
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMastering the potential of well-defined ML1L2 species in asymmetric catalysis through ligand immobilization (MLhetLhom). Use in highly enantioselective Pd-catalyzed spiroannulation
Maria Biosca†
a,
Pol de la Cruz-Sánchez† a,
Jorge Faiges†a,
Aitor Maestrobc,
C. Oliver Kappebc,
Sándor B. Ötvös*bc,
Miquel A. Pericàsad,
Oscar Pàmiesa and
Montserrat Diéguez*a
a,
Jorge Faiges†a,
Aitor Maestrobc,
C. Oliver Kappebc,
Sándor B. Ötvös*bc,
Miquel A. Pericàsad,
Oscar Pàmiesa and
Montserrat Diéguez*a
aUniversitat Rovira i Virgili, Departament de Química Física i Inorgànica, C/ Marcel lí Domingo, 1, 43007 Tarragona, Spain. E-mail: montserrat.dieguez@urv.cat
bInstitute of Chemistry, University of Graz, NAWI Graz, A-8010 Graz, Austria
cCenter for Continuous Flow Synthesis and Processing (CC FLOW), Research Center Pharmaceutical Engineering GmbH (RCPE), A-8010 Graz, Austria. E-mail: sandor.oetvoes@rcpe.at
dRoyal Academy of Sciences and Arts of Barcelona, Chemistry Section, La Rambla 115, 08002 Barcelona, Spain
First published on 12th May 2026
Abstract
Catalytic complexes involving different monodentate ligands coordinated to the same metal center offer considerable interest towards the development of new processes and the improvement of the characteristics of known ones. However, one of the major drawbacks in using mixtures of monodentate ligands in asymmetric metal catalysis lies in the difficulty of controlling the formation of active species containing both of the desired ligands coordinated at the same time to the metal center (ML1L2). Herein, we present a straightforward methodology to overcome this long-standing major limitation. Thus, we demonstrate that the formation of well-defined ML1L2 species can be attained by combining an immobilized ligand and a homogeneous ligand. This concept has been successfully applied to a synthetically relevant Pd-catalyzed [3 + 2] spiroannulation. The use of a polystyrene supported chiral binaphthyl-based phosphoramidite in combination with an achiral cheap biphenyl monophosphoramidite led to the sole formation of highly enantioselective active species containing both ligands coordinated to Pd, as demonstrated by gel-phase, high resolution 31P NMR. In addition, we have also studied the continuous flow spiroannulation, which further demonstrated that the reaction occurs in the heterogenous phase. The advantages associated to the use of well-defined PdLhetLhom in catalysis are further demonstrated in asymmetric allylic alkylation reactions, illustrating the potential of this methodology in asymmetric catalysis.
Introduction
Enantioselective metal catalysis has become pivotal in the synthesis of enantiomerically enriched compounds. Its success mainly relies on the use of chiral ligands able to transfer the chiral information from the catalysts to the product.1–3 In this realm, there are many relevant transformations whose enantiodetermining transition states involve two monodentated ligands (or a bidentate one) coordinated to the metal. The successful introduction of DIOP4 in the early 70's favored the use of chiral bidentate ligands instead of monodentate ligands. However, the development of DuPHOS-type monophospholane5 and binol-based mono-phosphonite,6,7 phosphite8 and phosphoramidite9,10 ligands in the late 90's and early 2000's paved the way for the use of monodentate ligands in asymmetric catalysis, outperforming bidentate ligands in certain cases. Nowadays, many reactions depend on them for excellent catalytic performance.1–3,11 Soon after, Reetz and coworkers went a step further by employing mixtures of chiral monodentate ligands (L1 and L2), thereby introducing a new concept in asymmetric catalysis that mimics the desymmetrization of C2-symmetric bidentate ligands into their C1-symmetric counterparts.12,13 An inherent limitation of this approach arises from the presence of three distinct catalytically active species in equilibrium: the two homocombinations (ML1L1 and ML2L2) and the desired heterocombination ML1L2 (Fig. 1a(1)). Using a combinatorial approach, Reetz and coworkers identified certain ligand heterocombinations that were more reactive and, in some cases, more enantioselective than the two possible homocombinations in Rh-catalyzed hydrogenation (Fig. 1a(1)). These findings are consistent with reports showing that certain C1-symmetric P–P′ bidentate ligands can outperform their C2-symmetric analogues.13 They further demonstrated that the sense of enantioselectivity can be reversed by selecting an appropriate mixture of chiral and achiral monodentate ligands (Fig. 1a(1)).14 However, the broader application of this strategy in asymmetric catalysis has been limited by the coexistence of multiple catalytically active species, the sensitivity of their equilibria to changes in reaction conditions and substrate, and the requirement that the heterocombination must be both more reactive and selective than the corresponding homocombinations to provide an improved catalytic system.15 Accordingly, since its first report in 2003, this approach has been explored in relatively few occasions, mainly within a relatively short time frame (2003–2008), and predominantly in asymmetric hydrogenation, with limited success in other asymmetric transformations.15 | ||
| Fig. 1 (a) Conceptual approach in the search of well-defined active species containing two distinct monodentate ligands (heterocombination); and (b) enantioselective spiroannulation towards structurally complex tetrahydrofuran-fused spirocyclic scaffolds as proof of concept. | ||
Based on our experience in covalent ligand immobilization16,17 as well as in the effective site isolation of immobilized ligands onto organic18,19 or inorganic20 supports, which prevents the coordination of two such immobilized ligands to a single metal atom, we envisaged exploring catalytic systems involving two different monodentate ligands in which one ligand remains in solution while the other is appropriately immobilized onto a solid support (Fig. 1a(2)). Whenever the condition of site isolation is fulfilled in the immobilized ligand, this should lead to the exclusive formation of a perfectly defined immobilized heterodimeric species MLHetLHom. It is worth noting that, since the liquid phase in these systems can be simply washed away by filtration, it should be possible to remove any excess of the homogeneous ligand prior to the catalytic use. Moreover, since a two-phase system is involved, the generation in the liquid phase of MLHomLHom species would only be possible subsequently to non-favorable metal leaching. Moreover, the exclusive formation of MLHetLHom species would easily pave the way towards the implementation of a heterogenized continuous flow protocol, with its concomitant advantages with respect to the still more common batch processing.21,22
To bring these ideas into practice, we hypothesized that ML1L2 species of the MLHetLHom type would be easily and selectively accessed by first coordinating the metal center from a convenient precursor to an immobilized ligand (Lhet) followed by the addition of the second, homogeneous ligand (Lhom) in solution and washing away any excess of it if necessary (Fig. 1a(2)). To avoid leaching of the valuable immobilized ligand, we selected a covalent immobilization strategy, while to preserve the selectivity characteristics of the parent homogeneous ligand we placed the immobilization point far away from the catalyst chiral pocket to avoid perturbation of the enantiodetermining transition states. As a support, we selected a Merrifield-type polystyrene (PS) resin with low cross-linking and low functionalization. This support offered several advantages. Firstly, the resulting resins swells efficiently in most common organic solvents and can therefore be easily characterized using standard NMR techniques, while providing a homogeneous-like environment for the catalytic process. In addition, the low functionalization of the resulting catalytic resins allows strict site isolation, so that no homocombination of ligands (MLHetLHet) is possible due to geometrical constraints.
To test our hypothesis, we selected the enantioselective Pd-catalyzed [3 + 2] spiroannulation and evaluated it under two scenarios: the usual batch process and in continuous flow, by taking advantage in both cases of the exclusive formation of well-defined PdLHetLHom active species (Fig. 1b). The reaction was chosen because its active species usually involves two monodentate ligands coordinated to Pd23–36 and because of the increasing interest of spirocyclic motifs which are present in many natural products, drugs and functional materials.37–42 In particular, we selected the reaction of 2-substituted-2-vinylepoxides with 5-benzylidene Meldrum's acid derivatives leading to spirocyclic tetrahydrofuran derivatives which remains comparatively underexplored.43 To further illustrate the synthetic value of this particular process, we straightforwardly converted one of the enantiopure spirocyclic products into rare and highly appealing enantiopure lactone motifs, which include enantiopure spirocyclic-β-, fused bicyclic γ- and α-benzylidene γ-lactones.
Results and discussion
Development of the spiroannulation reaction under homogeneous conditions using monodentate ligands
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, the major diastereoisomer 3aa could be easily secured in pure form by crystallization through simple addition of isopropyl alcohol to a concentrated dichloromethane solution of the crude reaction product. Thus, by taking advantage of this feature we could secure 3aa in good isolated yield (73%) and in excellent diastereo- and enantiopurity (entry 14) working at room temperature. Moreover, the reaction can be carried out in environmentally friendly solvents, such as 2-MeTHF, with no loss of selectivity.
:1 ratio, the major diastereoisomer 3aa could be easily secured in pure form by crystallization through simple addition of isopropyl alcohol to a concentrated dichloromethane solution of the crude reaction product. Thus, by taking advantage of this feature we could secure 3aa in good isolated yield (73%) and in excellent diastereo- and enantiopurity (entry 14) working at room temperature. Moreover, the reaction can be carried out in environmentally friendly solvents, such as 2-MeTHF, with no loss of selectivity.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T (°C) | % Convb | % Yieldc | 3aa:3′aab |
%eed |
| a Reaction conditions: Pd2(dba)3·CHCl3 (2.5 mol%), ligand (10 mol%), 1a (0.1 mmol), 2a (0.2 mmol), solvent (1.0 mL), 25 °C, overnight.b Measured by 1H-NMR.c Isolated yields as a mixture of diastereomers.d Referred to the major diastereomer and determined by HPLC using a chiral stationary phase.e Reaction carried out at 2 mmol scale.f After recrystallization. X-ray structure of enantiopure (S,R)-3aa is also shown. | |||||||
| 1 | L1 | THF | 25 | 100 | 98 | 8:1 |
93 (S,R) |
| 2 | L1 | CH3CN | 25 | 99 | 95 | 7:1 |
92 (S,R) |
| 3 | L1 | CH2Cl2 | 25 | 95 | 93 | 4:1 |
91 (S,R) |
| 4 | L1 | Toluene | 25 | 98 | 91 | 7:1 |
93 (S,R) |
| 5 | L1 | THF | 0 | 21 | 14 | 3:1 |
91 (S,R) |
| 6 | L2 | THF | 25 | <5 | — | — | — |
| 7 | L3 | THF | 25 | 27 | 19 | 1:1 |
22 (S,R) |
| 8 | L4 | THF | 25 | 100 | 97 | 7:1 |
91 (S,R) |
| 9 | L5 | THF | 25 | 100 | >99 | 3:1 |
>99 (S,R) |
| 10 | L6 | THF | 25 | 100 | >99 | 3:1 |
90 (S,R) |
| 11 | L7 | THF | 25 | 100 | >99 | 6:1 |
33 (S,R) |
| 12 | L8 | THF | 25 | 100 | 99 | 4:1 |
30 (S,R) |
| 13 | L9 | THF | 25 | 50 | 49 | 2:1 |
63 (S,R) |
| 14e | L5 | THF | 25 | 100 | 73f | >20:1f |
>99 (S,R) |

To verify that the species responsible for the catalytic activity contains two monodentate ligands coordinated to Pd through the catalytic cycle, we performed mechanistic investigations, including kinetic and NMR studies and Eyring plot analysis. We initially performed kinetic studies using the variable time normalization analysis (VTNA).59–61 To facilitate data collection by 19F-NMR, we chose the reaction of 5-(4-trifluorobenzylidene)-2,2-dimethyl-1,3-dioxane-4,6-dione (1b) with 2-phenyl-2-vinyloxirane (2a) leading to spirocyclic furan 3ba. We first determined the rate dependence on Meldrum's acid-type derivative (1b) and epoxide (2a) concentration. As shown in Fig. 2, a first-order dependence in 2a was determined, which indicates that this species participates in the rate-determining step of the reaction.
 | ||
| Fig. 2 Kinetic study of order in 2a (left) and 1b (right). | ||
In contrast, the reaction exhibited a negative order in Meldrum's acid-type derivative concentration ([1b]), indicating that 1b acts as an inhibitor. Accordingly, the presence of Pd species containing coordinated 1b were observed during the kinetic experiments in the 19F-NMR spectra as a broad signal at a −62.5 ppm (see SI).
We then determined the reaction order in catalyst and found a value higher than one (see SI), which is consistent with the off-cycle formation of catalytically inactive dimeric species containing coordinated 1b.62 In addition, the presence of free ligand was not observed in any of the 31P{1H} NMR spectra recorded at different stages of the catalytic reaction (see SI), which indicates that the two monophosphoramidite ligands remain coordinated to palladium through all the catalytic cycle.
Overall, our experimental data support a mechanism where coordination of multiple molecules of 1b originates off-cycle dimeric palladium species A that is in equilibrium with a monomeric Pd-active species B containing two coordinated monophosphoramidites (Scheme 1).
 | ||
| Scheme 1 Mechanistic proposal for the [3 + 2]-cycloaddition for the preparation of tetrahydrofuran-fused spirocyclic Meldrum's acid derivatives using Pd(S)-L5 catalytic system (Ar = p-CF3-C6H4). Rds = rate determining step. Eds = enantiodetermining step. | ||
As already mentioned, no free ligand was observed by 31P{1H}-MMR spectroscopy during the course of the reaction. Rate-determining step coordination of the vinyloxirane to the intermediate B is followed by oxidative addition to form the key zwitterionic Pd–allyl complex with two coordinated chiral monophosphoramidite ligands (C). Nucleophilic attack of the Pd–zwitterionic species on 1b, followed cyclization, leads to the formation of 3ba while closing the catalytic cycle.
We next calculated the activation parameters using the Eyring equations.63 Based on the Eyring plot (Fig. 3), we calculated the following values: ΔH‡ = 18.0 ± 2 kcal mol−1; ΔS‡ = −16.5 ± 8 cal mol−1 K−1; ΔG‡ = 23.0 ± 5 kcal mol−1. The negative value of ΔS‡ indicates that entropy decreases on forming the transition state, which is consistent with coordination of the vinyloxirane as the rate determining step.
 | ||
| Fig. 3 Eyring plot for the [3 + 2]-cycloaddition of 1b and 2a with PdL5 catalytic system. | ||
Development of well-defined PdL1L2 species by combining an immobilized ligand and a homogeneous ligand
 | ||
| Scheme 2 Synthesis of PS-(R)-L5. Reaction conditions: (a) DMF, THF, −50 °C, 3 h then NaBH4, MeOH, 0 °C, 3 h; (b) NaH, THF, Merrifield resin, rt, 16 h; (c) HCl (aq), THF, rt; (d) PCl3, Py, 90 °C, 16 h, then N-methyl-1,1-diphenylmethanamine, NEt3, DMAP, toluene, rt, 5 h (OEM = ethoxymethyl). | ||
Following the methodology reported by the Sasai group,64 PS-(R)-4 was easily prepared in three-steps from (R)-6-bromo-2,2′-bis(ethoxymethoxy)-1,1′-binaphthalene (R)-5. With only one more step, PS-(R)-4 was then transformed into the desired immobilized phosphoramidite PS-(R)-L5 by reaction with PCl3 followed by reaction with N-methyl-1,1-diphenylmethanamine. The catalytic resin PS-(R)-L5 was obtained with a functionalization of f = 0.41 mmol according to the elemental analysis of nitrogen. This corresponds to an immobilization yield of 89%.65
The performance of PS-(R)-L5 was initially studied in the model cycloaddition reaction of 1a with 2a under batch conditions (Table 2, entry 1), delivering 3aa in racemic form. This is a highly positive result because it confirms that the physical separation between the ligands on the resin satisfies the site isolation requirement, so that PdLHetLHet-type species cannot be generated, which is essential for our approach to succeed. To further confirm this interpretation, we added one equivalent of homogeneous (R)-L5, which resulted in a remarkable recovery of enantioselectivity (85% ee, entry 2). This finding agrees with the mechanistic investigations that show that two phosphoramidite units [PS-(R)-L5 and (R)-L5] are involved in the lowest energy, enantiodiscriminating transition state. As a further confirmation that the addition of monomer (R)-L5 is not simply promoting the spiroannulation in solution, the addition of the same amount of (S)-L5 led to a much lower enantioselectivity (33% ee; entry 3), pointing to the operation of a mismatched ligand combination.
|
|||||
|---|---|---|---|---|---|
| Entry | L1 | L2 | % Convb | 3aa:3′aab |
%eec |
| a Reaction conditions: Pd2(dba)3·CHCl3 (2.5 mol%), ligand (5 mol% of each ligand), 1a (0.1 mmol), 2a (0.2 mmol), THF (1.0 mL), 25 °C, overnight.b Conversions and diastereomeric ratios determined by 1H-NMR.c Enantiomeric excesses determined by HPLC. | |||||
| 1 | PS-(R)-L5 | — | 100 | 2.7:1 |
0 |
| 2 | PS-(R)-L5 | (R)-L5 | 100 | 3:1 |
85 (R,S) |
| 3 | PS-(R)-L5 | (S)-L5 | 54 | 3:1 |
33 (R,S) |
| 4 | PS-(R)-L5 | L10 | 60 | 3:1 |
82 (R,S) |
| 5 | PS-(R)-L5 | L11 | 100 | 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :1 :1 |
95 (R,S) |
| 6 | (S)-L5 | L10 | 46 | 3:1 |
7 (S,R) |
| 7 | (S)-L5 | L11 | 76 | 3:1 |
50 (S,R) |
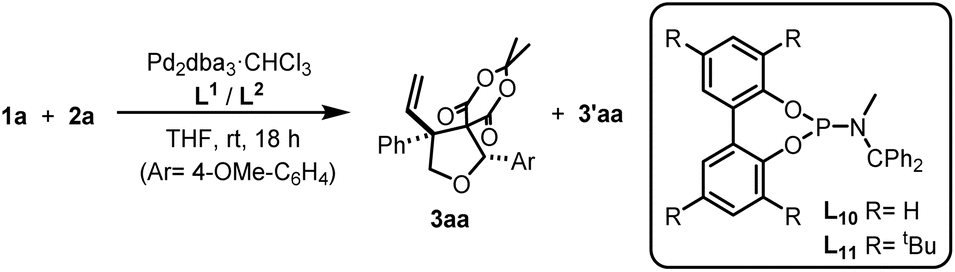
The results obtained so far clearly indicate that it is possible to generate well-defined PdLHetLHom species, opening the possibility to optimize the catalytic outcome by selecting the appropriate homogeneous ligand (LHom). For that purpose, we generated two different PdLHetLHom species by mixing PS-(R)-L5 with achiral ligands L10 or L11, which differ in the bulkiness of the biphenyl moiety. The use of species involving the less sterically congested ligand L10 (Pd(PS-(R)-L5)L10) led to a moderate conversion but a promising enantioselectivity (82% ee, entry 4). We were delighted to see that the use in the mixed species of the bulkier ligand L11 boosted the enantioselectivity to 95% ee (entry 5), with a complete conversion. This proves that under our conditions the formation of the heterocombination Pd(PS-(R)-L5)L11 is not only preferred over the homocombination (PdL11L11), but also it is highly enantioselective (entry 5 vs. 7). We further proved the formation of Pd(PS-(R)-L5)L11 species by performing a sol–gel 31P{1H} NMR of an equimolar mixture of supported PS-(R)-L5 and L11 that upon addition of Pd2(dba)3 showed the complete disappearance of the sharp signal corresponding to monomeric L11 and its rapid incorporation into the broad 31P band of the polymer-supported monophosphoramidite ligand (see SI). Moreover, the results obtained using Pd(PS-(R)-L5)L10 and Pd(PS-(R)-L5)L11 indicate that the tropoisomerism of the biphenyl moiety in inexpensive ligands L10 and L11 is efficiently controlled by a nearby chiral moiety – in our case, PS-(R)-L5.66 Accordingly, the results indicate that in both L10 and L11, the biphenyl moiety adopts an (R)-configuration in the active Pd species.67
In line with the spectroscopic evidence, the combination of equimolecular amounts of ligand (S)-L5 with achiral L11 under homogeneous conditions (Table 2, entry 7) led to much lower conversion and enantioselectivity than when using the Pd(PS-(R)-L5)L11 species (entry 5): 50 vs. 95% ee. The same behaviour is observed for the combinations involving L10 (entries 4 and 6). These results agree with the formation in solution of a mixture of active species containing both homodonor combinations and possibly the heterocombination, further confirming the difficulty of controlling the speciation of the active species in homogeneous phase.
We subsequently studied the scope of the reaction catalyzed by Pd(PS-(R)-L5)L11. Positively, the use of a range of 5-benzylidene Meldrum's acid derivatives (1a–l) and 2-aryl-2-vinyloxiranes (2a–j) led to the formation of the corresponding tetrahydrofurans in high yields and high-to-excellent enantioselectivities (Scheme 3), mean ee's 95.2%; 24 examples in total) regardless of the aryl substitution pattern and the electronic properties of the aryl substituents at both coupling units. The diastereoselectivities were affected by the substitution of the aryl group at the Meldrum's acid derivative, with the lowest diastereoselectivity observed when an electron withdrawing p-CF3 group was present, while the existence of substituents at the ortho position had a very positive impact on the diastereoselectivity, allowing to reach dr's (up to >20:1) without the need of further crystallization, see products 3gd, 3gf, 3gi). Notably, the reaction also tolerated well the presence of heteroarylmethylidene substituted (3ia and 3ja) and conjugated alkylidene Meldrum's acid derivatives (3la). Finally, the spiroannulation of Meldrum's acid derivatives with aliphatic substituents as well as with 2-alkyl-2-vinyloxiranes proceeded also well to form the corresponding tetrahydrofurans 3ka and 3bj in high yields and enantioselectivities (95–96% ee).
 | ||
| Scheme 3 Cycloaddition reactions using Pd(PS-(R)-L5)L11 with several 5-(R-ylidene) substituted 2,2-dimethyl-1,3-dioxane-4,6-diones (in red) and vinylepoxides (in blue). The yields and diastereoselectivities for the major diastereomer after crystallization of selected final compounds are shown in parentheses. | ||
Noteworthy, diastereomerically pure 3aa could be converted in a straightforward manner into enantiopure spirocyclic-β-, fused bicyclic γ- and α-benzylidene γ-lactones (see SI for details).
Recycling experiments in batch and toward continuous flow using well determined heterodonor Pd(PS-(R)-L5)L11 species
The heterogeneous nature of the catalytic system prompted us to study the possibility of catalyst recycling. Our first attempts were directed to the recovery of the catalyst by filtration and its reuse in batch. However, all our attempts in this direction failed. This was probably linked to the formation of inactive palladium black species, which was observed during work-up of every single catalytic experiment. This was not unexpected, since the formation of Pd black is typically reported after the use of palladium complexes in homogeneous catalysis. We then focused on the development of a more sustainable version of the annulation reaction, by developing a continuous flow version of the process in which separation and recycling of the heterogenized catalyst are integrated into the same process and would not require the breakdown of inert atmosphere, anhydrous conditions.For this purpose, a packed bed reactor was assembled by placing PS-(R)-L5 (0.25 g, 0.1 mmol) in a size-adjustable glass column. After swelling the resin with THF, the reactor was flushed with a stock solution of Pd2(dba)3·CHCl3 (6 mM in THF) for 20 minutes (0.2 mL min−1, 0.024 mmol). Then, a THF solution containing the second achiral ligand L11 was pumped for 20 minutes to generate in situ the active Pd complex (0.2 mL min−1, 0.05 mmol). Finally, a THF solution of 1a and 2a ([1a] = 0.025 M, [2a] = 0.033 M) was pumped at 0.2 mL min−1. Despite the initial good conversions and excellent enantioselectivity (e.g., 75% conversion and 95% ee), fast deactivation was observed, thus indicating that leaching of Pd complexes involving L11 was taking place.68–71 This observation prompted us to modify the continuous flow protocol by simultaneously pumping small amounts of the Pd-precursor and auxiliary ligand together with the substrates through the packed bed reactor containing ligand PS-(R)-L5 (Scheme 4). This could be easily performed by simultaneous pumping of two different solutions. One of them contains the Pd-precursor in THF, and the other one contains both substrates ([1a] = 0.050 M, [2a] = 0.07 M) and the auxiliary ligand (5 mM). Both THF solutions were pumped simultaneously at 0.1 mL min−1 and mixed just before entering the packed bed reactor by using a T-mixer. Initially, we pumped a highly concentrated Pd precursor solution (2.5 mM) and when high levels of conversion were achieved, the initial Pd solution was replaced by a more diluted one (0.75 mM). Positively, after optimization, 3aa was produced in good overall yield and enantioselectivity (up to 95% ee) for up to 3 h (Scheme 4). Interestingly, continuous addition of the auxiliary ligand and Pd-precursor did not diminish the high level of enantioselectivity, indicating that the reaction was happening solely at the resin, and there was no competition with the putative homogeneous complex bearing two homogeneous ligands.
 | ||
| Scheme 4 Set up of the optimized continuous flow protocol to perform Pd-catalyzed asymmetric cycloadditions using well defined Pd(PS-(R)-L5)L11 species. aIsolated as a 3:1 diastereomeric mixture. bReferred to 3aa and determined by HPLC. | ||
In summary, this heterogenous flow protocol, involving the cooperation of a homogeneous achiral ligand, that can be recovered after the reaction, enables to achieve high enantioselectivities in the preparation of 3 through simple and fast reactions (only 10 min of residence time). Moreover, it is a further confirmation that the reaction proceeds on the heterogeneous phase, and the observed catalytic activity has to be ascribed to well defined, immobilized Pd(PS-(R)-L5)L11 species and not to leached Pd species as in other Pd-catalytic systems reported in the literature.72–76
Conclusions
We have developed a new, simple methodology that allows the formation of well-defined highly enantioselective active species containing two different monodentate ligands coordinated at the same time to the metal center (ML1L2). This methodology takes advantage of covalently immobilizing one of the ligands onto a solid support with low functionalization, so that no homocombination of ligands (MLHetLHet) is possible, followed by its coordination to the metal and subsequently addition of a second different ligand in solution that can synergistically improve enantioselectivity. This methodology has been successfully applied in the Pd-catalyzed [3 + 2] spiroannulation for the synthesis of a wide range of tetrahydrofuran-fused spirocyclic Meldrum's acid derivatives (24 examples). The exclusive formation of the successful ML1L2-type species was achieved by combining a polystyrene supported chiral binaphthyl-based phosphoramidite with a cheap achiral biphenyl monophosphoramidite. Key to the success of this strategy have been (a) the use of a covalent immobilization to avoid leaching of the supported ligand (LHet), (b) the immobilization of the ligand through a position far away from the catalytic center to avoid perturbation of the enantiodifferentiating transition states by the polymer backbone, and (c) the use of a low cross-linked polymer backbone with low functionalization, which allows fast mass transfer, approaching homogeneous conditions, while preventing the formation of PdLHetLHet species. We have subsequently extended the use of this well-defined PdLHetLHom species to the spiroannulation in continuous flow. Positively, the selectivity of the cycloaddition reaction with the immobilized ligand is maintained when the system is operated in continuous flow mode. Leaching of Pd species during the continuous flow operation could be compensated by continuous addition of a solution of the Pd-precursor in THF. These flow experiments also demonstrated that Pd species in solution were not catalytically active, so that the catalytic performance of the spiroannulation reaction has to be ascribed to the heterogenous phase containing both types of monophosphoramidite ligands coordinated to Pd.Preliminary results indicate that this approach to control ML1L2 speciation can also be applied to enantioselective Pd-catalyzed allylic substitution reactions. Thus, the use of equimolar amounts of PS-(R)-L5 with achiral L11 provides higher enantioselectivity than that obtained under homogeneous conditions (62% vs. 40% ee) in the alkylation of 1,3-diphenylallyl acetate with dimethylmalonate (see SI for details). We are currently working on designing new catalytic systems of the general type MLHetLHom with increased metal–ligand (M–LHet) stability, with the aim of minimizing metal leaching and thus transforming the present proof of principle into finely tunable processes, suitable for the long-standing operation in continuous flow.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2474009 (3aa) and 2474010 (7) contain the supplementary crystallographic data for this paper.77a,bThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information: development and scope of the spiroannulation reaction, mechanistic investigations, experimental procedures for batch and flow spiroannulations, synthesis of immobilized ligand, synthesis of new ligands and substrates, post-modifications, characterization details and enantiomeric excess determination of products, copies of NMR spectra and of HPLC traces for ee determination of products, kinetic data and X-ray crystallographic data for compounds. See DOI: https://doi.org/10.1039/d6sc01392a.
Acknowledgements
This work was supported by grants from FEDER/Ministerio de Ciencia e Innovación (MICINN)/AEI (PID2022-139996NB-I00). Grant 2021SGR00163 funded by the Catalan Government are also gratefully acknowledged. J. F. also thanks Ministerio de Ciencia, Innovación y Universidades for FPU18/06352 fellowship. M. B. thanks to the “Ramón y Cajal” contract (RRYC-2023-045413-I) funded by MICIU/AEI/10.13039/501100011033 and ESF+.Notes and references
- Privileged Chiral Ligands and Catalysts, ed. Q.-L. Zhou, John Wiley& Sons Inc., New York, 2011 Search PubMed.
- Chiral ligands. Evolution of ligand libraries for asymmetric catalysis, ed. M. Diéguez, CRC Press, Boca Raton, 2021 Search PubMed.
- J. Margalef, M. Biosca, P. de la Cruz-Sánchez, J. Faiges, O. Pàmies and M. Diéguez, Evolution in heterodonor P-N, P-S and P-O chiral ligands for preparing efficient catalysts for asymmetric catalysis. From design to applications, Coord. Chem. Rev., 2021, 446, 214120 CrossRef CAS.
- T. P. Dang and H. B. Kagan, The asymmetric synthesis of hydratropic acid and amino-acids by homogeneous catalytic hydrogenation, J. Chem. Soc. Chem. Commun., 1971, 481 RSC.
- F. Guillen and J.-C. Fiaud, Enantiomerically pure 1, 2, 5-triphenylphospholane through the synthesis and resolution of the chiral trans-(2,5)-diphenylphospholanic acid, Tetrahedron Lett., 1999, 40, 2939–2942 CrossRef CAS.
- C. Claver, E. Fernandez, A. Gillon, K. Heslop, D. J. Hyett, A. Martorell, A. G. Orpen and P. G. Pringle, Biarylphosphonites: a class of monodentate phosphorus(III) ligands that outperform their chelating analogues in asymmetric hydrogenation catalysis, Chem. Commun., 2000, 961–962 Search PubMed.
- M. T. Reetz and T. Sell, Rhodium-catalyzed enantioselective hydrogenation using chiral monophosphonite ligands, Tetrahedron Lett., 2000, 41, 6333–6336 CrossRef CAS.
- M. T. Reetz and G. Mehler, Highly Enantioselective Rh-Catalyzed Hydrogenation Reactions Based on Chiral Monophosphite Ligands, Angew. Chem., Int. Ed., 2000, 39, 3889–3890 CrossRef CAS PubMed.
- G. Franció, F. Faraone and W. Leitner, Asymmetric catalysis with chiral phosphane/phosphoramidite ligands derived from quinoline (QUINAPHOS), Angew. Chem., Int. Ed., 2000, 39, 1428–1430 CrossRef.
- M. van den Berg, A. J. Minnaard, E. P. Schudde, J. van Esch, A. H. M. de Vries, J. G. de Vries and B. L. Feringa, Highly enantioselective rhodium-catalyzed hydrogenation with monodentate ligands, J. Am. Chem. Soc., 2000, 122, 11539–11540 CrossRef CAS.
- For a review illustrating the succesful use of monodentate phosphorus ligands, see: W. Fu and W. Tang, Chiral monophosphorus ligands for asymmetric catalytic reactions, ACS Catal., 2016, 6, 4814–4858 CrossRef CAS.
- Several metal catalytic asymmetric reactions have benefited from the use of nonsymmetrical bidentate P–P′ ligands since both functionalities can be independently boosted. See for example: A. Pfaltz and W. J. Drury III, Design of chiral ligands for asymmetric catalysis: From C2-symmetric P,P- and N,N-ligands to sterically and electronically nonsymmetrical P,N-ligands, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5723–5726 Search PubMed.
- M. T. Reetz, T. Sell, A. Meiswinkel and G. Mehler, A New Principle in Combinatorial Asymmetric Transition-Metal Catalysis: Mixtures of Chiral Monodentate P Ligands, Angew. Chem., Int. Ed., 2003, 42, 790–793 CrossRef CAS PubMed.
- M. T. Reetz and G. Mehler, Mixtures of chiral and achiral monodentate ligands in asymmetric Rh-catalyzed olefin hydrogenation: reversal of enantioselectivity, Tetrahedron Lett., 2003, 44, 4593–4596 CrossRef CAS.
- M. T. Reetz, Combinatorial Transition-Metal Catalysis: Mixing Monodentate Ligands to Control Enantio-, Diastereo-, and Regioselectivity, Angew. Chem., Int. Ed., 2008, 47, 2556–2588 CrossRef CAS PubMed.
- C. Rodríguez-Escrich and M. A. Pericàs, Organocatalysis on Tap: Enantioselective Continuous Flow Processes Mediated by Solid-Supported Chiral Organocatalysts, Eur. J. Org Chem., 2015, 1173–1188 CrossRef.
- C. Rodriguez-Escrich and M. A. Pericàs, Catalytic Enantioselective Flow Processes with Solid-Supported Chiral Catalysts, Chem. Rec., 2019, 19, 1872–1890 Search PubMed.
- X. Fan, C. Rodríguez-Escrich, S. Sayalero and M. A. Pericàs, Paraldehyde as an Acetaldehyde Precursor in Asymmetric Michael Reactions Promoted by Site-isolated Incompatible Catalysts, Chem.–Eur. J., 2013, 19, 10814–10817 CrossRef CAS PubMed.
- X. Fan, C. Rodríguez-Escrich, S. Wang, S. Sayalero and M. A. Pericàs, Highly Enantioselective Cross-Aldol Reactions of Acetaldehyde Mediated by a Dual Catalytic System Operating under Site Isolation, Chem.–Eur. J., 2014, 20, 13089–13093 Search PubMed.
- P. Borah, M. Fianchini and M. A. Pericàs, Assessing the Role of Site Isolation and Compartmentalization in Packed Bed Flow Reactors for Processes Involving Wolf-and-Lamb Scenarios, ACS Catal., 2021, 11, 6234–6242 CrossRef CAS.
- L. Capaldo, W. Zhenghui and T. Noël, A field guide to flow chemistry for synthetic organic chemists, Chem. Sci., 2023, 14, 4230–4247 RSC.
- F. Parveen, N. Watson, A. M. Scholes and A. G. Slater, Continuous flow as an enabling technology for sustainable supramolecular chemistry, Curr. Opin. Green Sustainable Chem., 2024, 48, 100935 CrossRef.
- M. Lautens, W. Klute and W. Tam, Transition Metal-Mediated Cycloaddition Reactions, Chem. Rev., 1996, 96, 49–92 CrossRef CAS PubMed.
- L. Yet, Metal-Mediated Synthesis of Medium-Sized Rings, Chem. Rev., 2000, 100, 2963–3008 CrossRef CAS PubMed.
- X. Xu and M. P. Doyle, The [3 + 3]-Cycloaddition Alternative for Heterocycle Syntheses: Catalytically Generated Metalloenolcarbenes as Dipolar Adducts, Acc. Chem. Res., 2014, 47, 1396–1405 CrossRef CAS PubMed.
- T. Hashimoto and K. Maruoka, Recent Advances of Catalytic Asymmetric 1,3-Dipolar Cycloadditions, Chem. Rev., 2015, 115, 5366–5412 CrossRef CAS PubMed.
- B. M. Trost, Z. Zuo and J. E. Schultz, Transition-Metal-Catalyzed Cycloaddition Reactions to Access Seven-Membered Rings, Chem.–Eur. J., 2020, 26, 15354–15377 CrossRef CAS PubMed.
- P. de la Cruz-Sánchez and O. Pàmies, Metal-π-Allyl Mediated Asymmetric Cycloaddition Reactions, Adv. Catal., 2021, 69, 103–180 Search PubMed.
- J. He, J. Ling and P. Chiu, Vinyl Epoxides in Organic Synthesis, Chem. Rev., 2014, 114, 8037–8128 CrossRef CAS PubMed.
- D. W. B. Allen, C. P. Lakeland and J. P. A. Harrity, Utilizing Palladium-Stabilized Zwitterions for the Construction of N-Heterocycles, Chem.–Eur. J., 2017, 23, 13830–13857 CrossRef PubMed.
- N. De and E. J. Yoo, Recent Advances in the Catalytic Cycloaddition of 1,n-Dipoles, ACS Catal., 2018, 8, 48–58 CrossRef CAS.
- Y. Liu, J. Oble, A. Pradal and G. Poli, Catalytic Domino Annulations through η3-Allylpalladium Chemistry: A Never-Ending Story, Eur. J. Inorg. Chem., 2020, 942–961 CrossRef.
- B. M. Trost and G. Mata, Forging Odd-Membered Rings: Palladium-Catalyzed Asymmetric Cycloadditions of Trimethylenemethane, Acc. Chem. Res., 2020, 53, 1293–1305 CrossRef CAS PubMed.
- J. Wang, S. A. Blaszczyk, X. Li and W. Tang, Transition Metal-Catalyzed Selective Carbon–Carbon Bond Cleavage of Vinylcyclopropanes in Cycloaddition Reactions, Chem. Rev., 2021, 121, 110–139 CrossRef CAS PubMed.
- C. Yang, Z.-X. Yang, C.-H. Ding, B. Xu and X.-L. Hou, Development of Dipolarophiles for Catalytic Asymmetric Cycloadditions through Pd-π-Allyl Zwitterions, Chem. Rec., 2021, 21, 1442–1454 CrossRef CAS PubMed.
- Even though the use of monophosphoramidite ligands for the enantioselective Pd-catalyzed [3 + 2] cycloaddition has been previously disclosed, there are examples for which the use of Pd/monophosphoramidite catalyst precursors are not active. See for example: J.-J. Suo, J. Du, Q.-R. Liu, D. Chen, C.-H. Ding, Qi. Peng and X.-L. Hou, Highly Diastereo- and Enantioselective Palladium-Catalyzed [3 + 2] Cycloaddition of Vinyl Epoxides and α,β-Unsaturated Ketones, Org. Lett., 2017, 19, 6658 CrossRef CAS PubMed.
- Spirocycles have a unique balance between structural rigidity and individual ring-related conformational flexibility, which for instance in drug discovery are key to overcome many liabilities of typical drug scaffolds based on flat (hetero)aromatics.
- Y. Zheng, C. M. Tice and S. B. Singh, The Use of Spirocyclic Scaffolds in Drug Discovery, Bioorg. Med. Chem. Lett., 2014, 24, 3673–3682 CrossRef CAS PubMed.
- E. M. Carreira and T. C. Fessard, Four-Membered Ring-Containing Spirocycles: Synthetic Strategies and Opportunities, Chem. Rev., 2014, 114, 8257–8322 CrossRef CAS PubMed.
- E. Chupakhin, O. Babich, A. Prosekov, L. Asyakina and M. Krasavin, Spirocyclic Motifs in Natural Products, Molecules, 2019, 24, 4165 CrossRef CAS PubMed.
- P. Yadav, R. Pratap and V. Ji Ram, Natural and Synthetic Spirobutenolides and Spirobutyrolactones, Asian J. Org. Chem., 2020, 9, 1377–1409 CrossRef CAS.
- K. Acosta-Quiroga, C. Rojas-Peña, L. S. Nerio, M. Gutiérrez and E. Polo-Cuadrado, Spirocyclic Derivatives as Antioxidants: a Review, RSC Adv., 2021, 11, 21926–21954 RSC.
- To the best of our knowledge the use of Meldrum's acid derivatives for the formation of spirocyclic compounds via Pd-catalyzed asymmetric [3 + 2] cycloaddition have not been reported. A single racemic example can be found in J.-G. Shim and Y. Yamamoto, Palladium-Catalyzed Regioselective [3 + 2] Cycloaddition of Vinylic Oxiranes with Activated Olefins. A Facile Synthesis of Tetrahydrofuran Derivatives, J. Org. Chem., 1998, 63, 3067–3071 CrossRef CAS.
- L. Panizzi, L. Mangoni and M. Belardini, The Structure of Grindelic Acid, a New Diterpene Acid, Tetrahedron Lett., 1961, 2, 376–381 CrossRef.
- F. J. Schmitz, F. J. McDonald and D. J. Vanderah, Marine Natural Products: Sesquiterpene Alcohols and Ethers from the Sea Hare Aplysia Dactylomela, J. Org. Chem., 1978, 43, 4220–4225 CrossRef CAS.
- E. Lee, D. S. Lee, Y. W. Choi and K. H. Lee, Stereoselective Reduction of α-Iodospirolactones. Total Synthesis of (±)-Liguloxide, Tetrahedron Lett., 1992, 33, 6673–6676 CrossRef CAS.
- Y. K. Booth, W. Kitching and J. J. De Voss, Biosynthesis of Insect Spiroacetals, Nat. Prod. Rep., 2009, 26, 490–525 RSC.
- A. Miyawaki, D. Kikuchi, M. Niki, Y. Manabe, M. Kanematsu, H. Yokoe, M. Yoshida and K. Shishido, Total Synthesis of Natural Enantiomers of Heliespirones A and C via the Diastereoselective Intramolecular Hosomi-Sakurai Reaction, J. Org. Chem., 2012, 77, 8231–8243 CrossRef CAS PubMed.
- D. R. Bhatia, P. Dhar, V. Mutalik, S. K. Deshmukh, S. A. Verekar, D. C. Desai, R. Kshirsagar, P. Thiagarajan and V. Agarwal, Anticancer Activity of Ophiobolin A, Isolated from the Endophytic Fungus Bipolaris Setariae, Nat. Prod. Res., 2016, 30, 1455–1458 CrossRef CAS PubMed.
- D. Q. Thach, Z. G. Brill, H. K. Grover, K. V. Esguerra, J. K. Thompson and T. J. Maimone, Total Synthesis of (+)-6-epi- Ophiobolin A, Angew. Chem., Int. Ed., 2020, 59, 1532–1536 CrossRef CAS PubMed.
- C. Ma, Y. Huang and Y. Zhao, Stereoselective 1,6-Conjugate Addition/Annulation of para-Quinone Methides with Vinyl Epoxides/Cyclopropanes, ACS Catal., 2016, 6, 6408–6412 CrossRef CAS (dr's: 5/1–>20/1 and ee's: 74–>99%)..
- J.-A. Xiao, Y.-C. Li, Z.-J. Luo, X.-L. Cheng, Z.-X. Deng, W.-Q. Chen, W. Su and H. Yang, Construction of Bispirooxindole Heterocycles via Palladium-Catalyzed Ring-Opening Formal [3+ 2]-Cycloaddition of Spirovinylcyclopropyl Oxindole and 3-Oxindole and 3-Oxindole Derivatives, J. Org. Chem., 2019, 84, 2297–2306 CrossRef CAS PubMed (dr: 5/1 and 71% ee)..
- J. Wang, L. Zhao, Q. Rong, R. Lv, Y. Lu, X. Pan, L. Zhao and L. Hu, Asymmetric synthesis of 3, 3′-tetrahydrofuryl spirooxindoles via palladium-catalyzed [3+ 2] cycloadditions of methyleneindolinones with vinylethylene carbonates, Org. Lett., 2020, 22, 5833–5838 CrossRef CAS PubMed (>20/1 dr's and ee's: 45–99%)..
- H. Zhang, X. Gao, F. Jiang, W. Shi, W. Wang, Y. Wu, C. Zhang, X. Shi and H. Guo, Palladium-Catalyzed Asymmetric [3+2] Cycloaddition of Vinylethylene Carbonates with 2-Arylidene-1,3-Indandiones: Synthesis of Tetrahydrofuran-Fused Spirocyclic 1,3-Indandiones, Eur. J. Org Chem., 2020, 4801–4804 CrossRef CAS (dr's: 1/1–4/1 and ee's: 75–99% at 0 °C)..
- H. J. Jeon, S. M. Park, Y. M. Lee and S.-G. Lee, Divergent Asymmetric Synthesis of Chiral Spiroheterocycles through Pd-Catalyzed Enantio- and Diastereoselective [3 + 2] Spiroannulation, Org. Lett., 2022, 24, 9189–9193 CrossRef CAS PubMed (dr's: 1.1/1–16/1 and ee's: 62–99% at 45 °C)..
- E. Pair, T. Cadart, V. Levacher and J.-F. Brière, Meldrum's Acid: A Useful Platform in Asymmetric Organocatalysis, ChemCatChem, 2016, 8, 1882–1890 CrossRef CAS.
- S. N. A. Bukhari, M. A. Abdelgawad, N. Ahmed, M. W. Amjad, M. A. Hussain, M. A. Elsherif, H. Ejaz, N. H. Alotaibi, I. Filipović and N. Janković, Synthesis, Characterization, and Biological Evaluation of Meldrum's Acid Derivatives: Dual Activity and Molecular Docking Study, Pharmaceuticals, 2023, 16, 281 CrossRef CAS PubMed.
- F. Brosge, P. Singh, F. Almqvist and C. Bolm, Selected Applications of Meldrum's Acid – a Tutorial, Org. Biomol. Chem., 2021, 19, 5014–5027 RSC.
- J. Burés, Variable Time Normalization Analysis: General Graphical Elucidation of Reaction Orders from Concentration Profiles, Angew. Chem., Int. Ed., 2016, 55, 16318–16321 CrossRef.
- J. Burés, A Simple Graphical Method to Determine the Order in Catalyst, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- C. D.-T. Nielsen and J. Burés, Visual kinetic analysis, Chem. Sci., 2019, 10, 348–353 Search PubMed.
- C. Jaramillo-Ferrer, G. Hutchinson and J. Burés, Mechanistic interpretation of orders in catalyst greater than one, Nat. Rev., 2023, 7, 26–34 Search PubMed.
- H. Eyring, The Activated Complex in Chemical Reactions, J. Chem. Phys., 1935, 3, 107–115 Search PubMed.
- D. Jayaprakash and H. Sasai, Synthesis and catalytic applications of soluble polymer-supported BINOL, Tetrahedron: Asymmetry, 2001, 12, 2589–2595 CrossRef CAS.
- A. Vidal-Ferran, N. Bampos, A. Moyano, M. A. Pericàs, A. Riera and J. K. M. Sanders, High catalytic activity of chiral amino alcohol ligands anchored to polystyrene resins, J. Org. Chem., 1998, 63, 6309–6318 CrossRef CAS PubMed.
- M. Diéguez, O. Pàmies and C. Moberg, Self-Adaptable Tropos Catalysts, Acc. Chem. Res., 2021, 54, 3252–3263 CrossRef PubMed.
- The higher enantioselectivity observed with L11 compared to that obtained with L10 can be explained either by the steric effect of the tert-butyl moieties at the ortho positions of the biphenyl moiety, by a slightly different dihedral angle of the biphenyl group in L10/L11, or by a combination of both factors.
- Note that during the reaction, the palladium species gets oxidized to Pd(II), which are known to favour the Pd-leaching.
- B. Gutmann, D. Cantillo and C. O. Kappe, Continuous-Flow Technology—A Tool for the Safe Manufacturing of Active Pharmaceutical Ingredients, Angew. Chem., Int. Ed., 2015, 54, 6688–6728 CrossRef CAS PubMed.
- D. Cantillo and C. O. Kappe, Immobilized Transition Metals as Catalysts for Cross-Couplings in Continuous Flow—A Critical Assessment of the Reaction Mechanism and Metal Leaching, ChemCatChem, 2014, 6, 3286–3305 CrossRef CAS.
- R. Greco, W. Goessler, D. Cantillo and C. O Kappe, Benchmarking immobilized di-and triarylphosphine palladium catalysts for continuous-flow cross-coupling reactions: efficiency, durability, and metal leaching studies, ACS Catal., 2015, 5, 1303–1312 CrossRef CAS.
- N. T. S. Phan, M. Van Der Sluys and C. W. Jones, On the Nature of the Active Species in Palladium Catalyzed Mizoroki–Heck and Suzuki–Miyaura Couplings–Homogeneous or Heterogeneous Catalysis, a Critical Review, Adv. Synth. Catal., 2006, 348, 609–679 CrossRef CAS.
- A. F. Shmidt and L. V. Mametova, Main Features of Catalysis in the Styrene Phenylation Reaction, Kinet. Catal., 1996, 37, 406–408 Search PubMed.
- R. G. Heidenreich, J. G. E. Krauter, J. Pietsch and K. Kohler, Control of Pd Leaching in Heck Reactions of Bromoarenes Catalyzed by Pd Supported on Activated Carbon, J. Mol. Catal. A: Chem., 2002, 182, 499–509 CrossRef.
- K. Kohler, R. G. J. Heidenreich, G. E. Krauter and M. Pietsch, Highly Active Palladium/Activated Carbon Catalysts for Heck Reactions: Correlation of Activity, Catalyst Properties, and Pd Leaching, Chem.–Eur. J., 2002, 8, 622–631 Search PubMed.
- R. K. Arvela, N. E. Leadbeater, M. S. Sangi, V. A. Williams, P. Granados and R. D. A. Singer, Reassessment of the Transition-Metal Free Suzuki-Type Coupling Methodology, J. Org. Chem., 2005, 70, 161–168 CrossRef CAS PubMed.
- (a) CCDC 2474009: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p1drx.; (b) CCDC 2474010: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2p1dsy.
Footnote |
| † These authors contributed equally, and they are arranged alphabetically. |
| This journal is © The Royal Society of Chemistry 2026 |