
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAchiral benzoic acid appended helical polymers for the copper-catalyzed asymmetric Diels–Alder reaction and Michael addition with excellent enantioselectivity and recyclability
Qi-Hui
Ti
a,
Ming-Yang
Mo
a,
Run-Tan
Gao
a,
Na
Liu
*b and
Zong-Quan
Wu
 *a
*a
aState Key Laboratory of Supramolecular Structure and Materials, College of Chemistry, Jilin University, 2699 Qianjin Street, Changchun 130012, China
bThe School of Pharmaceutical Sciences, Jilin University, 1266 Fujin Road, Changchun, Jilin 130021, China
First published on 6th March 2026
Abstract
One-handed helical polyisocyanides bearing achiral benzoic acid pendants were readily synthesized via the asymmetric polymerization of achiral isocyanides using chiral Pd(II) catalysts. Despite the inexistence of any stereogenic centers, these polymers showed significant optical activity owing to their one-handed helicity. Two adjacent carboxyl groups on the polymer backbone coordinate with a single Cu2+, producing helical polyisocyanide–Cu complexes that act as highly active and recyclable chiral catalysts for asymmetric Diels–Alder and Michael addition reactions. Under optimized conditions, the block copolymer S-poly(350-b-230-b-350)/CuCl2 delivers products in high yield (>77%) with outstanding stereocontrol (up to 98% ee). For Michael addition reactions, the same polymer gives up to 98% ee with yields >80%. Employing the enantiomeric backbone under the same conditions afforded the opposite R-configured product. Regulating the backbone helicity could control the direction of enantioselectivity and enantiomeric products could be facilely obtained using helical polyisocyanide with opposite handedness. Importantly, catalysts can be readily precipitated from the reaction mixture by addition of a poor solvent and reused for five consecutive cycles without significant loss of yield and enantioselectivity. After copper leaching and re-coordination, they can be cyclically applied to distinct asymmetric reactions.
Introduction
Chirality is a fundamental property of living systems.1–3 The basic molecular structures of biomolecules, such as amino acids and sugars, are all homochiral.4 All the amino acids in biological macromolecules are of the L-configuration, while the sugars are exclusively of the D-configuration.5,6 As a result, biological macromolecules further exhibit high-order homochirality by twisting into a single one-handed helix, such as the right-handed double-stranded helix in DNA and α-helix in proteins and saccharides, because left- and right-handed helices are mirror-image enantiomers.7 These chiralities play crucial roles in life processes, such as unique molecular recognition, stereospecific catalysis by enzymes, and the replication and inheritance of genetic information.8,9 Due to these chiralities, enantiomeric molecules often exhibit distinct or even opposite biological activities in living organisms. The development of highly efficient methods for preparing chiral molecules with high optical purity is of great demand, in pharmaceuticals, agrochemicals, and fine chemical synthesis.Among the various reported strategies for obtaining chiral molecules, enantioselective synthesis has attracted widespread interest owing to its mild conditions, high efficiency, and operational simplicity and is the focus of intense research in chemistry, materials, pharmaceuticals, and interdisciplines.10,11
The essence of enantioselective synthesis lies in the design and preparation of chiral catalysts with high activity and excellent enantioselectivity.12 The reported homogeneous small-molecular chiral catalysts may deliver outstanding catalytic efficiency and enantioselectivity. However, they often suffer from complex synthetic procedures, high costs, and difficulties in recovery and recycling, which severely constrain their large-scale industrial application.13–16 Immobilizing chiral catalysts onto polymers can significantly improve their recyclability but they suffer from lower catalytic activity and enantioselectivity due to polymer chain aggregation and entanglement, leading to lower catalytic activity and enantioselectivity.17,18 Inspired by the helical structures of biomacromolecules and stereospecific enzymatic catalysis, incorporating catalysts onto helical polymers has attracted considerable interest. The inherent helical chirality shows great potential for enhancing the enantioselectivity of asymmetric reactions, e.g., incorporating aminoacridine, dimethyl bipyridine and phenanthroline onto DNA could catalyze the Diels–Alder reaction, Friedel–Crafts alkylation, Michael addition reactions, and hydration reactions.19–21 However, since the modification of the helical scaffolds in natural macromolecules poses significant synthetic challenges, the design and synthesis of chiral catalysts based on artificial helical polymers are of great interest. Regulating the helicity could control the enantioselectivity and may result in products with desired configuration. According to the helix-inversion barrier, synthetic helical polymers are classified into dynamic and static helices.22–24 Dynamic helices usually have a low helix-inversion barrier and easily undergo helix-inversion between left- and right-handed helices, while static helices have a relatively high helix-inversion barrier and can retain the helical sense in polar solvents and at high temperature. Owing to the tunability of dynamic helices, the currently developed helical polymer catalysts are primarily based on dynamic helical polymers, which offer the advantages of facile control over the helical sense and the ability to obtain enantiomeric products.25–28 However the instability of dynamic helicity makes the asymmetric reactions susceptible to solvent polarity, reaction temperature, functional groups of substrates and so on, posing challenges in practical applications.29–33 In contrast, static helical polymers exhibit greater structural stability and are less affected by environmental factors, thus holding more promising prospects for real-world applications.34–36 While there are challenges in precise synthesis of one-handed static helical polymers, the incorporation of catalysts based on static helical polymers has rarely been investigated.
Stereoregular polyisocyanides are a kind of static helical polymer. Their main chains are connected by C–C single bonds, allowing for the introduction of functional groups on each main-chain carbon.37–39 Compared with other helical polymers, diverse catalytic moieties can be functionalized onto every main chain atom of polyisocyanide. The closely adjacent catalytic groups are expected to exhibit a synergistic catalytic effect and provide the helical polymer catalyst with high catalytic activity. Moreover, the rigid polyisocyanide backbone can prevent chain entanglement, facilitating solvent dispersion and enabling homogeneous catalysis. The high molecular weight of the polymer catalysts also facilitates their recovery and recycling.39–41 However, the control of the helical sense in static polyisocyanides is relatively challenging, typically requiring the use of chiral monomers in asymmetric polymerization, which consumes a significant amount of chiral starting materials. Therefore, the development of polyisocyanide catalysts based on non-chiral precursors is of great importance. It is noteworthy that one-handed helical polyisocyanides, prepared via homopolymerization of chiral catalytic monomers or copolymerization of chiral monomers with achiral catalytic monomers, have been realized and applied in organocatalyzed asymmetric reactions.39–41 In contrast, helical polyisocyanides derived solely from achiral monomers for use in organometal-catalyzed asymmetric reactions remain largely unexplored.
Chiral carboxylic acid catalysts have become an important research direction in asymmetric catalysis due to their excellent catalytic performance. Beyond serving as organocatalysts in their own right, they can also function as ligands to cooperate with metals or other organic species in synergistic catalytic systems.42–44 A variety of chiral carboxylic acid catalysts have been reported, demonstrating outstanding activity and enantioselectivity in numerous asymmetric transformations, such as Diels–Alder cycloadditions and Michael additions (Fig. 1a).45–47 The Diels–Alder reaction is an important tool in synthetic organic chemistry, forming the key step in the preparation of many six-membered rings.48 The Michael addition reaction holds significant importance in the field of organic chemistry, particularly among numerous reactions involving carbon–carbon and carbon–heteroatom bond formation.49 Cu2+ complexes bearing chiral ligands can efficiently catalyze these reactions; however, upon reaction completion the structurally complex and synthetically demanding chiral ligands and the associated Cu2+ are difficult to recover and recycle, resulting in environmental pollution and waste of resources. Moreover, individual small-molecule catalysts lack a cooperative catalytic effect; therefore, the reaction proceeds relatively slowly. Subsequently, the helical chirality of the polymer backbone is employed to achieve high enantioselectivity in asymmetric reactions, while the cooperative effects of adjacent side-chain functionalities serve to enhance catalytic activity. Very recently, Suginome et al. reported that helical poly(quinoxaline-2,3-diyl) bearing L-lactic acid derived side chains enables highly stereoselective Diels–Alder reactions.50 However, the enantioselectivity may be deteriorated by solvents and substrate functionalities to some extent due to the dynamic character of the helical poly(quinoxaline-2,3-diyl) backbone.
 | ||
| Fig. 1 Structural diagrams of (a) carboxylic acids reported for asymmetric catalysis and (b) benzoic acid appended polyisocyanides developed in this work. | ||
In this work, we designed and synthesized one-handed preferred helical polyisocyanides bearing achiral benzoic acid pendants directly from achiral monomers (Fig. 1b). Two adjacent carboxyl groups on the polymer backbone coordinate with a single Cu2+, thereby catalyzing both asymmetric Diels–Alder reactions and Michael addition reactions with high yields and excellent enantioselectivities. The enantioselectivity of the reactions is governed entirely by the helicity of the polymer backbone. The catalyst is readily recovered and retains high activity and enantioselectivity over five consecutive runs. The cooperative catalytic effect between the catalytic groups on adjacent pendants significantly enhances the catalytic efficiency. The polymeric catalyst demonstrates a two-fold increase in efficiency compared to analogous small-molecule structures.
Results and discussion
Synthesis and characterization
The tert-butyl-protected carboxyl isocyanide 1 was polymerized using an alkyne–Pd(II) catalyst bearing chiral bidentate phosphine ligands (LR and LS) with different ratios of [1]/Pd(II) in THF at 55 °C (Fig. 2). The number-average molecular weight (Mn) and its distribution (Mw/Mn) of the isolated R- or S-poly-1ns (R- and S- indicate the chirality of the catalyst and the footnote n denotes the ratio of [1]/Pd(II)) were estimated by size exclusion chromatography (SEC) (Fig. 3a). It was revealed that the Mn was linearly correlated with the initial feed ratios of [1]/Pd(II) and all the isolated polyisocyanides exhibit low distribution with Mw/Mn < 1.14 (Fig. 3b), confirming the living nature of the polymerization. Thus, a series of helical polyisocyanides with different Mn values and low Mw/Mn were facilely prepared (Table S1, SI). To control the helical sense, S-poly-1ns were prepared following the same procedure using an S-Pd(II) catalyst. On treating the isolated polymers with trifluoroacetic acid (TFA), both the tert-butyl protecting groups on the pendants and residual Pd(II)-moiety on the chain-end were completely removed, yielding R- and S-poly-2ns. SEC analysis revealed that the Mn of the deprotected polymers was lower than that of the precursor while retaining the low Mw/Mn (Fig. 3c). For instance, the Mn and Mw/Mn were 2.1 kDa and 1.11 for S-poly-110, which were changed to 43.5 kDa and 1.14 for S-poly-1200, respectively. Similarly, R-poly-2ns were facilely prepared through the deprotection of R-poly-1ns (Table S1, SI). | ||
| Fig. 2 Synthesis of helical polyisocyanide-supported chiral catalysts. | ||
 | ||
| Fig. 3 (a) SEC curves for R- and S-poly-1ns. (b) Plots of Mn and Mw/Mn values of S-poly-1ns with initial feed ratios of 1 and Ar–Pd(II). (c) SEC curves for R- and S-poly-2ns. (d) SEC curves for S-poly-350, S-poly(350-b-130), S-poly(350-b-130-b-350) and S-poly(350-b-230-b-350). SEC curves for (e) S-poly(3m-b-230-b-3m)s and (f) S-poly(350-b-2n-b-350)s with different lengths of poly-3ms and poly-2ns segments. | ||
To eliminate the chain-end effect on the asymmetric catalysis, triblock copolymers were prepared via the sequential copolymerization of noncatalytic isocyanide monomer 3 with 1 and then with monomer 3. Owing to the living nature of the polymerization, a family of R- and S-poly(3m-b-1n-b-3m)s was prepared, and the structures were first confirmed by SEC analysis. The first block S-poly-350 was prepared via the living polymerization of 3 initiated by the S-Pd(II) catalyst. The isolated S- poly-350 showed a single modal SEC trace with an Mn and Mw/Mn of 12.1 kDa and 1.11, respectively. The copolymerization of S-poly-350 with monomer 1 led to the formation of diblock copolymer S-poly(350-b-130), which shifted to the short retention time region on SEC, and the Mn was increased to 16.8 kDa, and retained low distribution with an Mw/Mn of 1.16 (Fig. 3d). The triblock copolymer S-poly(350-b-130-b-350) was prepared via the block copolymerization of 3 with the Pd(II)-terminated diblock copolymer S-poly(350-b-130). The successful chain extension was confirmed by the SEC analysis; the elution peak of the resultant S-poly(350-b-130-b-350) shifted to a higher Mn region with an Mw/Mn of 1.19. Taking advantage of this synthetic strategy, a family of S-poly(3m-b-2n-b-3m)s was prepared just by varying the feed ratio of monomers to catalysts (Fig. 3e and f). For comparison, R-poly(3m-b-2n-b-3m)s possessing the opposite helical sense were prepared in the same way just using an R-Pd(II) catalyst instead of S-Pd(II).
In addition to SEC, the structures of the obtained polymers R- and S-poly-1ns and R- and S-poly-2ns were further characterized by 1H NMR. The 1H NMR spectrum of S-poly-130 showed characteristic resonance of the phenyl ring and the tert-butyl moiety (Fig. S1, SI). The degree of polymerization (DP) deduced from the 1H NMR spectrum via end-group analysis was 30, agreeing well with the theoretic value. After the deprotection, the resonance of the tert-butyl moiety completely disappeared on the 1H NMR spectrum of S-poly-230 (Fig. S1, SI). The FT-IR spectrum of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretch at ∼1600 cm−1 confirms the intact polyisocyanide backbone, while absorptions at 2500–3400 cm−1 (–OH) verify the formation of the targeted block copolymer (Fig. S2, SI). Similarly, the structures of triblock copolymers R- and S-poly(350-b-130-b-350) and the deprotected R- and S-poly(350-b-230-b-350) were also verified by 1H NMR and FT-IR as well (Fig. S1 and S2, SI). In the 1H NMR spectrum of the TFA-treated polymer, the disappearance of tert-butyl signals at 1–2 ppm indicates complete deprotection. The aromatic proton region (5–8 ppm) shows resonances characteristic of both S-poly-2ns and S-poly-3ms, confirming successful copolymerization (Fig. S1, SI). In the FT-IR spectrum, the CN stretch at ∼1600 cm−1 confirms the intact polyisocyanide backbone, while absorptions at 1750 cm−1 (NHCO) and 2500–3400 cm−1 (–OH) verify the formation of the targeted block copolymer (Fig. S2, SI).
N stretch at ∼1600 cm−1 confirms the intact polyisocyanide backbone, while absorptions at 2500–3400 cm−1 (–OH) verify the formation of the targeted block copolymer (Fig. S2, SI). Similarly, the structures of triblock copolymers R- and S-poly(350-b-130-b-350) and the deprotected R- and S-poly(350-b-230-b-350) were also verified by 1H NMR and FT-IR as well (Fig. S1 and S2, SI). In the 1H NMR spectrum of the TFA-treated polymer, the disappearance of tert-butyl signals at 1–2 ppm indicates complete deprotection. The aromatic proton region (5–8 ppm) shows resonances characteristic of both S-poly-2ns and S-poly-3ms, confirming successful copolymerization (Fig. S1, SI). In the FT-IR spectrum, the CN stretch at ∼1600 cm−1 confirms the intact polyisocyanide backbone, while absorptions at 1750 cm−1 (NHCO) and 2500–3400 cm−1 (–OH) verify the formation of the targeted block copolymer (Fig. S2, SI).
Chiroptical properties
The chiroptical properties of the synthetic polymers were investigated by circular dichroism spectroscopy. The homopolymer S-poly-140 showed intense negative CD in 280–460 nm, the absorption region of the helical backbone. The molecular CD intensity at 364 nm was estimated to be −0.79 and increased with DP, increasing to the maximum when the DP reached 200 (Fig. 4a). As anticipated, R-poly-1ns showed a similar absorption profile to S-poly-1ns while the CD spectra were positive and mirror images of those of S-poly-1ns. The CD intensity of S-poly-1ns was retained in solvents with varying polarities and over a temperature range of −78 °C to 35 °C (Fig. S3a and b, SI). | ||
| Fig. 4 CD and UV-vis spectra of (a) R- and S-poly-1ns, (b) R- and S-poly-2ns, (c) S-poly(350-b-2n-b-350)s and (d) R- and S-poly(3m-b-230-b-3m)s of different components. The CD and UV-vis spectra were recorded in THF with c = 0.2 mg mL−1. CD and UV-vis spectra of S-poly(350-b-230-b-350) measured in different solvents at room temperature (e) and in THF at different temperatures (f). | ||
Interestingly, the CD spectra of deprotected R- and S-poly-2ns also exhibited distinct CD signals. Specifically, the CD intensity of the homopolymer S-poly-240 at a wavelength of 364 nm was determined to be −1.22. This intensity increased with the DP, reaching a maximum value when the DP approached 200 (Fig. 4b). Similarly, R-poly-2ns showed an absorption profile similar to that of S-poly-2ns, and the CD spectra of R- and S-poly-2ns are mirror symmetric. Furthermore, the circular dichroism intensity of both R- and S-poly-2ns remains stable across different polar solvents and within a temperature range of 35 °C to −78 °C (Fig. S3c and d, SI). Quantitative fitting of the CD intensity at 364 nm (Δε364(n)) of S-poly-1ns and S-poly-2ns as a function of DP was performed using the literature-reported loose-end domain model by nonlinear least-squares analysis.51 For S-poly-1ns, the fitting results indicated that the intrinsic CD intensity Δε364(max) (CD intensity at 364 nm of an ideal infinitely long, perfectly one-handed helix) of an ideal one-handed helix was 1.002 ± 0.008 and the number of repeating units (m) forming the loose-end domain at a single chain terminus was 1.78 ± 0.15, whereas that for S-poly-2ns was 1.603 ± 0.012 for Δε364(max) and the m was 1.85 ± 0.18 (Fig. S4, SI).
Similarly, the triblock copolymers R- and S-poly(350-b-130-b-350) also exhibit strong CD signals. The CD intensity of S-poly(350-b-130-b-350) at a wavelength of 364 nm is estimated to be −7.09, while the deprotected S-poly(350-b-230-b-350) also shows a CD intensity of −8.26 (Fig. 4c). The CD intensity of S-poly(3m-b-2n-b-3m)s increases with the DP of both chain segments. Similarly, R-poly(3m-b-2n-b-3m)s showed an absorption profile similar to that of S-poly(3m-b-2n-b-3m)s, and the CD spectra of R- and S-poly(3m-b-2n-b-3m)s are mirror symmetric (Fig. 4d). Furthermore, the CD profile of S-poly(350-b-230-b-350) remains essentially unchanged across solvents of differing polarity and also shows negligible variation between −78 °C and 25 °C (Fig. 4e and f), demonstrating the robustness of its rigid helical structure under these conditions.
Catalyzing the asymmetric Diels–Alder reaction
The norbornene framework is a common skeleton in drug molecules and natural products, and it can be conveniently constructed via an asymmetric Diels–Alder reaction. Coordination of a carboxylate group to Cu is a well-established strategy for catalyzing the asymmetric Diels–Alder reaction.52,53 Having established the chirality of S-poly-2ns, we employed it as a chiral ligand by coordinating the appended carboxyl groups with CuCl2 to catalyze the asymmetric Diels–Alder reaction of 2-alkenoylpyridines with cyclopentadiene. Under the conditions of 25 °C in THF using S-poly-2200 and CuCl2 as the catalyst, the reaction afforded the target product in 79% yield but with a low enantiomeric excess (ee) of 14% and 90![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 diastereomeric ratio (dr) (run 3, Table S2, SI); furthermore, the ee decreased similarly when the DP of S-poly-2ns was reduced (Fig. S5, SI). In contrast, using the 4-nitrobenzoic acid coordinated to CuCl2 afforded yields of 80%, but with no enantioselectivity (run 3, Table 1). The polymer alone, without CuCl2, also failed to catalyze the reaction. These studies revealed that the Diels–Alder reaction was catalyzed by the coordination of CuCl2 with the carboxyl pendants. Subsequently, we optimized the reaction conditions by exploring various solvents, temperatures, copper salts, and the DP of the polymers. Ultimately, using S-poly-2200 and CuCl2 as catalysts, with THF/H2O in a 7:3 ratio as the solvent at −10 °C, we achieved the best reaction results, yielding 80%, 17% ee and 90:10 dr (run 11, Table S2, SI). Despite optimizing the reaction conditions, the ee remained relatively low.
:10 diastereomeric ratio (dr) (run 3, Table S2, SI); furthermore, the ee decreased similarly when the DP of S-poly-2ns was reduced (Fig. S5, SI). In contrast, using the 4-nitrobenzoic acid coordinated to CuCl2 afforded yields of 80%, but with no enantioselectivity (run 3, Table 1). The polymer alone, without CuCl2, also failed to catalyze the reaction. These studies revealed that the Diels–Alder reaction was catalyzed by the coordination of CuCl2 with the carboxyl pendants. Subsequently, we optimized the reaction conditions by exploring various solvents, temperatures, copper salts, and the DP of the polymers. Ultimately, using S-poly-2200 and CuCl2 as catalysts, with THF/H2O in a 7:3 ratio as the solvent at −10 °C, we achieved the best reaction results, yielding 80%, 17% ee and 90:10 dr (run 11, Table S2, SI). Despite optimizing the reaction conditions, the ee remained relatively low.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Solvent | T (°C) | Yieldb (%) | eec (%) | drc |
| a All reactions were performed with 4a (0.1 mmol), polymer (2 mol% relative to pendant carboxyl groups), CuCl2 (0.001 mmol), and 5 (0.2 mmol) in 2 mL of solvent. b Isolated yield. c ee and dr were determined by HPLC. | ||||||
| 1 | S-poly-230 | THF | 25 | 82 | 9 | 90:10 |
| 2 | S-poly-2100 | THF | 25 | 75 | 13 | 89:11 |
| 3 | 4-Nitrobenzoic acid | THF | 25 | 80 | 0 | 85:15 |
| 4 | S-poly-330 | THF | 25 | 83 | 0 | 77:23 |
| 5 | S-poly(330-b-230-b-330) | THF | 25 | 85 | 47 | 90:10 |
| 6 | S-poly(330-b-230-b-330) | THF/H2O (v/v = 7/3) | 25 | 85 | 71 | 90:10 |
| 7 | S-poly(320-b-230-b-320) | THF/H2O (v/v = 7/3) | 25 | 86 | 61 | 90:10 |
| 8 | S-poly(350-b-230-b-350) | THF/H2O (v/v = 7/3) | 25 | 84 | 79 | 92:10 |
| 9 | S-poly(350-b-220-b-350) | THF/H2O (v/v = 7/3) | 25 | 68 | 77 | 90:10 |
| 10 | S-poly(350-b-240-b-350) | THF/H2O (v/v = 7/3) | 25 | 86 | 72 | 90:10 |
| 11 | S-poly(350-b-230-b-350) | THF/H2O (v/v = 7/3) | –10 | 87 | 98 | 90:10 |
| 12 | R-poly(350-b-230-b-350) | THF/H2O (v/v = 7/3) | –10 | 85 | –96 | 91:9 |

We speculated that the low ee values might arise both from the chain ends' inability to adopt a well-defined helix and from the overall weak helicity of the polymer backbone, which together generate only a feeble chiral environment. Therefore, the triblock copolymers S-poly(330-b-230-b-330) were employed in the asymmetric Diels–Alder reaction. After coordinating S-poly(330-b-230-b-330) with CuCl2, it was used to catalyze the asymmetric Diels–Alder reaction of 2-alkenoylpyridines with cyclopentadiene. When the reaction was conducted using S-poly(330-b-230-b-330) and CuCl2 as catalysts in a 7:3 THF/H2O mixture at 25 °C, it gave the desired product in 85% yield, 47% ee and 90:10 dr (run 5, Table 1).
Having validated the efficacy of our block-copolymer catalytic system, the reaction conditions were further optimized to improve both yield and ee (runs 12–23, Table S2). Since it has been reported in the literature that water exerts a beneficial effect on the Diels–Alder reaction of 2-alkenoylpyridines with cyclopentadiene, the mixed solvent of THF and water was then evaluated.54 It was found that THF/H2O = 7:3 was optimal solvent for the reaction, which delivered 85% yield, 71% ee and 90:10 dr (run 6, Table 1). After carefully screening the anion of copper salts (CuBr2, Cu(NO3)2, and Cu(OTf)2), it was revealed that CuCl2 is the best choice, which delivered the target product with 85% yield, 71% ee and 90:10 dr (run 23, Table S2, SI). The minimum required loading of the polymer is 2 mol% relative to pendant carboxyl groups and that of CuCl2 is 0.001 mmol; further reduction of either results in a decrease in yield (runs 28–29, Table S2, SI). Lowering the temperature further improved enantioselectivity: at –10 °C, we obtained 87% yield, 98% ee and 90:10 dr (run 11, Table 1). Collectively, the optimal conditions for these helical polyisocyanide-catalyzed Diels–Alder reactions are performing the reaction in THF/H2O (7/3, v/v) at –10 °C using CuCl2.
We then examined the effect of polymer composition on catalysis. Increasing the degree of polymerization of monomer 3 enhanced the CD signal and improved the product ee from 59% to 67%. Increasing the DP of monomer 2, which raises the number of catalytic sites, increased the yield from 68% to 86% at identical reaction times, but an overly long 2-segment slightly reduced ee from 77% to 72% (runs 6–10, Table 1). Balancing yield and selectivity, we identified S-poly(350-b-230-b-350) as optimal, giving 84% yield, 79% ee and 92:8 dr (run 8, Table 1). Finally, using the enantiomeric backbone R-poly(350-b-230-b-350) under the same conditions gave the opposite R-configured product in 85% yield, –96% ee and 91:9 dr (run 12, Table 1), confirming the high stereoselectivity of these helical polyisocyanide-based chiral catalytic systems arising from the one-handed helicity of the polyisocyanide backbone. Regulating the backbone helicity could control the direction of enantioselectivity, and enantiomeric products could be readily obtained by using helical polyisocyanides with opposite handedness.
With the optimal reaction conditions in hand, substrate scope was evaluated by performing the asymmetric Diels–Alder reaction of various substituted 2-alkenoylpyridines with cyclopentadiene (Table 2). Para-halogenated dienophiles gave the desired products 6b–6d in high yields (83–85%) with 97–98% ee and dr (>87:13) (Table 2). Ortho-halogenated substrates, due to greater steric hindrance, showed slight dr but retained high ee (93–94%), e.g., 6e, 6f, and 6g (Table 2). The substrates with strong electron-withdrawing groups (–CN, –NO2, and –CF3) also delivered expected products 6h, 6i, and 6j with excellent enantioselectivities (97–98% ee), albeit with a modestly lower dr (>72:28) (Table 2). The substrates with electron-donating substituents (–Me and –OMe) afforded the products, e.g., 6k, 6l, 6m, 6n and 6o, with good ee (91–97%) and dr (>81:19) (Table 2). A naphthyl-substituted pyridine 6p similarly provided 94% ee with a 12:88 dr (Table 2). These results demonstrate that the catalytic system based on the one-handed helical polyisocyanide possesses broad substrate compatibility; regardless of the electronic nature and substitution pattern, it consistently delivers the desired products in high yield and excellent enantioselectivity and diastereoselectivity. Employing the enantiomeric R-poly(350-b-230-b-350) as the catalyst, the reaction similarly afforded products 6a′, 6b′, 6i′, and 6j′ in high yields (83–85%) with –97% to –98% ee and dr (>61:39) (Table 2).
|
a The reactions were performed using 4 (0.1 mmol) and 5 (0.2 mmol) in the presence of S-poly(350-b-230-b-350) (2 mol% relative to pendant carboxyl groups), CuCl2 (0.001 mmol) in 2 mL of a THF/H2O (7:3) solvent mixture, –10 °C, 48 h.
b The reaction was catalyzed by R-poly(350-b-230-b-350) under the same conditions.
|
|---|
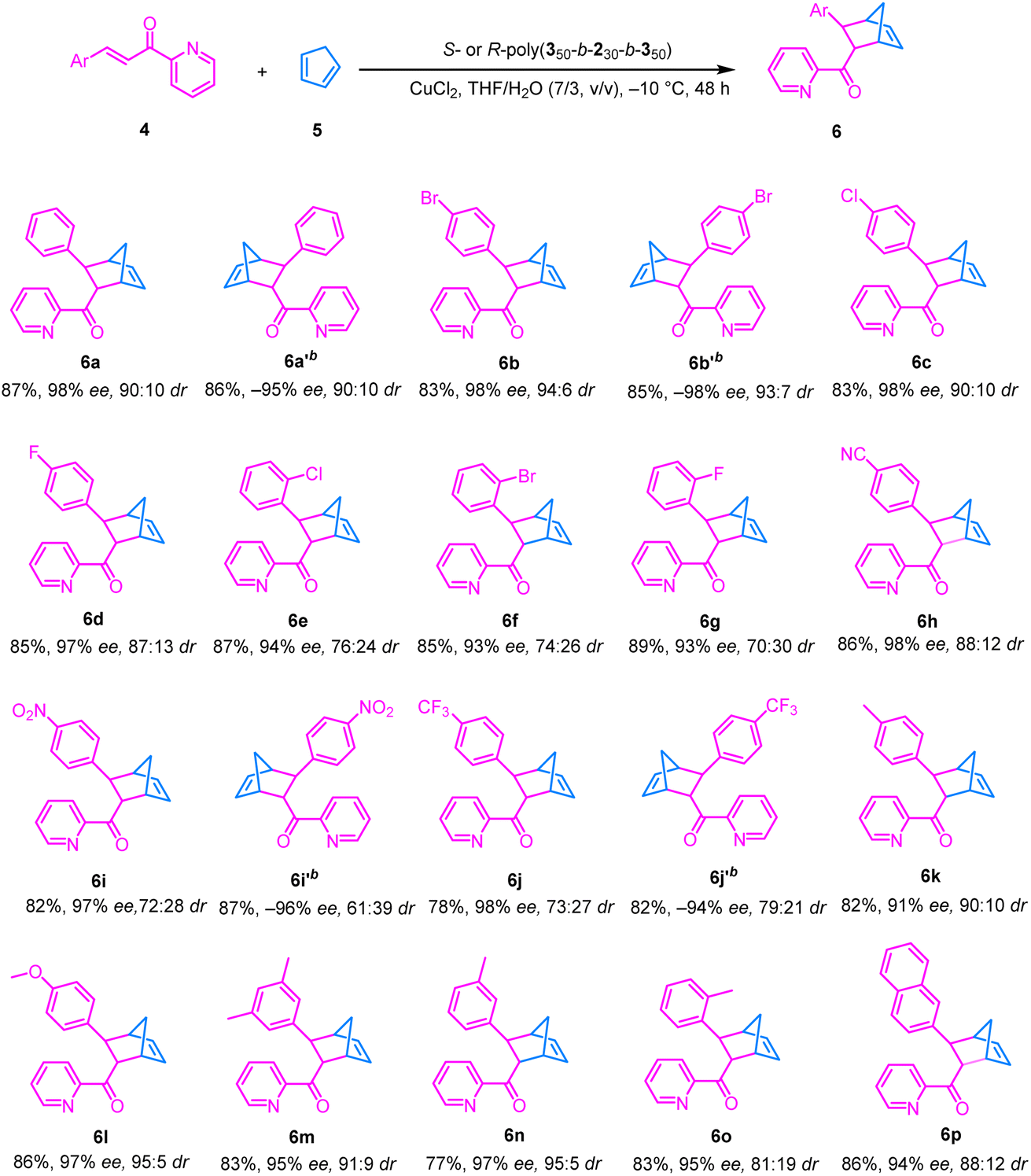
|
In addition to the excellent stereoselectivity, these helical polyisocyanide-based chiral catalytic systems also showed improved catalytic activity as compared to the small-molecule analogues. The reactions of 4a with 5 catalyzed by 4-nitrobenzoic acid, S-poly-230, and S-poly(350-b-230-b-350) were conducted in a THF/H2O (7:3) solvent mixture at −10 °C under identical experimental conditions and with the same catalyst loading. The reaction rate was systematically investigated by monitoring the conversion of 4a using 1H NMR and HPLC (Fig. 5a). As outlined in Fig. 5b, within 48 h, about 64%, 97%, and 98% of 4a was consumed using 4-nitrobenzoic acid, S-poly-230, and S-poly(350-b-230-b-350) as catalysts. Kinetic studies revealed the apparent rate constants for 4-nitrobenzoic acid, S-poly-230, and S-poly(350-b-230-b-350) were 0.021 h−1, 0.040 h−1 and 0.041 h−1 (Fig. 5b). The high catalytic activity of the helical polyisocyanide-based catalytic systems was due to the synergistic catalytic effect provided by the adjacent catalytic groups. Additionally, to verify whether the autocatalytic behavior was involved, the reaction of 4k with 5 was followed by HPLC to calculate the 4k conversion and ee of the yielded 6k (Fig. S6, SI). It was found that >99% of 4k was consumed within 48 hours, while the ee of 6k almost remained constant throughout the reaction process with variation <1%, suggesting that almost no autocatalysis effect was involved. This is probably because the product 6k does not possess sufficient chelating ability to compete with the carboxylate pendants.
 | ||
| Fig. 5 Plots of conversion (a) and −ln([M]/[M]0) values (b) of 4a as a function of reaction time. | ||
Catalyst recycling
Because the helical polyisocyanide has a high molecular weight while both substrates and products are small molecules, their differing solubilities allow easy separation of the polymer from the reaction mixture. During the catalytic reaction, the block copolymer is soluble in the reaction solution; after the reaction, addition of methanol precipitates the block copolymer while leaving the small-molecular products in solution. Centrifugation then recovers the polymer without affecting downstream processing of the products (Fig. S7a, SI). The recovered S-poly(350-b-230-b-350) was reused to catalyze the Diels–Alder reaction of 4a again under the abovementioned conditions and verify the catalytic activity. The reaction gave the desired product 6a in 83% yield and 96% ee, suggesting that the recovered S-poly(350-b-230-b-350) catalyst retained excellent activity and enantioselectivity. The catalyst was recycled 5 times without significant losses in selectivity and activity (Fig. S7b, SI). The ee dependence on substrate concentration experiments shows that the ee of product 6a slightly decreased as the concentration of substrate 4a reduced (Table S3, SI), indicative of a cooperative effect in the helical polyisocyanide-catalyzed enantioselective Diels–Alder reaction.Scale-up reaction and product transformation
Due to the high catalytic activity and excellent stereoselectivity of these helical polyisocyanide-based chiral catalytic systems, a gram-scale reaction of 4a with 5 catalyzed by S-poly(350-b-230-b-350) could also achieve good results, affording the target product 6a in high yield (76%) and good enantioselectivity (95% ee) and dr (90:10) (Fig. 6a). These results underscore the practical utility of these polymeric chiral catalytic systems. The afforded 6a possesses excellent post-functionalization potential via both oxidation and reduction reactions. According to previous literature reports, treatment of the obtained 6a with m-CPBA for the epoxidation of the internal CC bond afforded product 7 in 73% yield.55 In contrast, reduction of 6a with NaBH4 furnished the secondary alcohol product 8 in 86% yield (Fig. 6b).
 | ||
| Fig. 6 (a) Scale-up reaction of 4a and 5 using S-poly(350-b-230-b-350)/CuCl2 in THF/H2O (7/3, v/v) at –10 °C. (b) Product transformation of 6a into 7 and 8. | ||
Catalyzing asymmetric Michael addition
To determine the generality of the S-poly(3m-b-2n-b-3m)/CuCl2 catalytic system, we explored its application to other asymmetric catalytic reactions. Nitrogen-containing heteroaromatics and their derivatives are ubiquitous in bioactive and functional small molecules, and asymmetric Michael addition is a highly efficient route to such compounds.56 In particular, the nitro-Michael addition between nitroalkanes and chalcones is notable: the nitro group can be readily converted to other functionalities, and the resulting compounds often exhibit antibacterial activity where the R-enantiomer has been reported to outperform the S-enantiomer.57 Thus, we used the enantiomeric polymer catalyst R-poly(330-b-230-b-330) coordinated to CuCl2 to mediate the Michael addition of 9a with 10a in THF at 25 °C. Under these conditions, we obtained the Michael addition product in 81% yield and 56% ee (run 1, Table 3). As in the Diels–Alder reactions, using chain-end uncapped R-poly-2200 in the presence of CuCl2 as a catalyst, the Michael addition of 9a with 10a gave an expected product 11a in 75% yield but only 11% ee, again demonstrating that the chain-end capping of the poly(carboxy isocyanide)-based block is critical for achieving high enantioselectivity in this system (run 1, Table S4, SI).|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand | Copper catalyst | T (°C) | Yieldb (%) | eec (%) |
| a All reactions were carried out with 9a (0.1 mmol), polymer (2 mol% relative to pendant carboxyl groups), copper salt (0.001 mmol), and 10a (0.2 mmol) in 2 mL of CHCl3. b Isolated yield. c ee values were determined by HPLC. | |||||
| 1 | R-poly(330-b-230-b-330) | CuCl2 | 25 | 78 | 56 |
| 2 | R-poly-2200 | CuCl2 | 25 | 75 | 11 |
| 3 | R-poly(330-b-230-b-330) | Cu(OTf)2 | 25 | 82 | 69 |
| 4 | R-poly(310-b-230-b-310) | Cu(OTf)2 | 25 | 86 | 62 |
| 5 | R-poly(350-b-230-b-350) | Cu(OTf)2 | 25 | 80 | 75 |
| 6 | R-poly(350-b-220-b-350) | Cu(OTf)2 | 25 | 77 | 71 |
| 7 | R-poly(350-b-240-b-350) | Cu(OTf)2 | 25 | 83 | 72 |
| 8 | R-poly(350-b-230-b-350) | Cu(OTf)2 | –30 | 83 | 98 |
| 9 | S-poly(350-b-230-b-350) | Cu(OTf)2 | –30 | 85 | –97 |

To improve the yield and stereoselectivity, the reaction conditions were further optimized. First, we screened various organic solvents and found that CHCl3 gave the best result (78% yield, 56% ee; runs 1, Table 3). Next, we evaluated different copper salts. Unlike the Diels–Alder reaction, Cu(OTf)2 proved optimal for the Michael addition, delivering target product 11a in 82% yield and 69% ee (run 8, Table S4, SI). Finally, lowering the temperature further enhanced enantioselectivity. Performing the Michael addition of 9a with 10a at –30 °C using R-poly(350-b-230-b-350)/Cu(OTf)2 delivered 11a in 83% yield with 98% ee (run 8, Table 3). Using the enantiomeric backbone S-poly(350-b-230-b-350) under these same conditions afforded the opposite S-configured product in 85% yield and –97% ee (run 9, Table 3).
With the optimal Michael-addition conditions established, the substrate scope was then explored (Table 4). It was found that nitrostyrenes bearing halogen substituents all underwent the reaction (11b, 11c and 11d) and delivered the targets in high yields (>82%) with excellent enantioselectivity (>92% ee). Electron-donating methyl and methoxy substituents—whether in the para, meta, or ortho positions of nitrostyrene (11e–h), all delivered the anticipated targets in high yields (80–85%) with excellent enantioselectivities (92–98% ee). Furthermore, this catalytic system proved equally effective for nitro-Michael additions involving other heteroaromatic substrates such as pyrazine 11k and furan 11l, affording 82–84% yields and 98–99% ee. Employing the enantiomeric S-poly(350-b-230-b-350) as the catalyst, the reaction similarly afforded products 11a′, 11b′ and 11e′ in high yields (83–85%) with –87% to –96% ee (Table 4).
| a All reactions were carried out with 9 (0.1 mmol) and 10 (0.2 mmol), in the presence of R- or S-poly(350-b-230-b-350) (2 mol% relative to pendant carboxyl groups) and Cu(OTf)2 (0.001 mmol) in 2 mL of CHCl3 at –30 °C for 48 h. b The reaction was catalyzed by S-poly(350-b-230-b-350) under the same conditions. |
|---|

|
Catalyst recycling
The recovered R-poly(350-b-230-b-350) catalyst from the previous reaction was reused under the aforementioned conditions to catalyze the Michael addition reaction of 9a. The reaction afforded the target product 11a in 82% yield with 97% ee, demonstrating that the recovered catalyst retained excellent activity and enantioselectivity. Furthermore, even after five consecutive recycling cycles, no significant attenuation in either the selectivity or activity of the catalyst was observed (Fig. 7a). Stirring the R-poly(350-b-230-b-350) recovered from the Diels–Alder reaction in 0.1 M trifluoroacetic acid removed the CuCl2 coordinated to the carboxyl groups. After re-coordinating R-poly(350-b-230-b-350) with Cu(OTf)2, the material was used directly to catalyze the asymmetric Michael addition of 9a, affording the desired product 11a′ in high yield with the corresponding selectivity (Fig. 7b). | ||
| Fig. 7 (a) Yields and ee values of the Michael addition products obtained using the recovered R-poly(350-b-230-b-350). (b) Recycling and successively catalyzing two different asymmetric reactions of 4a and 9a using the S-poly(350-b-230-b-350) catalyst. | ||
Discussion of the mechanism
Building on previous studies, we propose a mechanism for helical polyisocyanide promoted enantioselectivity and activity for asymmetric transformations (Fig. 8). Taking the asymmetric Diels–Alder reaction as an example, when CuCl2 is added to the copolymer solution, the two adjacent carboxylic acid groups of the polymer pendants coordinate with one Cu2+. Due to the rigid helical structure of polyisocyanide, the coordinated Cu2+ complexes are helically aligned along the polymer chain, following the handedness of the polymer backbone. When the substrate azachalcone was introduced, its CO carbonyl oxygen and the pyridine N coordinate to the Cu2+ center, which significantly lowers the LUMO energy level of azachalcone, activating it as a stronger electrophilic acceptor. The resulting metal–ligand complex likewise adopts the helical arrangement following the polymer backbone. Concurrently, cyclopentadiene can be captured and localized in the vicinity of the metal active site by the polymer side chains through weak noncovalent interactions such as π–π stacking, CH–π contacts, and hydrophobic aggregation. This capture markedly increases the local effective concentration of the diene at the active site and restricts its conformational freedom, thereby reducing the entropic penalty for bimolecular encounter and facilitating transition-state formation. This combined effect rationalizes the observed higher reaction rate with S-poly(350-b-230-b-350) relative to using 4-nitrobenzoic acid as the ligand/activator. In the favored transition state, the Cu-activated azachalcone is approached from its Re-face by the HOMO of cyclopentadiene in an endo [4 + 2] fashion. Secondary orbital interactions—particularly the alignment between the carbonyl and the developing π* orbitals—further stabilize this endo trajectory and account for the observed dr. After C–C bond formation, the cyclohexene adduct dissociates, regenerating the catalytic Cu2+ site for the next cycle.
 | ||
| Fig. 8 Mechanistic study of the Diels–Alder reaction catalyzed by helical polymers. | ||
These combined effects collectively drive the reaction selectivity. That is, the combined effects of LUMO lowering via substrate coordination, favorable secondary π–π interactions in the endo transition state, higher occupancy-induced minor cooperative enhancement from local substrate accumulation, and spatial shielding of the Si-face by the helical backbone collectively bias S-poly(350-b-230-b-350)/CuCl2 toward Re-face endo addition in this asymmetric Diels–Alder reaction, affording high catalytic efficiency, excellent endo/exo selectivity, and outstanding enantioselectivity.
Conclusions
In conclusion, we have designed and synthesized a family of one-handed helical polyisocyanide homopolymers and block copolymers bearing achiral benzoic acids on the pendants, via the helix-sense selective living polymerization of the corresponding achiral monomers with chiral Pd(II)-catalysts. The one-handed helicity of the polymers was confirmed by CD analyses. The helical sense was quite stable and can tolerate various solvents with different polarities and functional groups and high temperatures. Remarkably, helical polymers appended with achiral benzoic acids showed great promise in asymmetric Diels–Alder reactions and Michael additions in the presence of copper salts, delivering target products in high yields (up to 87%) with excellent stereoselectivity (up to 98% ee and 90:10 dr). The enantioselectivity of the asymmetric reactions was completely determined by the handedness of the polyisocyanide backbone, regulating the helicity of polyisocyanide. Enantiomeric products could be facilely obtained in comparable yield and ee values. Compared to small-molecular analogues, both the catalytic activity and stereoselectivity were pronouncedly improved, because the unique C1 backbone of polyisocyanide provides the synergistic catalytic effect of the closely adjacent catalytic units on the pendants of the unique structure of polyisocyanide. The polymer catalyst can be recovered by simple precipitation and centrifugation and retains its high activity and enantioselectivity upon five consecutive recycles. Furthermore, the recycled polymer could sequentially catalyze both the Diels–Alder reaction and the Michael addition reaction by simply switching the copper salt. These studies confirm that highly optically active helical polymers can be readily prepared from racemic monomers. The obtained polymers combine the advantages of both homogeneous and heterogeneous catalysts. This study provides a framework for designing future recyclable chiral catalysts by introducing novel loading systems and key design principles.
Author contributions
Zong-Quan Wu and Na Liu designed and directed the project. Qi-Hui Ti, Ming-Yang Mo and Run-Tan Gao performed the experiments and analyzed the data. Zong-Quan Wu and Na Liu wrote the manuscript with input from all other authors.Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available in the supplementary information (SI) of this article. Supplementary information: experimental details, materials, synthesis, NMR and FT-IR spectra, SEC curves, and figures and tables. See DOI: https://doi.org/10.1039/d6sc01369d.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 22571117, 52573008, and 52273006), Major Research Plan of the National Natural Science Foundation of China (No. 92256201 and 92356302), and Science and Technology Development Plan of Jilin Province (20240101181JC).Notes and references
- M. Liu, L. Zhang and T. Wang, Chem. Rev., 2015, 115, 7304–7397 CrossRef CAS PubMed.
- E. Yashima, N. Ousaka, D. Taura, K. Shimomura, T. Ikai and K. Maeda, Chem. Rev., 2016, 116, 13752–13990 CrossRef CAS PubMed.
- K. Parkatzidis, H. S. Wang, N. P. Truong and Co-workers, Chem, 2020, 6, 1575–1588 CAS.
- M. Lago-Silva, M. Fernández-Míguez, R. Rodríguez, E. Quiñoa and F. Freire, Chem. Soc. Rev., 2024, 53, 793–852 RSC.
- H. Wu, J. Yang, B. B. C. Peters, L. Massaro, J. Zheng and P. G. Andersson, J. Am. Chem. Soc., 2021, 143, 20377–20383 CrossRef CAS PubMed.
- J. Grosskopf, C. Gopatta, R. T. Martin, A. Haseloer and D. W. C. MacMillan, Nature, 2025, 641, 112–121 CrossRef CAS PubMed.
- V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2022, 122, 1752–1829 CrossRef CAS PubMed.
- J. Cao, O. T. Zaremba, Q. Lei, E. Ploetz, S. Wuttke and W. Zhu, ACS Nano, 2021, 15, 3900–3926 CrossRef CAS PubMed.
- L. L. Zheng, J. Z. Li, M. Wen, D. Xi, Y. Zhu, Q. Wei, X.-B. Zhang, G. Ke, F. Xia and Z. F. Gao, Sci. Adv., 2023, 9, eadf5868 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem.–Eur. J., 2011, 17, 8514–8523 CrossRef CAS PubMed.
- V. M. Lechner, M. Nappi, P. J. Deneny, S. Folliet, J. C. K. Chu and M. J. Gaunt, Chem. Rev., 2022, 122, 1752–1829 CrossRef CAS PubMed.
- N. Luo, M. Turberg, M. Leutzsch, B. Mitschke, S. Brunen, V. N. Wakchaure, N. Nothling, M. Schelwies, R. Pelzer and B. List, Nature, 2024, 632, 795–801 CrossRef CAS PubMed.
- M. Reggelin, M. Schultz and M. Holbach, Angew. Chem., Int. Ed., 2002, 41, 1614–1617 CrossRef CAS PubMed.
- T. Yamamoto, T. Yamada, T. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef CAS PubMed.
- R. P. Megens and G. Roelfes, Chem.–Eur. J., 2011, 17, 8514–8523 CrossRef CAS PubMed.
- H. Iida, S. Iwahana, T. Mizoguchi and E. Yashima, J. Am. Chem. Soc., 2012, 134, 15103–15113 CrossRef CAS PubMed.
- T. Yamamoto, R. Murakami, S. Komatsu and M. Suginome, J. Am. Chem. Soc., 2018, 140, 3867–3870 CrossRef CAS PubMed.
- A. Shrotri, H. Kobayashi and A. Fukuoka, Acc. Chem. Res., 2018, 51, 761–768 CrossRef CAS PubMed.
- A. J. Boersma, D. Coquière, D. Geerdink, F. Rosati, B. L. Feringa and G. Roelfes, Nat. Chem., 2010, 2, 991–995 CrossRef CAS PubMed.
- D. Coquière, B. L. Feringa and G. Roelfes, Angew. Chem., Int. Ed., 2007, 46, 9308–9311 CrossRef PubMed.
- J. H. Yum, H. Sugiyama and S. Park, Chem. Rec., 2022, 22, e202100333 CrossRef CAS PubMed.
- M. Reggelin, S. Doerr, M. Klussmann, M. Schultz and M. Holbach, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5461–5466 CrossRef CAS PubMed.
- H. Wang, N. Li, Z. Yan, J. Zhang and X. Wan, RSC Adv., 2015, 5, 2882–2890 RSC.
- L. Zhou, K. He, N. Liu and Z.-Q. Wu, Polym. Chem., 2022, 13, 3967–3974 RSC.
- N. Kamiya, T. Kuroda, Y. Nagata, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2025, 147, 8534–8547 CrossRef CAS PubMed.
- T. Yamamoto, R. Murakami and M. Suginome, J. Am. Chem. Soc., 2017, 139, 2557–2560 CrossRef CAS PubMed.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, ACS Macro Lett., 2017, 6, 705–710 CrossRef CAS PubMed.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, Angew. Chem., Int. Ed., 2020, 59, 7251–7255 CrossRef CAS PubMed.
- Y. Yoshinaga, T. Yamamoto and M. Suginome, J. Am. Chem. Soc., 2020, 142, 18317–18323 CrossRef CAS PubMed.
- H. Wang, N. Li, Z. Yan, J. Zhang and X. Wan, RSC Adv., 2015, 5, 52410–52419 RSC.
- J. Deng and J. Deng, Polymer, 2017, 125, 200–207 CrossRef CAS.
- H. Zhang and J. Deng, Macromol. Chem. Phys., 2016, 217, 880–888 CrossRef CAS.
- H. Zhang, W. Yang and J. Deng, J. Polym. Sci., Part A:Polym. Chem., 2015, 53, 1816–1823 CrossRef CAS.
- M. Ando, R. Ishidate, T. Ikai, K. Maeda and E. Yashima, J. Polym. Sci., Part A:Polym. Chem., 2019, 57, 2481–2490 CrossRef CAS.
- L. M. Takata, H. Iida, K. Shimomura, K. Hayashi, A. A. dos Santos and E. Yashima, Macromol. Rapid Commun., 2015, 36, 2047–2054 CrossRef CAS PubMed.
- Z. Tang, H. Iida, H. Y. Hu and E. Yashima, ACS Macro Lett., 2012, 1, 261–265 CrossRef CAS PubMed.
- N. Liu, X. Zhou, L. Zhou and Z.-Q. Wu, Catalysts, 2021, 11, 1369 CrossRef CAS.
- L. Shen, L. Xu, X.-H. Hou, N. Liu and Z.-Q. Wu, Macromolecules, 2018, 51, 9547–9554 CrossRef CAS.
- Z.-Q. Wu, X. Song, Y.-X. Li, L. Zhou, Y.-Y. Zhu, Z. Chen and N. Liu, Nat. Commun., 2023, 14, 566 CrossRef CAS PubMed.
- L. Xu, L. Zhou, Y.-X. Li, R.-T. Gao, Z. Chen, N. Liu and Z.-Q. Wu, Nat. Commun., 2023, 14, 7287 CrossRef CAS PubMed.
- L. Zhou, B.-F. Chu, X.-Y. Xu, L. Xu, N. Liu and Z.-Q. Wu, ACS Macro Lett., 2017, 6, 824–829 CrossRef CAS.
- Y. Hirata, D. Sekine, Y. Kato, L. Lin, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2022, 61, e202205341 CrossRef CAS PubMed.
- L. T. Huang, Y. Kitakawa, K. Yamada, F. Kamiyama, M. Kojima, T. Yoshino and S. Matsunaga, Angew. Chem., Int. Ed., 2023, 62, e202305480 CrossRef CAS PubMed.
- Z. Zhu, M. Odagi, N. Supantanapong, W. Xu, J. Saame, H.-U. Kirm, K. A. Abboud, I. Leito and D. Seidel, J. Am. Chem. Soc., 2020, 142, 15252–15258 CrossRef CAS PubMed.
- Y. Kato, L. Lin, M. Kojima, T. Yoshino and S. Matsunaga, ACS Catal., 2021, 11, 4271–4277 CrossRef CAS.
- Y. Li, Y. C. Liou, J. C. A. Oliveira and L. Ackermann, Angew. Chem., Int. Ed., 2022, 61, e202212595 CrossRef CAS PubMed.
- G. Zhou, T. Zhou, A. L. Jiang, P. F. Qian, J. Y. Li, B. Y. Jiang, Z. J. Chen and B. F. Shi, Angew. Chem., Int. Ed., 2024, 63, e202319871 CrossRef CAS PubMed.
- S. Otto, F. Bertoncin and J. B. F. N. Engberts, J. Am. Chem. Soc., 1996, 118, 7702 CrossRef CAS.
- T. Hamlin, I. Fernández and F. M. Bickelhaupt, Angew. Chem., Int. Ed., 2019, 58, 8922–8926 CrossRef CAS PubMed.
- N. Kamiya, T. Yamamoto and M. Suginome, Chem.–Eur. J., 2025, 31, e02697 CrossRef CAS PubMed.
- N. Suzuki, T. Suzuki and H. Minami, J. Phys. Chem. B, 2025, 129, 5077–5081 CrossRef CAS PubMed.
- J. Jiang, Y. Meng, L. Zhang and M. Liu, J. Am. Chem. Soc., 2016, 138, 15629–15635 CrossRef CAS PubMed.
- S. Shen, Y. Sang, T. Wang, J. Jiang, Y. Meng, Y. Jiang, K. Okuro, T. Aida and M. Liu, Nat. Commun., 2019, 10, 3976 CrossRef PubMed.
- S. Otto, G. Boccaletti and J. B. F. N. Engberts, J. Am. Chem. Soc., 1998, 120, 4238 CrossRef CAS.
- H. Wei, Y. Zhang, S. Jin, Y. Yu, N. Chen, J. Xu and Z. Yang, Molecules, 2024, 29, 2978 CrossRef CAS PubMed.
- A. J. Simpson and H. W. Lam, Org. Lett., 2013, 15, 2586–2589 CrossRef CAS PubMed.
- G. Zhang, C. Zhu, D. Liu, J. Pan, J. Zhang, D. Hu and B. Song, Tetrahedron, 2017, 73, 129 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |