials for photocatalytic H2O2 production
Received
27th February 2026
, Accepted 3rd May 2026
First published on 5th May 2026
Abstract
Hydrogen peroxide (H2O2) is one of the most essential chemicals; it has a high oxidising ability (1.763 V at pH 0) and the highest active oxygen content (47.1 wt%). Current synthesis techniques are energy-intensive, generate harmful waste, and pose the potential danger of explosions. Photocatalytic H2O2 production via O2 reduction is a promising, sustainable, and eco-friendly method that harnesses solar energy to drive the reaction while also requiring low energy input and ensuring safety. Interestingly, advanced functional porous materials (AFPMs) like covalent organic frameworks (COFs), metal–organic frameworks (MOFs), porous organic polymers (POPs), covalent triazine frameworks (CTFs), metal–organic polyhedra (MOP) and hydrogen bonded organic frameworks (HOFs) have surfaced as promising photocatalysts for effective photocatalysis. These materials exhibit a tunable pore size, high surface area, and robust framework, all of which facilitate mass transfer, adequate active sites, and efficient recycling. In this review, we have discussed the different strategies applied in recent literature to improve the photocatalytic efficiency of these materials towards photocatalytic H2O2 synthesis. This review provides an extensive summary and discussion of the latest applications of AFPM-based photocatalysts in H2O2 photosynthesis. The historical background and fundamental principles underlying photocatalysts for photocatalytic H2O production are briefly outlined. This is followed by a detailed classification and discussion of the strategies reported to enhance photocatalytic performance. The review addresses the challenges and future prospects of AFPM-based photocatalysts for light-driven H2O2 production.
Introduction
Hydrogen peroxide (H2O2), first prepared by Thenard in 1818, is a product of a reaction between barium peroxide and nitric acid.1 Also known as oxygenated water, H2O2 is a non-planar molecule with C2 symmetry and enantiomerism, attributed to its 95° dihedral angle between its two O–H bonds. Besides being the smallest and simplest chiral molecule, H2O2 can act as both a strong oxidant across a wide pH range and a reducing agent capable of reducing stronger oxidising compounds such as Ce(SO4)2, KMnO4, Cl2, NaClO, etc.1 Additionally, the derivatives of H2O2, such as hydroxyl radicals (˙OH), can break down nearly all types of organic pollutants or damage essential components within microbial cells, leading to the removal of organic contaminants or the elimination of harmful microorganisms.2,3 Initially, H2O2 was known for bleaching natural dyes and was synthesised from barium peroxide. Gradually, researchers found applications of H2O2 in various fields. The significant timeline of H2O2 towards various applications includes the following: (a) 1888: its first medical use for treating numerous diseases, (b) 1891: first report as a disinfectant,4 (c) 1920: reports of using an intravenous injection of H2O2 for treating influenza pneumonia during the first World War,5 (d) 1967: protective action of H2O2 against ischemia-reperfusion (IR) injury,6 (e) 1980: intravenous infusion of H2O2 for oxidative therapy,7 (f) H2O2 as an alternative to chlorine as a bleaching agent in paper production,8 and (g) as a terminal oxidant towards industrial synthesis of propene epoxide (PO).8 Today, H2O2 has been established as one of the most essential chemicals globally.9 It is recognised as a versatile and sustainable reagent. With the improvement of technologies, it is more dedicatedly applied in chemical synthesis, paper and pulp bleaching, food processing, biomedical therapy, disinfection, wastewater treatment, metal mining and processing, etc. (Scheme 1a).10–12 Furthermore, H2O2 is an eco-friendly and safe energy storage medium, offering an energy density of 2.1 MJ kg−1 at 60 wt%, making it ideal for applications in H2O2 fuel cells and cutting-edge power technologies. The H2O2 fuel cell, with a voltage of +1.09 V vs. NHE (standard hydrogen electrode), is similar in performance to the methanol fuel cell (+1.21 V) and the H2 fuel cell (+1.23 V), making it an environmentally benign alternative as an energy carrier.13 The increasing use of H2O2 has led to a rise in global demand, reaching a market volume of 270![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons in 2023, with projections to grow to 400000 tons by 2032 (Scheme 1b).14 Due to the various intriguing properties and versatile applications, the market value of H2O2 was estimated at $5.2 billion in 2023. According to the report, this is expected to grow to $8.8 billion by 2032 (Scheme 1b).15
000 tons in 2023, with projections to grow to 400000 tons by 2032 (Scheme 1b).14 Due to the various intriguing properties and versatile applications, the market value of H2O2 was estimated at $5.2 billion in 2023. According to the report, this is expected to grow to $8.8 billion by 2032 (Scheme 1b).15
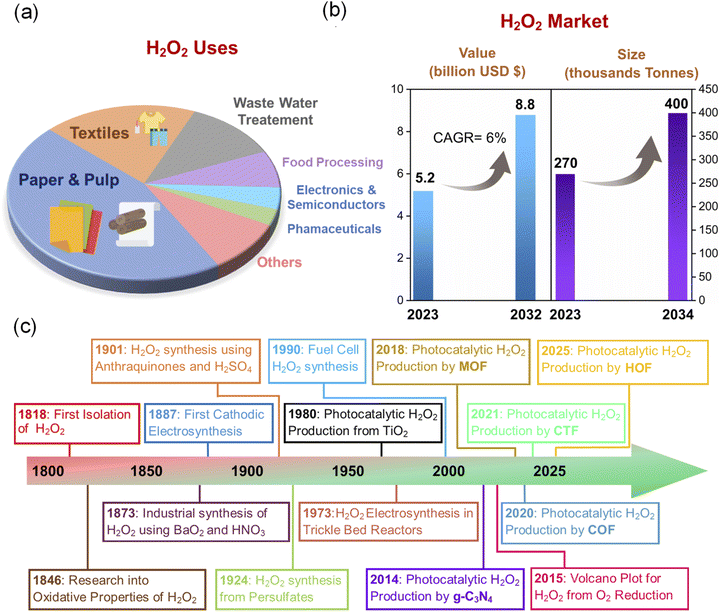 |
| | Scheme 1 (a) Pi diagram showing various uses of H2O2.14 (b) Summary of the expected market value.14,15 (c) Schematic timeline for the synthesis of H2O2 using different techniques.16 | |
The first commercial production of H2O2 was achieved by 1909 via an electrochemical method using H2SO4. A major advancement in H2O2 production emerged in the 1930s with the development of oxidation-based synthetic routes, which were subsequently scaled up for industrial application during the 1940s. The oxidation process was proven to be economically more preferred over the electrolytic process, and two processes, namely isopropyl oxidation and anthraquinone oxidation (AO), were developed for the large-scale production of H2O2. Today, the rising global demand for H2O2 is mainly satisfied using the AO process developed by IG Farben (now BASF).1,8 The AO process primarily involves the hydrogenation of 2-ethylanthraquinone to form 2-ethylanthrahydroquinone and its oxidation further using a Pd or Ni catalyst. The AO process demands hydrogen in high purity and oxygen or air for non-catalytic oxidation. Consequently, the AO process could produce H2O2 with up to 60–70% w/w strength. However, the AO process is less atom-economic and energy-intensive, with an energy requirement of ∼13000 Btu lb−1 (8.1 kWh kg−1).17 On the other hand, the isopropyl oxidation process has significant drawbacks for industrial H2O2 production, as it requires large quantities of raw materials and faces challenges in isolating H2O2 with high purity.18 Apart from the oxidation process, the direct synthesis of H2O2 is also well known. It can produce ∼61 moles per kilogram of catalyst (kg cat) per hour by reacting H2 and O2 at high pressure and using a Pd–Sn catalyst.19–21 However, the explosive nature of H2 and O2 under high pressure and the requirement for novel metal catalysts have limited the industrial-scale synthesis.19,22 Therefore, with the significant limitations of conventional methods, there is significant focus and research to explore and develop eco-friendly alternatives for producing H2O2, addressing both the rising demand and the need for environmentally benign H2O2 production methods. A schematic of the timeline for the production of H2O2 using different techniques is provided in Scheme 1c.16 Towards developing sustainable alternative processes for H2O2 production, electrochemical and photocatalytic techniques have gained significant interest. In electrochemical techniques, H2O2 can be produced by the reduction of O2 in H2O under ambient conditions, also well known as a two-electron oxygen reduction reaction (2e− ORR). It offers numerous benefits, including safe operation, a clean production process, simple reactions under ambient conditions, and low energy consumption. However, the electrochemical 2e− ORR is often hindered by a high overpotential due to slow kinetics and is also influenced by the competing four-electron pathway that leads to the production of H2O. Significant research is going on for its development towards H2O2 production. Various efficient electrocatalysts have been reported that include metal oxides,23,24 single-atom catalysts (SACs),25–27 carbon materials,28,29 porous materials,30 etc. On the other hand, the photocatalytic production of H2O2 using solar energy is one of the most sustainable methods of producing H2O2. Sunlight, a clean and abundant source, provides more than 1.19 × 1017 J of energy to the Earth per second. In nature, green plants and certain organisms are well known to harness solar energy to convert carbon dioxide into carbohydrates through photosynthesis. Motivated by the conversion of solar energy to chemical energy is well-known chemistry today and is applied towards the production of valuable chemicals such as ammonia,31 water splitting,32–34 organic chemicals,35–37 CO2 reduction,38,39 etc. The photocatalytic production of H2O2 has also gained significant interest recently and is developing as a promising alternative to conventional methods, especially targeting the decentralised production and in situ utilisation. The photocatalytic H2O2 production from abundant H2O and O2 as raw materials offers high product selectivity, high atom economy, and less waste production. The photosynthesis of H2O2 proceeds through an oxygen reduction reaction (ORR) and a water oxidation process (WOR). However, the photocatalytic production of H2O2 using water and oxygen is thermodynamically not feasible as it has a high ΔG value of ∼117 kJ mol−1. Therefore, developing an efficient photocatalyst is essential for the photocatalytic production of H2O2, and research in this area is gaining significant interest.
Mechanistic pathway towards photocatalytic H2O2
The photocatalytic production of H2O2 over functional porous materials typically involves three key steps: (i) light absorption, which excites electrons and generates photoinduced charge carriers, (ii) migration of these electrons from the valence band (VB) to the conduction band (CB), leaving behind holes—the energy gap between the VB and CB is also known as bandgap energy (BE)—and (iii) redox reactions between the remaining photogenerated carriers and surface-adsorbed O2 and H2O molecules, leading to H2O2 formation. For a photocatalyst to be efficient towards the desired photocatalytic reaction, the energy of the incident light must be equal to or exceed that of the photocatalyst, so that the electron and hole pairs can be photoexcited and separated into the CB and VB, respectively.
The production of H2O2 could be achieved through the ORR and WOR concomitantly. The ORR, leading to the formation of H2O2, typically proceeds through either a direct one-step 2e− pathway or an indirect two-step, single-electron reduction (eqn (1), Scheme 2) or via a direct one-step, two-electron reduction (eqn (2), Scheme 2).40 In the direct one-step ORR, the adsorbed oxygen is reduced by two electrons, and its combination with two protons produces H2O2 directly (eqn (1), Scheme 2). The thermodynamic potential for procuring the direct 2e− ORR pathway is 0.68 V. In the indirect two-step ORR pathway, the absorbed oxygen is first reduced with one electron to form the superoxide radical (˙O2−) (eqn (2), Scheme 2). This is also the rate-determining step that requires a potential of −0.33 V. The superoxide formed then combines with one electron and two protons to produce H2O2 (eqn (3), Scheme 2). Additionally, the 2e− ORR for H2O2 production competes with the 4e− ORR pathway to H2O (eqn (4), Scheme 2), which is thermodynamically more favourable due to its more positive potential (+1.23 V) compared to the 2e− ORR (+0.68 V). Since the ORR pathway depends on photogenerated electrons, their rapid recombination with holes can significantly reduce the efficiency of H2O2 formation. The use of sacrificial agents such as methanol, ethanol, or benzyl alcohol can selectively scavenge photogenerated holes, suppress electron–hole recombination, and thereby enhance the overall photocatalytic performance. Similar to the ORR pathway, H2O2 can also be generated via the WOR route, in which photogenerated holes at the VB participate in the oxidation process leading to H2O2 formation. The WOR pathway for H2O2 production can be categorized into two mechanisms: (i) a direct one-step 2h+ oxidation process, in which H2O is oxidized by two holes to generate H2O2 at a potential of 1.76 V (eqn (5), Scheme 2) and (ii) an indirect two-step pathway, where oxidation of H2O at the valence band (eqn (6), Scheme 2) produces ˙OH radicals that subsequently dimerize to form H2O2 (eqn (6), Scheme 2). However, the indirect route requires a much more positive potential (2.73 V), making it thermodynamically less favourable. In addition, the WOR can also proceed via 4h+ oxidation of H2O to form O2 at the VB (eqn (7), Scheme 2), and the generated O2 may subsequently participate in a cascade 2e− ORR process to yield H2O2.
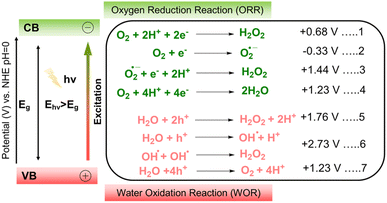 |
| | Scheme 2 Schematic illustration and energy diagram of the photocatalytic production of H2O2 over a photoactive COF-based material via the ORR pathway or WOR pathway, where potentials are given against the normal hydrogen electrode (NHE) at pH = 0. | |
Advanced functional porous materials (AFPMs) as photocatalysts
An ideal photocatalyst exhibits a broad light absorption range, a suitable band gap, efficient charge separation, long-term stability, abundant active sites, and low cost. The pioneering work on photocatalysis was reported by Fujishima and Honda in 1972, where titanium dioxide was demonstrated as an n-type semiconductor for water splitting under UV irradiation.41 Since then, great efforts have been devoted to this research area, and various photocatalytic reactions have been carried out. A primary focus has been devoted to developing inorganic semiconductors such as metal oxides,23 metal sulfides,42,43 metal selenide quantum dots,44 organic dyes,45 etc. However, the practical applications of these photocatalysts are limited by the toxicity, high cost of rare metal components, and the inherent limitation of being non-recyclable. Additionally, many of them have a large band gap of approximately 3.2 eV and exhibit efficiency only under UV-light irradiation, which limits their use under visible light, resulting in low photocatalytic efficiency and photocurrent quantum yield. Metal-free organic semiconductors such as graphitic carbon nitride (g-C3N4) have shown promising efficiency under visible light irradiation. Apart from being metal-free, these materials also possess intriguing properties, such as being earth-abundant, having an appropriate band gap of approximately 2.7 eV, and exhibiting high thermal and chemical stability.46,47 The suitable band structure also makes them ideal for constructing a type II heterojunction combined with other photoactive materials with large band gaps.48 However, g-C3N4 has limited structural diversity and is typically synthesised through thermal polycondensation, which involves calcinating nitrogen-containing molecules at a temperature exceeding 600 °C.49 In the quest for better photocatalysts, researchers are becoming more interested in exploring the photocatalytic performance of advanced functional porous materials (AFPMs). AFPMs refer to materials constructed from organic and/or inorganic building block units through various forces, such as coordination bonds, covalent and hydrogen bonds, etc. They exhibit standard features, including a high specific surface area, a periodic nanoporous structure, and tunable supramolecular structures. Following reticular chemistry, there have been reports of new types of porous materials that include metal–organic frameworks (MOFs), covalent-organic frameworks (COFs), porous organic polymers (POPs), covalent triazine frameworks (CTFs), metal–organic polyhedra (MOPs), and hydrogen-bonded organic frameworks (HOFs). Owing to their large surface area, these materials have been widely explored for separation and gas storage. However, research has also been propelled towards demonstrating their potential as efficient photocatalysts particularly towards H2O2 production (Scheme 3).
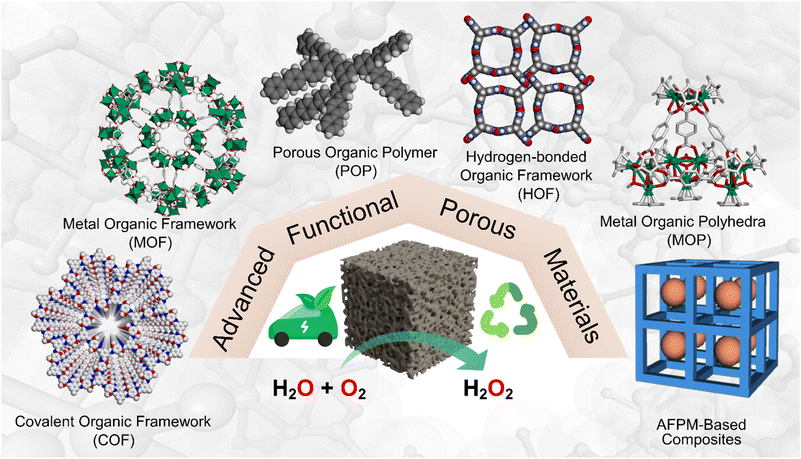 |
| | Scheme 3 Representation of various advanced functional porous materials discussed in this review for photocatalytic H2O2 production. | |
Advantages of AFPM-based photocatalysts
In contrast to traditional semiconductors, functional porous materials exhibit common characteristics and several distinct advantages that make them particularly beneficial for photocatalysis. The main advantages of AFPMs are: (1) a high surface area, which enables efficient mass transport, and their large pore channels, which allow molecules to pass through, resulting in more accessible active sites where substrates can bind and react. (2) Enhanced light-harvesting capability, where π-conjugated frameworks and chromophoric or semiconductive units enhance light harvesting by enabling broad visible-light absorption. (3) Tunable pore structure and morphology, where hierarchical porosity could be introduced to enhance selective diffusion of solvents, mass transport, and better accommodation of reactants/products. (4) Tunable electronic structure, which can be tuned through functional groups or donor–acceptor units to adjust the bandgap, charge separation, and redox properties. (5) High functionalisation flexibility that enables diverse post-synthetic modifications for easy incorporation of catalytic sites, creation of polar environments, and tuning of surface charge. (6) Structural stability and reusability of many AFPMs arising from their chemical robustness and thermal stability, enabling long-term cycling without loss of activity. With all these intriguing properties, AFPMs have been extensively applied to photocatalysis, especially MOFs and COFs. The pioneering work of Zecchina's group in 2004 showed Zn4O13 clusters in MOF-5 to behave similarly to ZnO quantum dots (QDs).50 In 2007, Garcia and coworkers reported the first application of MOFs in photocatalysis using MOF-5 as a photocatalyst for phenol degradation.51 Extensive research has been carried out after that, and the progress of MOFs as a photocatalyst has been summarised in many reviews.52–54 Similarly, the first report of COFs towards photocatalysis was recorded in 2010 by Müllen and coworkers, where COFs have been identified as a novel poly(azomethine) network (ANW) and explored towards photocatalytic H2 evolution.55 Recent advances in using COFs as photocatalysts have been summarised in various reviews.56,57 Regarding photocatalysis, MOFs that are constructed via metal nodes coordinated to organic ligands have large band gaps, which are not feasible for photocatalytic reactions or require high energy to generate electron–hole pairs. Conversely, COFs present a significant opportunity to address the aforementioned challenges at the photocatalyst level. Furthermore, the metal-free nature makes it a more favourable catalyst in terms of environmental considerations. However, the synthesis complexity and photostability are major challenges in using COFs as photocatalysts. POPs are also popular photocatalysts for a wide range of photocatalytic applications.58,59 However, their amorphous nature is a major challenge, as non-crystalline materials typically exhibit poorer photocatalytic performance due to the defects in the structure which disrupt conjugation and hinder efficient charge separation and migration. Various types of AFPMs exhibit distinct characteristics in terms of monomer structure, preparation methods, and optical properties, and each presents its own set of challenges. Different approaches enhancing the performance of AFPMs have been proposed, which include methods such as ligand functionalization, mixed-metal/linker strategies, metal ion/ligand immobilisation, metal nanoparticle loading, semiconductor coupling, composite synthesis, etc. In this review, we discuss the photocatalytic applications of various AFPMs, with a focus on H2O2 production, and relate their photocatalytic performance to the structural and electronic properties of their compositional components.
AFPM-based photocatalysts for H2O2 production
As the demand for H2O2 continues to rise and the search for environmentally friendly production methods intensifies, several well-known semiconductors, such as metal oxides and g-C3N4, have garnered significant attention towards photocatalytic H2O2 production.23,60 Metal oxides such as TiO2 serve as a benchmark photocatalyst for H2O2 production through two-electron oxygen reduction under UV/visible light irradiation. Due to the wide band gap of TiO2, it has often been combined with other materials to form a composite appropriate for H2O2 photosynthesis. Modifications such as Au–Ag or Pd cocatalysts enhance charge separation and selectivity for H2O2 production on TiO2 and achieve yields up to 3.6 mM.61 Similarly, BiVO4 has also shown efficiency for H2O2 production through 2e− ORR pathways due to the favourable alignment of its conduction band edge.62 Similar to metal oxides, modified metal sulfides have also shown promising efficiency for H2O2 production. 2D layered materials such as g-C3N4 also serve as the standard metal-free benchmark for photocatalytic H2O2 production, achieving ∼20–30 µmol h−1 (pure water/O2) via the 2e− ORR due to its suitable conduction band (−0.4 to −1.0 V vs. NHE) for ˙O2- formation, though limited by the poor WOR (VB ∼+1.7 V) and recombination, yielding an apparent quantum yield (AQY) of ∼0.5–2% at 420 nm.63 Beyond conventional photocatalysts, lignin, one of the most abundant renewable aromatic biopolymer resources, has also recently been reported as an effective photocatalyst for H2O2 production.64 In the past few years, there has been a notable increase in research focused on using porous materials for photocatalytic H2O2 production. The statistical data obtained by searching by using phrases “Photocatalytic Production of Hydrogen Peroxide” or “H2O2” and “Porous” or “Frameworks” as the title in the Web of Science database are shown in Fig. 1. Tremendous developments have been made in enhancing the photochemical efficiency of AFMPs towards light-driven H2O2 production, and are well documented in various reviews.30,65–69 While previous reviews have extensively documented and discussed the progress of porous materials in photocatalytic H2O2 production, they typically focused on a single type of AFPM. In contrast, detailed reviews that collectively examine the progress and advances across all functional porous materials remain limited. Capitalising on this opportunity, the present review provides a unified and critical discussion of photocatalytic H2O2 production using a broad range of porous frameworks, including COFs, MOFs, POPs, CTFs, HCPs, HOFs, MOPs, and their composite systems. Considering the large volume of publications in this field and the fact that many earlier advances have already been comprehensively covered in previous reviews, this manuscript focuses on literature published between 2024 and 2025. Furthermore, the review highlights the key opportunities and challenges associated with AFPM-based photocatalysts, with particular emphasis on the synthetic techniques of AFPM photocatalysts, scalability toward practical applications, long-term photostability, long-term H2O2 production, and efficiency limitations. All AFPMs have been discussed as individual topics/subtopics for the readers. The review also provides perspectives on and recommendations for the future progress of this field.
 |
| | Fig. 1 Bar chart showing the number of articles per year concerning photocatalytic hydrogen peroxide production by advanced functional porous materials (accessed 21 January 2026). The statistical data were obtained by searching using the phrases “Photocatalytic Production of Hydrogen Peroxide” or “H2O2” and “Porous” or “Frameworks” in the Web of Science database, and the numbers correspond to the final publication year. | |
Covalent organic framework (COF)-based photocatalysts for H2O2 production
COFs are porous materials composed of organic building blocks connected by strong covalent bonds. The covalent bonds are formed due to reactions like condensation, resulting in highly ordered, crystalline structures with large surface areas and tunable pore sizes (Fig. 2a). Since the pioneering work by Yaghi and co-workers70 in 2005, they have attracted significant interest in terms of synthesis and applications.71–73 Owing to their well-defined74 structures, high crystallinity, permanent porosity, unlimited tenability, metal-free nature, and appreciable stability, COFs have been established as a promising material for catalysis,75,76 capture and separation,77,78 energy materials,79,80 bioapplication,81 sensing,82,83 pollutant degradation,84 CO2 reduction,85 etc. In particular, enhanced charge separation, excellent light-harvesting capabilities, conjugated structures, semiconductor nature, and an opportunity to tune the band gap have made COFs an interesting photocatalytic material. The semiconductor behaviour of COFs was first reported by Jiang et al. for a boronic ester COF.86 Later, in 2014, Stegbauer et al. explored the efficiency of a hydrazine-based COF in producing hydrogen from pure water upon irradiation with visible light, marking the first application of a COF as a photocatalyst.74 COFs exhibit various intrinsic features that make them ideal for photocatalytic applications, including their crystalline, porous structure for efficient charge transport, extended π-conjugation for enhanced light absorption, and a high surface area that provides abundant active sites. Their modular design enables the precise tuning of electronic properties through the incorporation of functional groups, while excellent chemical and thermal stability ensure durability during photocatalytic reactions. To date, extensive research has reported the use of COFs as efficient metal-free heterogeneous photocatalysts for various applications, including CO2 reduction, organic transformations, H2 evolution, ammonia synthesis, and pollutant degradation.87,88 The photocatalytic application of COFs towards H2O2 production was pioneered by Van Der Voort and coworkers in 2020, with a series of 2D imine-based COFs.89 Since then, various strategies have been implemented to improve the photocatalytic efficiency of COFs. To be precise, donor–acceptor optimisation, functional group incorporation, linkage modification, topological and structural modification and post-synthetic modification (PSM) are some of the important areas that can severely impact the photocatalytic efficiency of COFs (Fig. 2b). These strategies have been meticulously discussed as subtopics in this review.
 |
| | Fig. 2 (a) Model reaction for COF formation through a condensation reaction. (b) Application of COFs, discussed in this review, to enhance photocatalytic H2O2 production. | |
Donor–acceptor (D–A) COFs for photocatalytic H2O2 production
The donor–acceptor (D–A) architecture is an inherent property of COFs. The monomers, often comprising aromatic and π-electron-rich moieties, typically act as donors, while functionalities containing electron-deficient groups generally serve as acceptors. The alternating arrangement of D–A units in COFs enables band gap tunability, enhances charge transport, and promotes efficient electron–hole separation, collectively leading to improved photocatalytic performance.90 D–A moieties within the framework also create a strong push–pull effect that promotes efficient charge separation and transfer. This effect lowers the electrostatic attraction between photogenerated carriers, thereby improving the overall charge-separation efficiency. Furthermore, COFs designed with D–A configurations exhibit a markedly lower exciton binding energy compared to their symmetric counterparts.91 The strong intraframework charge-transfer interactions shift the electron density from the donor units toward the acceptor domains upon photoexcitation, generating a built-in electric field that spatially separates charge carriers. This separation reduces the coulombic attraction between electrons and holes, thereby facilitating rapid exciton dissociation into free carriers. Consequently, D–A COFs display enhanced charge mobility, suppressed recombination, and improved photocatalytic efficiencies. Various D–A COFs have been reported recently that possess superior activity towards photocatalytic H2O2 production. For instance, considering the large π-conjugated structure, excellent light-harvesting abilities, and electron donor capabilities of pyrene moieties, Van Der Voort and their group reported the synthesis of four novel pyrene-based COFs.92 The π-electron-rich pyrene acts as a donor, and its combination with an appropriate electron acceptor monomer has been found to be efficient towards H2O2 production, with a rate of 1242 µmol g−1 h−1 in the biphasic system (benzyl alcohol and water). The biphasic system has also been advantageous as it successfully inhibited the breakdown of H2O2 and facilitated efficient photocatalytic generation of H2O2.
Modulating the D–A property of COFs through linkage chemistry toward H2O2 production has been thoroughly examined by Jiang et al. Using a hexaphenyl triphenylene donor and a benzothiadiazole acceptor, they constructed hexavalent COFs featuring hydrazone, imine, or cyano-vinylene linkages (Fig. 3a).93 These materials possess sites that enable both the 2e− ORR and 4e− WOR, allowing concurrent formation of H2O2 and O2. Among the linkages, the hydrazone bridge provided the most favourable characteristics, including superior stability, higher polarity, and abundant adsorption sites for water and oxygen (Fig. 3b). As a result, the Hz-TP-BT-COF delivered the highest H2O2 production rate (5.7 mmol g−1 h−1), significantly outperforming the imine-linked (0.6 mmol g−1 h−1) and vinylene-linked (2.3 mmol g−1 h−1) analogues (Fig. 3c) without sacrificial agents alongside an apparent quantum yield (AQY) of 17.5% and a TOF of 4.2 h−1 at 420 nm. All synthesised COFs also show better performance towards photocatalytic H2O2 production compared to the benchmark g-C3N4 catalyst (Fig. 3c). The hexavalent hydrazone-linked COF also demonstrated the continuous photosynthesis of H2O2 in flow reactors with air-saturated water, reporting a record-high yield of 6480 mL of H2O2 over 144 h (Fig. 3d). Building on their earlier success with hydrazone-linked hexavalent COFs, the same group advanced their H2O2 photosynthesis strategy by designing a new series of hexavalent D–A COFs through precise tuning of π-frameworks and pore environments.94 In this design, diphenylbutadiyne units replaced benzothiadiazole as the electron-accepting component. Besides serving as an active site for the 2e− oxygen-reduction process, the hydrophobic diphenylbutadiyne motif discourages water adsorption and promotes spontaneous oxygen enrichment within the pores. The overall hydrophobicity of the COFs was further modulated by extending the ether side chains on the linker (Fig. 3e). Under visible light in pure water, the resulting COFs delivered H2O2 at rates of 7.2, 5.9, and 3.4 mmol g−1 h−1 for the TP-DPBD30-COF, TP-DPBD20-COF, and TP-DPBD10-COF, respectively, consistent with the 2e− ORR mechanism. When exposed to natural sunlight, the TP-DPBD30-COF maintained a high activity of 3.4 mmol g−1 h−1. Their practical potential was demonstrated in a continuous-flow reactor (Fig. 3f), which produced 15.1 L of H2O2 solution (0.16 mM) over two weeks/336 hours, highlighting the scalability of this COF-based system (Fig. 3g). The overall performance of TP-DPBD-COF has been shown through a radar plot (Fig. 3h), which highlights the production, solar-to-chemical conversion (SCC), AQY, stability, and turnover frequency (TOF) during photocatalytic H2O2 production. Tuning the D–A properties of a COF by introduction of a heteroatom is one of the well-known strategies. The thiophene-S units are well known for the ORR, and its increased density within the COF enhances the ORR efficiencies.95 The study performed by Wang et al. demonstrates the photocatalytic efficiency of the benzenetrithiophene (Btt)-based COF towards H2O2 production.96 Three D–A COFs (TpaBtt, TapbBtt, and TaptBtt) were obtained by combining sulphur-rich Btt-based aldehyde with different types of triphenylamine derivatives (Fig. 4a). The synthesised COFs acquired a dual D–A structure that contributes an appropriate energy band position and a feasible intermediate anion towards H2O2 photosynthesis. Under visible light irradiation, TaptBtt with a triazine core produced H2O2 at a high rate of 2111 µM h−1 in pure water and air with an SCC of 0.296%. Similarly, Cao et al. reported that BBT-ACN COF-1, synthesised via Knoevenagel condensation between benzotristhiophene (BTT) and benzo[1,2-d;4,5-d′]bisthiazole (BBT), exhibited higher efficiency for photocatalytic H2O2 production than BBT-ACN COF-2, which was formed by coupling conjugated pyrene aldehyde with BBT.97 Both COFs are sp2-carbon-linked and have distinct D–A properties. BBT-ACN COF-1 features a D–A structure with BBT as the acceptor and BTT as the donor, whereas BBT-ACN COF-2 adopts a π–A architecture with pyrene as the π-unit and BBT as the acceptor. The use of thiophene-containing COFs for photocatalytic H2O2 production has also been reported by Yue et al.98 By regulating the N-heteroatom in the framework, they have synthesised two thiophene COFs (TD-COF and TT-COF). The regulation of N atoms has brought differences in their wettability, as the TD-COF is superhydrophilic and the TT-COF has a hydrophobic nature. Consequently, the TD-COF demonstrated H2O2 production of 4060 and 3364 µmol g−1 h−1 in deionised water and seawater, respectively, both of which are higher than those of the TT-COF. The mechanistic pathway revealed the photocatalytic production of H2O2 through the indirect 2e− ORR and direct 2e− WOR dual pathways, where thiophene is likely the photo-reduction unit, while benzene is the probable photo-oxidation unit. Beyond simple D–A assemblies in COFs, incorporating multiple D–A moieties has been shown to further enhance the photochemical efficiency. As shown by Shu et al., the multiple donor–acceptor–acceptor (D–A–A) type FS-OHOMe-COF shows high efficiency for H2O2 production compared to FS-OH-COF and FS-Ome-COF, which have simple D–A units (Fig. 4a).99 The sulfone group present within the COF linker serves as an active site and enhances stability by strengthening interlayer forces. Furthermore, compared to the pristine COF mixed ligand strategy, the synthesised FS-OHOMe-COF structure features extended π–π conjugation, long excited-state lifetime, low exciton binding energy, and high photostability. The H2O2 production rate of FS-OHOMe-COF was measured to be 1.0 mM h−1 in O2-saturated water under visible light irradiation, which is higher than that of its pristine counterpart (Fig. 4b). Furthermore, an AQY of 9.6% at 420 nm and an SCC of 0.58% were also observed. Most reported photocatalysts generate only very low H2O2 concentrations, typically below 2 mM, far from the levels needed for practical use. In contrast, the FS-OHOMe-COF demonstrated steady and long-term accumulation of H2O2, achieving 19 mM in pure water after 72 h of irradiation. When benzyl alcohol was added, the concentration further increased to 120 mM after 153 hours, although part of this enhancement may arise from autocatalytic H2O2 formation associated with BA (Fig. 4c). Furthermore, the recyclability up to four cycles has been demonstrated by the FS-OHOMe-COF in both pure water and BA (Fig. 4d). The importance of the S-atom towards improving various properties of the COF photocatalyst has also been demonstrated by Cheng and coworkers.100 They constructed a heteropolyaromatic TTT-COF by introducing thiazole units through a post-cyclisation reaction (Fig. 5a), which resulted in three key improvements (Fig. 5b): (i) markedly higher chemical stability, (ii) a fully extended π-conjugated framework that supports efficient single-electron and energy-transfer processes, and (iii) increased intramolecular polarity that strengthens the D–A character and promotes charge separation. Owing to these features, the TTT-COF shows far superior stability, conjugation, and photocatalytic performance compared to its imine-linked counterpart, TTI-COF. Consequently, using 10% benzyl alcohol, the TTT-COF produces H2O2 at a rate of 29.9 mmol g−1 h−1, which is about 20 times higher than that of the imine-linked COF and superior to many state-of-the-art photocatalysts (Fig. 5c and d). The TTT-COF shows excellent durability, operating continuously for over 200 h with no significant loss in photocatalytic activity (Fig. 5e). Thiophene-based COFs are generally effective for H2O2 production. Notably, when the thiophene density was tuned using bis- and tris(thiazole) amines, the bis(thiazole)-based COF showed higher H2O2 production
efficiency as reported by Qiao et al.101 Two COFs—Tp-BTz and Tp-TTz (Fig. 5f)—were assessed their H2O2 production using a mass-transfer-gating (MTG) (Fig. 5g) strategy based on sacrificial agents. The MTG approach improves interfacial reactions by easing mass-transport constraints, increasing access to catalytic sites, scavenging photogenerated holes, and providing protons for downstream coupling steps. Introducing benzyl alcohol (BA) into the system activated a catalytic mode-switching behaviour, dramatically boosting performance. Under 10% BA and O2-saturated conditions, the Tp-BTz COF delivered a record H2O2 production rate of 100.9 mmol g−1 h−1, outperforming state-of-the-art photocatalysts and exhibiting 1.8-fold higher activity than the Tp-TTz COF (Fig. 5h). Also, the AQY of these COFs at 420 nm were calculated to be 18.0% and 16.5%, and the SCC efficiency evaluated using a solar simulator (AM 1.5G), yielded values of 1.47% and 0.72% for Tp-BTz COF and Tp-TTz COF, respectively. The sulfur-containing thiophene unit has moreover served as an electron-accepting site for the reduction of oxygen during H2O2 production. However, the electron donor strength of sulfur-containing moieties with the COF has also been explored and found to be efficient for H2O2 production. Van Der Voort and coworkers102 report the synthesis of three D–A COFs (TMT-TT, TMT-N, and TMT-BT) by combining the triazine (TMT) unit with an aldehyde containing either thienothiophene (TT), naphthalene (N), or benzothiazole (BT) moieties, where the TT unit, with its two sulfur groups, exhibited the highest donor strength, followed by N and BT, with the electron-donating ability following the order: TT > N > BT. Therefore, with the D–A property in synthesised COFs, the H2O2 production rates in the O2-purged pure water system were measured to be 1952, 1742, and 748 µmol g−1 h−1 for the TMT-TT-, TMT-N-, and TMT-BT-COF, respectively. The presence of nitrogen can also play a significant role in improving charge-carrier transport. As shown by Zhang et al., the systematic regulation of nitrogen atoms within the framework modulates the electron-accepting capacity of COFs.103 Regulating the nitrogen content in COFs leads to a well-defined D–π–A architecture. A higher density of nitrogen atoms in the heterocyclic units strengthens charge migration along the π-conjugated pathway. As a result, these COFs can promote the ORR through both the π-bridge and acceptor centres, while the WOR proceeds at the donor sites. This synergistic division of functions greatly enhances their overall efficiency in photocatalytic H2O2 generation. Notably, the Tf-TAPT-COF with a triazine unit can produce H2O2 at a rate of 2700 mmol g−1 h−1 from water and O2. The major achievements of the D–A COF photocatalyst for H2O2 production has been highlighted in Table 1.
 |
| | Fig. 3 (a) Structural representation of Hz-TP-BT-COF, Im-TP-BT-COF, and sp2c-TP-BT-COF. (b) and (c) Comparison of photocatalytic H2O2 production of the synthesised catalyst with the benchmark g-C3N4. (d) Continuous H2O2 production up to 6480 mL within 140 h with Hz-TP-BT-COF. (Reproduced and reprinted with permission from ref. 93. Copyright 2024, Springer Nature.) (e) Structural representation of TP-DPBD10-COF, TP-DPBD20-COF, and TP-DPBD30-COF. (f) Schematic representation of continuous photocatalysis in a flow reactor set up and WOR and ORR active sites in TP-DPBD30-COF. (g) Schematic illustration of the production of H2O2 over 336 h through continuous photocatalysis in a flow reactor. (h) Graph for overall performance of TP-DPBD-COF towards H2O2 production. (Reproduced and reprinted with permission from ref. 94. Copyright 2024, Springer Nature.) | |
 |
| | Fig. 4 (a) Synthesis scheme and structure of FS-COFs. (b) H2O2 production comparison of different FS-COFs. (c) Long-term stability of FS-OHOMe-COF in benzyl alcohol (BA) and water over 160 h. (d) Performance of recycled FS-OHOMe-COF in benzyl alcohol (BA) and water over four cycles. (Reproduced and reprinted with permission from ref. 99. Copyright 2024, Wiley-VCH GmbH.) | |
 |
| | Fig. 5 (a) Schematic illustration of synthesis of thiazole-based homologous heteropolyaromatic COFs. (b) Representation of D–A moieties and the structural advantages of homologous heteropolyaromatic COFs. (c) Time-dependent photocatalytic activity of TTI-COF and TTT-COF for H2O2 production. (d) Photocatalytic H2O2 production comparison of TTI-COF and TTT-COF under different conditions. (e) Long-term H2O2 production of TTT-COF (inset: daily yield over eight days). (Reproduced and reprinted with permission from ref. 100. Copyright 2025, Springer Nature.) (f) Schematic illustration of the synthesis of Tp-BTz COF and Tp-TTz COF. (g) H2O2 production through mass-transfer gating (MTG). (h) Photocatalytic H2O2 production with Tp-BTz COF in different solvents. (Reproduced and reprinted with permission from ref. 101. Copyright 2025, Wiley-VCH GmbH.) | |
Table 1 Summary of representative D–A COF photocatalysts for H2O2 photosynthesisa
| Sl. no. |
Catalyst |
Reaction conditions |
Cat. dosage (mg) & solvent volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC |
Ref. |
| Light irradiation: λ > 420 nm; BA: benzyl alcohol; BAm: benzyl amine; ACN: acetonitrile. |
| 1 |
Py–Py COF |
H2O:BA (9:1) |
10 mg & 20 mL |
1242 |
4.5% @ 420 nm |
— |
92 |
| 2 |
Hz-TP-BT-COF |
H2O |
3 mg & 15 mL |
5700 |
17.5% @ 420 nm |
— |
93 |
| 3 |
TP-DPBD30-COF |
H2O |
2.5 mg & 15 mL |
7200 |
18% @ 420 nm |
0.91% |
94 |
| 4 |
FS-OHOMeCOF |
H2O |
10 mg & 20 mL |
1100 µM h−1 |
9.6% @ 420 nm |
0.58% |
99 |
| 5 |
TaptBtt-COF |
H2O |
15 mg & 50 mL |
1407 |
4.6% @ 450 nm |
0.296% |
96 |
| 6 |
BBT-ACN-COF-1 |
H2O |
5 mg & 50 mL |
2500 |
3.12% @ 420 nm |
— |
97 |
| 7 |
TD-COF |
H2O and O2 |
1 mg & 4 mL |
4620 |
— |
0.15% |
98 |
| 8 |
TTT-COF |
H2O:BA (9:1) |
5 mg & 45 mL |
29905 |
12% @ 400 nm |
0.32% |
100 |
| 9 |
Tp-BTz-COF |
H2O:BA (9:1) |
5 mg & 20 mL |
100864 |
18% @ 420 nm |
1.47% |
101 |
| 10 |
COF-JLU92 |
H2O:BA (1:1) |
5 mg & 50 mL |
39283.3 |
15.4% @ 420 nm |
2.35% |
104 |
| 11 |
TT-T-COF |
H2O:BA (9:1) |
5 mg & 25 mL |
10066 |
19.2% @ 420 nm |
— |
105 |
| 12 |
TMT-TT-COF |
H2O and O2 |
10 mg & 30 mL |
1952 |
— |
— |
102 |
| 13 |
Tf-TAPT-COF |
H2O and O2 |
10 mg & 50 mL |
2700 |
2.48% @ 420 nm |
— |
103 |
| 14 |
TTF-BT-COF |
H2O and O2 |
5 mg & 10 mL |
2.76 mM g−1 h−1 |
11.19%@ 420 nm |
0.49% |
108 |
| 15 |
JUC-165 |
BAm, O2 and ACN |
5 mg & 10 mL |
22800 |
15.7% |
1.09% |
109 |
| 16 |
COF-2 |
Seawater |
5 mg & 50 mL |
6930 |
2.5% @ 420 nm |
0.26% |
111 |
| 17 |
TACOF-1-COOH |
H2O and O2 |
2.5 mg & 18 mL |
3542 |
5.7% @ 420 nm |
0.55% |
112 |
Apart from sulfur- and nitrogen-containing COFs, oxygen-rich frameworks have also demonstrated notable efficiency in H2O2 photosynthesis. In particular, benzotrifuran-based COF-JLU92 (Fig. 6a) featuring electron D–A characteristics with a hydrazone linkage has been reported by Zhang et al. for photocatalytic H2O2 production.104 The partially separated D–A π-stacks in the benzotrifuran-based COF promote efficient charge separation and provide active sites for O2 reduction and H2O oxidation. Also, the 1D heteroatom-rich pores within the framework enhance water transport and lower the energy barrier for oxidation, enabling high H2O2 production rates of up to 9800 µmol g−1 h−1 from water and oxygen with an AQY of 8.7% at 450 nm. Varying the ratio of water to benzyl alcohol from 9:1 to 1:1, the H2O2 production rate drastically increased from 17089 to 39284 µmol g−1 h−1 (Fig. 6b). The high yield of H2O2 in BA was also recently reported with the nitroxyl radical decorated TT-T-COF.105 Owing to the presence of an unpaired electron in 2,2,6,6-tetramethylpiperidine-N-oxyl (TEMPO), it has been used as a nitroxyl radical mediator within the COF (Fig. 6c). The TEMPO-based COF TT-T-COF showed good crystallinity and had a Brunauer–Emmett–Teller (BET) surface area of 88.48 m2 g−1. The radical-based D–A architecture in the TT-T-COF promoted efficient separation of photogenerated electron–hole pairs, extends charge lifetimes, and enhances overall electron utilisation. However, COFs incorporated with TEMPO have band edges suitable for the ORR but not for the WOR (2e− or 4e−). Femtosecond transient absorption (fs-TA) studies reveal that the TT-T-COF undergoes ultrafast charge-separated state (CSS) formation (<5 ps) upon 365 nm excitation, as evidenced by characteristic ground-state bleaching (GSB, ∼461 nm) and excited-state absorption (ESA, ∼536 nm) signals (Fig. 6c). The rapid ESA evolution indicates efficient intramolecular electron transfer from the TAPP donor to the TEMPO acceptor within the D–A framework. Notably, incorporation of the redox-active TEMPO unit significantly prolongs the photogenerated electron lifetime (∼671 ps), highlighting enhanced charge separation and carrier utilisation compared to conventional COFs. Time-resolved kinetic measurements excited at 365 nm over a timescale of 0.5 ps to 1 ns revealed an intramolecular charge transfer process from TAPP to TPDA-TEMPO (Fig. 6d). The photocatalytic H2O2 production rate of 10066 and ∼34000 µmol g1 h1 in 10% and 20% BA under light irradiation >420 nm was noted for the TT-T-COF (Fig. 6d). In situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) showed an increase in the intensity at 1178 cm−1, 809 cm−1, and 850 cm−1 corresponding to *OOH and HO–OH bonds. Combined theoretical and experimental (in situ DRIFTS) investigations demonstrate that the enhanced ORR activity and stability of the TT-T-COF arise from the participation of the nitroxyl radical (N–O˙). The redox-active site modulates the local electron density and facilitates selective O2 adsorption via a Yeager-type direct 2e− ORR pathway.
 |
| | Fig. 6 (a) Schematic illustration of the synthesis of oxygen-rich COF-JLU. (b) Photocatalytic H2O2 production with COF-JLU in pure water and BA. (Reproduced and reprinted with permission from ref. 104 Copyright 2025, Wiley-VCH GmbH). (c) Schematic illustration of the synthesis of TEMPO decorated TT-T-COF. (d) Time-resolved kinetic measurements over a timescale of 0.5 ps to 1 ns revealing an intramolecular charge transfer process from TAPP to TPDA-TEMPO. (e) Photocatalytic H2O2 production with COF-JLU under different conditions. (Reproduced and reprinted with permission from ref. 105, Wiley-VCH GmbH). | |
COFs with different functional groups for photocatalytic H2O2 production
The profound opportunity to synthesise COFs with various functional groups represents one of the most important advancements in the field. Functional group modification in COFs involves altering the chemical groups attached to the framework to enhance its properties or tailor its functionality. Customisation of desired functional groups within the framework can enhance the electronic characteristics of COFs, boosting their ability to absorb visible light and improving the charge separation efficiency. The nature of substituents on COF linkers plays a decisive role in dictating both framework characteristics and catalytic behaviour. Fluorinated groups, for instance, can reshape the crystallinity, pore environment, and acid–base properties, ultimately influencing oxygen-reduction pathways and light-driven activity.106 In contrast, hydroxyl functionalities help capture photogenerated holes and suppress charge-recombination events, thereby improving the durability.107 Cyanide units steer the oxygen activation pathway by favouring the generation of singlet oxygen (1O2) instead of superoxide species.113 Additionally, the functional modifications can increase the stability of COFs under various operational conditions, ensuring reliable performance over time. Moreover, the strategic addition or removal of groups facilitates efficient charge transfer, minimising the recombination of electron–hole pairs and increasing the production of H2O2.
The functionalization of COFs with heteroatoms (N, O, S, etc.) has a profound effect that enhances the photocatalytic activity.110 The presence of heteroatoms alters the electronegativity of adjacent carbon atoms, potentially making these carbon atoms catalytically active as well. Moreover, the inherent hierarchy in electronegativity among the three heteroatoms significantly influences the resulting framework characteristics. In addition to the type of heteroatom present, its density and spatial arrangement within the framework also play critical roles in modulating the charge-transfer dynamics of COFs.114 Some studies show that COFs containing nitrogen-rich triazine, heptazine, can rationally induce the electrophilicity and results in more efficient ORR, forming H2O2.115 The effect of different amounts of nitrogen in COFs on photocatalytic H2O2 production has been demonstrated by Zhang et al.116 Two photoactive COFs (COF-JLU51 and COF-JLU52) were synthesised via a Schiff base condensation reaction between [1,2,4]-triazolo-[1,3,5]-triazine (TTT) and triphenylbenzene as a building block. COF-JLU51 and COF-JLU52 have a nitrogen content of 23.01 wt% and 26.79 wt%, respectively. TTT moieties have strong electron-withdrawing ability, and their combination with electron-donating triphenylbenzene imparts optimum electron D–A characteristics to COF-JLU51. Consequently, the photochemical activity of COF-JLU51 is higher than that of COF-JLU52. The H2O2 production rates of 4260.3 µmol g−1 h−1 and 3033.3 µmol g−1 h−1 were observed for COF-JLU51 and COF-JLU52, respectively. However, in a biphasic system containing water and benzyl alcohol in a ratio of 9:1, COF-JLU52 could produce H2O2 at a rate of 7624.7 µmol g−1 h−1 with an AQY of 18.2% at 420 nm. The SCC efficiency of COF-JLU51 and COF-JLU52 was reported to be 0.19% and 0.12%, respectively. Similarly, the influence of thiophene and high nitrogen content in the COF towards enhanced photocatalytic activity has been reported by Zeng and co-authors by demonstrating the N-site engineering strategy on the thiophene-containing COF.117 Through the Schiff base condensation of various N-heterocycle (pyridine, pyrimidine, and triazine) units with a thiophene building unit, five COFs were synthesised. Among them, the TTA-BT-COF and TTA-TF-COF were multicomponent COFs as both triphenyltriazine aldehyde and triphenyl triazine-NH2 reacted with the thiophene moiety. The multicomponent TTA-TF-COF, which has more N-sites, was efficient towards photocatalytic H2O2 production with a maximum rate of 3343 µmol g−1 h−1 in pure water and air. These results suggest a direct correlation between the nitrogen atom density in the COF and photocatalytic H2O2 production. However, recent studies indicate that not only the nitrogen density but also its spatial positioning within the framework plays a crucial role in governing the efficiency of H2O2 formation. The effect of adjacent N in the COF skeleton towards photocatalytic H2O2 production has been demonstrated by Zhao and co-authors.118 Three COFs, namely, BD-COF, Bpy-COF and PyIm-COF, were synthesised via a Schiff base condensation reaction by reacting pyrene-NH2 with various aldehydes that contain either 1,1′ biphenyl (BD) or 2,2′ bipyridine (Bpy) or 3,3′ bipyridine (PyIm). The photocatalytic H2O2 production was evaluated using pure water under Xe light irradiation without any sacrificial agent. The H2O2 production with the PyIm-COF was found to be 5850 µmol g−1 h−1, which is the highest amongst Bpy-COFs (3060 µmol g−1 h−1) and BD-COFs (670 µmol g−1 h−1). The AQY and SCC of PyIm-COF were 3.70% at 420 nm and 0.28% under AM 1.5 G simulated sunlight. The theoretical investigation shows that the pyrimidine unit is responsible for the enhanced separation of the photogenerated electron–hole pair, which prevents the recombination and strong adsorption of O2 on the adjacent N-atoms.
It is well known that H2O2 formation proceeds through a 2e− ORR pathway; the electrophilicity of the COF and the nature of its oxygen-adsorption sites are crucial determinants of its photocatalytic efficiency. Rationally introducing electrophilic functional groups such as fluorine, vinylene, and cyanide has also been shown to improve the photocatalytic H2O2 generation performance. For instance, Zhao et al.119 have synthesised pyridyl-imine functionalized D–A COFs (PyIm-B and PyIm-BT_X, where X = H, F or 2F), by regulating the electronegative fluorine atoms (Fig. 7a). The different F-atoms within the framework regulated the electron-deficient nature of the acceptor moiety. The electrochemical impedance spectra (Fig. 7b) show the lowest electrochemical impedance and smallest charge-transfer resistance for PyIm-BT_F, followed by PyIm-BT_2F, PyIm-BT_H, and PyIm-B. These results demonstrate that tuning the D–A configuration effectively modulates photoinduced charge separation and transport in PyIm-COFs, with PyIm-BT_F showing superior optical and electrochemical performances due to its monofluorinated benzothiadiazole unit. Consequently, PyIm-BT_F achieved the highest H2O2 production rate of 5342 µmol g−1 h−1, surpassing both PyIm-BT_H (3577 µmol g−1 h−1) and PyIm-BT_2F (4061 µmol g−1 h−1) (Fig. 7c). The COF exhibited good structural integrity and sustained photoefficiency during stability tests conducted for up to 10 h. The photoexcited carrier dynamics were evaluated through femtosecond transient absorption spectroscopy (fs-TAS), where all PyIm-COFs displayed ultrafast excited-state absorption (ESA) signals after photoexcitation (Fig. 7d). PyIm-B exhibited a strong ESA band at 580 nm, indicating a high excited-state population. In contrast, BT-functionalized COFs (PyIm-BT_H, PyIm-BT_F, and PyIm-BT_2F) showed negative peaks at 740 nm, attributed to photoinduced emission from interfacial charge transfer between Py and BT units. These results confirm that the D–A architecture enhances charge transfer and suppresses electron–hole recombination. Although PyIm-BT_2F contains more fluorine atoms, the superior activity of PyIm-BT_F suggests that an optimal, rather than excessive, electrophilic character is essential for efficient H2O2 generation. Similarly, regulation of the cyanide moiety within the framework has been reported by Tong et al., who synthesised three ketoenamine-based D–A COFs by regulating the number of CN groups (COF-xCN, where x = 0, 1, 2) (Fig. 7e).120 The H2O2 photosynthesis performance of synthesised COFs in pure water and O2 under visible light irradiation was studied, which showed an increasing production with increasing number of CN functional groups. Consequently, the highest production for COF-2CN (1601 mmol g−1 h−1) was observed, followed by COF-1CN (573 mmol g−1 h−1), and lastly COF-0CN (194 mmol g−1 h−1). The remarkable efficiency of COF-2CN can be attributed to the presence of two CN groups within the framework, which activate the adjacent carbon atom, facilitating the formation of the key *OH intermediate that lowers the energy barrier for the rate-determining two-electron water oxidation step in H2O2 photosynthesis. H2O2 was also produced efficiently and stably at a rate of ≈716 µmol g−1 h−1 under visible-light irradiation over a 20 h photocatalytic operation in a continuous-flow reactor (Fig. 7f). The effect of regulating the number and position of hydroxyl (–OH) groups on photocatalytic H2O2 production has been investigated in porphyrin-based COFs by Liu et al. (Fig. 7g).121 Three COFs, namely 2-DhaTph, 2,5-DhaTph, and 2,3-DhaTph, were synthesised, in which the porphyrin unit acts as the electron acceptor, while the hydroxyl-functionalized moiety serves as the electron donor. An increase in the number of –OH groups enhance the donor ability of the COFs, leading to improved photoefficiency. However, compared to 2,3-DhaTph with para-positioned hydroxyl groups, 2,5-DhaTph with para-positioned –OH groups exhibit higher crystallinity and better exposure of catalytically active sites. Consequently, 2,5-DhaTph showed a H2O2 production rate of 2103.1 µmol h−1 g−1, 1.1 times higher than that of 2,3-DhaTph. DFT analysis revealed that 2,5-DhaTph exhibits the lowest energy barrier (0.541 eV) for the formation of the *OOH intermediate, which is the rate-determining step, compared to the other COFs (Fig. 7h). Similarly, incorporating thiophene and furan isomeric units into the side chains of COFs enables precise tuning of COF electronic structures, and photocatalytic activity was demonstrated by Tang et al.122 Four COFs, namely DT2TA-TAPB, DT3TA-TAPB, DF2TA-TAPB, and DF3TA-TAPB, were synthesised by reacting 1,3,5-tris(4-aminophenyl)benzene (TAPB) and terephthaldehyde substituted with 2 or 3 thiophene or furan moieties (Fig. 8a). Thiophene isomer-functionalized COFs exhibited superior performance relative to their furan isomer-based counterparts, which can be attributed to a more favourably matched D–A architecture. Consequently, DT2TA-TAPB achieved the H2O2 production of 10972 and 8587 µmol g−1 h−1 in 10% ethanol and pure water, which are ∼23 and ∼41 times greater than those of benchmark g-C3N4 (Fig. 8b). The DT2TA-TAPB-COF also showed stable photocatalytic H2O2 production for 24 h under simulated natural conditions, followed by a steady plateau up to 28 h, indicating a dynamic formation-decomposition equilibrium (Fig. 8c).
 |
| | Fig. 7 (a) Synthetic pathway of the pyridyl-imine COF and the structure of synthesised COFs with regulated F-atoms. (b) Nyquist plots of PyIm-COFs. (c) Time-dependent photocatalytic H2O2 production with the pyridyl-imine COF. (d) Fs-TSA plots of PyIm-COFs. (Reproduced and reprinted with permission from ref. 119. Copyright 2025, Wiley-VCH GmbH.) (e) Structure of cyanide decorated COFs, highlighting their D–A moieties. (f) Long-term photocatalytic H2O2 production using COF-2CN via flow catalysis, and the inset shows the reaction setup for flow catalysis. (Reproduced and reprinted with permission from ref. 120. Copyright 2023, Wiley-VCH GmbH.) (g) Schematic illustration of the synthesis of three hydroxyl-functionalized D–A COFs. (h) The Gibbs free energy spectra of the 2e− ORR process on hydroxyl-functionalized D–A COFs. Reproduced and reprinted with permission from ref. 121. Copyright 2025, American Chemical Society. | |
 |
| | Fig. 8 (a) Schematic route for the synthesis of thiophene- and furan-decorated imine COFs. (b) Photocatalytic H2O2 production comparison of DT2TA-TAPB and g-C3N4 photocatalysts. (c) Long-term photocatalytic H2O2 production plot with DT2TA-TAPB. (Reproduced and reprinted with permission from ref. 122. Copyright 2025, Springer Nature.) (d) Schematic route for the synthesis of vinylene decorated COFs. (Reproduced and reprinted with permission from ref. 123. Copyright 2024, Wiley-VCH GmbH.) (e) Synthetic scheme for the synthesis of cyanide-functionalized D–A COFs. (Reproduced and reprinted with permission from ref. 124. Copyright 2024, Wiley-VCH GmbH.) | |
The vital challenges in COFs, such as low charge-generation and -transfer efficiencies, as well as rapid charge recombination that severely hindered the H2O2 production, have also been addressed by anchoring the vinyl moieties within the COF skeleton. The recent work from Chen and group demonstrates the synthesis of two imine-based COFs, one decorated with a vinyl group (DVA-COF) and another without any functionalization (PDA-COF) (Fig. 8d).123 Owing to the excellent π–π conjugated structure, abundant electronics, non-polar nature, and other characteristics of the vinyl group, the role of intralayer electron delocalisation and interlayer π–π stacking in charge generation and separation has been rationally studied. The photocatalytic performance of the synthesised COF towards H2O2 production has been studied in the two-phase water/benzyl alcohol (9:1, v/v) system. The vinyl containing DVA-COF could produce H2O2 photocatalytically at a rate of 84.5 µmol per h per 10 mg photocatalyst, which is higher than that of the non-functionalised PDA-COF (8.6 µmol per h per 10 mg photocatalyst). The AQY and SCC efficiency values for the DVA-COF were calculated to be 2.84% at 420 nm and ∼0.08% under AM 1.5G simulated sunlight, respectively. On investigation of the catalytic pathway, the vinyl group present in the COF was found to inhibit photogenerated electron–hole recombination. The DVA-COF showed excellent cycling stability over five irradiation cycles, with no noticeable changes in PXRD patterns. Continuous H2O2 production was maintained under light irradiation for 16 h, accompanied by nearly equimolar formation of H2O2 and benzaldehyde. The photo efficiency of COFs could also be rationally enhanced by functionalizing with various D–A moieties. For instance, Luo et al.124 synthesised COFs by integration of the π-accepting cyanide functional group that acts as a π electron acceptor. Its combination with donor thiophene and acceptor triazine monomers yielded D–A–π–D (ECUT-COF-50) and D–A–π–A (ECUT-COF-51) frameworks, respectively (Fig. 8e). Interestingly, the D–A–π–D COF demonstrated significantly higher efficiency than the D–A–π–A counterpart, achieving a remarkable H2O2 production rate of 4742 µmol h−1 g−1 using only air and water. Furthermore, ECUT-COF-50 maintained consistent H2O2 production over 80 h of continuous operation without performance loss. Similarly, as hydroxyl moieties are known to enhance the hydrophilicity of COFs, Shen et al. demonstrated that synergistic functional group modification, which tunes electronic conjugation and intramolecular polarity, significantly enhances light absorption, charge separation/transfer, and interfacial water–oxygen affinity.125 This was achieved by synthesising a series of imine-linked COFs (TT-COF-X) with electronically tunable substituents (X = –H, –OMe, –OH, –Br) (Fig. 9a). Photocatalytic H2O2 production proceeds via a dual-channel pathway involving 2e− ORR and WOR pathways, highlighting 100% atom utilisation. Among the series, hydroxyl-functionalized TT-COF-OH delivered the highest H2O2 production rate (3406.25 µmol g−1 h−1) (Fig. 9b) and an impressive AQY of 8.1%. It also exhibited efficient performance over a wide pH range and in real water matrices. fs-TA spectroscopy (460–800 nm) was used to probe the ultrafast exciton dissociation dynamics. The results show that a polarity-induced built-in electric field (BIEF) in TT-COF-OH enables a BIEF-mediated shallow trap state, facilitating exciton dissociation and suppressing electron–hole recombination, leading to a much longer average lifetime (τavg = 475.76 ps) compared to TT-COF-H (τavg = 76.32 ps). These results further reveal that the exciton binding energy (Eb) and charge-transfer resistance (Rct) decrease with increasing BIEF intensity (Fig. 9c). A stronger BIEF introduces a trap state that promotes exciton dissociation and charge transport, thereby enhancing the exciton dissociation efficiency (σd). Mechanistic studies reveal that hydroxyl-induced electronic microenvironment regulation enhances conjugation for improved light harvesting, generates a 6.01-fold stronger BIEF to accelerate exciton dissociation and charge transport, and promotes oxygen/water adsorption with faster interfacial electron transfer. The Schiff base condensation reaction between amines and aldehydes remains one of the most widely utilised techniques for synthesising COFs. However, a significant challenge encountered by researchers is the frequent hydrolysis of amine bonds formed through the condensation reaction, which leads to structural collapse. Notably, Bannerjee and co-workers addressed this challenge and markedly enhanced the stability of Schiff base-condensed COFs by using triformylphloroglucinol (Tp) as an aldehyde linker.126 They demonstrated that the hydroxyl group adjacent to the formyl group in Tp facilitates irreversible keto–enol tautomerism, thereby inhibiting the hydrolysis of the amine bond. Although the availability of three hydroxyl groups in Tp provides stability to the COF, its irreversible tautomerism decreases the crystallinity of the COF and can hinder photocatalytic activity. Due to this, the decrease of the number of hydroxyl groups in Tp aldehyde has allowed for the partly reversible keto–enol tautomerism that enhances both the stability and crystallinity of the COF.127,128 The effect of the number of hydroxyl groups on its photocatalytic activity has been recently explored by Kong et al., and they reported the synthesis of four imine-based COFs (0H, 1H, 2H, and 3H-COF, Fig. 10a) with different numbers of hydroxyl functional groups (0–3).129 The degree of keto–enol tautomerism in imine-linked COFs can be controlled by varying the number of –OH groups attached to the aldehyde precursors. This adjustment significantly influences the crystallinity and photocatalytic properties of the COFs. Among the four COFs studied, 1H-COF, which contains a single –OH group, exhibits the most ordered structure and the highest conjugation due to the predominant tautomerization. Additionally, the rearrangement between enol–imine and keto–amine linkages optimises its optical absorbance, electron push–pull effect, and water stability. This dynamic charge redistribution suppresses electron–hole recombination, resulting in a longer excited-electron lifetime for the 1H-COF, as confirmed by time-resolved photoluminescence (TRPL) measurements (Fig. 10b). As a result, the 1H-COF can produce H2O2 with a rate of 44.5 µmol h−1 in the presence of 10% IPA solution (Fig. 10c). The effect of keto–enol tautomerism on the photocatalytic efficiency of imine-based COFs has also been studied by Long and co-workers.130 Intrigued by the role of anthraquinone moieties in mass production of H2O2, they have synthesised an imine-based COF (TpAQ-COF) by the Schiff base condensation reaction between 2,6-diaminoanthraquinone (AQ) and triformylphloroglucinol (Tp) (Fig. 10d). The anthraquinone moieties serve as the active sites for the selective two-electron ORR, promoting the separation and migration of photogenerated charges. Interestingly, the keto–amine form of the COF was found to be more efficient for H2O2 production than the enol–imine form, and by adjusting the condensation time, researchers could vary the ratio of the two forms, with the keto–amine form promoting better D–A coupling structures that favour the migration of photogenerated carriers. Consequently, the TpAQ-COF could produce H2O2 at the rate of 420 µmol g−1 h−1 with an AQY of 7.2% at 420 nm. The COF also showed good stability upon prolonged irradiation for 12 h, with steady production of H2O2. Motivated by the result, the same group has extended the research through synthesising a keto-form anthraquinone COF (Kf COF) via a mechanochemical route (Fig. 10e).131 The Kf-COF could form H2O2 in alkaline medium (pH = 13) at a rate of 4784 µmol g−1 h−1. At pH 13, Kf-AQ exhibited the highest H2O2 formation rate constant (kf = 31.39 µM min−1), while maintaining a moderate decomposition rate constant (kd = 0.031 µM min−1) (Fig. 10f). The highly alkaline environment promoted the formation of OH−(H2O)n clusters, which were adsorbed onto the keto moieties of the framework. This adsorption significantly lowered the energy barrier for hydrogen abstraction, facilitating their dissociation into O2 and active hydrogen species. The generated hydrogen subsequently reduced anthraquinone to anthrahydroquinone, which was then re-oxidised by O2 to yield H2O2. The integration of the quinone unit in the COF has also demonstrated H2O2 production in the full pH range (pH 1–13), as demonstrated by Li et al.132 Two COFs, Por-HQ-COF and Por-BQ-COF, have been synthesised from p-hydroquinone (HQ) and p-benzoquinone (BQ) moieties. Interestingly, under alkaline conditions, the Por-HQ-COF undergoes conversion to the Por-BQ-COF (Fig. 11a). In this transformed structure, the imidazole nitrogen in the Por-BQ-COF facilitates water molecule dissociation to generate protons and enhances the ORR, resulting in a notable H2O2 production rate of up to 1525 µmol g−1 h−1. Furthermore, the Por-BQ-COF showed the kf of 13.55 µM min−1 and the lowest kd of 0.0165 µM min−1 at pH 13. The efficiency of quinoline-linked COFs has also been widely explored by Dong et al. Benzo[f]quinoline-linked COFs have been synthesised through a one-pot, three-component [4 + 2] cyclic condensation strategy.133 This approach involves the reaction of aldehydes and aromatic amines using triethylamine as a readily accessible vinyl source, resulting in the formation of stable and highly photoactive benzo[f]quinoline-linked COFs (Fig. 11b). The high crystallinity, improved stability, and strong light-harvesting ability of these COFs contributed to their excellent performance in H2O2 photosynthesis. As a result, B[f]QCOF-1 exhibited exceptional photocatalytic activity, reaching an H2O2 production rate of 9025 µmol g−1 h−1 in pure water (Fig. 10e) without any sacrificial agent, accompanied by nearly equimolar formation of H2O2 and benzaldehyde. The major achievements of the functionalised COF photocatalyst for H2O2 production have been highlighted in Table 2.
 |
| | Fig. 9 (a) Synthetic route for the synthesis of different functionalized imine COFs. (b) Photocatalytic H2O2 production comparison of functionalized COFs. (c) Built-in electric field (BIEF) plot of functionalized imine COFs. (Reproduced and reprinted with permission from ref. 125. Copyright 2025, Wiley-VCH GmbH.) | |
 |
| | Fig. 10 (a) Illustration of iminol-to-ketoenamine tautomerism in XH-COFs via proton transfer. (b) TRPL spectra of ketoamine COFs. (c) Comparison of photocatalytic H2O2 production of ketoamine COFs. (Reproduced and reprinted with permission from ref. 129. Copyright 2023, Elsevier.) (d) Structure of the TpAQ-COF in its keto and enol forms. (Reproduced and reprinted with permission from ref. 130. Copyright 2023, Elsevier.) (e) Mechanochemical synthetic route for the Kf-AQ COF. (f) H2O2 formation and decomposition plot using the Kf-AQ COF in different pH solutions. (Reproduced and reprinted with permission from ref. 131. Copyright 2024, Springer Nature.) | |
 |
| | Fig. 11 (a) Synthetic route for the Por-HQ-COF and Por-BQ-COF. (Reproduced and reprinted with permission from ref. 132. Copyright 2024, Wiley-VCH GmbH.) (b) Synthesis scheme and structure of benzo[f]quinoline-linked COFs. (c) Photocatalytic H2O2 production with benzo[f]quinoline-linked COFs using different water samples. (Reproduced and reprinted with permission from ref. 133. Copyright 2024, Springer Nature.) | |
Table 2 Summary of the representative functionalization of COF photocatalysts for H2O2 photosynthesisa
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC |
Ref. |
| Light irradiation: λ > 420 nm, sunlight. LED light; BA: benzyl alcohol; IPA: isopropanol. |
| 1 |
HEP-TAPT-COF |
H2O and O2 |
50 mg & 100 mL |
87.50 µmol h−1 |
— |
0.65% |
115 |
| 2 |
COF-JLU51 |
H2O and O2 |
5 mg & 50 mL |
3388.3 |
6.4% |
0.19% |
116 |
| 3 |
TTA-TF-COF |
H2O |
1 mg & 8 mL |
3343 |
4.1% @ 420 nm |
0.13% |
117 |
| 4 |
PyIm-COF |
H2O |
10 mg & 50 mL |
5850 |
3.70% @ 420 nm |
0.28% |
118 |
| 5 |
PyIm-BT_F |
H2O:IPA = 9:1 |
10 mg & 50 mL |
5342 |
2.7% @ 420 nm |
— |
119 |
| 6 |
COF-2CN |
H2O and O2 |
12.5 mg & 50 mL |
1601 |
6.8% @ 459 nm |
0.6% |
120 |
| 7 |
2,5-DhaTph |
H2O:EtOH = 9:1 |
10 mg & 50 mL |
2103.1 |
0.15% @ 450 nm |
— |
121 |
| 8 |
DT2TA-TAPB |
H2O and O2 |
5 mg & 25 mL |
8587 |
3.3% @ 380 nm |
— |
122 |
| 9 |
DVA-COF |
H2O:BA = 9:1, O2 |
10 mg & 20 mL |
8450b |
2.84 @ 420 nm |
0.08% |
123 |
| 10 |
ECUT-COF-50 |
H2O |
5 mg & 50 mL |
5382 |
11.2% @ 400 nm |
0.68% |
124 |
| 11 |
TT-COF-OH |
H2O |
5 mg & 50 mL |
3406 |
8.1% @ 400 nm |
0.58 |
125 |
| 12 |
1H-COF |
H2O:IPA (9:1), O2 |
30 mg & 30 mL |
44.5 |
5.4% @ 420 nm |
— |
129 |
| 13 |
TpAQ-COF |
H2O and O2 |
10 mg & 30 mL |
420 |
7.4% @ 420 nm |
— |
130 |
| 14 |
Kf-AQ |
H2O (pH = 13), O2 |
5 mg & 30 mL |
4784 |
15.8% @ 400 nm |
0.87% |
131 |
| 15 |
POR-BQ-COF |
H2O (pH = 13), O2 |
10 mg & 30 mL |
1525 |
5.05% @ 420 nm |
— |
132 |
| 16 |
B[f]QCOF-1 |
H2O |
10 mg & 50 mL |
9025 |
8.9% @ 450 nm |
0.23% |
133 |
Tuning of linkage in COFs for photocatalytic H2O2 production
Linkage engineering has emerged as a powerful strategy to enhance the photocatalytic performance of COFs for H2O2 production. The chemical nature of the linkages, such as imine, β-ketoenamine, hydrazone, azine, or imidazole, directly influences the COFs' electronic structure, charge separation efficiency, and interaction with oxygen molecules. By precisely tuning the linkage, researchers can modulate bandgaps, charge transfer pathways, redox potentials, and surface adsorption sites, all of which are critical for the two-electron ORR producing H2O2. Electron-rich linkages improve light absorption and facilitate exciton dissociation, whereas electron-deficient linkages enhance O2 adsorption and promote selective 2e− reduction over the competing 4e− pathway. Additionally, robust linkages can also improve chemical stability, allowing sustained catalytic activity under aqueous and oxidative conditions. To elucidate how linkage positioning governs the photocatalytic performance and stability of COFs toward H2O2 production, Yang et al. systematically investigated the influence of ortho and para linkages in COFs. Six COFs were synthesised from ortho/para-substituted diamines (Bda, Pda, and Pzda) and Tp.134 Pyridinic nitrogen favoured the 2e− ORR, while β-ketoenamine carbons acted as 4e− WOR active sites, making Pzda-based COFs the most efficient. Although p-COF-TpBda showed a high initial H2O2 rate (6434 µmol g−1 h−1), it rapidly degraded, whereas o-COF-TpPzda maintained stable H2O2 production (4396 µmol g−1 h−1) for 48 h. DFT revealed a lower ΔG for the rate-determining WOR step in o-COF-TpPzda, enabling faster hole consumption and enhanced stability, while largely preserving 2e− ORR activity. Linkage chemistry also plays a key role in achieving crystalline, robust COFs while enabling functionalities such as band-gap modulation and redox activity. Mou et al. reported azole-linked COFs synthesised from pyrene aldehyde and heteroatom-substituted phenylenediamines, yielding thiazole (TZ-COF), oxazole (OZ-COF), and imidazole (IZ-COF) frameworks.135 These azole linkages introduce electron-donating conjugation within a D–π–A architecture, enhancing visible-light absorption and charge transport. Femtosecond transient absorption revealed the longest excited-state lifetime for the TZ-COF (5.94 ps), followed by the OZ-COF (3.74 ps) and IZ-COF (1.86 ps), indicating more efficient charge separation with thiazole linkages. Accordingly, the O2 production activity follows the order: TZ-COF > OZ-COF > IZ-COF. A distinct difference in efficiency and stability between reversible imine-linked COFs (AICOF-1) and irreversible amide-linked COFs (AmCOF-1) has been demonstrated by Li et al. for photocatalytic H2O2 production.136 They have synthesised the self-standing membrane of crystalline COFs for photocatalytic H2O2 production. In brief, AICOF-1 was synthesised using an ingenious autocatalytic interfacial polymerisation (AIP) strategy. Subsequently, AICOF-1 was converted into a fully amide-linked crystalline membrane (AmCOF-1) via a traditional irreversible linker renovation (ILR) reaction with acyl chlorides, yielding COFs with well-defined Turing structures (Fig. 12a). This approach offers several advantages, including broad applicability, a simple and catalyst-free synthesis at ambient temperature, and the ability to produce large, free-standing membranes with diverse Turing patterns. Given the robust nature, efficient electron-withdrawing ability, and potential π-electron segregation effect of the amide units, the authors have demonstrated the photocatalytic efficiency of AmCOF-1 membrane towards H2O2 production in various media, including pure water, seawater, and solutions with high pH and low pH (Fig. 12b). The photocatalytic H2O2 production reached up to 4353 µmol g−1 h−1 in O2-saturated H2O. Furthermore, a large-area (16 cm × 13 cm) self-standing AmCOF-1 membrane showed stable H2O2 production for 4 h under natural sunlight (Fig. 12c). The AmCOF-1 membrane also shows remarkable stability over 150 h under sunlight without sacrificial agents (Fig. 12d), which was further verified by PXRD of recovered samples. In a similar direction, Zhao and co-workers compared the photocatalytic performance of azo-linked and imine-linked COFs for H2O2 production.137 They rationally synthesised two COFs, an imine-linked COF (COF-TPT-TPA) and an azo-linked COF (COF-TPT-Azo) via Schiff-base polycondensation (Fig. 12e). Remarkably, despite differing by only a single atom (–N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–vs.–CN–) COF-TPT-Azo shows a much narrower band gap, improved charge transport, and enhanced photoactivity compared to its imine counterpart. Consequently, COF-TPT-Azo was found to produce H2O2 at a rate of 1498 µmol g−1 h−1 in O2-saturated alkaline solution of pH = 11 under visible light irradiation (λ > 420 nm), which is 7.9 times higher than that of imine-based COF-TPT-TPA (Fig. 12f). COF-TPTAzo showed remarkable H2O2 production in alkaline solution both under O2 and air (Fig. 12g). DFT studies show that the HOMO is distributed in the NN group, while the LUMO is on the same azobenzene units. Therefore, the NN groups in COF-TPT-Azo and CN groups in COF-TPT-TPA play an important role in light absorption and promote the photocatalytic oxidation.
N–vs.–CN–) COF-TPT-Azo shows a much narrower band gap, improved charge transport, and enhanced photoactivity compared to its imine counterpart. Consequently, COF-TPT-Azo was found to produce H2O2 at a rate of 1498 µmol g−1 h−1 in O2-saturated alkaline solution of pH = 11 under visible light irradiation (λ > 420 nm), which is 7.9 times higher than that of imine-based COF-TPT-TPA (Fig. 12f). COF-TPTAzo showed remarkable H2O2 production in alkaline solution both under O2 and air (Fig. 12g). DFT studies show that the HOMO is distributed in the NN group, while the LUMO is on the same azobenzene units. Therefore, the NN groups in COF-TPT-Azo and CN groups in COF-TPT-TPA play an important role in light absorption and promote the photocatalytic oxidation.
 |
| | Fig. 12 (a) Illustration of synthesis of AmCOF-1 from AlCOF-1. (b) Photocatalytic H2O2 production of AmCOF-1 under different conditions. (c) Time course of H2O2 production using the membrane of AlCOF-1 and AmCOF-1 under natural light (inset: AmCOF-1 membrane). (d) Long term production H2O2 over 150 h with AmCOF-1. (Reproduced and reprinted with permission from ref. 136. Copyright 2025 Wiley-VCH GmbH.) (e) Synthesis scheme and structure of imine- and azo-linked COFs. (f) Photocatalytic H2O2 production comparison under different pH conditions. (g) Continuous H2O2 production with COF-TPT-Azo over 2 h under different conditions at pH 11. (Reproduced and reprinted with permission from ref. 137. Copyright 2024, Wiley-VCH GmbH.) | |
The effect of hydrazone linkages and their density has recently been studied by Pachfule et al.138 They synthesised a series of D–A thiophene-based COFs (BTT DAB, BTT-H1 and BTT-H2) and regulated the hydrazone density (0–2) (Fig. 13a). This gradual variation enabled a direct comparison of how hydrazone density affects photocatalytic behaviour. The introduction of hydrazone linkages decreased e–h recombination, as indicated by the decreased PL (Fig. 13b). The production of H2O2 was observed to vary directly in proportion to the number of hydrazone linkages. As a result, BTT-DAB, BTT-H1, and BTT-H2 produced H2O2 at a rate of 854, 1096, and 1359 µmol g−1 h−1 from water and air under irradiation of 467 nm light, respectively. BTT-H2 also delivered a dramatically higher output of 21641 µmol g−1 h−1 in the presence of benzyl alcohol as a sacrificial agent (Fig. 13c). Motivated by the results, the authors also synthesised the BTT-H3 COF with three hydrazone linkages (Fig. 13a). An increase in hydrazone density increased the lifetime of the photogenerated electron, as indicated by the TRPL plot (Fig. 13d). BTT-H3 delivered H2O2 production of 1588 µmol g−1 h−1 from water and air (Fig. 13e), higher than that of BTT-H2. COF BTT-H3 also produced up to 550 µmol g−1 h−1 H2O2 under natural sunlight, highlighting its potential for practical solar-driven applications. It is well known that COFs have traditionally been built from aromatic linkers because their π–π stacking promotes high crystallinity and provides continuous π-channels for efficient exciton and charge transport.
 |
| | Fig. 13 (a) Structures of benzotrithiophene-based COFs with regulation of the hydrazone linkage. (b) PL spectra of benzotrithiophene-based COFs with and without hydrazone linkage. (c) Comparison of photocatalytic H2O2 production of benzotrithiophene-based COFs with different sacrificial electron agents. (d) TRPL data of BTT-H2 and BTT-H3 COFs. (e) Photocatalytic H2O2 production with BTT-H3 in different environments. (Reproduced and reprinted with permission from ref. 138. Copyright 2025, Springer Nature.) (f) Synthesis scheme and structure of COFs with an aliphatic linker. (g) Photocatalytic H2O2 production with aliphatic linker-based COFs. (h) Long-term photocatalytic H2O2 production with the TAH-COF. (Reproduced and reprinted with permission from ref. 139. Copyright 2025, American Chemical Society.) | |
Despite these advantages, aromatic linkers are often hydrophobic, which reduces water affinity and limits the efficiency of COFs in aqueous photocatalytic systems. To overcome this challenge, Zhu and co-workers demonstrated that aliphatic linkers can also form crystalline COFs and show subsequent enhancement in their hydrophilicity by reacting with aliphatic linkers such as tartronic acid dihydrazide (TAH) or butanedioic acid dihydrazide (BAH) with 1,3,5-triformylphloroglucinol (TP) that resulted in two hydrazone-linked COFs, termed TAH-COF and BAH-COF (Fig. 13e).139 These two COFs are crystalline and have a BET surface area of 703 m2 g−1 and 530 m2 g−1 for the TAH-COF and BAH-COF, respectively. These aliphatic linkers create dense hydrogen-bonding networks that stabilise the framework, enhance crystallinity, and ensure good compatibility with water while maintaining strong visible-light absorption. As a result, the COFs display narrow optical band gaps (∼1.9 eV) and high photocatalytic activity, with the TAH-COF delivering an impressive H2O2 production rate of 6003 µmol g−1 h−1 without sacrificial agents (Fig. 13f). The aliphatic TAH-COF also showed notable structural integrity following photocatalysis, as highlighted by the nominal difference in PXRD before and after photocatalysis (Fig. 13g). The influence of linkage orientation on photocatalytic H2O2 production has also been clearly illustrated through the work of Yue et al.140 Two isomeric COFs: TB-COF and TA-COF, were constructed with identical building blocks but opposite imine-bond orientations (Fig. 14a). Although the structural difference between the two frameworks is subtle, their photocatalytic behaviours diverge sharply. The orientation present in the TB-COF generates a larger dipole moment and promotes more efficient charge separation, which together account for its markedly higher H2O2 production compared to the TA-COF. Both COFs show notable H2O2 production both in O2 and air-saturated water (Fig. 14b). The H2O2 production of TB-COF was measured to be 5186 and 4111 µmol g−1 h−1 in air-saturated water and natural seawater, respectively, which are higher than the values of TA-COF (2387 and 1640 µmol g−1 h−1). A similar study was also reported by Zhou et al., wherein they synthesised constitutional-isomerised COFs (PB-PT-COF and PT-PB-COF) by swapping monomers around imine bonds.141 The two isomeric COFs have similar composition and pore sizes but different optoelectronic properties, leading to distinct photocatalytic efficiency. Consequently, under O2-saturated conditions, the PB-PT-COF and PT-PB-COF generated 1076 and 2169 µmol g−1 h−1 H2O2, respectively. In contrast, when the reaction was carried out in N2-saturated water, the PB-PT-COF delivered a higher activity (645 µmol g−1 h−1) than the PT-PB-COF (349 µmol g−1 h−1) (Fig. 14c). DFT calculations revealed that the PT-PB-COF has a stronger affinity for O2 and activates it more effectively, thereby favouring H2O2 formation through the ORR pathway. The PB-PT-COF, on the other hand, interacts more strongly with H2O, leading to H2O2 production primarily via the WOR pathway (Fig. 14d). The major achievements of the linkage modified COF photocatalyst for H2O2 production have been highlighted in Table 3.
 |
| | Fig. 14 (a) Illustration of synthesis of isomeric TA-COF and TB-COF. (b) Time-dependent H2O2 synthesis with isomeric COFs in different atmospheres. (Reproduced and reprinted with permission from ref. 140. Copyright 2025, American Chemical Society.) (c) Comparison of photocatalytic H2O2 production with PB-PT-COF and PT-PB COF. (d) Structures of isomeric PB-PT-COF and PT-PB COF, highlighting their pathways for the production of H2O2. (Reproduced and reprinted with permission from ref. 141. Copyright 2024, Wiley-VCH GmbH.) | |
Table 3 Summary of the representative linkage modification on COF photocatalysts for H2O2 photosynthesis
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC |
Ref. |
| Light irradiation: λ > 420 nm, sunlight; BA: benzyl alcohol. |
| 1 |
o-COFTpPzda |
H2O and O2 |
5 mg & 40 mL |
4396 |
— |
0.46% |
134 |
| 2 |
TZ-COF |
H2O:BA (1:1) and O2 |
15 mg & 30 mL |
4951 |
0.6% @ 475 nm |
0.036% |
135 |
| 3 |
AmCOF-1 |
H2O and O2 |
Membrane based |
4353 |
8.72% @ 420 nm |
— |
136 |
| 4 |
COF-TPT-Azo |
H2O (pH 11) and O2 |
10 mg & 40 mL |
1498 |
— |
— |
137 |
| 5 |
BTT-H3 |
H2O and O2 |
5 mg & 10 mL |
1770 |
17.7% @ 467 nm |
2.02% |
138 |
| 6 |
TAH-COF |
H2O |
5 mg & 50 mL |
6003 |
7.72% @ 500 nm |
0.66% |
139 |
| 7 |
TB-COF |
H2O |
1 mg & 8 mL |
5186 |
3.45% @ 420 nm |
1.08% |
140 |
| 4111a |
| 8 |
PT-PB-COF |
H2O and O2 |
5 mg & 20 mL |
2169 |
0.74% |
0.055% |
141 |
| 9 |
COF-Tfp-BDDA |
H2O and O2 |
10 mg & 20 mL |
880 |
— |
— |
142 |
Tuning the structural topology of COFs for photocatalytic H2O2 production
Topological structures, such as lattices, aggregations, and textural configurations, are fundamental in natural and synthetic systems.143 In nature, these structures can be found in various biological, geological, and chemical systems.144 The pursuit of designing and engineering topological structures at the molecular and atomic scales is driven by the potential to create materials with unprecedented precision and performance. At these small scales, the properties of materials can be radically different from those at the macro scale, often exhibiting enhanced or entirely new functionalities, such as improved strength, electrical conductivity, or catalytic activity. In reticular chemistry,145 COFs with various topologies can be designed by manipulating the arrangement of building blocks through geometric control. This method enables the creation of highly ordered structures by linking organic molecules with covalent bonds, offering precise tuning of properties such as surface area, stability, and functionality. The impact of various factors, such as changes in linkage, functional groups, and D–A incorporation, on the optical and electronic properties of COFs for H2O2 production has been extensively studied. Recently, there has been growing interest in exploring how topological changes in COFs influence their performance in H2O2 production. For instance, Yue et al. investigated the impact of topology on the photocatalytic production of H2O2 in 2D COFs.146 They synthesised two 2D COFs, TBD-COF and TBC-COF, which exhibit similar chemical components but different topologies (Fig. 15a). The TBD-COF exhibited a cpt topology with six-arm cores, while the TBC-COF displayed an hcb topology with three-arm cores. The topological differences have brought distinct variations in the photophysical properties of these two COFs. In addition, it has also influenced mass transport and charge transfer modulation, enhancing their photocatalytic performance for H2O2 production (Fig. 15b). On evaluating the catalytic activity towards light-driven H2O2 production, the TBD-COF outperforms the TBC-COF, producing H2O2 at a rate of 5448 and 4049 µmol g−1 h−1, respectively, from air and pure water. When evaluated for photocatalytic H2O2 production, the TBD-COF exhibited superior performance to the TBC-COF, showing a steady increase in the H2O2 yield with prolonged irradiation, particularly beyond 6 h (Fig. 15c). The TBD-COF achieved a production rate of 5448 µmol g−1 h−1, higher than that of TBC-COF (4049 µmol g−1 h−1) using air and pure water. The mechanistic investigation revealed the formation of H2O2 through three channels: two distinct 2e− ORR routes involving superoxide (O2–O2˙−·H2O2) and singlet oxygen species (O2–O2˙−–O21–H2O2), along with one 2e− WOR pathway (H2O–H2O2) (Fig. 15d–f). The photocatalytic generation of H2O2 is generally accompanied by its decomposition, typically following zero-order and first-order kinetics, respectively. For example, the TBD-COF and TBC-COF exhibited H2O2 formation rate constants (kf) of 0.84 and 0.64 mM h−1, respectively, while the corresponding decomposition rate constants (kd) were 0.49 and 0.54 h−1, respectively. Long-term irradiation studies further illustrate a characteristic production-decomposition interplay. The concentration of H2O2 increases steadily during the initial reaction period (up to 10 h), followed by a gradual decline as decomposition becomes dominant. This behaviour is often associated with photocatalyst fatigue or partial structural degradation under extended light exposure, induced by reactive oxygen species and accumulated charge carriers, which leads to reduced crystallinity and consequently shifts the balance toward faster H2O2 breakdown. The effect of topology on H2O2 production has also been meticulously studied by Xu et al., where they have synthesised three 2D sp2 carbon-linked π-conjugated COFs, such as QP-TPB-COF, QP-BPT-COF, and QP-HPTP-COF, having hcb, sql, and hxl topologies, respectively (Fig. 15g).147 Compared to COFs adopting hcb or sql lattices, the hxl-topology QP-HPTP-COF stands out by exhibiting the lowest exciton binding energy (Eb), markedly reduced carrier effective masses, and the highest charge-transfer efficiency (Fig. 15h). To probe the topology-dependent electronic characteristics of COFs, the effective carrier masses (m*) were evaluated through DFT calculations. Notably, the QP-HPTP-COF displayed remarkably lower electron and hole effective masses (me = 0.52me; mh = 0.54me) relative to the QP-TPB-COF (me = 312.5me; mh = 104.17me) and QP-BPT-COF (me = 16.34me; mh = 8.96me) (Fig. 15i), suggesting a substantially higher intrinsic carrier mobility and, consequently, enhanced photocatalytic potential for the QP-HPTP-COF. Consequently, the photocatalytic tests conducted in O2-saturated pure water under visible light irradiation revealed the highest efficiency for the QP-HPTP-COF with the hxl topology, followed by the QP-BPT-COF with the sql topology, and finally the QP-TPB-COF with the hcb topology. The respective H2O2 production rates for these materials were 4388, 1902, and 921 µmol g−1 h−1 (Fig. 15j). The QP-HPTP-COF also exhibited an AQE of 22.78% and an SCC of 1.41%, the highest reported SCC value among other COFs. The synthesis of 3D COFs with high surface areas, exposed active sites, and tunable frameworks has gained interest among researchers. However, synthesising 3D COFs overcomes various challenges, such as low-connectivity building units, low crystallinity, low yield, etc. With the use of a tetrahedral linkage, Omar M. Yaghi reported the first 3D COF in 2007.148 Since then, various 3D COFs have been synthesised, and different types of topologies have been reported for 3D COFs.149 The construction of 3D COFs generally demands linkers with predefined spatial orientation and high-connectivity nodes. In contrast to 2D COFs, only a limited number of frameworks have been realised, primarily due to the synthetic complexity, harsh reaction conditions, scarcity of suitable 3D linkers, such as tetrahedral, hexavalent triangular prisms, anti-triangular prisms, and octavalent cubic units, and the inherent crystallisation difficulty arising from multiple degrees of rotational freedom. Recently, progress has been made through strategies that exploit the torsional characteristics of linkers, where modulation of the torsion angle enables the formation of stable 3D COF networks.150 In line with this approach, Zhu et al. demonstrated a successful synthesis of a 3D COF by utilising controlled molecular torsion to drive the 3D framework.151 They deliberately chose 2,2′,6,6′-tetramethylbenzidine (TMB) as a linker where the four methyl groups introduce steric hindrance between the two phenyl rings, generating a stereoscopic disc-like conformation that preserves local planarity while promoting antiparallel stacking (Fig. 16a). Four COFs were subsequently synthesised by coupling TMB with a series of trialdehydes containing varying numbers of hydroxyl groups (ranging from 0 to 3) (Fig. 16a). The steric hindrance in TMB introduces an additional force that alters the torsion angles of the peripheral triangular units, directing the linear unit to connect with the trigonal unit and adapted 3D srs topology. By exploiting steric hindrance effects, these 3D COFs exhibit exceptionally high surface areas and well-defined pore architectures, which collectively promote greater exposure of catalytic sites and facilitate efficient mass transport. A clear structure–activity trend is observed: as the number of ketone-based active sites increases, the photocatalytic H2O2 production performance improves accordingly. Under pure water and O2-saturated conditions, TMB-COF-1, TMB-COF-2, and TMB-COF-3 deliver H2O2 formation rates of 218, 1293, and 2621 µmol g−1 h−1, respectively, while TMB-COF-4 achieves an impressive 5106 µmol g−1 h−1. TMB-COF-4 also exhibited H2O2 photocatalytic production rate of 1396 µmol g−1 h−1 under pure water and air conditions. In comparison, the analogous 2D BZD-COFs demonstrate considerably lower activity under identical conditions. Collectively, these findings highlight the strong correlation between 3D framework design, active-site incorporation, and photocatalytic efficiency, emphasising that deliberate structural optimisation is a powerful strategy for advancing COF-mediated H2O2 production. Since topology strongly influences parameters such as pore arrangement, charge-transfer pathways, and crystallinity, different 3D COFs inherently exhibit diverse property profiles and performance behaviours. The photocatalytic performance of two topologically distinct 3D COFs for H2O2 production was recently reported by Liao et al. Two 3D COFs (TAA/TABCTP-COF) were obtained by reacting a trigonal antiprism (C3) monomer, denoted as CTP, with a planar amine linker monomer, denoted as TAA or TAB (Fig. 16b).152 CTP has N, O, and P that can act as active sites for the WOR, whereas the amine linker has triazine (TAA) or benzene (TAB) in its core that acts as an acceptor to photogenerated electrons during the ORR. The triazine-containing COF, denoted as TAACTP-COF, produced H2O2 at a rate of 1041 µM h−1 from H2O and O2. In the presence of isopropanol as a sacrificial agent, the production rate increased to 2221 µM h−1. The theoretical calculation showed that the distribution of the HOMO, mostly on the CTP ring, and the LUMO on TAA or TAB indicates CTP as an oxidising site for the WOR and TAA/TAB as a reducing site for the ORR. The unprecedented efficiency of 3D COFs over 2D COFs has also been recently reported by Lan et al. Two 3D COFs (XJU-1 and XJU-2) using a hexadentate triptycene-based monomer and a tridentate triazine amine linker were synthesised (Fig. 16c).153 The two COFs are isomeric, having a difference in the linkage position. The 3D COFs exhibit high crystallinity with a ceq topology. The two COFs have distinct photochemical properties, and in the presence of benzyl alcohol, XJU-1 and XJU-2 demonstrated impressive photocatalytic activity for H2O2 production, achieving rates of 34777 µmol g−1 h−1 and 11922 µmol g−1 h−1, respectively. In the absence of a sacrificial agent, the production reached up to 7453 and 8124 µmol g−1 h−1 for XJU-1 and XJU-2, respectively. The larger pore aperture of XJU-1 relative to XJU2 promotes faster mass diffusion and more efficient O2 activation, resulting in enhanced H2O2 output. Mechanistic studies (radical quenching, EPR, and RDE) confirmed ORR-dominated H2O2 formation in both COFs, where XJU-1 follows a mixed 1e−/2e− ORR process, while XJU-2 mainly adopts a direct 2e− pathway. DFT results further attributed the performance differences to topology-dependent charge separation and strong O2 binding at multiple active sites. The reduced singlet–triplet energy gap (ΔES–T) and orthogonal triptycene structure additionally favour 1O2 formation, collectively accelerating H2O2 evolution. The study underscores how the 3D COF topology governs ORR selectivity and boosts the photocatalytic H2O2 production efficiency. The synthesis of COFs with connectivity up to four is well known, and its synthesis procedure has been generalised. However, designing 3D COFs with high connectivity (≥8) is challenging and scarce. The higher number of connectivity often correlates with high stability of the framework; therefore, it is desirable to synthesise highly connected 3D COFs. Porphyrin units, known for their excellent light-harvesting capability, have been strategically employed as linkers in COF design for photocatalytic H2O2 generation. Upon protonation of the pyrrole nitrogen, the porphyrin macrocycle undergoes pronounced geometric distortion both within and out of the molecular plane, which can significantly influence charge separation and exciton dynamics. Leveraging this structural tunability, Jiang et al. synthesised two three-dimensional porphyrin-based COFs (Por-COF-cya) featuring vinyl linkages through an aldol condensation reaction.154 Por-COF-cya adopts a first-ever-reported cya topology that has the interconnectedness of 8-connected and 3-connected nodes (Fig. 17a). The incorporation of porphyrin units into the Por-COF-cya framework imparts favourable photophysical and chemical stability. Moreover, the electron-deficient triazine moieties enable efficient encapsulation of the electron-donating tetrathiafulvalene (TTF) species within the hierarchical pores via electrostatic attraction and host–guest interactions. This results in the formation of a donor–acceptor architecture (TTF@Por-COF-cya) that promotes rapid charge transfer and ultimately enhances photocatalytic H2O2 production. Consequently, the H2O2 yield in pure aqueous solution reached 2209 and 6994 µmol g−1 h−1 with Por-COF-cya and TTF@Por-COF-cya, respectively (Fig. 17b). Analysis of H2O2 yields under different atmospheres, supported by isotope-labelling experiments, confirmed that 84% of the oxygen atoms in the generated H2O2 originated from the ORR pathway, while the remaining 16% were derived from the WOR route. DFT calculations identified two potential electrophilic sites in Por-COF-cya: the non-protonated porphyrin nitrogen and the triazine ring, where O2 showed preferential adsorption at the triazine site, consistent with in situ DRIFTS results and confirming it as the primary ORR-active centre. Moreover, TTF@Por-COF-cya displayed lower O2 adsorption free energy and reduced intermediate-conversion barriers compared to Por-COF-cya, facilitating ˙OOH formation and thereby enhancing the H2O2 production efficiency. A highly connected 3D COF for photocatalytic H2O2 production has also been reported by Wang et al.155 They synthesised highly connected phthalocyanine-based (Pc-based) two 3D COFs MPc-THHI-COFs (M = H2, Ni) with 12 connecting sites (Fig. 17c). These COFs were found to adopt a shp topology, featuring 1D triangular channels with diameters of 12 Å and 15 Å along the c axis, which are further interconnected by rhombohedral windows, each with a size of 9 Å along the a and b axes (Fig. 17d). The triangular channels similar to the open channel of the SHP networks of the COF can be visualised through HRTEM images, indicating the high crystallinity of the synthesised 3D COF (Fig. 17e). The synthesised 3D COFs exhibit a broadened visible light absorption range, a narrow optical band gap, and 3D interconnected nanochannels, which collectively enhance their photocatalytic performance. These features enable efficient H2O2 generation at a rate of 4511 and 4589 µmol g−1 h−1 from O2-saturated H2O via 2e− ORR and WOR pathways, respectively, under visible-light irradiation (Fig. 17f). The major achievements of the topology and structural modified COF photocatalyst for H2O2 production have been highlighted in Table 4.
 |
| | Fig. 15 (a) Structures of TBD-COF and TBC-COF. (b) Photocurrent plot of the TBD-COF and TBC-COF. (c) Photocatalytic H2O2 production comparison between the TBD-COF and TBC-COF. (d–f) Gibbs free energy diagrams of TBD-COF and TBC-COF for H2O2 photoproduction via the (d) O2–˙O2⋯H2O2 pathway, (e) O2–˙O2−–1O2–H2O2 pathway, and (f) 2e− WOR pathway. (Reproduced and reprinted with permission from ref. 146. Copyright 2024, Wiley-VCH GmbH.) (g) Structures of COFs with different topologies. (h) Exciton dissociation rates in COFs. (i) Schematic illustration of the correlation between the topology and effective mass ( and and 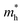 ) in different COFs. (j) Long-term H2O2 photoproduction over 8 h using the P-TPB-COF, QP-BPT-COF, and QP-HPTP-COF. (Reproduced and reprinted with permission from ref. 147. Copyright 2024, Wiley-VCH GmbH.) ) in different COFs. (j) Long-term H2O2 photoproduction over 8 h using the P-TPB-COF, QP-BPT-COF, and QP-HPTP-COF. (Reproduced and reprinted with permission from ref. 147. Copyright 2024, Wiley-VCH GmbH.) | |
 |
| | Fig. 16 (a) Schematic route to formation of 2D COFs using planar and linear monomers. (Reprinted with permission from ref. 151. Copyright 2024, Wiley-VCH GmbH.) (b) Schematic route to synthesis of 3D COFs. (Reprinted with permission from ref. 152. Copyright 2024, American Chemical Society.) (c) Synthetic scheme of 3D COFs XJU-1 and XJU-2. (Reprinted with permission from ref .153. Copyright 2025, Wiley-VCH GmbH.) | |
 |
| | Fig. 17 (a) Synthetic scheme for cya topology-based Por-COF-cya. (b) Reaction kinetics for H2O2 production. (Reprinted with permission from ref. 154 Copyright 2024 Wiley-VCH GmbH). (c) Synthetic scheme of MPc-THHI-COF (M = 2H, Ni) with 12 connecting sites. (d) Simulated packing structures in MPc THHI-COF (M = 2H, Ni) along the c axis (left) and a or b axis (right). (e) HRTEM image of the H2Pc-THHI-COF. (f) Comparison of photocurrent of different 3D THHI-COFs. (g) Kinetics for H2O2 production with the H2Pc-THHI-COF and NiPc-THHI-COF. (Reprinted with permission from ref. 155. Copyright 2024, Wiley-VCH GmbH.) | |
Table 4 Summary of representative topological and structural modifications on COF photocatalysts for H2O2 photosynthesisa
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (mmol g−1 h−1) |
AQY |
SCC |
Ref. |
| Light irradiation: Xe lamp (λ > 420 nm); BA: benzyl alcohol. |
| 1 |
TBD-COF |
H2O |
1 mg & 8 mL |
5448 |
5.95% @ 420 nm |
0.90% |
146 |
| 2 |
QP-HPTP-COF |
H2O and O2 |
30 mg & 10 mL |
4388 |
22.78% @ 420 nm |
1.41% |
147 |
| 3 |
TMB-COF-4 |
H2O and O2 |
— |
5106 |
— |
— |
151 |
| 4 |
TAA-CTP COF |
H2O and O2 |
10 mg & 5 mL |
1041 mM h−1 |
— |
— |
152 |
| 5 |
XJU-1 |
H2O:BA EtOH (9:1) and O2 |
5 mg & 20 mL |
34777 |
9.7% @ 380 nm |
— |
153 |
| 6 |
TTF@Por-COF-cya |
H2O |
5 mg & 50 mL |
6994 |
14.98% @ 420 nm |
— |
154 |
| 7 |
H2Pc-THHI-COF |
H2O and O2 |
5 mg & 50 mL |
4511 |
— |
— |
155 |
Microenvironment tuning in COFs for efficient H2O2 production
The term microenvironment usually refers to the local chemical and structural surroundings within and around the framework where catalytic reactions occur. The concept of microenvironment in COFs has emerged as a critical determinant of their functional behaviour, particularly in catalysis. Beyond framework composition, the confined pore space, including its topology, chemical polarity, electronic distribution, and host–guest interactions, collectively shapes how reactants diffuse, adsorb, and activate within the lattice. Despite being a well-defined and widely applied strategy in zeolite catalysis, microenvironment control in COF-based catalysts is only beginning to emerge, offering substantial room for advancement.156 The modulation in the microenvironment dictates molecular diffusion, O2 adsorption, charge separation, and intermediate stabilisation. By tailoring these confined reaction spaces, COFs can selectively promote the 2e− ORR/WOR pathway while suppressing side reactions, enabling efficient H2O2 generation under mild conditions. In a recent report, Guo and co-workers revealed that Pauli repulsion combined with electron interactions leads to uneven electron distribution in COFs, creating selective O2 binding sites that promote efficient photocatalytic H2O2 generation.157 Two COFs were synthesised for further study, namely, TAPT-FTPB and TAPT-BPDB, with asymmetric and symmetric local electron distributions (Fig. 18a). Their findings indicate that TAPT-FTPB COFs, featuring a locally asymmetric electron distribution, exhibit enhanced oxygen adsorption capacity and stronger adsorption interactions, contributing to improved photocatalytic performance. Consequently, a H2O2 production of 3780 µmol g−1 h−1 was achieved (Fig. 18b) with an SCC of 1.22%. The TAPT-FTPB also shows long-term H2O2 production over 30 h and exhibits no degradation after ten continuous cycles (Fig. 18c). Similarly, Ding et al. reported a one-pot “grafting-to” approach to modulate the local electronic environment within a series of isoreticular multicomponent COFs (Fig. 18d).158 By incorporating electron-donating or electron-withdrawing groups into the pore channels, they successfully converted photo-inert COFs into photoactive ones through the formation of intramolecular D–A architectures. Remarkably, the COF functionalized with electrophilic 2-aminothiazole delivered an impressive H2O2 production rate of 3701 µmol g−1 h−1 and attained an SCC efficiency of 0.13%. Recently, Zhang et al. reported the impact of microenvironment regulation in COFs on the overall photocatalytic activity by varying the number of nitrogen atoms within the framework.159 An increased number of nitrogen atoms effectively modulated the local microenvironment and enhanced the electrophilicity of the COF. Among the series, the COF with the highest nitrogen content demonstrated superior performance, achieving efficient photogenerated charge separation and maximising H2O2 production from natural resources like sunlight and seawater. A remarkable H2O2 generation rate of 4881 µmol g−1 h−1 was attained. Typically, organic polymeric materials, including COFs, exhibit an exciton diffusion distance of only 5–10 nm, which is significantly smaller than the overall particle size of COFs.160 As a result, it is believed that modifying the local structure within small domains of the COF, rather than across the entire particle, can more effectively enhance charge transfer and thereby improve the photocatalytic performance. To elucidate the structure–activity relationship governing H2O2 generation in COFs, Guo et al. synthesised a series of quinoline-based COFs bearing different functional groups—COFs-R (R = –OH, –OMe, –H, –Br, –CN)—using a multicomponent reaction (MCR) strategy (Fig. 18e).161 Inspired by natural enzyme systems, where catalytic efficiency is finely tuned by microenvironmental regulation, the authors introduced these diverse functional groups into the COF. The quinoline backbone, resembling that of glucose oxidase, allows its catalytic behaviour to be modulated by the attached R-groups. The aldehyde and amine building blocks used to construct the framework both contain a triazine core, making these COFs inherently highly electrophilic. Thus, as the electron-donating strength of the substituents increases, the overall donor character of the framework is enhanced, leading to improved D–A charge-separation efficiency. As a result, H2O2 generation reaches up to 4458 µmol g−1 h−1, with most electron-donating functional groups (COF-OH), and with most electron-withdrawing functional groups (COF-CN), it reaches only 2032 µmol g−1 h−1 (Fig. 18f). This was further evidenced by a similar performance trend in pure water, showing a negative correlation with the Hammett σp values of the –R group, irrespective of the presence or absence of a sacrificial agent (Fig. 18g). A change in the local structure of COFs was also achieved by Jiang et al. by conducting a three-component solvothermal reaction between diamines, thiadiazole-based tetraaldehydes, and sulfur (Fig. 18h).162 The reaction yielded mixed USTB-x-S (x = 7–10) COFs, which are composed of approximately 90% imine and 10% thiazole linkages. Compared to their fully imine-linked D–A counterparts (USTB-7-10), the USTB-x-S series demonstrated significantly enhanced photocatalytic performance, with H2O2 production rates increased by 77–95% in pure water under O2. Notably, USTB-10-S achieved a rate of 5041 µmol g−1 h−1, which further increased to 16152 µmol g−1 h−1 upon the addition of benzyl alcohol as a sacrificial agent. This work highlights that the minor local structural modifications in COFs can markedly improve charge separation, thereby boosting photocatalytic performance. The major achievements of the microenvironment tuned COF photocatalyst for H2O2 production have been highlighted in Table 5.
 |
| | Fig. 18 (a) Structures of TAPT-FTPB-COF and TAPT-BPDB-COF highlighting symmetric and asymmetric local electron distributions. (b) Kinetics of the two COFs for H2O2 production. (c) Recyclability experiment of the TAPT-FTPB-COF over 30 h. (Reprinted with permission from ref. 157. Copyright 2025, Springer Nature.) (d) Schematic representation of the structure of the COF synthesised through the one-pot ‘grafting-to’ approach and its different functional groups. (Reprinted with permission from ref. 158. Copyright 2025, American Chemical Society.) (e) Structure of COFs-R synthesised through a multivariate reaction. (f) Photocatalytic H2O2 production comparison of COFs-R. (g) Hammett plot showing the relation between the photocatalytic performance of COFs-R and Hammett parameters. Reproduced and reprinted with permission from ref. 161. (h) Schematic representation of the synthesis of USTB-COF. (Reprinted with permission from ref. 162. Copyright 2024, Wiley-VCH GmbH.) | |
Table 5 Summary of representative microenvironment tuning of COF photocatalysts for H2O2 photosynthesisa
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (mmol g−1 h−1) |
AQY |
SCC |
|
| Light irradiation: Xe lamp (λ > 420 nm); BA: benzyl alcohol. |
| 1 |
TAPT-FTPB |
H2O and O2 |
10 mg & 10 mL |
3780 |
— |
1.22% |
157 |
| 2 |
COF-Thz |
H2O |
2 mg & 40 mL |
3701 |
22.78% @ 420 nm |
1.41% |
158 |
| 3 |
N3-COF |
H2O and O2 |
10 mg & 50 mL |
4820 |
1.5% @ 420 nm |
0.413% |
159 |
| 4 |
COF-OMe |
H2O:BA (9:1) |
5 mg & 18 mL |
5497 |
6.5% @ 420 nm |
0.16% |
161 |
| 5 |
USTB-10-S |
H2O:BA (1:1) |
2.5 mg & 25 mL |
16152 |
4.98% @ 420 nm |
0.06% |
162 |
Metal–organic framework (MOF)-based photocatalysts for H2O2 production
MOFs, significant class of crystalline porous materials, comprise metal ions or clusters and organic ligands. They have a large specific surface area, regular and controllable pore structures, and various chemical compositions and functionalities, including the ability to tune the metal centre.163–166 By altering the kinds of ligands and/or metal nodes, it is possible to rationally tailor the chemical reaction conditions and electrical structures of active centres for specific purposes.167,168 The increased porosity speeds up mass movement throughout the network of MOFs and encourages the exposure and accessibility of active sites. The high metal content, ordered crystallinity, large surface area, and adjustable pore size of MOFs have made them attractive candidates for various applications, such as catalysis,169–171 gas storage and separation,172–174 environmental remediation,175,176 sensing,177–179 biomedical applications,180 etc. Towards photocatalysis, the intrinsic photocatalytic properties of MOFs arise from their tunable band structure, photoactive metal nodes, and π-conjugated organic linkers. These features enable efficient light harvesting via the antenna effect of linkers, which transfer energy to metal sites through linker-to-metal charge transfer (LMCT), mimicking semiconductor behaviour with adjustable band gaps for visible-light response. Their high porosity and large surface area provide abundant accessible active sites and facilitate mass transport of reactants, while the ordered crystalline framework promotes directional charge transport. Additionally, internal electric fields originating from polar structures or donor–acceptor architectures enhance charge separation and suppress electron–hole recombination. Strong metal–ligand coordination imparts chemical and photostability, and selective adsorption of substrates such as O2, CO2, or H2O further improves catalytic efficiency and selectivity, making MOFs intrinsically well-suited for photocatalytic applications. Recently, Park et al. have synthesised copper-based electrically conductive MOFs and introduced a D–A moiety by integrating spatially separated electron D–A moieties via a click reaction (Fig. 19a).181 After the click reaction in MOFs with phenylacetylene (PA) and 1-ethynyl-4-fluorobenzene (EFB), the researcher observed the tuning in the band positions that further aligned with the redox potential of photocatalytic H2O2 production. Fluorine incorporation facilitates efficient charge separation by suppressing electron–hole recombination, thereby maximising photocatalytic efficiency. The photosynthesis of H2O2 with synthesised MOFs has been explored in pure water and O2 irradiated with a 300 W Xe lamp. The maximum yield was obtained for the clicked MOF with EFB, and the H2O2 production rate of 1676 µmol g−1 h−1 was reported with an AQY of 1.03% at 410 nm. Toward developing the pristine MOF as an efficient photocatalyst, Wang et al. developed an uranyl-organic framework named YTU-W-2 using a mixed ligand strategy where thiophene and pyridine ligands were used (Fig. 19b).182 The introduction of mixed ligands has increased the photon absorption and carrier transfer into the framework. Furthermore, the H2O2 rate of 345 µmol h−1 g−1 has been reported in a 9:1 water and ethanol mixture. The structural effect of pristine MOF towards photocatalytic synthesis of hydrogen peroxide has also been recently studied by Zhang et al. by synthesising two isomeric Ti-based MOFs [NH2-UiO-66(Ti) and NH2-MIL-125(Ti)] (Fig. 19c).183 The researchers reported the efficient electron transport of the Ti–O oxygen cluster as NH2-MIL-125(Ti) has a shorter bond length than NH2-UiO-66(Ti). Consequently, the photocatalytic activity of NH2-MIL-125(Ti) was better than that of NH2-UiO-66(Ti). Furthermore, the defective structure in the MOF was found to acquire more active sites that promote the photocatalytic activity, and the H2O2 production of defective NH2-MIL-125 was found to be efficient among all, with a production rate of 101 µmol g−1 h−1. Similarly, Zhou and co-workers engineered MIL-125-NH2, including coexisting (111) and (001) facets, facilitating the formation of a Z-scheme structure to enhance spatial charge separation.184 A dual-channel pathway for synthesising H2O2 was identified, including the ORR on the (111) facets and OH− oxidation on the (001) facet. The exposure degree of the (111) facet of MIL-125-NH2 was systematically adjusted by varying the quantities of acetic acid modulator during the preparation process; an increase in acetic acid resulted in a greater exposure modulator during the preparation process; an increase in acetic acid resulted in a greater exposure of (111) facets.185 The (111) facets may reduce charge transport resistance and extend the lifespan of photo-induced electrons and holes, resulting in an ideal H2O2 production rate of 925.8 µmol g−1 h−1 (visible light, triethanolamine as a sacrificial agent, and pure O2), in the indirect ORR. In another study, Zhu and co-workers discovered self-assembled tetrakis(4-carboxyphenyl) porphyrin supramolecular photocatalysts that generate H2O2 solely from H2O and O2, achieving a production rate of 1.72 mM h−1 at 293 K, with quantum efficiencies of 14.9% at 420 nm and 1.1% at 940 nm.186 The catalyst attains an SCC efficiency of 1.2% at 328 K under irradiation and heating with simulated sunlight. The findings indicated that photogenerated electrons and holes facilitate H2O2 generation by interacting with distinct active sites, such as the pyrrole N–H ring and carboxyl groups. They specifically suggest a hole-induced H2O2 generation method, which entails the photoconversion of –COOH to –CO3H groups on the catalyst, succeeded by thermal breakdown. Furthermore, after 20 cycles, the buildup of photocatalytic H2O2 reached about 50 mM, indicating the materials' strong recyclability. The regulation of D–A interaction mode in Zr-based UiO-67 MOF has been recently reported by Guo et al.187 The author reports the synthesis of a family of UiO-67-type D–A MOFs, in which pyrene, benzothiadiazole, and naphthalene-benzothiadiazole linkers of identical length are co-assembled with Zr4+ nodes (Fig. 19d). The precisely tuned short-range (through-bond) and long-range (through-space) D–A interactions were appropriate for the photocatalytic H2O2 production from water and O2 in a water/benzyl alcohol biphasic system. The mixed-linker UiO-67-PENB, the first example of a MOF combining both DA modes in one framework, exhibits an optimised band structure, enhanced charge separation, and suppressed recombination. Consequently, through an indirect two-step 2e− ORR pathway, a high H2O2 formation rate of 5141 µmol g−1 h−1 and an AQY of 4.24% at 400 nm were achieved. The MOF further demonstrated excellent structural and operational stability over 48 h and H2O2 accumulation up to ∼32 mM. Comprehensive spectroscopic and electrochemical studies (UV-vis DRS, Mott–Schottky, EIS, photocurrent, PL, fs-TA, EPR, and in situ DRIFTS analyses) underpin the mechanistic pathway for H2O2 photoproduction as the synergistic photoinduced electron transfer from pyrene to the NB-based DA linker, followed by efficient O2 reduction and effective hole scavenging by benzyl alcohol (Fig. 19e). This cooperative process enables UiO-67-PENB to outperform the single-mode D–A analogues UiO-67-NB and UiO-67-PEBB in H2O2 production and collectively accounts for the superior H2O2 photosynthesis performance to both single-mode D–A analogues (UiO-67-NB and UiO-67-PEBB). Toward sustainable H2O2 generation and its practical utilisation, Pal et al. demonstrated the use of MOFs as an emerging class of thermocatalysts capable of producing H2O2 by harvesting small temperature gradients under ambient conditions.188 In their study, MOFs were immobilised onto carbon fibre fabrics (MOF@CFF) to construct antibacterial filtration systems activated by temperature cycling. Three representative MOFs (CuBDC, MOF-303, and ZIF-8) were evaluated and benchmarked against bismuth telluride (Bi2Te3), a well-known
thermocatalytic material. Among them, ZIF-8 exhibited the highest H2O2 production efficiency under low temperature differences, leading to ∼96% antibacterial efficacy against Escherichia coli and Staphylococcus aureus. This work highlights the potential of MOF-based thermocatalysts for thermoelectric-assisted H2O2 generation, offering a promising strategy for real-time water purification and disinfection. Tailoring the morphology and size of photocatalysts has emerged as an effective strategy to enhance H2O2 generation by improving charge carrier dynamics and tuning band energetics. For instance, Li and co-workers developed an ultrathin 2D Al-MOF nanosheet of 1 nm (Al-P(AQ)) through the co-assembly of porphyrin (TCPP) as an electron donor and anthraquinone-based ligands (AQCA) as an electron acceptor.189 The resulting ultrathin framework significantly reduces the diffusion distance for photogenerated charge carriers, thereby facilitating rapid e− and h+ transport to the catalyst surface and improving the photocatalytic efficiency. Moreover, incorporation of AQCA alters the electronic structure of the MOF, inducing a downward shift in the LUMO level, narrowing the band gap, and enhancing the thermodynamic driving force for the ORR. The major achievements of the MOF photocatalyst for H2O2 production have been highlighted in Table 6.
 |
| | Fig. 19 (a) Schematic route for the synthesis of an electrically conductive MOF and its post-synthetic click reaction. (Reprinted with permission from ref. 181. Copyright 2024, American Chemical Society.) (b) Crystal structure of YTU-W2 and its band gap position towards H2O2 production. (Reprinted with permission from ref. 182. Copyright 2024, American Chemical Society.) (c) Isomeric structures of the Ti-based MOF: NH2-UiO-66(Ti) and NH2-MIL-125(Ti). (Reprinted with permission from ref. 183. Copyright 2024, Royal Society of Chemistry.) (d) Illustration of the strategic installation of different donor and acceptor interaction modes and the structure of synthesised UiO-67-type D–A MOFs. (e) Catalytic pathway for H2O2 photoproduction via the two-step 2e− ORR on UiO-67-PE/NB. (Reprinted with permission from ref. 187. Copyright 2025, American Chemical Society.) | |
Table 6 Summary of representative metal–organic framework (MOF) photocatalysts for H2O2 photosynthesis
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC |
Ref. |
| Light irradiation: Xe lamp (λ > 420 nm) except visible light; BA = benzyl alcohol; TEA: triethanolamine. |
| 1 |
EFB-MOF |
H2O and O2 |
5 mg & 30 mL |
1676 |
1.03 @ 410 nm |
— |
181 |
| 2 |
YTU-W-2 |
H2O:EtOH (9:1) |
10 mg & 50 mL |
345 |
0.073 @ 420 nm |
— |
182 |
| 3 |
NH2-MIL-125 |
H2O and O2 |
15 mg & 50 mL |
101 |
— |
— |
183 |
| 4 |
MIL-125-NH2a |
H2O/TEA and O2 |
5 mg & 20 mL |
925.8 |
— |
— |
184 |
| 5 |
SA-TCPPa |
H2O and O2 |
25 mg & 50 mL |
0.66 mM h−1 |
14.9% @ 420 nm |
1.2% |
186 |
| 6 |
UiO-67-PE/BB |
H2O:IPA (9:1) |
10 mg & 50 mL |
5141 |
4.24% @ 400 nm |
— |
187 |
| 7 |
Al-P(20AQ) |
H2O and O2 |
0.5 g L−1 |
163.2 |
4.8% @ 400 nm |
0.11% |
189 |
Porous organic polymer (POP)-based photocatalysts for H2O2 production
Porous organic polymers (POPs) are a diverse class of materials known for their highly porous structures, which are constructed from organic monomers that primarily contain light elements such as C, H, O, N, B, etc.190 POPs are primarily synthesised through the polymerisation of organic building blocks into macromolecular frameworks. POPs possess various properties that make them ideal for photocatalysis such as high porosity, large surface areas, and tunable extended π-conjugated structures that enable broad visible-light absorption and efficient exciton generation. Their lightweight nature, excellent chemical/thermal stability, and pre-designable D–A building blocks facilitate superior charge separation, reduced recombination, and precise modulation of band gaps and redox potentials for targeted photocatalytic reactions. Unlike COFs, POPs can be synthesised under milder conditions, feature irreversible covalent bonds that impart greater robustness, and avoid harsh crystallisation steps. POPs can be further classified into two categories based on their long-range order, such as crystalline and amorphous POPs.191 COFs and covalent triazine frameworks (CTFs) are well-known crystalline POPs, and some examples of amorphous POPs are porous aromatic frameworks (PAFs), hypercrosslinked polymers (HCPs), polymers of intrinsic microporosity (PIMs), and conjugated microporous polymers (CMPs), etc. The high crystallinity in a material can significantly promote electron/hole separation, reduce their possible recombination, and enhance the photocatalytic activity.192,193 Therefore, various reports could be seen regarding the development of crystalline POPs for the photosynthesis of H2O2, such as COFs, as discussed above. CTF is also a crystalline POP, distinguished by its aromatic C–N linkage (triazine unit) and lack of weak bonds. Strong aromatic covalent connections mainly impart CTFs with high nitrogen content and excellent chemical stability, which are valuable for various real-world uses and the intriguing heteroatom effect (HAE). Because of their unique qualities, CTFs are appealing for several uses, including energy storage, photocatalysis, heterogeneous catalysis, and gas separation and storage. CTFs may be divided into two groups based on the state of research: crystalline and amorphous CTFs. The kind of monomers and the synthesis procedure have a significant impact on the characteristics of CTFs. In recent years, significant research has been done on synthesis techniques such as aldehyde–amidine condensation, trifluoromethanesulfonic acid-catalysed synthesis, amino-aldehyde synthesis, and ionothermal polymerisation.194,195 CTFs are promising semiconductor candidates for photocatalysis, including the photodegradation of organic pollutants, the conversion of chemical molecules, the splitting of water, the reduction of CO2, and more, due to their abundance of nitrogen, strong conjugated structure, and absorption of visible light.196–198 By altering the phenyl and thiophene moieties, J. Byun and colleagues have synthesised two CTFs with adjustable D–A character, demonstrating that regulating the oxidative reaction medium significantly affects photocatalytic H2O2 generation (Fig. 20a).199 The photocatalytic H2O2 generation ability of CTFs was assessed under both aqueous and non-aqueous conditions. The O2 reduction process, in conjunction with toluene oxidation, is specifically made possible by CTF-Th, which has more thiophene donors. This results in a large generation of H2O2 up to 105 µmol, which is 4.7 and 2.5 times higher than those of H2O (22.3 µmol) and H2O/alcohol (42.4 µmol) oxidation, respectively (Fig. 20b). According to the theoretical computation, benzaldehyde and H2O2 can be produced simultaneously on CTF surfaces due to the binding affinities of toluene and O2, respectively. Toluene and water layers form a dual-phase system that makes it easy to separate the two very pure products. Inspired by the sequential energy- and charge-transfer processes from PS II to PS I in natural photosynthesis, K. A. I. Zhang and colleagues developed and synthesised three CTF photocatalysts with stepwise charge transfer (Fig. 20c).200 They created effective CTFs for high-efficiency photosynthesis of H2O2 from water and O2 without the use of sacrificial agents by carefully designing linkers with different degrees of conjugation to act as electron transfer spacers and photocatalytic ORR centres between the four molecular donor–acceptor domains (M1–M4) in the asymmetric CTF framework. The acetenyl moiety improved the spontaneous O2 adsorption in the CTFs compared to the heavily conjugated thiophene moiety. Because Ace-asy-CTF's acetylene moiety effectively lowers the energy barrier for the stepwise two-electron ORR, it has a higher photocatalytic H2O2 production efficiency than Th-asy-CTFs with strong conjugated thiophene units or asy-CTF without a spacer. This results in an enhanced photocatalytic H2O2 yield (2594 µmol g−1 h−1, Fig. 20d) with an SCC of 0.48% without sacrificial agents. By creating and manufacturing two highly crystalline layered CTFs using 1,1′-biphenyl-4,4′-dicarbonitrile (BP-CN) and 9H-fluorene-2,7-dicarbonitrile (FL-CN) as the building blocks for the first time, Xu and colleagues created high-performance metal-free photocatalysts (Fig. 20e).200,201 The direct photosynthesis of H2O2 generation on the as-prepared crystalline CTFs with a locked-in coplanar conformation has been significantly improved by utilising logical control of the torsion angle in the basal plane of the CTFs (Fig. 20f). The exfoliated few-layer crystalline 2D triazine polymer nanosheets from the bulk CTF exhibit a remarkable SCC efficiency of 0.91% at a low catalyst concentration (0.5 g L−1) and a noteworthy AQY of 16.8% at 420 nm, demonstrating an excellent photocatalytic H2O2 production rate of up to 5007 µmol g−1 h−1 from oxygen and water without sacrificial agents. A series of CTFs with different numbers of acetylene groups were reported by H. Xu and colleagues as metal-free photocatalysts for the generation of H2O2. Acetylene or diacetylene moieties may be added to CTFs to increase photocatalytic H2O2 production significantly.202 The integrated carbon–carbon triple bonds, which are crucial for adjusting the electrical structures of CTFs and preventing charge recombination, impart this natural improvement. Additionally, the acetylene and diacetylene moieties can drastically lower the energy required to generate OH*, opening up a novel two-electron oxidation route that produces H2O2. After 24 h, the H2O2 generation of CTF-BDDBN with two acetylene centres was greater than that of CTF-EDDBN with one acetylene moiety (39 µmol) and CTF-BPDCN without an acetylene moiety (21 µmol). Li and colleagues demonstrated how to increase the electrical conductivity of CTF by ten times for excellent, highly conductive carbon quantum dots (CQDs) as an electron transport medium inside the CTF pores.203 In pure water, without any sacrificial agent, the optimised CQD-CTF (0.5 wt%) shows a high H2O2 production rate of up to 1036 µmol g−1 h−1 under visible light, 4.6 times that of CTF. According to this study, the presence of CQDs will improve the performance of charge separation and migration with higher electronic conductivity. The greater negative zeta potential, which can significantly trigger the ORR process, may also increase the affinity of H+. Additionally, photocatalysis increases the oxidation capacity, which benefits the WOR process. This work offers a novel approach to the practical H2O2 synthesis of low-conductivity organic semiconductors. Despite the challenges associated with amorphous POPs, such as low crystallinity, weak light absorption in the visible spectrum, poor separation of photogenerated carriers, and limited structural diversity, which hinder photocatalytic performance, there are only a few reports focusing on their development for photocatalytic H2O2 production. For instance, Nie et al. have reported the photocatalytic efficiency of four HCP photocatalysts with different arene monomers towards photocatalytic H2O2 production.204 A series of HCPs have been synthesised by a facile Friedel–Crafts alkylation reaction of different arene monomers, which include benzene (BE), diphenyl (DP), p-terphenyl (TP), and p-quaterphenyl (QP) (Fig. 21a). Amongst all, HCP with a QP group has the optimal separation and transfer efficiency of photogenerated e−–h+ due to the higher degree of conjugation of the QP monomer. The efficiency
of synthesised HCPs has been evaluated towards photocatalytic H2O2 production in acetonitrile and benzylamine under blue LED irradiation. The H2O2 production rate for BE, DP, TP, and QP was observed to be 6.0, 6.8, 7.8, and 9.3 mmol g−1 h−1, respectively. Recently, Wang et al. have reported the synthesis of two alkynyl-containing POP (POP-DT and POP-DF) via the Sonogashira cross-coupling reaction between the acetylene-based monomer and thiophene or furan monomer (Fig. 21b).205 The authors report that the acetylene moieties in the framework acted as the oxygen-reducing active site, while the thiophene or furan ring functioned as the electron donor unit. Furthermore, the integration of acetylene and thiophene facilitated faster charge separation and improved charge transfer efficiency, thereby greatly enhancing the kinetic performance of the oxygen reduction reaction. Therefore, the H2O2 production rate of POP-DT, under visible light irradiation, reached 2422.2 µmol g−1 h−1, which was 4.3 times higher than that of POP-DF, representing one of the best performances ever observed for polymeric photocatalysts. The coordination of the pyridine moiety with the acetylene-based monomer has also proved to be efficient for H2O2 photosynthesis. In this context, Wen et al. have synthesised TEADP-POF by reacting tris(4-ethynylphenyl)amine (TEA) with 2,5-dibromopyridine (DP) and TEADB-POF with dibromobenzene.206 Triphenylamine functions as the electron-donating unit, while pyridine serves as the electron acceptor, enabling a push–pull effect within the 2D π-conjugated framework that facilitates charge separation and transfer. Consequently, H2O2 production at a rate of 2136 µmol g−1 h−1 was obtained with TEADB-POF from H2O and O2. Similarly, the same group investigated photocatalytic H2O2 production in conjugated organic polymers through active-site position engineering. In this approach, 2,5-dibromopyrazine and 3,6-dibromopyrazine were employed in place of DP to synthesise TEAPYRA-COP and TEAPYRI-COP, respectively.207 TEAPYRI-COP, containing adjacent pyridazine nitrogen sites, exhibits stronger hydrogen-bonding with water compared to TEAPYRA-COP, where nitrogen atoms are positioned oppositely. This configuration facilitates O–H bond activation, enhances water oxidation to generate protons, and promotes efficient H2O2 formation via O2 reduction. Additionally, the adjacent nitrogen sites in TEAPYRI-COP stabilise key intermediates through a low-energy six-membered transition state, improving the 2e− ORR pathway. As a result, TEAPYRI-COP achieves a significantly higher H2O2 production rate (2884 µmol g−1 h−1), approximately 3.5 times greater than that of TEAPYRA-COP under visible light without additives. The major achievements of the POP photocatalyst for H2O2 production have been highlighted in Table 7.
 |
| | Fig. 20 (a) Donor–acceptor type covalent triazine frameworks, CTF-Ph and CTF-Th and (b) photocatalytic production of H2O2 in 50% toluene in acetonitrile under saturated O2 conditions. (Reprinted with permission from ref. 199. Copyright 2023 Elsevier). (c) Schematic illustration of the stepwise charge transfer within the CTF and (d) time course of H2O2 evolution over CTFs (300 W xenon lamp with a 400 nm cutoff filter; light intensity: 100 mW cm−2). (Reprinted with permission from ref. 200. Copyright 2024, American Chemical Society.) (e) 13C CP-MAS NMR spectra of CTF-BP and CTF-FL and (f) time course of photocatalytic H2O2 production (λ > 420 nm, 300 W xenon lamp; 10 mg of catalyst in 100 mL of pure water; O2-saturated, 25 °C). (Reprinted with permission from ref. 201. Copyright 2024, American Chemical Society.) | |
 |
| | Fig. 21 (a) Schematic illustration of the synthesis of different HCPs for benzylamine oxidation and H2O2 production. (Reprinted with permission from ref. 204. Copyright 2024, Wiley-VCH GmbH.) (b) Schematic of the synthesis of POP-DT and POP-DF. (Reprinted with permission from ref. 205. Copyright 2024, Royal Society of Chemistry.) (c) Structural representation of TpMaTAE. (d) Surface potential values of TpMa, TpTAE, and TpMaTAE. (e) Photocatalytic H2O2 production with TpMa, TpTAE, and TpMaTAE when light is on and off. (f) Mechanism of H2O2 production and the built-in an electric field and the schematic of the synthesis of POP-DT and POP-DF. (Reprinted with permission from ref. 208. Copyright 2024, Wiley-VCH GmbH.) | |
Table 7 Summary of representative porous organic polymer (POP) photocatalysts for H2O2 photosynthesis
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC (%) |
Ref. |
| Light irradiation: Xe lamp (λ > 420 nm) except visible light; IPA: isopropyl alcohol; BAm: benzylamine. |
| 1 |
CTF-Th |
H2O:IPA (9:1) and O2 |
50 mg & 30 mL |
43 µmol in 3 h |
— |
— |
199 |
| 2 |
Ace-asy-CTF |
H2O and O2 |
5 mg & 10 mL |
2594 |
4.4% @ 420 nm |
0.48 |
200 |
| 3 |
CTF-NSs |
H2O and O2 |
10 mg & 100 mL |
5007 |
16.8% @ 420 nm |
0.91 |
201 |
| 5 |
0.5 CQD-CTFa |
H2O and O2 |
5 mg & 20 mL |
1036 |
1.03% @ 420 nm |
0.21 |
203 |
| 6 |
QP-HCP |
CH3CN and BAm |
10 mg & 20 mL |
9300 |
— |
— |
204 |
| 7 |
POP-DT |
H2O and O2 |
10 mg & 50 mL |
2422.2 |
2.67% @ 400 nm |
— |
205 |
| 8 |
TEADP-POF |
H2O and O2 |
30 mg & 50 mL |
2136 |
2.39% @ 400 nm |
— |
206 |
| 9 |
TEAPYRI-COP |
H2O and O2 |
30 mg & 50 mL |
2884 |
3.27% @ 400 nm |
— |
207 |
| 10 |
TpMaTAE |
H2O and O2 |
5 mg & 50 mL |
6280 |
— |
1.03 |
208 |
| 11 |
CTF-BTT |
H2O/BA and O2 |
5 mg & 50 mL |
74956 |
7.61% @ 420 nm |
0.66 |
209 |
| 12 |
CTF-DA-film-3a |
H2O and O2 |
Thin film |
8920 |
1.242% @ 450 nm |
0.12 |
210 |
| 13 |
BPYTEA-POF |
H2O and O2 |
30 mg & 20 mL |
3446 |
— |
— |
211 |
In a study, Zhu et al. reported the synthesis of TpMaTAE by multicomponent Schiff-base polycondensation of Tp-aldehyde, melamine (Ma), and 1,3,5-tris(4-aminophenyl)benzene (TAE).208 The reaction resulted in a covalently integrated acetylene donor with dual acceptors (triazine and carbonyl) and yields an amorphous, high-surface-area, fibrous COP with a strong built-in dipole and enhanced O2 adsorption (Fig. 21c). Band-structure analysis reveals visible-light absorption, suitable band edges for the 2e− ORR/WOR, and reduced exciton binding energy. Also, Kelvin probe force microscopy (KPFM) and surface charge measurements confirm a much stronger internal electric field than in D–A analogues TpTAE and TpMa (Fig. 21d). Consequently, TpMaTAE achieves an H2O2 production rate of 5860 µmol g−1 h−1 from air and 7830 µmol g−1 h−1 (Fig. 21e) under AM 1.5G with SCC values of 1.03% and 93.1% for O2 utilisation. Mechanistic analyses supported by experimental data reveal a stepwise single-electron 2e− ORR via O2- and one-step 2e− WOR pathways (Fig. 21f). Spectroscopy (fs-TAS and TRPL), together with EIS, OCVD, and light-induced EPR, shows that the D–A1–A2 architecture significantly improves exciton dissociation and reduces charge-transfer resistance. These measurements also reveal prolonged charge-carrier lifetimes and a higher density of stored electrons in TpMaTAE. In addition, in situ FTIR and dark H2O2 generation experiments confirm that the carbonyl groups function as electron reservoirs. This stored charge enables continued H2O2 formation even after the light is switched off.
Metal–organic polyhedra (MOP)-based photocatalysts for H2O2 production
Metal–organic cages (MOC) or metal–organic polyhedra (MOP) are an old but rarely explored subclass of AFPMs.212,213 MOPs are attractive for their structural similarity to MOFs, except that they have to cap terminal groups that prevent indefinite extension into networks, making them discrete molecules.214 From the standpoint of the metal cation or cations and the organic linkers used in their self-assembly, MOP has demonstrated synthetic flexibility. The interior cavity, allowing guest molecules to be absorbed, is a distinguishing feature of these materials. Consequently, coordination cages have found utility in a wide range of applications, including molecular recognition and separation, host–guest chemistry, catalysis, and biological applications.215–218 In contrast to polymeric materials, MOP photocatalysts with well-defined active sites enable clear elucidation of structure–mechanism–performance relationships. Their photophysical properties can be precisely tuned through atomic-level modifications, leading to exceptional catalytic activity and attracting growing attention in recent years. MOPs are particularly advantageous due to their precisely controllable geometries, well-defined pore sizes, tunable electronic structures, and broad photoresponse windows. Furthermore, MLCT enhances the reactivity of the photoexcited states, while synergistic metal–ligand interactions promote efficient charge separation. Despite this, the photocatalytic applications of MOPs are less explored compared to other AFPMs. The first work with MOP for photocatalytic H2O2 production was reported in 2023, where Y. Q. Lan and co-workers reported two Co-MOCs (Co14(L-H)24 and Co14(L-CH3)24) integrated with an imidazole-based ligand (Fig. 22a and b).219 The photocatalyst's light absorbance is enhanced, and the recombination efficiency of photogenerated electrons and holes is decreased due to MOC functionalization. According to theoretical simulations, the CH3 functionalized ligand is better suited to adsorb the important *OH and *HOOH intermediates produced by the WOR and ORR. (Co14(L-CH3)24) exhibits a higher H2O2 generation rate of 146.60 µmol g−1 h−1 in O2-saturated water, and subsequently, the MOP photocatalyst could produce a significant amount of H2O2 under different conditions such as air, MeOH and IPA (Fig. 22c). The catalyst can be recycled up to three cycles with a nominal decrease in the yield (Fig. 22d). Isotope tracing experiments and comparative studies show that for these two catalysts, the Co site acts as the reduction site, allowing the synthesis of H2O2 via the 2e− two-step ORR route, while the imidazole site from the ligand acts as the oxidation site, producing H2O2 via the 2e− two-step WOR pathway (Fig. 22e). More significantly, in these Co(II)-based supramolecular cages, the imidazole site directly coordinates with the Co site, reducing the spatial separation between the oxidation and reduction sites and increasing the charge migration efficiency, which in turn increases the catalytic activity of photocatalytic H2O2 synthesis. Similarly, Xiao et al. present a novel supramolecular approach for photocatalytic H2O2 production using discrete, non-noble MOCs, overcoming key limitations of conventional polymeric and framework-based photocatalysts.220 Unlike MOFs or COFs, these cages offer atomic-level control over active sites, enabling direct correlation between the structure, photophysical properties, and catalytic performance. Two Zn-based metallo-cages (S1 and S2) were rationally designed and synthesised with distinct electronic and photophysical properties (Fig. 22f). The atomic-level control over active sites and charge-transfer pathways, with S2, exhibits high structural stability, low exciton binding energy, ultrafast intramolecular electron transfer, and prolonged excited-state lifetimes, all of which promote efficient charge separation. A bottom-up nanosizing strategy was further employed by dispersing MOCs in DMF to generate ultrasmall, well-dispersed catalytic particles, significantly enhancing substrate accessibility and reaction kinetics. The photocatalytic H2O2 production was carried out in a H2O:DMF (9:1) solution using Na2C2O4 as a sacrificial agent, and an exceptionally high H2O2 production rate of 77401 µmol g−1 h−1 with an SCC efficiency of 0.97% was achieved. Mechanistic studies reveal that the cage architecture facilitates simultaneous 2e− ORR and WOR pathways, while Na2C2O4 suppresses charge recombination and provides a kinetically favourable route for superoxide radical formation, collectively overcoming bottlenecks for the ORR and enabling efficient and stable H2O2 production.
 |
| | Fig. 22 (a) Crystal structure of (Co14(L-H)24), (b) polyhedral representation of (Co14(L-H)2), (c) photocatalytic performances of H2O2 production by Co14(L-CH3)24 in pure water and different atmospheres, and (d) recyclability of Co14(L-CH3)24 after photosynthesis of H2O2. (e) Catalytic sites in Co14(L-CH3)24 for the ORR and OER (Reprinted with permission from ref. 219. Copyright 2023, Wiley-VCH GmbH.) (f) Schematic representation for the synthesis of S1, S2, and their photocatalytic H2O2 production. (Reprinted with permission from ref. 220. Copyright 2025, Wiley-VCH GmbH.) | |
Hydrogen-bonded organic framework (HOF)-based photocatalysts for H2O2 production
Hydrogen-bonded organic frameworks (HOFs) are a class of porous crystalline materials formed by organic molecules linked through hydrogen bonding and other intermolecular forces.221,222 Although HOFs were proposed as porous materials in the 1990s, it was not until 2010 that their permanent porosity was confirmed through gas sorption studies.223,224 Most HOFs, constructed through weak intermolecular forces, generally exhibit lower stability compared to MOFs and COFs, which are built via coordination or covalent bonds. Nevertheless, the overall stability of HOFs can be significantly enhanced through synergistic effects of hydrogen bonding, π–π stacking, electrostatic interactions, and framework interpenetration.225 The π–π stacking interactions in HOFs not only enhance structural stability but also play a functional role in facilitating efficient electron transfer. As a result, HOFs have emerged as promising photocatalysts for various applications, including H2 production226 and CO2 reduction.227 Recently, Zhong et al. have reported the D–A HOF (TTF-Bpy-HOF) towards photocatalytic H2O2 production from H2O and O2 by reacting electron-rich tetrathiafulvalene tetracarboxylic acid (TTF) and electron-deficient 4,4′-bipyridine (Bpy) as the electron-accepting units (Fig. 23a and b).228 The TTF-Bpy-HOF demonstrated a significantly enhanced photocatalytic performance, achieving a maximum H2O2 production rate of 681.2 µmol g−1 h−1 (Fig. 23c). In contrast, the Bpy-free counterpart, TTF-HOF, exhibited much lower efficiency, with a production rate of only 74.4 µmol g−1 h−1. The HOF demonstrated excellent recyclability, retaining its photocatalytic efficiency over 10 consecutive cycles, showcasing its structural integrity over prolonged H2O2 production. The photodecomposition of H2O2 over the TTF-HOF and TTF-Bpy-HOF was evaluated under light irradiation. The H2O2 concentration remained essentially unchanged over 2 h, confirming its stability in the presence of these materials under illumination. Mechanistic studies reveal that H2O2 formation in the HOF system proceeds via both the 2e− ORR and WOR pathways. As illustrated in Fig. 23d, under visible-light irradiation, the TTF units in TTF-Bpy-HOF absorb photons, generating separated electron–hole pairs. The photogenerated electrons migrate to the Bpy moieties, while holes remain on the TTF units, enabling spatially separated redox reactions. Water oxidation proceeds on the TTF units via a direct 2e− pathway to produce H2O2, whereas O2 reduction occurs on the Bpy units through a two-step 2e− process involving ˙O2− intermediates, ultimately yielding H2O2.
 |
| | Fig. 23 (a) Schematic representation of D–A HOF synthesis. (b) Time-dependent production of H2O2 by the TTF-Bpy-HOF in an O2 and air-saturated solution. (c) H2O2 decomposition study by the TTF-Bpy-HOF in reaction medium. (d) Recycle experiment of the TTF-Bpy-HOF for H2O2 photosynthesis over 10 cycles. (e) Schematic diagram of the photosynthesised H2O2 through the ORR and WOR pathways. (Reprinted with permission from ref. 228. Copyright 2025, Springer Nature.) | |
AFPM-based composites for photocatalytic H2O2 production
One of the key advantages of functional porous materials is the vast potential for post-synthetic modification (PSM), thanks to their rigid framework and large surface area.229 Functionality that cannot be directly incorporated into functional porous materials during de novo synthesis can be introduced through post-synthetic modification (PSM) strategies. In the case of COFs, there are current strategies for the PSM COF involving (i) incorporation of a variety of active metal species by using metal complexation through coordination chemistry, (ii) covalent bond formation between existing pendant groups and incoming constituents and (iii) chemical conversion of linkages. (iv) A monomer truncation strategy for the internal functionalization of COFs sometimes assists post-synthetic modification. Current strategies for post-synthetic modification (PSM) of COFs include (i) encapsulation of various active species without covalent threading, (ii) formation of covalent bonds between functional groups of COFs and active species, (iii) chemical conversion of linkages into desired active sites and (iv) the use of a monomer truncation strategy to assist in the functionalization of COFs. In PSMs, COFs can be utilised as a host or template to encapsulate or wrap other substances, such as metal nanoparticles, organic molecules, or other porous materials, and the material synthesised is generally referred to as COF composites. These composites leverage the unique properties of COFs while enhancing the functionality by including complementary substances. The resulting COF composites often exhibit synergistic properties beneficial for desired applications.229 Cong et al. have reported the synthesis of two novel polyimide COF (MPa-COFs/CQDs and MNd-COFs/CQDs) composites using different types of dianhydrides and carbon quantum dots (CQDs) via a one-pot hydrothermal method.230 The CQDs on COFs were loaded by treating the synthesised COF with D(+) glucose and heating it in a Teflon-lined stainless-steel autoclave at 160 °C for 3 h. After the introduction of CQDs, both photocatalytic and electronic properties of polyimide COFs were boosted, and suppression of photoinduced electrons and holes recombination was also noticed. The photocatalytic activity of two COF composites for H2O2 production was evaluated using pure water and O2 under visible light irradiation, and the highest production yield of up to 540 µmol g−1 h−1 was observed for MPa-COFs/CQDs-2. The synthesis of the COF composite with the desired heterojunction has also been a topic of interest for many researchers. As organic semiconductors, COFs can be combined with suitable inorganic semiconductors to form various types of heterojunctions, significantly enhancing redox capacity regarding thermodynamics and -kinetics.231 In that context, Wong et al. have combined the D–A conjugated TpMA-COF with an inorganic semiconductor CdS by the hydrothermal method and report the formation of S-scheme heterojunctions.232 The formation of the heterojunction enhanced the photocatalytic activity of the composites. To optimise the catalyst, various amounts of CdS were loaded onto the COF, with the resulting catalysts being named CT-5, CT-10, CT-15, CT-20, and CT-30. The photocatalytic activity of the COF composite material towards H2O2 production was evaluated using pure water under simulated sunlight. The H2O2 production yield of CT-10 was found to be the highest, with a production rate of up to 608.41 µmol L−1 in 90 min. Similar work has also been reported by Xia et al. by constructing a heterojunction on the TpPa-1 COF through the incorporation of ZnIn2S4 nanosheets on the COF.233 The COF composite, ZnIn2S4/TpPa-1, with a hierarchical heterostructure, could produce H2O2 at a rate of 516 µmol L−1 under visible light irradiation, which is almost three times higher than that of individual COF and ZnIn2S4 catalysts. In a quest for synthesising COF composites with improved photocatalytic activity, Wang et al. have incorporated tungsten oxide (WO3) into the Tp-TAPB-COF and report the formation of the WO3/Tp-TAPB composite bearing a Z-scheme heterojunction.234 The Z-scheme heterojunction has promoted the separation of useful e−–h+ pairs, and the overall composites have enhanced light absorption properties. To get the optimum catalyst, the amount of WO3 was varied from 5 mg to 40 mg and named XWO3/Tp-TAPB, where X = 10, 20, 30, and 40, respectively. The photocatalytic activity of different COF composites was evaluated in pure water and oxygen under irradiation of LED light with a wavelength >420 nm. Amongst all the COF composites, the catalytic activity of 20WO3/Tp-TAPB was found to be optimum towards light-driven H2O2 production with a rate of 1488.4 µmol h−1 g−1. Towards the PSM of COFs and encapsulation of metal nanocrystals (NCs), Shang et al. have reported a novel strategy by generating anchoring sites within the framework of COFs that bind targeted NCs.235 To achieve this, the TAPT-COF was first synthesised solvothermally and then decorated with carbonyl groups using the well-known Doebner–Miller reaction (Fig. 24a). The carbonyl groups acted as anchoring sites, and upon treatment of the functionalised COF with HAuCl4·3H2O, gold NCs (Au NCs) were incorporated into the pores of the COF (Fig. 24a). Additionally, the carbonyl groups within the COF served as a bridge. The Au NCs incorporated into the COF enhanced charge transfer and catalytic activity, while the COF provided structural stability and a high surface area for efficient H2O2 production. The Au modified COF (Au@COF) has shown the photocatalytic H2O2 production rate of 18933.58 µmol g−1 h−1 at pH = 3 (Fig. 24b). Also, the decomposition rate of H2O2 over Au@COF was merely 5.5% at pH = 3, which was lower than that at pH = 7 (35.2%) (Fig. 24c). The PSM of porous materials often uses toxic reagents or metal catalysts under harsh conditions, risking framework instability, metal contamination, and environmental hazards. Recently, Xu et al. have reported an eco-friendly route for PSM of COFs through an enzymatic reaction.236 Through horseradish peroxidase (HRP)-catalysed click reactions, 2-hydroxyethylthio (–S–EtOH) and ethylthio (–S–Et) groups were covalently introduced into the COF pores under mild aqueous conditions (Fig. 25a). The modified COFs (ACOF–S–EtOH and ACOF–S–Et) retained their crystallinity relative to the pristine COF, as confirmed by PXRD (Fig. 25b) and HRTEM (Fig. 25c). After the introduction of the functional group, the H2O2 production was escalated up to 27.9 times, reaching a maximum production rate of 5440 µmol g−1 h−1 with COF–S–EtOH from H2O and O2 (Fig. 25d). Interestingly, the thermally modified sample showed a lower H2O2 production rate than the enzymatically modified route, confirming the advantage of the enzymatic PSM method. Furthermore, a continuous-flow photocatalytic reactor packed with ACOF–S–EtOH (Fig. 3d, S24) sustained a steady H2O2 concentration of 0.56 mM over 58 h, achieving a cumulative output of 2.5 L (Fig. 25e). The enhanced polarity after PSM has been verified through temperature-dependent photoluminescence (PL) studies. The altered PL emission of COFs was attributed to aggregation effects and enhanced charge polarisation induced by functionalization. Furthermore, the exciton binding energies (Eb) derived from Arrhenius fitting were observed to be 68.4 meV (ACOF), 58.6 meV (ACOF–S–Et), and 34.8 meV (ACOF–S–EtOH) (Fig. 25f). The significantly lower Eb of ACOF–S–EtOH indicates more efficient electron–hole separation, explaining its superior photocatalytic performance. Mechanistic investigation highlights the formation of H2O2 via both ORR and WOR pathways. In situ DRIFTS further verified the ˙OOH intermediate by the intensified peak at 1176 cm−1 (Fig. 25g). The growing band at 892 cm−1 indicated O2 adsorption on ACOF–S–EtOH, while increased signals at 3330 cm−1 (O–H stretching) and at 1402, 1442, and 1471 cm−1 (H–O–O bending) confirmed the formation and accumulation of H2O2. The major achievements of the AFPM-based composites for H2O2 production have been highlighted in Table 8.
 |
| | Fig. 24 (a) Synthetic route for the covalently treated Au@COF composite. (b) Photocatalytic H2O2 production of the Au@COF at different pHs. (c) The photocatalytic decomposition of H2O2 (200 mM) over the Au@COF at different pHs. (Reprinted with permission from ref. 235. Copyright 2023, Royal Society of Chemistry.) | |
 |
| | Fig. 25 (a) Schematic route for PSM of the ACOF through an enzymatic click reaction. (b) PXRD of pristine & modified ACOFs. (c) HRTEM image of ACOF–S–EtOH. (d) Comparative H2O2 production efficiency of pristine & modified ACOFs. (e) Long-term photocatalytic H2O2 production through continuous flow catalysis. (f) Integrated PL emission intensity as a function of temperature for ACOF–S–Et and ACOFS–EtOH. (g) In situ DRIFTS spectra of ACOF–S–EtOH during photocatalytic H2O2 production (Reprinted with permission from ref. 236. Copyright 2025, American Chemical Society.) | |
Table 8 Summary of representative AFPM-based composites for photocatalytic H2O2 production
| Sl. no. |
Catalyst |
Reaction conditions |
Dosage (mg) & volume (mL) |
H2O2 yield (µmol g−1 h−1) |
AQY |
SCC |
Ref. |
| 1 |
Au@COF |
H2O:BA (4:1) and O2 |
5 mg & 50 mL |
18933.58 |
— |
— |
235 |
| 2 |
MPa-COFs/CQDs-2 |
H2O and O2 |
20 mg & 100 mL |
540 |
13.27% @ 400 nm |
0.8% |
230 |
| 3 |
CdS/TpMA |
H2O and O2 |
20 mg & 50 mL |
511.54 µmol L−1 h−1 |
— |
— |
232 |
| 4 |
ZnIn2S4/TpPa-1 |
H2O, EtOH and O2 |
20 mg & 40 mL |
516 µmol L−1 |
— |
— |
233 |
| 5 |
WO3/Tp-TAPB |
H2O and O2 |
5 mg & 10 mL |
1488.4 |
1.22% @ 420 nm |
— |
234 |
| 6 |
ZnIn2S4/UiO-66-NH2 |
H2O and air |
20 mg & 40 mL |
799 |
— |
— |
237 |
| 7 |
Al-TCPP (10-X)-TBAPyX |
H2O and O2 |
5 mg & 5 mL |
127 |
— |
— |
238 |
| 8 |
Al-MIL-101-NH2 |
H2O and O2 |
5 mg & 5 mL |
295 |
— |
— |
239 |
| 9 |
Pd1/A-aUiO |
H2O and O2 |
Membrane based |
1.74 mmol gcat−1 h−1 |
— |
— |
240 |
| 10 |
COF@H3PO4 |
H2O and O2 |
10 mg & 20 mL |
5214 |
— |
0.69% |
241 |
| 11 |
Fe SAS-TpPP-COF |
H2O:BA (9:1) and O2 |
5 mg & 50 mL |
4130 |
6.4% @ 420 nm |
0.20% |
242 |
Similar to COFs, the ubiquitous opportunity of MOF materials for PSM has been widely explored, especially for electrocatalytic applications.243–246 Due to their distinctive advantages, MOF-based materials have become promising electrocatalysts for the 2e− ORR in H2O2 generation.247,248 Accordingly, MOF composites, pure MOFs, and bimetallic MOFs have been demonstrated to be promising materials for photocatalytic H2O2 generation. Yao and the team recently synthesised several ZnIn2S4/UiO-66-X (X = H, OH, (OH)2, and NH2) and discovered that amino or hydroxyl functionalisation may activate the effective photocatalytic production of H2O2 under visible light (Fig. 26a).237 The H2O2 yields for ZnIn2S4/UiO-66-NH2, ZnIn2S4/UiO-66-(OH)2, and ZnIn2S4/UiO-66-OH are 799, 733, and 165 µmol L−1, respectively, whereas the yield for ZnIn2S4/UiO-66 is just 84 µmol L−1 (without a sacrificial agent, in ambient air, and indirect ORR). The enhanced visible-light absorption and Z-scheme heterojunctions of ZnIn2S4/UiO-66-NH2 and ZnIn2S4/UiO-66-(OH)2 led to their significant H2O2 generation (Fig. 26b). The Yamashita research group synthesised an Al-MOF utilising mixed ligands of 1,3,6,8-tetrakis(p-benzoic acid) pyrene and tetrakis(4-carboxyphenyl) porphyrin for photocatalytic H2O2 generation under visible light (Fig. 26c).238 Al-MOFs have the capacity to inhibit the breakdown of H2O2 and have introduced a novel pathway for the generation of H2O2 utilising 1O2 as the intermediate.239 The production of H2O2 via both the ORR (1O2 + 2H+ + 2e− → H2O2) and WOR (1O2 + 2H2O → 2H2O2) utilizing 1O2 was deduced. Wang et al. deposited Pd single atoms over faulty UiO-66-NH2 and then fabricated gas-permeable Pd/UiO-66-NH2 membranes for photocatalytic H2O2 production from humid O2 at gas–solid interfaces.240 The positively charged Pd single atoms can enhance H2O2 generation and simultaneously impede H2O2 breakdown, as shown in the prior work.249 The defect sites facilitated light absorption and interaction with metal single atoms, whereas the UiO-66-NH2 molecular structure functioned as several linked non-reactors. A notable H2O2 production rate of 10.4 mmol g−1 h−1 was achieved using Pd/UiO-66-NH2 membranes in a gas–membrane–gas configuration, which is 4.9 times more than that produced with Pd/UiO-66-NH2 particles in a particle-in-solution setup (utilising visible light, isopropanol as a sacrificial agent, and pure O2 for the ORR) (Fig. 26f).
 |
| | Fig. 26 (a) Preparation diagram of ZnIn2S4/UiO-66-X hybrids, (b) the proposed mechanism for photocatalytic H2O2 evolution using Z-UN and Z-U(OH)2. (Reprinted with permission from ref. 237. Copyright 2023, Royal Society of Chemistry.) (c) schematic of Al-TCPP(10-X)-TBAPyX, (d) comparison of photocatalytic H2O2 production over Al-TCPP(10-X)-TBAPyX samples, and (e) schematic showing that humidified gases (e.g., CO2 and O2) can be fed through the gas-permeable MOF/PTFE membranes and photocatalytically reduced to value-added chemicals (e.g., HCOOH and H2O2) under visible light irradiation and ambient conditions and with controllable defect-engineering, specific metal SAs (e.g., Ir and Pd) can be precisely anchored on the edges of the Zr6O4(OH)4(–CO2)12−x octahedral to act as programable catalytic centers for catalyzing different reactions, such as photocatalytic CO2-to-HCOOH and O2-to-H2O2 conversion. The open and interconnected MOF pores serve as the nanoreactors facilitating the diffusion and conversion of gas reactants. (Reprinted with permission from ref. 238. Copyright 2023, Royal Society of Chemistry.) (f) Time course of H2O2 generation on pristine A-aUiO and PdNPs-, Pdx-, and Pd1-decorated A-aUiO catalysts. (Reprinted with permission from ref. 240. Copyright 2023, Springer Nature.) | |
Conclusion and future perspectives
In recent years, H2O2 has received gradually increasing attention, and it is recognised not only as an efficient oxidant in various fields but also as a prospective energy carrier. The photosynthesis of H2O2 over AFPM has attracted growing interest, and efforts are being made to regulate the efficiency of AFPM-based photocatalysts to achieve remarkable H2O2 production performance. In this review, we have elaborated on the recent strategies applied to increase the efficiency of various AFPM-based photocatalysts for H2O2 photosynthesis. More importantly, the modification strategies reported thus far for achieving improved photocatalytic H2O2 production performance over AFPM-based photocatalysts have been comprehensively summarised and discussed. Most of the reported articles have tried to understand the effect of linkage modification, heteroatom doping, topology and structural modification, functional group incorporation, post-synthetic modification, etc., towards improving the D–A property, enhanced charge separation, and effect on the band gap. The active site for H2O2 production has been meticulously studied in metal and metal-free AFPM using experimental and computational techniques. This has further enabled the better design of materials for practical implementation. Similarly, the range of applications has expanded, offering possibilities for photocatalytic H2O2 production that is not only efficient but also selective and recyclable for multiple cycles. The capability to adjust functionality in metal-free AFPMs has further provided an opportunity to tune band positions for efficient H2O2 production. The synthesis of composites using AFPMs has also showcased its photocatalytic efficiency by creating heterojunctions appropriate for photocatalytic H2O2 production. Though tremendous research is going on to improve the photocatalytic production of H2O2, there are still several challenges that need to be addressed to translate the potential of materials from laboratory to practical, real-world applications (Fig. 27). This involves tackling the following aspects:
 |
| | Fig. 27 Representative diagram highlighting the future perspectives and challenges in AFPM-based photocatalytic H2O2 production. | |
(i) AFPM diversification with hierarchical porosity: among the various types of metal-free AFPM materials, there are numerous reports on COFs, while other AFPM materials have received less attention. Although COFs offer an ordered structure and a larger surface area than other organic frameworks like POPs and CTFs, more detailed research on other AFPMs to enhance photocatalytic activity would be beneficial. The photocatalytic performance is strongly dependent on the crystallinity of COFs; however, crystallinity can deteriorate over time during long-term catalytic operation. Therefore, the development of porous materials that can sustain high photocatalytic performance independent of long-range crystalline order, while effectively leveraging porous architectures, is highly desirable. In this regard, the development of porous material-based aerogels for H2O2 photosynthesis represents a promising breakthrough, as such systems can exploit hierarchical macro–mesoporosity to enhance mass transport and catalytic efficiency.
(ii) ORR-WOR engineering: the rational design of porous materials for photocatalytic H2O2 production must be tailored to the targeted reaction pathway: the ORR or WOR, as each imposes distinct electronic and structural requirements. After photoinduced electron–hole separation in a photocatalyst, the ORR proceeds via the excited electrons, while the WOR is driven by the photogenerated holes. The electron-deficient moieties like triazine, benzothiadiazole, and cyano groups function as ORR-active centres for the efficient 2e− ORR, while electron-rich sulfur-containing units such as pyrene, thiophene, and triarylamines proved to be effective WOR-active sites for the 2e− WOR. COFs, with tunable π-conjugation and ordered porosity, are well suited for ORR-driven H2O2 generation through D–A engineering, band-edge alignment toward the O2/H2O2 potential, and pore environments that enhance O2 diffusion and H2O2 desorption while suppressing the 4e− pathway. POPs, owing to their high chemical and oxidative stability, are particularly promising for WOR-driven H2O2 production, where functionalisable backbones and hydrophilic pores facilitate water adsorption, intermediate stabilisation, and proton-coupled electron transfer. MOFs offer site-isolated metal nodes (Zn, Zr, or single-site Fe/Co under controlled coordination) and modular coordination environments, enabling selective ORR via weak O–O activation or WOR through stable water-oxidation centres, provided oxidative robustness is ensured. MOPs and HOFs with an appropriate D–A site have shown H2O2 production through the simultaneous ORR–WOR pathway.
(iii) NIR photocatalysis: the most practical and sustainable route for H2O2 synthesis relies on solar energy; however, nearly 53% of the solar spectrum lies in the near-infrared (NIR, λ > 760 nm) region. In contrast, photocatalytic H2O2 production over AFPM-based catalysts remains largely restricted to visible-light excitation (λ < 760 nm). To date, only a handful of studies have focused on the rational design of NIR-responsive catalysts, highlighting a significant research gap. Therefore, greater emphasis should be placed on the development of NIR-active porous photocatalysts to more effectively utilise the full solar spectrum. Furthermore, as the AQY and SCC are key metrics for evaluating photocatalytic performance, photoefficiency improvement should not be overlooked. The highest AQYs of 22.78%, 14.9%, and 16.8% have been reported for COFs, MOFs, and CTFs, respectively, at ∼420 nm. Similarly, SCC efficiencies of 1.41%, 0.91%, and 1.2% were reported for COFs, MOFs, and CTFs under artificial sunlight simulation. While SCC values above 1.0% have been reported under laboratory conditions, these are typically achieved through extensive parameter optimisation rather than intrinsic catalyst efficiency. Under natural sunlight, the SCC efficiencies of photocatalysts for H2O2 production remain well below the ∼0.10% efficiency of natural photosynthesis, posing a major barrier to industrial application.
(iv) Facile and scalable synthetic route: from the viewpoint of practical photocatalytic H2O2 production, the scalability and facile synthesis of AFPM-based materials are of paramount importance. Although these materials have demonstrated impressive activity and selectivity toward the 2e− ORR, most reported synthetic strategies remain complex, low-yielding, and difficult to translate to large-scale manufacturing. Future efforts should therefore focus on developing cost-effective, environmentally benign, and scalable fabrication routes, while maintaining precise control over active sites and electronic structures. Furthermore, achieving batch-to-batch uniformity and reproducibility remains a key challenge for the commercialisation of porous materials. Therefore, the development of well-controlled and scalable synthetic routes, such as melt polymerisation and continuous-flow COF synthesis, to enable reliable bulk production is necessary. Addressing these challenges will be essential for advancing AFPM-based photocatalysts from laboratory studies toward industrially viable H2O2 production technologies.
(v) Artificial intelligence (AI) and machine learning (ML) guided photocatalytic screening: AI and ML provide transformative high-throughput strategies for the discovery of efficient COF-based photocatalysts for H2O2 production. By learning structure–property–performance relationships from existing experimental and computational datasets, ML models should be employed to rapidly predict key descriptors such as band-edge alignment, charge-carrier separation efficiency, pore polarity, and adsorption energies of ORR intermediates (*OOH, H2O2, etc.). These data-driven predictions enable virtual screening of vast COF chemical spaces, significantly reducing reliance on trial-and-error synthesis. Coupled with automated synthesis and high-throughput photocatalytic evaluation, AI-guided frameworks can iteratively optimise COF architectures to selectively promote the 2e− ORR pathway while suppressing competing reactions. Such AI-assisted discovery platforms are expected to accelerate the development of high-performance, solar-driven COF photocatalysts with improved AQY and SCC values. Furthermore, the integration of AI/ML automation and robotics into COF synthesis can also offer a powerful approach for rapidly screening reaction parameters and discovering new chemistries, thereby accelerating the transition of porous material from laboratory-scale materials to industrially viable platforms.
(vi) Decentralised H2O2 production and industrial validation: although the anthraquinone process remains the benchmark for large-scale H2O2 production, porous material-based photocatalytic systems are not yet positioned to compete directly at the industrial scale. While the anthraquinone process remains the established benchmark for large-scale, centralised H2O2 production. Rather than serving as direct replacements, AFPM-based systems are better suited for decentralised and on-demand H2O2 generation at low-to-moderate concentrations (10–100 mM), where transportation, storage, and safety considerations become critical. Operating under mild conditions and enabling integration with flow reactors and in situ utilisation, these systems are well-positioned to complement existing technologies in applications such as water treatment, disinfection, and selective oxidation.
(vii) Extraction and utilisation of in situ produced H2O2: the extraction and utilisation of generated H2O2 remain critical challenges for its practical deployment. While ex situ extraction and purification are often necessary for off-site applications, they introduce additional energy and separation costs and can accelerate H2O2 decomposition during handling. In contrast, in situ utilisation strategies, where H2O2 is consumed immediately after generation, offer a more efficient and sustainable alternative by minimising accumulation, suppressing decomposition, and reducing safety concerns. Oxidation reactions, antimicrobial processes, pollutant degradation, and heavy metal capture and sequestration offer practical strategies to consume H2O2 immediately, suppress degradation pathways, and extend the catalyst lifetime. Future advances will depend on reactor designs and porous catalyst architectures that simultaneously facilitate controlled H2O2 release, selective mass transport, and seamless integration with end-use reactions, thereby bridging the gap between laboratory-scale synthesis and real-world applications.
(viii) Continuous flow catalysis: flow catalysis presents a compelling strategy for advancing porous materials toward stable and scalable H2O2 synthesis. Continuous-flow reactors enable precise control over mass transport, reactant residence time, and photon utilisation, which are critical for regulating the 2e− ORR or WOR pathways while minimising H2O2 over-reduction and catalytic decomposition. Importantly, flow operation alleviates local H2O2 accumulation and concentration gradients that often accelerate framework degradation in batch systems, thereby enhancing the structural stability and operational durability of porous catalysts. When coupled with robust frameworks and optimised pore environments, flow catalysis offers a practical platform for sustained H2O2 production, bridging the gap between laboratory-scale demonstrations and long-term, industrially relevant operation. By enabling decentralised, on-site generation, flow catalysis eliminates challenges associated with H2O2 storage, decomposition, and transport. For real-world implementation, photocatalytic systems should therefore be designed to deliver H2O2 on demand at concentrations in the range of 10–100 mM.
(ix) Suppression of H2O2 decomposition: direct exposure of the photocatalyst to the generated H2O2 frequently promotes its undesired decomposition, thereby restricting achievable yields. To address this challenge, several catalyst-product separation strategies have been explored. Among them, biphasic reaction systems (e.g., water–benzyl alcohol) are widely employed, wherein the hydrophobic character of certain MOFs, combined with density-driven phase separation, enables the preferential transfer of H2O2 into the aqueous phase. Although sacrificial agents effectively suppress charge recombination, their use complicates H2O2 separation and purification. A more sustainable strategy is to employ sacrificial agents that selectively consume photogenerated holes while simultaneously enabling the synthesis of value-added chemicals.
(x) Structure–activity relationships and mechanistic insights: clarifying reaction mechanisms and structure–activity relationships remains a central challenge for AFPM-based photocatalysts in H2O2 photosynthesis. Although AFPM offers rich structural diversity through versatile bonding motifs, abundant building blocks, and tunable functional groups, their structure–performance correlations in H2O2 generation are still poorly understood due to complex architectures and reaction environments. Substantial efforts are therefore required to elucidate the roles of electronic structure, pore hydrophilicity/hydrophobicity, and nano-confinement effects in governing photocatalytic activity and selectivity. Beyond rational material design, the integration of advanced in situ characterisation techniques with density functional theory (DFT) calculations is essential to unravel charge-transfer pathways and free-radical mechanisms involved in AFPM-driven H2O2 synthesis, which will be critical for accelerating progress in this field.
Author contributions
Sagarmani Rasaily: conceptualisation, literature survey, writing original draft, figure preparation, and funding acquisition. Gourab K. Dam: literature survey, writing draft and figure preparation. Sujit K. Ghosh: review & editing, supervision, and funding acquisition.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Acknowledgements
SMR acknowledges ANRF (formerly SERB) for funding through the National Postdoctoral Fellowship (NPDF) Scheme (project no. PDF/2023/001234). GKD thanks CSIR for the fellowship. SKG acknowledges DST-SERB (project no. CRG/2022/001090) and CSIR, India (project no. 30523670 (01/3124/23/EMR-II)), for funding.
References
- J. M. Campos-Martin, G. Blanco-Brieva and J. L. G. Fierro, Angew. Chem., Int. Ed., 2006, 45, 6962–6984 CrossRef CAS PubMed.
- H. J. H. Fenton, J. Chem. Soc. Trans., 1894, 65, 899–909 RSC.
- M. A. Voinov, J. O. S. Pagán, E. Morrison, T. I. Smirnova and A. I. Smirnov, J. Am. Chem. Soc., 2011, 133, 35–41 CrossRef CAS PubMed.
- B. W. Richardson, Lancet, 1891, 137, 707–709 CrossRef.
- T. H. Oliver and D. V. Murphy, Lancet, 1920, 195, 432–433 CrossRef.
- J. W. Finney, H. C. Urschel, G. A. Balla, G. J. Race, B. E. Jay, H. P. Pingree, H. L. Dorman and J. T. Mallams, Ann. N. Y. Acad. Sci., 1967, 141, 231–241 CrossRef CAS PubMed.
- M. Armogida, R. Nisticò and N. B. Mercuri, Br. J. Pharmacol., 2012, 166, 1211–1224 CrossRef CAS PubMed.
- R. Ciriminna, L. Albanese, F. Meneguzzo and M. Pagliaro, ChemSusChem, 2016, 9, 3374–3381 CrossRef CAS PubMed.
- R. L. Myers, The 100 Most Important Chemical Compounds: A Reference Guide, Greenwood Press, 2007 Search PubMed.
- A. B. Trench, C. M. Fernandes, J. P. C. Moura, L. E. B. Lucchetti, T. S. Lima, V. S. Antonin, J. M. de Almeida, P. Autreto, I. Robles, A. J. Motheo, M. R. V. Lanza and M. C. Santos, Chemosphere, 2024, 352, 141456 CrossRef CAS.
- Y. Tang, W. Wang, J. Ran, C. Peng, Z. Xu and W. Chu, Energy Environ. Sci., 2024, 17, 6482–6498 RSC.
- Y. Sun, L. Han and P. Strasser, Chem. Soc. Rev., 2020, 49, 6605 RSC.
- Y. Yamada, M. Yoneda and S. Fukuzumi, Energy Environ. Sci., 2015, 8, 1698–1701 RSC.
- Hydrogen Peroxide Market – By Function (Disinfectant, Bleaching, Oxidant), By Application (Pulp & Paper, Chemical Synthesis, Wastewater Treatment, Mining, Food & Beverages, Personal Care, Healthcare, Textiles), & Forecast, 2024–2032, 2024.
- India Hydrogen Peroxide Market Analysis: Industry Market Size, Plant Capacity, Production, Operating Efficiency, Demand & Supply, End-User Industries, Sales Channel, Regional Demand, Company Share, Foreign Trade, Manufacturing, 2024.
- X. Zhang, X. Zhao, P. Zhu, Z. Adler, Z. Y. Wu, Y. Liu and H. Wang, Nat. Commun., 2022, 13, 2880 CrossRef CAS.
- S. C. Perry, D. Pangotra, L. Vieira, L. I. Csepei, V. Sieber, L. Wang, C. Ponce de León and F. C. Walsh, Nat. Rev., 2019, 3, 442–458 CAS.
- X. Liang, Z. Mi, Y. Wang, L. Wang, X. Zhang and T. Liu, Chem. Eng. Technol., 2004, 27, 176–180 CrossRef CAS.
- C. Xia, Y. Xia, P. Zhu, L. Fan and H. Wang, Science, 2019, 366, 226–231 CrossRef CAS.
- R. Ernstorfer, M. Harb, C. T. Hebeisen, G. Sciaini, T. Dartigalongue and R. J. D. Miller, Science, 2009, 323, 1033–1037 CrossRef CAS PubMed.
- J. H. Lunsford, J. Catal., 2003, 216, 455–460 CrossRef CAS.
- Oxygen-enhanced combustion, ed. C. E. Baukal Jr, CRC Press, 2nd edn, 2013 Search PubMed.
- L. Wang, J. Zhang, Y. Zhang, H. Yu, Y. Qu and J. Yu, Small, 2022, 18, 2104561 CrossRef CAS.
- H. Tada, M. Teranishi and S. I. Naya, J. Phys. Chem. C, 2023, 127, 5199–5209 CrossRef CAS.
- H. Huang, M. Sun, M. Li, L. Tang and S. Zhang, Green Energy Resour., 2023, 1, 10003125 Search PubMed.
- W. Wang, Q. Song, Q. Luo, L. Li, X. Huo, S. Chen, J. Li, Y. Li, S. Shi, Y. Yuan, X. Du, K. Zhang and N. Wang, Nat. Commun., 2023, 14, 2493 CrossRef CAS PubMed.
- Z. Li, Z. Yu, C. Guan, K. Xu and Q. Xiang, J. Materiomics, 2024, 11, 100982 CrossRef.
- Z. Jiang, C. Li, F. Qi, Z. Wang, Y. Liu, F. Li, H. Wang, Z. Bian, M. Zhu, J. Kumirska and E. M. Siedlecka, Appl. Mater. Interfaces, 2025, 17, 42–66 CrossRef CAS PubMed.
- H. He, S. Liu, Y. Liu, L. Zhou, H. Wen, R. Shen, H. Zhang, X. Guo, J. Jiang and B. Li, Green Chem., 2023, 25, 9501–9542 RSC.
- Y. Yang, C. Wang, Y. Li, K. Liu, H. Ju, J. Wang and R. Tao, J. Mater. Sci. Technol., 2024, 200, 185–214 CrossRef CAS.
- Y. Shi, Z. Zhao, D. Yang, J. Tan, X. Xin, Y. Liu and Z. Jiang, Chem. Soc. Rev., 2023, 52, 6938–6956 RSC.
- Q. Wang and K. Domen, Chem. Rev., 2020, 120, 919–985 CrossRef CAS PubMed.
- X. Tao, Y. Zhao, S. Wang, C. Li and R. Li, Chem. Soc. Rev., 2022, 51, 3561 RSC.
- R. Marschall, Eur. J. Inorg. Chem., 2021, 2021, 2435–2441 CrossRef CAS.
- Y. N. Gong, X. Guan and H. L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- Y. Hao, Y. L. Lu, Z. Jiao and C. Y. Su, Angew. Chem., Int. Ed., 2024, 63, e202317808 CrossRef CAS.
- B. Gurung, S. Pradhan, D. Sharma, D. Bhujel, S. Basel, S. Chettri, S. Rasaily, A. Pariyar and S. Tamang, Catal. Sci. Technol., 2022, 12, 5891–5898 RSC.
- Ž. Kovačič, B. Likozar and M. Huš, ACS Catal., 2020, 10, 14984–15007 CrossRef.
- W. Gao, H. Li, J. Hu, Y. Yang, Y. Xiong, J. Ye, Z. Zou and Y. Zhou, Chem. Sci., 2024, 15, 14081–14103 RSC.
- S. Wu and X. Quan, ACS ES&T Eng., 2022, 2, 1068–1079 Search PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- G. Zhang, Z. Guan, J. Yang, Q. Li, Y. Zhou and Z. Zou, Sol. RRL, 2022, 6, 2200587 CrossRef CAS.
- V. Soni, M. Malhotra, P. Singh, A. A. P. Khan, W.
A. Bawazir, V. H. Nguyen, T. Ahamad, Q. Van Le, P. Raizada and K. A. Alzahrani, J. Environ. Chem. Eng., 2025, 13, 115151 CrossRef CAS.
- W. Ji, Z. Xu, S. Zhang, Y. Li, Z. Bao, Z. Zhao, L. Xie, X. Zhong, Z. Wei and J. Wang, Catal. Sci. Technol., 2022, 12, 2865–2871 RSC.
- M. Gryszel, R. Rybakiewicz and E. D. Głowacki, Adv. Sustainable Syst., 2019, 3, 1900027 CrossRef.
- Y. Zheng, J. Liu, J. Liang, M. Jaroniec and S. Z. Qiao, Energy Environ. Sci., 2012, 5, 6717 RSC.
- S. Cao, J. Low, J. Yu and M. Jaroniec, Adv. Mater., 2015, 27, 2150–2176 CrossRef CAS.
- J. Fu, J. Yu, C. Jiang and B. Cheng, Adv. Energy Mater., 2018, 8, 1701503 CrossRef.
- J. Zhu, P. Xiao, H. Li and S. A. C. Carabineiro, ACS Appl. Mater. Interfaces, 2014, 6, 16449–16465 CrossRef CAS PubMed.
- S. Bordiga, C. Lamberti, G. Ricchiardi, L. Regli, F. Bonino, A. Damin, K. P. Lillerud, M. Bjorgen and A. Zecchina, Chem. Commun., 2004, 2300–2301 RSC.
- M. Alvaro, E. Carbonell, B. Ferrer, F. X. Llabrés I Xamena and H. Garcia, Chem.—Eur. J., 2007, 13, 5106–5112 CrossRef CAS.
- S. Navalón, A. Dhakshinamoorthy, M. Álvaro, B. Ferrer and H. García, Chem. Rev., 2023, 123, 445–490 CrossRef PubMed.
- J. D. Xiao and H. L. Jiang, Acc. Chem. Res., 2019, 52, 356–366 CrossRef CAS.
- R. Chen, G. Liu, B. Xia, T. Liu, Y. Xia, S. Liu, A. Talebian-Kiakalaieh and J. Ran, Chem. Commun., 2024, 60, 10989 RSC.
- M. G. Schwab, M. Hamburger, X. Feng, J. Shu, H. W. Spiess, X. Wang, M. Antonietti and K. Müllen, Chem. Commun., 2010, 46, 8932–8934 RSC.
- S. Liu, M. Wang, Y. He, Q. Cheng, T. Qian and C. Yan, Coord. Chem. Rev., 2023, 475, 214882 CrossRef CAS.
- H. Ran, Q. Xu, Y. Yang, H. Li, J. Fan, G. Liu, L. Zhang, J. Zou, H. Jin and S. Wang, ACS Catal., 2024, 14, 11675–11704 CrossRef CAS.
- M. Debruyne, P. Van Der Voort, V. Van Speybroeck and C. V. Stevens, Chem.–Eur. J., 2024, 30, e202400311 CrossRef CAS PubMed.
- T. X. Wang, H. P. Liang, D. A. Anito, X. Ding and B. H. Han, J. Mater. Chem. A, 2020, 8, 7003 RSC.
- T. Gao, D. Zhao, S. Yuan, M. Zheng, X. Pu, L. Tang and Z. Lei, Carbon Energy, 2024, 6, e596 CrossRef CAS.
- D. Tsukamoto, A. Shiro, Y. Shiraishi, Y. Sugano, S. Ichikawa, S. Tanaka and T. Hirai, ACS Catal., 2012, 2, 599–603 CrossRef CAS.
- H. L. Tan, C. H. T. Chai, J. Z. X. Heng, Q. V. Thi, X. Wu, Y. H. Ng and E. Ye, Adv. Sci., 2025, 12, 2407801 CrossRef CAS.
- Y. Shiraishi, S. Kanazawa, Y. Sugano, D. Tsukamoto, H. Sakamoto, S. Ichikawa and T. Hirai, ACS Catal., 2014, 4, 774–780 CrossRef CAS.
- X. Xiao, H. Wang, X. Wang, C. Liu, Y. Han, S. Zhai and H. Du, Carbon Energy, 2025, 7, e666 CrossRef CAS.
- D. Tan, R. Zhuang, R. Chen, M. Ban, W. Feng, F. Xu, X. Chen and Q. Wang, Adv. Funct. Mater., 2024, 34, 2311655 CrossRef CAS.
- J. Qiu, D. Dai and J. Yao, Coord. Chem. Rev., 2024, 501, 215597 CrossRef CAS.
- W. Wang, X. Wang, M. Gao, Z. Li and W. Zhou, Coord. Chem. Rev., 2024, 506, 215694 CrossRef CAS.
- D. Tan and X. Fan, Polymers, 2024, 16, 659 CrossRef CAS PubMed.
- K. H. Xie, G. B. Wang, F. Zhao, M. C. Wang, H. Y. Zhang, H. R. Ma, Z. Z. Chen, L. Jiang, Y. Geng and Y. Bin Dong, Inorg. Chem. Front., 2024, 11, 1322 RSC.
- A. P. Cote, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1164–1166 CrossRef.
- N. L. Campbell, R. Clowes, L. K. Ritchie and A. I. Cooper, Chem. Mater., 2009, 21, 204–206 CrossRef CAS.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- Y. N. Gong, X. Guan and H. L. Jiang, Coord. Chem. Rev., 2023, 475, 214889 CrossRef CAS.
- L. Stegbauer, K. Schwinghammer and B. V. Lotsch, Chem. Sci., 2014, 5, 2789–2793 RSC.
- Y. Yusran, H. Li, X. Guan, Q. Fang and S. Qiu, J. Energy Chem., 2020, 2, 100035 Search PubMed.
- Z. Alsudairy, N. Brown, A. Campbell, A. Ambus, B. Brown, K. Smith-Petty and X. Li, Mater. Chem. Front., 2023, 7, 3298–3331 RSC.
- H. Li, A. Dilipkumar, S. Abubakar and D. Zhao, Chem. Soc. Rev., 2023, 52, 6294–6329 RSC.
- Z. Wang, S. Zhang, Y. Chen, Z. Zhang and S. Ma, Chem. Soc. Rev., 2020, 49, 708–735 RSC.
- X. Zhao, P. Pachfule and A. Thomas, Chem. Soc. Rev., 2021, 50, 6871–6913 RSC.
- D. G. Wang, T. Qiu, W. Guo, Z. Liang, H. Tabassum, D. Xia and R. Zou, Energy Environ. Sci., 2021, 14, 688–728 RSC.
- J. Guo, S. Kong, Y. Lian and M. Zhao, Chem. Commun., 2024, 60, 918–934 RSC.
- X. Liu, D. Huang, C. Lai, G. Zeng, L. Qin, H. Wang, H. Yi, B. Li, S. Liu, M. Zhang, R. Deng, Y. Fu, L. Li, W. Xue and S. Chen, Chem. Soc. Rev., 2019, 48, 5266–5302 RSC.
- R. Xue, Y. S. Liu, S. L. Huang and G. Y. Yang, ACS Sens., 2023, 8, 2124–2148 CrossRef CAS PubMed.
- J. Wang and S. Zhuang, Coord. Chem. Rev., 2019, 400, 213046 CrossRef CAS.
- C. I. Yeo, Y. S. Tan, H. T. A. Awan, A. Hanan, W. P. Wong, R. Walvekar, B. H. Goh and M. Khalid, Coord. Chem. Rev., 2024, 521, 216167 CrossRef CAS.
- S. Wan, J. Guo, J. Kim, H. Ihee and D. Jiang, Angew. Chem., Int. Ed., 2008, 47, 8826–8830 CrossRef CAS PubMed.
- I. H. Chowdhury, S. Gupta and V. G. Rao, ChemCatChem, 2023, 15, e202300243 CrossRef CAS.
- H. Wang, H. Wang, Z. Wang, L. Tang, G. Zeng, P. Xu, M. Chen, T. Xiong, C. Zhou, X. Li, D. Huang, Y. Zhu, Z. Wang and J. Tang, Chem. Soc. Rev., 2020, 49, 4135–4165 RSC.
- C. Krishnaraj, H. Sekhar Jena, L. Bourda, A. Laemont, P. Pachfule, J. Roeser, C. V. Chandran, S. Borgmans, S. M. J. Rogge, K. Leus, C. V. Stevens, J. A. Martens, V. Van Speybroeck, E. Breynaert, A. Thomas and P. Van Der Voort, J. Am. Chem. Soc., 2020, 142, 20107–20116 CrossRef CAS PubMed.
- G. Zhang, M. Zhao, L. Su, H. Yu, C. Wang, D. Sun and Y. Ding, ACS Appl. Mater. Interfaces, 2023, 15, 20310–20316 CrossRef CAS PubMed.
- H. Liu, S. Zhu, Y. Zhi, H. Yue and X. Liu, Chem. Sci., 2025, 16, 12768 RSC.
- J. Sun, H. Sekhar Jena, C. Krishnaraj, K. Singh Rawat, S. Abednatanzi, J. Chakraborty, A. Laemont, W. Liu, H. Chen, Y. Y. Liu, K. Leus, H. Vrielinck, V. Van Speybroeck and P. Van Der Voort, Angew. Chem., Int. Ed., 2023, 62, e202216719 CrossRef CAS PubMed.
- R. Liu, Y. Chen, H. Yu, M. Položij, Y. Guo, T. C. Sum, T. Heine and D. Jiang, Nat. Catal., 2024, 7, 195–206 CrossRef CAS.
- Y. Chen, R. Liu, Y. Guo, G. Wu, T. C. Sum, S. W. Yang and D. Jiang, Nat. Synth., 2024, 3, 998–1010 CrossRef CAS.
- D. Li, C. Li, L. Zhang, H. Li, L. Zhu, D. Yang, Q. Fang, S. Qiu and X. Yao, J. Am. Chem. Soc., 2020, 142, 8104–8108 CrossRef CAS PubMed.
- C. Qin, X. Wu, L. Tang, X. Chen, M. Li, Y. Mou, B. Su, S. Wang, C. Feng, J. Liu, X. Yuan, Y. Zhao and H. Wang, Nat. Commun., 2023, 14, 5238 CrossRef CAS PubMed.
- X. Wang, H. Li, S. Zhou, J. Ning, H. Wei, X. Li, S. Wang, L. Hao and D. Cao, Adv. Funct. Mater., 2025, 35, 2424035 CrossRef CAS.
- J. Y. Yue, L. P. Song, Y. F. Fan, Z. X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2023, 62, e202309624 CrossRef CAS PubMed.
- C. Shu, X. Yang, L. Liu, X. Hu, R. Sun, X. Yang, A. I. Cooper, B. Tan and X. Wang, Angew. Chem., Int. Ed., 2024, 63, e202403926 CrossRef CAS PubMed.
- J. Yang, Z. Zhao, X. Liu, H. Yang, J. Yang, M. Zhou, S. Li, X. Liu, X. Xu and C. Cheng, Nat. Commun., 2025, 16, 7654 CrossRef CAS PubMed.
- Z. Xue, B. Zhang, Q. Guo, Y. Wang, Q. Li, K. Yang and S. Qiao, Adv. Mater., 2025, 37, e10201 CrossRef CAS.
- M. Deng, L. Wang, Z. Wen, J. Chakraborty, J. Sun, G. Wang and P. Van Der Voort, Green Chem., 2024, 26, 3239–3248 RSC.
- H. Ma, Y. Zhang, Y. Wu, Q. Gu, Z. Li and Q. Zhang, Chem. Sci., 2025, 16, 16668–16677 RSC.
- Z. Zhang, Y. Hou, S. Zhu, L. Yang, Y. Wang, H. Yue, H. Xia, G. Wu, S. wang Yang and X. Liu, Angew. Chem., Int. Ed., 2025, 64, e202505286 CrossRef CAS PubMed.
- J. Chen, S. Yan, F. Wang, F. Lin, J. Lin, R. A. Borse and Y. Wang, Angew. Chem., Int. Ed., 2025, 64, e202500924 CrossRef CAS PubMed.
- H. Wang, C. Yang, F. Chen, G. Zheng and Q. Han, Angew. Chem., Int. Ed., 2022, 61, e20220232 Search PubMed.
- Y. Yang, J. Kang, Y. Li, J. Liang, J. Liang, L. Jiang, D. Chen, J. He, Y. Chen and J. Wang, New J. Chem., 2022, 46, 21605–21614 RSC.
- J. N. Chang, Q. Li, J. W. Shi, M. Zhang, L. Zhang, S. Li, Y. Chen, S. L. Li and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202218868 CrossRef CAS PubMed.
- J.-C. Liu, C. Tuo, W.-Y. Xiao, M.-Y. Qi, Y. Yusran, Z.-T. Wang, H. Li, C.-S. Guo, J.-L. Song, S.-L. Qiu, Y.-J. Xu and Q. Fang, Angew. Chem., Int. Ed., 2025, 64, e202416240 CrossRef CAS PubMed.
- R. Wang, Z. Wang, L. Li, L. Zhang, Ji. Zhang, H. Jin, Q. Xu, Y. Wei, Y. Yang and S. Wang, J. Catal., 2025, 450, 116289 CrossRef CAS.
- Y. Xie, F. Mao, Q. Rong, X. Liu, M. Hao, Z. Chen, H. Yang, G. I. N. Waterhouse, S. Ma and X. Wang, Adv. Funct. Mater., 2024, 34, 2411077 CrossRef CAS.
- H. Xu, Y. Wang, Y. Xu, Q. Wang, M. Zhuang, Q. Liao and K. Xi, Angew. Chem., Int. Ed., 2024, 63, e202408802 CrossRef CAS PubMed.
- X. Di, X. Lv, H. Wang, F. Chen, S. Wang, G. Zheng, B. Wang and Q. Han, Chem. Eng. J., 2023, 455, 140124 CrossRef CAS.
- V. S. Vyas, F. Haase, L. Stegbauer, G. Savasci, F. Podjaski, C. Ochsenfeld and B. V. Lotsch, Nat. Commun., 2015, 6, 8508 CrossRef CAS PubMed.
- D. Chen, W. Chen, Y. Wu, L. Wang, X. Wu, H. Xu and L. Chen, Angew. Chem., Int. Ed., 2023, 62, e202217479 CrossRef CAS PubMed.
- Z. Zhang, Q. Zhang, Y. Hou, J. Li, S. Zhu, H. Xia, H. Yue and X. Liu, Angew. Chem., Int. Ed., 2024, 63, e202411546 CrossRef CAS PubMed.
- X. Yang, Z. X. Pan, J. Y. Yue, X. Li, G. Liu, Q. Xu and G. Zeng, Small, 2024, 20, 2405907 CrossRef CAS PubMed.
- W. Wu, Z. Li, S. Liu, D. Zhang, B. Cai, Y. Liang, M. Wu, Y. Liao and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202404563 CrossRef CAS PubMed.
- Z. Li, B. Cai, Y. Zou, D. Zhang, Y. Liang, Y. Zhou, Y. Ma, X. Wang, B. Shi, W. K. Chen, Y. Liu and X. Zhao, Adv. Energy Mater., 2025, 15, 2500341 CrossRef CAS.
- Y. Hou, P. Zhou, F. Liu, Y. Lu, H. Tan, Z. Li, M. Tong and J. Ni, Angew. Chem., Int. Ed., 2024, 63, e202318562 CrossRef CAS PubMed.
- D. Cao, J. Du, J. Li, Q. Sun, J. Guan and J. Liu, ACS Catal., 2025, 15, 3584–3594 CrossRef CAS.
- Y. Ju, H. Lin, G. Tan, P. Su, Z. Wang, C. Hu, R. Hou, T. Hao, F. Chen and Y. Tang, Nat. Commun., 2025, 16, 5658 CrossRef CAS PubMed.
- H. Yu, F. Zhang, Q. Chen, P. K. Zhou, W. Xing, S. Wang, G. Zhang, Y. Jiang and X. Chen, Angew. Chem., Int. Ed., 2024, 63, e202402297 CrossRef CAS PubMed.
- L. Guo, L. Gong, Y. Yang, Z. Huang, X. Liu and F. Luo, Angew. Chem., Int. Ed., 2025, 64, e202414658 CrossRef CAS PubMed.
- Y. Yao, C. Zhu, R. Liu, Q. Fang, S. Song, B. Chen and Y. Shen, Small, 2024, 20, 2404885 CrossRef CAS PubMed.
- S. Kandambeth, A. Mallick, B. Lukose, M. V. Mane, T. Heine and R. Banerjee, J. Am. Chem. Soc., 2012, 134, 19524–19527 CrossRef CAS PubMed.
- H. Wang, C. Qian, J. Liu, Y. Zeng, D. Wang, W. Zhou, L. Gu, H. Wu, G. Liu and Y. Zhao, J. Am. Chem. Soc., 2020, 142, 4862–4871 CrossRef CAS PubMed.
- Y. Kong, X. He, H. Wu, Y. Yang, L. Cao, R. Li, B. Shi, G. He, Y. Liu, Q. Peng, C. Fan, Z. Zhang and Z. Jiang, Angew. Chem., Int. Ed., 2021, 60, 17638–17646 CrossRef CAS PubMed.
- H. Hu, Y. Tao, D. Wang, C. Li, Q. Jiang, Y. Shi, J. Wang, J. Qin, S. Zhou and Y. Kong, J. Colloid Interface Sci., 2023, 629, 750–762 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, J. Miao, X. Wen, C. Chen, B. Zhou and M. Long, Chem. Eng. J., 2023, 466, 143085 CrossRef CAS.
- X. Zhang, S. Cheng, C. Chen, X. Wen, J. Miao, B. Zhou, M. Long and L. Zhang, Nat. Commun., 2024, 15, 2649 CrossRef CAS PubMed.
- H. Li, Y. Li, X. Lv, C. Liu, N. Zhang, J. Zang, P. Yue, Y. Gao, C. Liu and Y. Li, Adv. Mater., 2025, 37, 2415126 CrossRef CAS PubMed.
- K.-H. Xie, G.-B. Wang, F. Huang, F. Zhao, J.-L. Kan, Z.-Z. Chen, L. Cai, S.-L. Han, Y. Geng and Y.-B. Dong, Nat. Commun., 2025, 16, 3493 CrossRef CAS PubMed.
- T. Yang, D. Zhang, A. Kong, Y. Zou, L. Yuan, C. Liu, S. Luo, G. Wei and C. Yu, Angew. Chem., Int. Ed., 2024, 63, e202404077 CrossRef CAS PubMed.
- Y. Mou, X. Wu, C. Qin, J. Chen, Y. Zhao, L. Jiang, C. Zhang, X. Yuan, E. Huixiang Ang and H. Wang, Angew. Chem., Int. Ed., 2023, 62, e20230948 Search PubMed.
- L. Fang, H. Xu, S. Qiu, T. Ye, T. Wang, J. Shang, C. Gu, S. Kitagawa and L. Li, Angew. Chem., Int. Ed., 2025, 64, e202423220 CrossRef CAS PubMed.
- H.-H. Sun, Z.-B. Zhou, Y. Fu, Q.-Y. Qi, Z.-X. Wang, S. Xu and X. Zhao, Angew. Chem., Int. Ed., 2024, 63, e202409250 CrossRef CAS PubMed.
- A. Chakraborty, A. Alam, U. Pal, A. Sinha, S. Das, T. Saha-Dasgupta and P. Pachfule, Nat. Commun., 2025, 16, 503 CrossRef CAS PubMed.
- T. Xu, Z. Wang, W. Zhang, S. An, L. Wei, S. Guo, Y. Huang, S. Jiang, M. Zhu, Y. B. Zhang and W. H. Zhu, J. Am. Chem. Soc., 2024, 146, 20107–20115 CrossRef CAS PubMed.
- J. Y. Yue, Y. F. Fan, Z. X. Pan, P. Yang, Y. Ma, Q. Xu and B. Tang, ACS Catal., 2024, 14, 4728–4737 CrossRef CAS.
- W. Wang, R. Zhang, H. Chu, Z. Zhan, Q. Huang, Z. Li, X. Wang, F. Bai and W. Zhou, Small, 2025, 21, 2406527 CrossRef CAS PubMed.
- P. Li, H. Zhao, R. Ji, W. Chi, X. Sun, Y. Dong and Y. Zhu, Catal. Sci. Technol., 2024, 14, 2470–2478 RSC.
- K. Prakash, R. Deka and S. M. Mobin, Inorg. Chem. Front., 2024, 11, 6711–6752 RSC.
- C. Pan, Y. Han and J. Lu, Appl. Sci., 2020, 10, 6374 CrossRef CAS.
- R. Freund, S. Canossa, S. M. Cohen, W. Yan, H. Deng, V. Guillerm, M. Eddaoudi, D. G. Madden, D. Fairen-Jimenez, H. Lyu, L. K. Macreadie, Z. Ji, Y. Zhang, B. Wang, F. Haase, C. Wöll, O. Zaremba, J. Andreo, S. Wuttke and C. S. Diercks, Angew. Chem., Int. Ed., 2021, 60, 23946–23974 CrossRef CAS PubMed.
- J. Y. Yue, J. X. Luo, Z. X. Pan, R. Z. Zhang, P. Yang, Q. Xu and B. Tang, Angew. Chem., Int. Ed., 2024, 63, e202405763 CrossRef CAS PubMed.
- J. Cheng, Y. Wu, W. Zhang, L. Wang, X. Wu and H. Xu, Adv. Mater., 2025, 37, 2410247 CrossRef CAS PubMed.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortés, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268–272 CrossRef CAS PubMed.
- X. Guan, F. Chen, Q. Fang and S. Qiu, Chem. Soc. Rev., 2020, 49, 1357–1384 RSC.
- K. Yu, P. He, D. Zhang, H. Wu, K. Shu, H. Long, Y. Sun, X. Pei, B. Jiang, K. Cao, Y. Li and L. Ma, Angew. Chem., Int. Ed., 2025, 64, e202511501 CrossRef CAS PubMed.
- R.-M. Zhu, Y. Liu, W.-K. Han, J.-D. Feng, J. Zhang, H. Pang, J. Zhang and Z.-G. Gu, Angew. Chem., Int. Ed., 2025, 64, e202412890 CrossRef CAS PubMed.
- J. P. Liao, M. Zhang, P. Huang, L. Z. Dong, T. T. Ma, G. Z. Huang, Y. F. Liu, M. Lu, S. L. Li and Y. Q. Lan, ACS Catal., 2024, 14, 3778–3787 CrossRef CAS.
- W. Lan, B. Wei, Y. Jin, S. Xu, H. Zhou, Y. Wu, Q. Liu, P. Chen, J. Wang, X. Zhao, H. Meng, L. Liu, D. Wang, H. Huang, Y. Wei, Q. Zhu and Y. Yu, Small, 2025, 21, 2501327 CrossRef CAS PubMed.
- Z. Gong, Y. Gao, J. Li, Z. Cai, N. Liu and J. Jiang, Angew. Chem., Int. Ed., 2025, 64, e202423205 CrossRef CAS PubMed.
- X. Wang, Y. Jin, N. Li, H. Zhang, X. Liu, X. Yang, H. Pan, T. Wang, K. Wang, D. Qi and J. Jiang, Angew. Chem., Int. Ed., 2024, 63, e202401014 CrossRef CAS PubMed.
- Z. S. Zhu, S. Zhong, C. Cheng, H. Zhou, H. Sun, X. Duan and S. Wang, Chem. Rev., 2024, 124, 11348–11434 CrossRef CAS PubMed.
- Y. Liu, L. Li, Z. Sang, H. Tan, N. Ye, C. Sun, Z. Sun, M. Luo and S. Guo, Nat. Synth., 2025, 4, 134–141 CrossRef CAS.
- Z. Ding, J. Yang, Z. Wu, M. Adeli, X. Luo, X. Wang, X. Xie, X. Xu, C. Cheng and C. Zhao, Chem. Mater., 2025, 37, 1972–1982 CrossRef CAS.
- Y. Zhang, Y. Wu, H. Ma, Y. Gao, X. Fan, Y. Zhao, F. Kang, Z. Li, Y. Liu and Q. Zhang, Small, 2025, 21, 2500674 CrossRef CAS PubMed.
- Q. Zhou, Y. Guo and Y. Zhu, Nat. Catal., 2023, 6, 574–584 CrossRef CAS.
- M. Guo, C. He, Z. Wu, Y. Tian, J. Yang, Y. Wang, H. Wu, J. Yang, M. Xu, W. Xue, C. Cheng, S. Li and C. Zhao, Adv. Sci., 2025, 12, e05794 CrossRef CAS PubMed.
- X. Ding, T. Wang, B. Yu, Q. Zhi, H. Wang, H. Liu, P. A. Stuzhin and J. Jiang, Adv. Funct. Mater., 2025, 2422291 CrossRef CAS.
- X. F. Lu, Y. Fang, D. Luan and X. W. D. Lou, Nano Lett., 2021, 21, 1555–1565 CrossRef CAS PubMed.
- W. Xia, A. Mahmood, R. Zou and Q. Xu, Energy Environ. Sci., 2015, 8, 1837–1866 RSC.
- J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- W. Cheng, X. F. Lu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2020, 59, 18234–18239 CrossRef CAS PubMed.
- S. Zhao, Y. Wang, J. Dong, C. T. He, H. Yin, P. An, K. Zhao, X. Zhang, C. Gao, L. Zhang, J. Lv, J. Wang, J. Zhang, A. M. Khattak, N. A. Khan, Z. Wei, J. Zhang, S. Liu, H. Zhao and Z. Tang, Nat. Energy, 2016, 1, 16184 CrossRef CAS.
- J. Q. Shen, P. Q. Liao, D. D. Zhou, C. T. He, J. X. Wu, W. X. Zhang, J. P. Zhang and X. M. Chen, J. Am. Chem. Soc., 2017, 139, 1778–1781 CrossRef CAS PubMed.
- N. Diyali, S. Rasaily and B. Biswas, Coord. Chem. Rev., 2022, 469, 214667 CrossRef CAS.
- S. Rasaily, D. Sharma, S. Pradhan, N. Diyali, S. Chettri, B. Gurung, S. Tamang and A. Pariyar, Inorg. Chem., 2022, 61, 13685–13699 CrossRef CAS PubMed.
- D. Sharma, S. Rasaily, S. Chettri, D. Sureka, S. Tamang and A. Pariyar, Inorg. Chem., 2023, 62, 4540–4549 CrossRef CAS PubMed.
-
(a) L. J. Murray, M. Dincă and J. R. Long, Chem. Soc. Rev., 2009, 38, 1294–1314 RSC;
(b) H. Li, L. Li, R. B. Lin, W. Zhou, Z. Zhang, S. Xiang and B. Chen, J. Energy Chem., 2019, 1, 100006 Search PubMed.
- R. B. Lin, S. Xiang, W. Zhou and B. Chen, Chem, 2020, 6, 337–363 CAS.
- S. Dutta, S. Mukherjee, O. T. Qazvini, A. K. Gupta, S. Sharma, D. Mahato, R. Babarao and S. K. Ghosh, Angew. Chem., Int. Ed., 2022, 61, e202114132 CrossRef CAS PubMed.
- S. Mollick, S. Saurabh, Y. D. More, S. Fajal, M. M. Shirolkar, W. Mandal and S. K. Ghosh, Energy Environ. Sci., 2022, 15, 3462–3469 RSC.
- S. Fajal, D. Ghosh, W. Mandal and S. K. Ghosh, Chem. Commun., 2024, 60, 1884–1887 RSC.
- H. Sohrabi, S. Ghasemzadeh, Z. Ghoreishi, M. R. Majidi, Y. Yoon, N. Dizge and A. Khataee, Mater. Chem. Phys., 2023, 299, 127512 CrossRef CAS.
- P. Samanta, S. Let, W. Mandal, S. Dutta and S. K. Ghosh, Inorg. Chem. Front., 2020, 7, 1801–1821 RSC.
- A. Karmakar, N. Kumar, P. Samanta, A. V. Desai and S. K. Ghosh, Chem.–Eur. J., 2016, 22, 864–868 CrossRef CAS PubMed.
- H. S. Wang, Y. H. Wang and Y. Ding, Nanoscale Adv., 2020, 2, 3788–3797 RSC.
- J. Y. Choi, B. Check, X. Fang, S. Blum, H. T. B. Pham, K. Tayman and J. Park, J. Am. Chem. Soc., 2024, 146, 11319–11327 CAS.
- X. Wang, J. Li, X. Wei, J. Song, J. Xie, Z. Li, M. Yuan, L. Jiang, Y. Wang, C. Liang and W. Liu, ACS Omega, 2024, 9, 33671–33678 CrossRef CAS PubMed.
- Y. Zhang, Z. Luo, T. Zhou, H. Huang and H. Tang, Chem. Commun., 2024, 60, 1920–1923 RSC.
- Y. Zheng, H. Zhou, B. Zhou, J. Mao and Y. Zhao, Catal. Sci. Technol., 2022, 12, 969–975 RSC.
- X. Y. Ji, Y. Y. Wang, Y. Li, K. Sun, M. Yu and J. Tao, Nano Res., 2022, 15, 6045–6053 CrossRef CAS.
- Y. Zhang, C. Pan, G. Bian, J. Xu, Y. Dong, Y. Zhang, Y. Lou, W. Liu and Y. Zhu, Nat. Energy, 2023, 8, 361–371 CrossRef CAS.
- M. Shao, R. Bao, H. Guo, C. Yao, S. Wang, Y. Song and X. Guo, Inorg. Chem., 2025, 64, 18567–18576 CrossRef CAS PubMed.
- A. Pal, S. Suresh, A. Khan, L. Huai Kuo, L. Tang Chi, A. Ganguly, C.-Y. Kao, M. Kumar Sharma, T.-S. Andrew Wang, D.-Y. Kang and Z.-H. Lin, Sci. Adv., 2025, 11, 4711 CrossRef PubMed.
- Y.-X. Li, Y. Hu, H.-S. Bae, J. Du, S. Zhao, D. Pan and W. Choi, ACS Nano, 2024, 42, 29233–29247 CrossRef PubMed.
- S. Fajal, S. Dutta and S. K. Ghosh, Mater. Horiz., 2023, 10, 4083–4138 RSC.
- A. Trewin and A. I. Cooper, Angew. Chem., Int. Ed., 2023, 2010(49), 1533–1535 Search PubMed.
- B. Mishra, A. Alam, B. Kumbhakar, D. Díaz Díaz and P. Pachfule, Cryst. Growth Des., 2023, 23, 4701–4719 CrossRef CAS.
- C. Liu, M. Chen, H. Li, Q. Shi, Y. Feng and B. Zhang, Cryst. Growth Des., 2022, 22, 4745–4756 CrossRef CAS.
- L. Liao, M. Li, Y. Yin, J. Chen, Q. Zhong, R. Du, S. Liu, Y. He, W. Fu and F. Zeng, ACS Omega, 2023, 8(5), 4527–4542 CrossRef CAS PubMed.
- M. Liu, L. Guo, S. Jin and B. Tan, J. Mater. Chem. A, 2019, 7, 5153–5172 RSC.
- S. R. Zhu, Q. Qi, Y. Fang, W. N. Zhao, M. K. Wu and L. Han, Cryst. Growth Des., 2018, 18, 883–891 CrossRef CAS.
- F. Niu, L. Tao, Y. Deng, H. Gao, J. Liu and W. Song, New J. Chem., 2014, 38, 5695–5699 RSC.
- C. Yang, W. Huang, L. C. da Silva, K. A. I. Zhang and X. Wang, Chem.–Eur. J., 2018, 24, 17454–17458 CrossRef CAS PubMed.
- S. Kumar, B. Bayarkhuu, H. Ahn, H. Cho and J. Byun, Nano Trends, 2023, 4, 100023 CrossRef.
- H. Zhang, W. Wei, K. Chi, Y. Zheng, X. Y. Kong, L. Ye, Y. Zhao and K. A. I. Zhang, ACS Catal., 2024, 17654–17663 CrossRef CAS.
- L. Zhang, C. Wang, Q. Jiang, P. Lyu and Y. Xu, J. Am. Chem. Soc., 2024, 146, 29943–29954 CrossRef CAS PubMed.
- L. Chen, L. Wang, Y. Wan, Y. Zhang, Z. Qi, X. Wu and H. Xu, Adv. Mater., 2020, 32, 1904433 CrossRef CAS PubMed.
- Y. Yang, Q. Guo, Q. Li, L. Guo, H. Chu, L. Liao, X. Wang, Z. Li and W. Zhou, Adv. Funct. Mater., 2024, 34, 2400612 CrossRef CAS.
- X. Nie, Y. Zhao, W. Gao, W. Liu, X. Cheng, Y. Gao, N. Shang, S. Gao and C. Wang, Chem.–Eur. J., 2023, 29, e202203607 CrossRef CAS PubMed.
- Y. Wang, W. Chi, R. Zhang, Y. Guo, X. Sun, H. Zhao, J. Zhang, Y. Dong and Y. Zhu, New J. Chem., 2024, 48, 9043–9051 RSC.
- X. Xu, D. Jiang, K. Xie, Y. Sui, W. Chen, Y. Li, H. Zhong and H.-R. Wen, ACS Appl. Polym. Mater., 2025, 7, 6920–693 CrossRef CAS.
- X. Xu, L. Chai, Y. Li, W. Chen, X. Li, W. Huang, G. Zhou, H. Zhong and Y. Sui, Polymer, 2025, 340, 129236 CrossRef CAS.
- G. Lv, X. Yu, J. Wang, J. Qiu, D. Yang and Y. Zhu, Adv. Funct. Mater., 2026, 36, e17393 CrossRef CAS.
- R. Sun, X. Yang, X. Hu, Y. Guo, Y. Zhang, C. Shu, X. Yang, H. Gao, X. Wang, I. Hussain and B. Tan, Angew. Chem., Int. Ed., 2025, 64, e202416350 CrossRef CAS PubMed.
- Y. Guo, X. Yang, R. Sun, X. Hu, C. Shu, X. Yang, H. Gao, X. Wang and B. Tan, Small, 2024, 20, 2403743 CrossRef CAS PubMed.
- X. Xu, Y. Sui, W. Chen, S. Xiong, Y. Li, X. Li, W. Huang, H. Zhao, G. Zhou, H. Chen and H. Zhong, ACS Sustain. Chem. Eng., 2024, 12, 11409–11418 CrossRef CAS.
- S. Lee, H. Jeong, D. Nam, M. S. Lah and W. Choe, Chem. Soc. Rev., 2021, 50, 528–555 RSC.
- A. J. Gosselin, C. A. Rowland and E. D. Bloch, Chem. Rev., 2020, 120, 8987–9014 CrossRef CAS PubMed.
- E. S. M. El-Sayed, Y. Di Yuan, D. Zhao and D. Yuan, Acc. Chem. Res., 2022, 55, 1546–1560 CrossRef CAS PubMed.
- Y. P. Wang, Y. Zhang, X. H. Duan, J. J. Mao, M. Pan, J. Shen and C. Y. Su, Coord. Chem. Rev., 2024, 501, 215570 CrossRef CAS.
- D. Majumder, S. Fajal, M. M. Shirolkar, A. Torris, Y. Banyla, K. Biswas, S. Rasaily and S. K. Ghosh, Angew. Chem., Int. Ed., 2025, 64, e202419830 CrossRef CAS PubMed.
- Y. Liu, L. Huang, L. Qin and T. Zhou, ChemCatChem, 2025, 17, e202401487 CrossRef CAS.
- Y. L. Lu, Y. P. Wang, K. Wu, M. Pan and C. Y. Su, Acc. Chem. Res., 2024, 57, 3277–3291 CrossRef CAS PubMed.
- J. N. Lu, J. J. Liu, L. Z. Dong, J. M. Lin, F. Yu, J. Liu and Y. Q. Lan, Angew. Chem., Int. Ed., 2023, 62, e202308505 CrossRef CAS PubMed.
- L. Xiao, H. Zhao, F. Fu, Y. Han, J. Wang, Q. Dong, X. Hu, N. Wang, Y. Wei, P. Wang, D. Liu and Y. Li, Angew. Chem., Int. Ed., 2025, 64, e202516678 CrossRef CAS PubMed.
- R. B. Lin, Y. He, P. Li, H. Wang, W. Zhou and B. Chen, Chem. Soc. Rev., 2019, 48, 1362–1389 RSC.
- B. Wang, R. B. Lin, Z. Zhang, S. Xiang and B. Chen, J. Am. Chem. Soc., 2020, 142, 14399–14416 CrossRef CAS PubMed.
- W. Yang, A. Greenaway, X. Lin, R. Matsuda, A. J. Blake, C. Wilson, W. Lewis, P. Hubberstey, S. Kitagawa, N. R. Champness and M. Schröder, J. Am. Chem. Soc., 2010, 132, 14457–14469 CrossRef CAS PubMed.
- Y. He, S. Xiang and B. Chen, J. Am. Chem. Soc., 2011, 133, 14570–14573 CrossRef CAS PubMed.
- Y. Li, S. Tang, A. Yusov, J. Rose, A. N. Borrfors, C. T. Hu and M. D. Ward, Nat. Commun., 2019, 10, 4477 CrossRef PubMed.
- C. J. Lu, W. J. Shi, Y. N. Gong, J. H. Zhang, Y. C. Wang, J. H. Mei, Z. M. Ge, T. B. Lu and D. C. Zhong, Angew. Chem., Int. Ed., 2024, 63, e202405451 CrossRef CAS PubMed.
- Q. Yin, E. V. Alexandrov, D. H. Si, Q. Q. Huang, Z. Bin Fang, Y. Zhang, A. A. Zhang, W. K. Qin, Y. L. Li, T. F. Liu and D. M. Proserpio, Angew. Chem., Int. Ed., 2022, 61, e202115854 CrossRef CAS PubMed.
- J. H. Zhang, Z. M. Ge, J. Wang, D. C. Zhong and T. B. Lu, Nat. Commun., 2025, 16, 2448 CrossRef CAS PubMed.
- J. L. Segura, S. Royuela and M. Mar Ramos, Chem. Soc. Rev., 2019, 48, 3903–3945 RSC.
- Y. Cong, X. Li, S. Zhang, Q. Zheng, Y. Zhang and S. W. Lv, ACS Appl. Mater. Interfaces, 2023, 15, 43799–43809 CrossRef CAS.
- L. Ma, J. Xu, S. Zhao, L. Li and Y. Liu, Chem.–Eur. J., 2021, 27, 15795–15805 CrossRef CAS PubMed.
- Y. He, J. Zhao, Y. T. Sham, S. Gao, M. Pan, Q. Chen, G. Huang, P. K. Wong and J. Bi, ACS Sustain. Chem. Eng., 2023, 11, 17552–17563 CrossRef CAS.
- G. Xia, J. Qiu, L. Zhang, D. Dai and J. Yao, Colloids Surf., A, 2023, 664, 131124 CrossRef CAS.
- Y. Yang, Y. Li, X. Ma, L. Xie, D. Lv, L. Jiang, J. He, D. Chen and J. Wang, Catal. Sci. Technol., 2023, 13, 5599–5609 RSC.
- Q. Shang, Y. Liu, J. Ai, Y. Yan, X. Yang, D. Wang and G. Liao, J. Mater. Chem. A, 2023, 11, 21109–21122 RSC.
- Q. Zuo, B. Chu, X. Ye, F. Li, L. Li and Q. Xu, J. Am. Chem. Soc., 2025, 147, 34681–34689 CrossRef CAS.
- J. Qiu, L. Zhang, G. Xia, D. Dai, Y. Tang and J. Yao, Catal. Sci. Technol., 2023, 13, 2101–2107 RSC.
- Y. Kondo, K. Hino, Y. Kuwahara, K. Mori and H. Yamashita, J. Mater. Chem. A, 2023, 11, 9530–9537 RSC.
- Y. Kondo, K. Hino, Y. Kuwahara, K. Mori, H. Kobayashi and H. Yamashita, Chem. Commun., 2022, 58, 12345–12348 RSC.
- Y. C. Hao, L. W. Chen, J. Li, Y. Guo, X. Su, M. Shu, Q. Zhang, W. Y. Gao, S. Li, Z. L. Yu, L. Gu, X. Feng, A. X. Yin, R. Si, Y. W. Zhang, B. Wang and C. H. Yan, Nat. Commun., 2021, 12, 2682 CrossRef CAS PubMed.
- Y. Lin, J. Zou, X. Wu, S. Tong, Q. Niu, S. He, S. Luo and C. Yang, Nano Lett., 2024, 24, 6302–6311 CrossRef CAS PubMed.
- Z. Li, X. Shi, H. Cheng, Y. Song, Y. Jiao, S. Shi, J. Gao and J. Hou, Adv. Energy Mater., 2024, 14, 2302797 CrossRef CAS.
- J. Zhu, X. F. Lu, D. Luan and X. W. Lou, Angew. Chem., Int. Ed., 2024, 63, e202408846 CrossRef CAS PubMed.
- S. Rasaily, K. Baruah, D. Sharma, P. Lepcha, S. Biswas, A. N. Biswas, S. Tamang and A. Pariyar, Inorg. Chem., 2023, 62, 3026–3035 CrossRef CAS PubMed.
- S. Rasaily, S. Chettri, D. Sharma, K. Baruah, R. Dewan, S. Tamang and A. Pariyar, Inorg. Chem., 2024, 63, 17846–17855 CrossRef CAS PubMed.
- Y. T. Zheng, S. Li, N. Y. Huang, X. Li and Q. Xu, Coord. Chem. Rev., 2024, 510, 215858 CrossRef CAS.
- M. I. Gonzalez, A. B. Turkiewicz, L. E. Darago, J. Oktawiec, K. Bustillo, F. Grandjean, G. J. Long and J. R. Long, Nature, 2020, 577, 64–68 CrossRef CAS PubMed.
- Z. R. Tao, J. X. Wu, Y. J. Zhao, M. Xu, W. Q. Tang, Q. H. Zhang, L. Gu, D. H. Liu and Z. Y. Gu, Nat. Commun., 2019, 10, 2911 CrossRef PubMed.
- C. D. Gheewala, B. E. Collins and T. H. Lambert, Science, 2016, 351, 961–965 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Gourab K. Dam
a,
Gourab K. Dam

















 and
and  ) in different COFs. (j) Long-term H2O2 photoproduction over 8 h using the P-TPB-COF, QP-BPT-COF, and QP-HPTP-COF. (Reproduced and reprinted with permission from ref. 147. Copyright 2024, Wiley-VCH GmbH.)
) in different COFs. (j) Long-term H2O2 photoproduction over 8 h using the P-TPB-COF, QP-BPT-COF, and QP-HPTP-COF. (Reproduced and reprinted with permission from ref. 147. Copyright 2024, Wiley-VCH GmbH.)










