
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Brønsted acid–base approach for the net monoselective C–F substitution of (trifluoromethyl)alkanes
Nicholas J.
Coradi
 and
Jeffrey S.
Bandar
*
and
Jeffrey S.
Bandar
*
Department of Chemistry, Colorado State University, Fort Collins, Colorado 80523, USA. E-mail: jeff.bandar@colostate.edu
First published on 17th March 2026
Abstract
We disclose a method for the net coupling of nucleophiles with a single fluorine of unactivated (trifluoromethyl)alkanes. The process occurs via an initial base-promoted dehydrofluorination/defluorinative nucleophilic addition cascade to generate vinylfluoride intermediates that undergo a rapid hydrofluorination second step to yield gem-difluorinated products. Thus, the aliphatic-CF3 group of commercial building blocks and complex medicinal compounds can now be transformed into numerous classes of valuable α,α-difluoro(thio)ether substructures in an efficient and modular manner.
Introduction
Selective C–F functionalization of multifluorinated compounds is a powerful synthetic strategy for the preparation of precisely fluorinated products.1 An especially useful application of this concept is the substitution of a single fluorine of trifluoromethyl (–CF3) groups to access gem-difluorinated motifs (Fig. 1).2,3 Such transformations not only require initial activation of a strong C–F bond but also must not functionalize the relatively weaker C–F bonds of the gem-difluorinated products.4 These challenges typically preclude monoselective –CF3 functionalization via C–F insertion or fluoride abstraction mechanistic strategies.5–7 Nonetheless, substantial recent effort has led to general solutions for monoselective functionalization of π-system-adjacent –CF3 groups, where the appended functionality can be reduced or substituted (e.g., SN2′) to initiate selective C–F removal (Fig. 1).2,8–10 The Young group has also established a frustrated Lewis pair approach to selectively activate aryl- and heteroatom-adjacent trifluoromethyl groups.11 However, there remains no method for the monoselective C–F coupling of simple (trifluoromethyl)alkanes, which is likely a consequence of the increased C–F bond strengths combined with the absence of adjacent functionality.12 We herein describe a simple protocol for the net C–F substitution of (trifluoromethyl)alkanes that extends the synthetic advantages of defluorofunctionalization methodology to this important substrate class. | ||
| Fig. 1 Motivation for a C–F substitution method of (trifluoromethyl)alkanes. NuH = pronucleophile. | ||
A method to substitute a single fluorine on an alkyl–CF3 group would introduce a variety of useful synthetic capabilities. First, numerous building-block (trifluoromethyl)alkanes are commercially available and could be used as attractive precursors to difluorinated compounds.13,14 This would offer a more modular and streamlined approach to gem-difluorinated substructures over existing multistep sequences that typically rely on difficult deoxyfluorination procedures.3,15 Second, because –CF3 groups are inert to most chemical reagents, they could be carried through multistep syntheses and later transformed into gem-difluorinated derivatives.16 Third, (trifluoromethyl)alkyl substructures are frequently featured in complex compounds used in medicinal, materials, and agrochemical research due to the beneficial properties of multifluorinated alkyl groups.17 Diversification of the –CF3 group in these contexts would enable the rapid study of gem-difluoroalkyl analogs for potentially improved function. Thus, we were motivated to develop a C–F functionalization protocol to realize these myriad opportunities.
We reasoned the electron-withdrawing nature of a –CF3 group could be leveraged for the reactivity of an adjacent α-C–H bond such that C–F substitution could be achieved via a base-promoted dehydrofluorination/nucleophilic hydrofunctionalization cascade (Fig. 2a).12 The initial elimination step is inspired by two reports of unactivated (trifluoromethyl)alkane dehydrofluorination, as initially documented in 2004 via a KO-t-Bu-promoted elimination of 6,6,6-trifluorohexanoic acid to the corresponding gem-difluoroalkene.18 Next, the electrophilicity of unactivated gem-difluoroalkenes (e.g., not Michael acceptor or styrene derivatives) is evidenced by prior studies on intermolecular alkyllithium and intramolecular alkoxide addition–elimination reactions that generate vinyl fluoride products.19,20 These literature precedents, while limited, suggested an opportunity for C–F substitution directly from unactivated (trifluoromethyl)alkanes.12,21 This reactivity could thus enable the use of alkyl–CF3 groups as synthons and would also complement radical-based thiol and phenol addition methods of independently prepared gem-difluoroalkenes.20a,22
 | ||
| Fig. 2 (a) Initially proposed and (b) revised method design for a net C–F coupling reaction of (trifluoromethyl)alkanes and alcohols. | ||
Our initial efforts to promote tandem dehydrofluorination and fluorine-retentive nucleophilic addition (using alcohols, thiols, and amines) led to over-reactivity, yielding α-substituted vinyl fluorides exclusively (Fig. 2a).23,24 Nonetheless, we were encouraged by the fact that dehydrofluorination and intermolecular addition of common nucleophiles to an unactivated (trifluoromethyl)alkane occurs readily. We then realized that development of a regioselective hydrofluorination protocol for α-substituted fluoroalkenes could afford the desired α,α-difluorosubstituted target.25 We thus revised our approach toward C–F substitution to a simple two-step substitution/hydrofluorination sequence.
We selected alcohols as coupling partners to develop this C–F substitution method due to the value of α,α-difluoroethers and the current limitations associated with their preparation (Fig. 2b).26 These substructures are prominently featured in molecules used for medicinal, veterinary, agrochemical, battery, and electronic materials applications.27 For example, the strong stereoelectronic conformational effects of α,α-difluoroethers regulate optical and physical properties that are critical for liquid crystal display (LCD) technology.28 Difluoromethylene units are also commonly incorporated in bioactive compounds to increase lipophilicity and oxidative/metabolic stability, as well as to electronically modulate the properties of nearby functional groups.17,29 The most practiced route to α,α-difluoroethers is a three-step acid coupling/thionation/fluorination sequence, a process that is difficult to scale and can be incompatible with medicinally relevant functional groups.30 Alternative recent approaches, such as the use of AgF for the fluorination of thionoesters or the hydrofunctionalization of gem-difluoroalkenes, can display a broader functional group tolerance.15b,22 Selective alcohol coupling with (trifluoromethyl)alkanes could address important remaining limitations and unlock the unique capabilities of –CF3 defluorofunctionalization methodology. Furthermore, successful implementation of this strategy could be the foundation for a generalized alkyl–CF3 C–F substitution platform.
Results and discussion
We began this method development by studying the base-promoted cascade reaction between (trifluoromethyl)alkane 1 and 2-ethylhexan-1-ol (2) to form fluorovinyl ether 3 (Fig. 3a). First, we found that use of 3 eq. of KO-t-Bu or bis(trimethylsilyl)amide (K/NaHMDS) bases in DMF promote high-yielding product formation at room temperature (rt).31 Weaker bases (e.g., KOMe) and those with lithium countercations (e.g., LiHMDS) do not promote the initial dehydrofluorination step. With KHMDS as a representative base, the yield of fluorovinyl ether 3 decreases in less polar solvents or when less base or alcohol is used. While the initial experiments were conducted with solid KHMDS base, the use of inexpensive and convenient KHMDS solution (in THF) also promotes the reaction in high yield, including when conducted under ambient atmosphere. | ||
| Fig. 3 Investigations of the two key steps to achieve net C–F substitution. Yields determined by 19F NMR spectroscopy. aOther entries use solid base reagents. bPhMe as solvent. | ||
The potential for regioselective hydrofluorination of fluorovinyl ether 3 was assessed using crude material obtained from the reaction shown in Fig. 3a. Of the common commercial HF sources, Olah's reagent (Pyr·HF) and DMPU·HF promote hydrofluorination to form α,α-difluoroether 4 (75% and 95% yield, respectively), while less reactive HF reagents (e.g., NEt3·3HF and HBF4) are ineffective (Fig. 3b).32 Despite their good yields, we were concerned about the practicality of this protocol given that Olah's reagent and DMPU·HF readily etch glass and evolve HF fumes.33 We therefore sought to find conditions that employ NEt3·3HF, which is more easily handled and measured as it does not etch glass or generate substantial HF vapors at rt.34 We reasoned the poor hydrofluorination reactivity of NEt3·3HF is due to its lower acidity and the corresponding difficulty of vinyl ether protonation. Paquin addressed a similar challenge for terminal alkene hydrofluorination via use of methanesulfonic acid (MsOH) as a coacid in conjunction with NEt3·3HF.35 In the case of fluorovinyl ether 3, use of MsOH (pKa = 1.6 in DMSO)36 with NEt3·3HF increases the hydrofluorination yield to 83%, but with substantial side-product formation. We speculated that a less acidic coacid could prevent undesired reactivity and found that trifluoroacetic acid (TFA, pKa = 3.5 in DMSO)36 with NEt3·3HF affords 99% yield of 4 in PhMe. Beyond practical advantages, the dual acid approach is useful as it allows the coacid strength to be adjusted to improve hydrofluorination when other pronucleophile classes are used (vide infra).
We next implemented the conditions identified in Fig. 3 for a simplified C–F substitution protocol with a substrate scope shown in Chart 1. During the optimization studies, we observed that addition of base to (trifluoromethyl)alkanes and alcohols in DMF is exothermic.37 To address this, preparative scale reactions were cooled to 0 °C prior to base addition and then warmed and monitored (by TLC) until full conversion was reached. This protocol employs a solvent switch between the two steps, although we note that the entire procedure is often complete in 6 h and can be conducted in a one-pot manner.38 Although KO-t-Bu and KHMDS bases can both promote the initial step (Fig. 3a), we found that KHMDS generally promotes full conversion of primary (trifluoromethyl)alkanes (Condition A) while KO-t-Bu is more effective for secondary substrates (Condition B).39 We found the α,α-difluoroether products to be readily isolable and stable during storage, although we note previous studies have described variants that are prone towards hydrolysis.15b
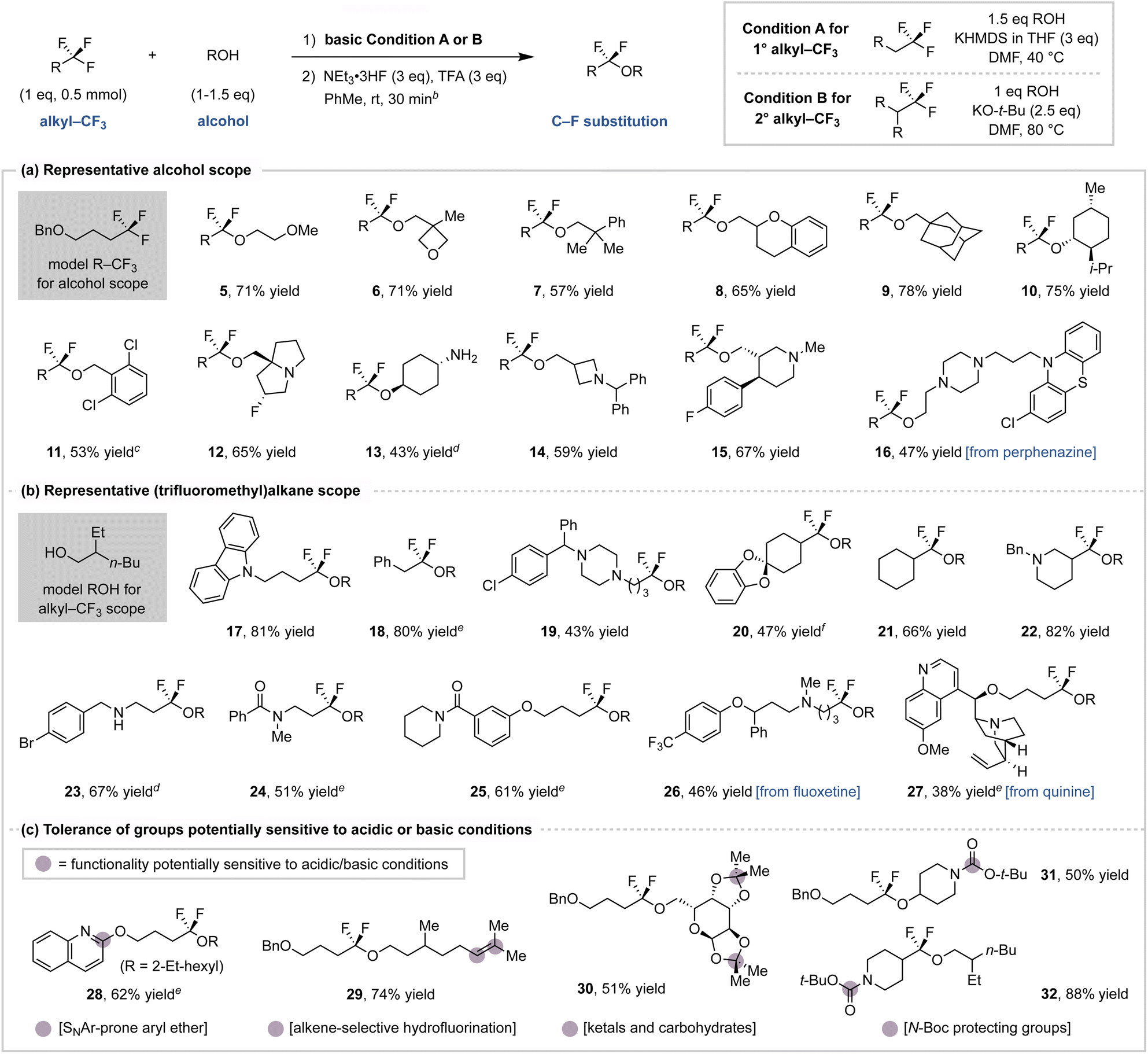 | ||
Chart 1 Substrate scope for (trifluoromethyl)alkane C–F substitution with alcohols.a aIsolated yields of 0.5 mmol scale reactions; the reaction progress for step 1 was monitored by TLC with times varying from 1–24 h. The use of racemic alcohol 2 results in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 dr for products that have multiple stereocenters (e.g., 19, 22, 26 and 27). bAn additional 2 eq. of each acid used if an amine is present in the compound. c1 eq. of alcohol and 2.5 eq. of base used. dKO-t-Bu used as base with DMA as solvent. eNaHMDS used as base. f4 eq. of base used at 50 °C. :1 dr for products that have multiple stereocenters (e.g., 19, 22, 26 and 27). bAn additional 2 eq. of each acid used if an amine is present in the compound. c1 eq. of alcohol and 2.5 eq. of base used. dKO-t-Bu used as base with DMA as solvent. eNaHMDS used as base. f4 eq. of base used at 50 °C. | ||
Chart 1a shows example alcohols that undergo C–F coupling with model (trifluoromethyl)alkane 1. Primary and secondary alcohols (5–10) provide α,α-difluoroethers in good yield, including those with oxetane (6) and azetidine (14) heterocycles that are prone to ring-opening under acidic conditions.40 Perphenazine (16) and other hydroxyl-bearing drug fragments (12, 15) also couple in moderate to good yield. In addition to tertiary-amine-bearing alcohols (14, 15), unprotected primary amines (13) are tolerated with exclusive O-coupling selectivity. Chemoselectivity for –CF3 defluorofunctionalization is observed in the presence of a secondary alkyl fluoride (12). Under the standard conditions, tertiary alcohols can form vinyl ether intermediates although the hydrofluorination step is low yielding.
Chart 1b demonstrates this protocol on commercially available (trifluoromethyl)alkane building blocks and their derivatives using 2-ethylhexan-1-ol (2) as the nucleophile. Substrates bearing diversifiable functionality such as aryl halides (19, 23), secondary amines (23), ketals (20), or alkenes (27) are well tolerated. The ability to activate secondary –CF3 groups allows for the functionalization of medicinally-relevant trifluoromethylated cyclohexanes (20, 21) and piperidines (22). This protocol also tolerates amide groups (24, 25) that would react under traditional deoxyfluorination sequences.15 (Trifluoromethyl)alkyl-appended drugs, such as fluoxetine (26) and quinine (27) derivatives, provide moderate C–F substitution yields. Moreover, fluoxetine derivative 26 demonstrates this protocol's chemoselectivity for alkyl–CF3 functionalization in contrast to many recently developed methods for Ar–CF3 activation.8
Chart 1a and b collectively demonstrate that this method tolerates diverse functionality despite the use of relatively strong bases and acids. This led us to further investigate the compatibility of groups that are known to be acid-sensitive, as shown in Chart 1c. The acidic hydrofluorination step is chemoselective for the fluorovinyl ether intermediate over other olefins (27, 29). Despite the use of excess TFA, ketals (20, 30) and Boc-protected amines (31, 32) remain intact.41 We also note that an SNAr-prone 2-substituted quinoline (28) gives good C–F coupling yield with only a minor amount of SNAr observed during fluorovinyl ether formation, indicating tolerance for certain base-sensitive groups.42
We next sought to engage phenols in this coupling reaction given that the α,α-difluoroalkyl aryl ether linkage is valued in LCD technology and represents an alkylated derivative of medicinally-relevant Ar–OCF3/CF2H substituents.26 The addition of 18-crown-6 to Condition A enables high-yielding aryl fluorovinyl ether formation, while the use of the stronger acid MsOH over TFA is needed to promote subsequent hydrofluorination.43Fig. 4a shows examples of C–F substitution on a carbazole-based (trifluoromethyl)alkane with electron-rich phenols (33, 34), the vitamin E compound α-tocopherol (35), and the secondary-amide-bearing drug paracetamol (36). This procedure is also applicable to β-aryl (37) and free amine-containing (38) (trifluoromethyl)alkanes. The generality of this C–F substitution protocol was further evaluated with thiol coupling partners. The use of the phenol coupling basic conditions enables formation of fluorovinyl alkylthioethers in good yields. However, due to the lower basicity of vinyl thioethers over vinyl ethers, we found that hydrofluorination requires the use of pyr·HF with CuCl as a Lewis acid additive.44Fig. 4b illustrates alkylthiol C–F coupling products that form in moderate to good yields (39–41). Collectively, the pronucleophile expansion in Fig. 4 provides an alkyl–CF3 coupling route that complements radical-based hydrothiolation and -phenolation reactions of gem-difluoroalkenes.45 The reactions shown in Fig. 4 are low yielding for electron-deficient phenols and thiophenols. We also note that arylamines and diarylphosphines undergo the first step of this method but do not undergo successful hydrofluorination under the optimized conditions; this information is described in the SI.
 | ||
| Fig. 4 Value and demonstration of the use of phenols and thiols for (trifluoromethyl)alkane C–F coupling. Isolated yields reported. aExcess (∼8 eq.) acid used. bUsed hydrofluorination conditions shown in Fig. 4b. cYield determined in crude reaction material by 19F NMR spectroscopy. | ||
This C–F coupling method could be particularly useful when applied to the derivatization of aryl trifluoroethoxy substituents (ArOCH2CF3) that are commonly incorporated in medicinal compounds (Fig. 5).46 Access to difluorinated derivatives (ArOCH2CF2–R) can be prohibitively difficult as this often requires the independent synthesis of each difluoroalkyl coupling partner (e.g., HOCH2CF2–R) and its successful attachment to the arene.47 In this regard, direct ArOCH2CF3 C–F diversification could substantially streamline difluoroalkyl analog study. Such a C–F substitution method must overcome several potential competing side reactions that are unique compared to the (trifluoromethyl)alkanes in Chart 1. This includes the potential for nucleophilic substitution of the aryloxy group over fluoride elimination, as well as the requirement of regioselective hydrofluorination of the 1,2-vinyldiether intermediate (Fig. 5a).48
 | ||
| Fig. 5 Utility, challenges, and demonstrations of C–F coupling of (2,2,2-trifluoroethoxy)aryl substructures. aROH = 2-ethylhexan-1-ol; yields determined by 19F NMR spectroscopy. bMsOH used instead of TFA in step 2. | ||
Despite these potential challenges, we found that subjection of 1-bromo-4-(2,2,2-trifluoroethoxy)benzene (42) to Condition A affords the desired vinyldiether 43 in 77% yield (Fig. 5b). Hydrofluorination of 43 with TFA as the co-acid gives poor yield of 44 (25%), while use of MsOH provides 44 in 72% yield as a single regioisomer. The regioselective hydrofluorination of the 1,2-vinyldiether intermediate 43 is likely due to the initial protonation being guided by the greater donating ability of the alkylether substituent. This protocol was then implemented on complex pharmaceuticals and drug-like compounds (Fig. 5c).49 A small library of lansoprazole sulfide difluoroalkyl analogs is shown through the coupling of i-PrOH (46), cyclobutanol (47), and methanol-d3 (48). C–F substitution of (±)-suvecaltamide with i-PrOH (49) is notable as such products would be inaccessible through late stage deoxyfluorination given the incompatibility of the amide group.15 Likewise, silodosin, which contains unprotected alcohol, amine, and amide groups, undergoes selective C–F coupling (50) to illustrate this method's broad potential for late-stage diversification.
Conclusions
In summary, (trifluoromethyl)alkanes are sufficiently reactive under basic conditions to generate functionalized fluorovinyl (thio)ether intermediates that undergo hydrofluorination to achieve net C–F substitution. This allows common (trifluoromethyl)alkane building blocks to serve as ideal retrons to functionalized gem-difluorinated units and provides new –CF3 diversification opportunities for complex molecules. More broadly, we anticipate the versatility of the fluoroalkene intermediates will provide additional opportunities for selective C–F functionalization sequences of (trifluoromethyl)alkanes.Author contributions
Both authors contributed to the conceptualization, analysis and writing of this project. N. J. C. was responsible for the methodology and J. S. B. was responsible for project administration and supervision.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: all experimental procedures and characterization data (e.g., NMR spectra, IR data, mass spectrometry data, and melting point analysis) for new compounds. See DOI: https://doi.org/10.1039/d6sc01042c.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. CHE-1944478. We also thank Colorado State University (CSU) for funding and the Analytical Resources Core (RRID: SCR_021758) at CSU for instrument access, training and assistance with sample analysis.Notes and references
- For general reviews on C–F bond functionalization, see: (a) L. V. Hooker and J. S. Bandar, Angew. Chem., Int. Ed., 2023, 62, e202308880 CrossRef CAS PubMed; (b) H. Amii and K. Uneyama, Chem. Rev., 2009, 109, 2119–2183 CrossRef CAS PubMed; (c) T. Ahrens, J. Kohlmann, M. Ahrens and T. Braun, Chem. Rev., 2015, 115, 931–972 CrossRef CAS PubMed; (d) Q. Shen, Y.-G. Huang, C. Liu, J.-C. Xiao, Q.-Y. Chen and Y. Guo, J. Fluorine Chem., 2015, 179, 14–22 CrossRef CAS; (e) J.-D. Hamel and J.-F. Paquin, Chem. Commun., 2018, 54, 10224–10239 RSC; (f) S. Li and W. Shu, Chem. Commun., 2022, 58, 1066–1077 RSC; (g) T. Fujita, K. Fuchibe and J. Ichikawa, Angew. Chem., Int. Ed., 2019, 58, 390–402 CrossRef CAS PubMed.
- For reviews on selective trifluoromethyl defluorofunctionalization, see: (a) F. Jaroschik, Chem.–Eur. J., 2018, 24, 14572–14582 CrossRef CAS PubMed; (b) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (c) G. Yan, K. Qiu and M. Guo, Org. Chem. Front., 2021, 8, 3915–3942 RSC.
- For reviews on synthetic routes to gem-difluorinated substructures, see: (a) D. R. Carvalho and A. H. Christian, Org. Biomol. Chem., 2021, 19, 947–964 RSC; (b) M.-C. Belhomme, T. Besset, T. Poisson and X. Pannecoucke, Chem.–Eur. J., 2015, 21, 12836–12865 CrossRef CAS PubMed; (c) A. Lemos, C. Lemaire and A. Luxen, Adv. Synth. Catal., 2019, 361, 1500–1537 CrossRef CAS PubMed; (d) D.-Q. Dong, H. Yang, J.-L. Shi, W.-J. Si, Z.-L. Wang and X.-M. Xu, Org. Chem. Front., 2020, 7, 2538–2575 RSC; (e) Z.-Y. Wang, Z. Zhang, W. Chen, Y. Liu, A. Sun and R. Xia, Org. Biomol. Chem., 2025, 23, 7383–7400 RSC; (f) Z. Feng, Y.-L. Xiao and X. Zhang, Acc. Chem. Res., 2018, 51, 2264–2278 CrossRef CAS PubMed.
- For discussions of C–F bond strengths, see: (a) D. O'Hagan, Chem. Soc. Rev., 2008, 37, 308–319 RSC; (b) D. M. Lemal, J. Org. Chem., 2004, 69, 1–11 CrossRef CAS PubMed; (c) K. B. Wiberg and P. R. Rablen, J. Am. Chem. Soc., 1993, 115, 614–625 CrossRef CAS.
- For discussions of the challenge of direct C–F bond activation processes, see ref. 1c and: (a) J. L. Kiplinger, T. G. Richmond and C. E. Osterberg, Chem. Rev., 1994, 94, 373–431 CrossRef CAS; (b) O. Eisenstein, J. Milani and R. N. Perutz, Chem. Rev., 2017, 117, 8710–8753 CrossRef CAS PubMed.
- For reviews of acid-promoted C–F activation and examples of over functionalization, see: (a) T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 CrossRef CAS; (b) J. Zhu, M. Pérez, C. B. Caputo and D. W. Stephan, Angew. Chem., Int. Ed., 2016, 55, 1417–1421 CrossRef CAS PubMed; (c) W. Gu, M. R. Haneline, C. Douvris and O. V. Ozerov, J. Am. Chem. Soc., 2009, 131, 11203–11212 CrossRef CAS PubMed.
- A recent method for ArCF3 C–F coupling operates via a rare example of fluorine atom abstraction; see: J. Koo, W. Kim, B. H. Jhun, S. Park, D. Song, Y. You and H. G. Lee, J. Am. Chem. Soc., 2024, 146, 22874–22880 CrossRef CAS PubMed.
- For selected examples of ArCF3 C–F coupling reactions, see: (a) D. B. Vogt, C. P. Seath, H. Wang and N. T. Jui, J. Am. Chem. Soc., 2019, 141, 13203–13211 CrossRef CAS PubMed; (b) S. E. Wright and J. S. Bandar, J. Am. Chem. Soc., 2022, 144, 13032–13038 CrossRef CAS PubMed; (c) Y.-C. Luo, F.-F. Tong, Y. Zhang, C.-Y. He and X. Zhang, J. Am. Chem. Soc., 2021, 143, 13971–13979 CrossRef CAS PubMed; (d) J. Xu, J.-W. Liu, R. Wang, J. Yang, K.-K. Zhao and H.-J. Xu, ACS Catal., 2023, 13, 7339–7346 CrossRef CAS.
- For examples of vinyl–CF3 functionalization, see: (a) F. Liu, Q. Wu, X. Wang, Q. Dong and K. Sun, Org. Chem. Front., 2025, 12, 3725–3737 RSC; (b) Q. Tian, B. Pei, C. Wang, C. Zhang and Y. Li, J. Org. Chem., 2022, 87, 10908–10916 CrossRef CAS PubMed; (c) F. Ye, Y. Ge, A. Spannenberg, H. Neumann, L.-W. Xu and M. Beller, Nat. Commun., 2021, 12, 3257 CrossRef CAS PubMed; (d) S.-S. Yan, D.-S. Wu, J.-H. Ye, L. Gong, X. Zeng, C.-K. Ran, Y.-Y. Gui, J. Li and D.-G. Yu, ACS Catal., 2019, 9, 6987–6992 CrossRef CAS.
- For examples of α-carbonyl CF3 C–F functionalization, see: (a) Y.-J. Yu, F.-L. Zhang, T.-Y. Peng, C.-L. Wang, J. Cheng, C. Chen, K. N. Houk and Y.-F. Wang, Science, 2021, 371, 1232–1240 CrossRef CAS PubMed; (b) M. W. Campbell, V. C. Polites, S. Patel, J. E. Lipson, J. Majhi and G. A. Molander, J. Am. Chem. Soc., 2021, 143, 19648–19654 CrossRef CAS PubMed; (c) R. Doi, M. Ohashi and S. Ogoshi, Angew. Chem., Int. Ed., 2016, 55, 341–344 CrossRef CAS PubMed; (d) H. Amii, T. Kobayashi, Y. Hatamoto and K. Uneyama, Chem. Commun., 1999, 1323–1324 RSC.
- (a) D. Mandal, R. Gupta, A. K. Jaiswal and R. D. Young, J. Am. Chem. Soc., 2020, 142, 2572–2578 CrossRef CAS PubMed; (b) S. Yoshida, K. Shimomori, Y. Kim and T. Hosoya, Angew. Chem., Int. Ed., 2016, 55, 10406–10409 CrossRef CAS PubMed; (c) R. Idogawa, Y. Kim, K. Shimomori, T. Hosoya and S. Yoshida, Org. Lett., 2020, 22, 9292–9297 CrossRef CAS PubMed; (d) K. Lye and R. D. Young, Chem. Sci., 2024, 15, 2712–2724 RSC.
- Net C–F functionalization reactions have been reported for polyhalogenated alkyl–CF3 substructures (e.g., hexafluoroisopropoxy derivatives) or through multistep sequences of specific substructures, see: (a) D. Chaudhary and M. R. Kuram, J. Org. Chem., 2024, 89, 7347–7351 CrossRef CAS PubMed; (b) X.-J. Tang and Q.-Y. Chen, Synlett, 2020, 31, 2046–2048 CrossRef CAS; (c) K. Wu and Q.-Y. Chen, Tetrahedron, 2002, 58, 4077–4084 CrossRef CAS; (d) J.-L. Xu, C. Yang, D.-T. Dai, W.-S. Hou and Y.-H. Xu, Adv. Synth. Catal., 2022, 364, 1228–1232 CrossRef CAS; (e) B. W. Metcalf, E. T. Jarvi and J. P. Burkhart, Tetrahedron Lett., 1985, 26, 2861–2864 CrossRef CAS.
- For reviews on synthetic routes to (trifluoromethyl)alkanes, see: (a) C. Alonso, E. Martínez de Marigorta, G. Rubiales and F. Palacios, Chem. Rev., 2015, 115, 1847–1935 CrossRef CAS PubMed; (b) X. Liu, C. Xu, M. Wang and Q. Liu, Chem. Rev., 2015, 115, 683–730 CrossRef CAS PubMed; (c) D. Mandal, S. Maji, T. Pal, S. K. Sinha and D. Maiti, Chem. Commun., 2022, 58, 10442–10468 RSC; (d) W. Domowki and R. A. Koliński, J. Fluorine Chem., 1972, 2, 210–213 CrossRef; (e) S. Barata-Vallejo, B. Lantaño and A. Postigo, Chem.–Eur. J., 2014, 20, 16806–16829 CrossRef CAS PubMed; (f) S. Kumawat, T. Bhatt, V. Goyal, H. Neumann, R. Zbořil, R. V. Jagadeesh, M. Beller and K. Natte, Coord. Chem. Rev., 2025, 542, 216885 CrossRef CAS; (g) H. Xiao, Z. Zhang, Y. Fang, L. Zhu and C. Li, Chem. Soc. Rev., 2021, 50, 6308–6319 RSC.
- Many (trifluoromethyl)alkyl-containing building blocks are available according to https://www.emolecules.com, such as those with halogen, alcohol, amine, and carbonyl groups for further synthetic diversification..
- (a) C. Ni, M. Hu and J. Hu, Chem. Rev., 2015, 115, 765–825 CrossRef CAS PubMed; (b) J. Newton, D. Driedger, M. B. Nodwell, P. Schaffer, R. E. Martin, R. Britton and C. M. Friesen, Chem.–Eur. J., 2019, 25, 15993–15997 CrossRef CAS PubMed.
- For the utility of late-stage C–F functionalization, see ref. 1a and: (a) N. J. Castellino, A. P. Montgomery, J. J. Danon and M. Kassiou, Chem. Rev., 2023, 123, 8127–8153 CrossRef CAS PubMed; (b) M. Wang, J. Ruskin, J. Marques, N. Garrison and T. Lectka, Chem. Rev., 2025, 125, 9382–9428 CrossRef CAS PubMed.
- For reviews that show the utility of alkyl–CF3 groups in medicinal, agricultural, and other commercial applications, see: (a) S. Jeanmart, A. J. F. Edmunds, C. Lamberth and M. Pouliot, Bioorg. Med. Chem., 2016, 24, 317–341 CrossRef CAS PubMed; (b) Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, iScience, 2020, 23, 101467 CrossRef CAS PubMed; (c) P. Jeschke, ChemBioChem, 2004, 5, 570–589 CrossRef CAS PubMed; (d) E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed; (e) N. A. Meanwell, J. Med. Chem., 2018, 61, 5822–5880 CrossRef CAS PubMed; (f) M. Kotthoff, J. Müller, H. Jürling, M. Schlummer and D. Fiedler, Environ. Sci. Pollut. Res., 2015, 22, 14546–14559 CrossRef CAS PubMed; (g) H. Mei, J. Han, R. Takeda, T. Sakamoto, T. Miwa, Y. Minamitsuji, H. Moriwaki, H. Abe and V. A. Soloshonok, ACS Omega, 2019, 4, 11844–11851 CrossRef CAS PubMed.
- (a) T. Pitterna, M. Böger and P. Maienfisch, Chimia, 2004, 58, 108–116 CrossRef CAS; (b) M. Jouffroy, P. Pye, S. Jerhaoui, W. Chen and M. Surkyn, J. Org. Chem., 2022, 87, 2136–2141 CrossRef CAS PubMed.
- (a) J. Ichikawa, Y. Wada, M. Fujiwara and K. Sakoda, Synthesis, 2002, 1917–1936 CAS; (b) S. Hayashi, T. Nakai and N. Ishikawa, Chem. Lett., 1980, 9, 935–938 CrossRef.
- For reviews on fluorine-retentive reactions of gem-difluoroalkenes, see: (a) J. P. Sorrentino and R. A. Altman, Synthesis, 2021, 53, 3935–3950 CrossRef CAS PubMed; (b) S. Koley and R. A. Altman, Isr. J. Chem., 2020, 60, 313–339 CrossRef CAS PubMed.
- For examples of defluorinative elimination/addition sequences on activated alkyl–CF3 substructures, such as those with acidifying β-aryl and -carbonyl groups, see: (a) M. Shigeno, Y. Shishido, A. Soga, K. Nozawa-Kumada and Y. Kondo, J. Org. Chem., 2023, 88, 1796–1802 CrossRef CAS PubMed; (b) M. Bilska-Markowska, M. Kaźmierczak, W. Jankowski and M. Hoffmann, Beilstein J. Org. Chem., 2024, 20, 2946–2953 CrossRef CAS PubMed; (c) M. Lecea, A. Grassin, L. Ferreiro-Mederos, S. Choppin, A. Urbano, M. C. Carreňo and F. Colobert, Eur. J. Org Chem., 2013, 2013, 4486–4489 CrossRef CAS; (d) A. T. Adam, F. R. Fronczek and D. A. Colby, Org. Lett., 2020, 22, 2630–2633 CrossRef CAS PubMed.
- For examples of radical-based heteroatom additions to unactivated gem-difluoroalkenes, see: (a) A. J. Intelli, R. T. Lee and R. A. Altman, J. Org. Chem., 2023, 88, 14012–14021 CrossRef CAS PubMed; (b) R. M. Herrick, M. K. A. El-Gaber, G. Coy and R. A. Altman, Chem. Commun., 2023, 59, 5623–5626 RSC; (c) J. P. Sorrentino, R. M. Herrick, M. K. Abd El-Gaber, A. Z. Abdelazem, A. Kumar and R. A. Altman, J. Org. Chem., 2022, 87, 16676–16690 CrossRef CAS PubMed; (d) Y.-Z. Liao, B.-Q. Ruan, H.-Y. Hu, H. Cheng, B.-Q. Wang and F. Pan, Org. Lett., 2025, 27, 9293–9298 CrossRef CAS PubMed; (e) Y.-Y. Zhang, Y. Zhang, X.-S. Xue and F.-L. Qing, Angew. Chem., Int. Ed., 2024, 63, e202406324 CrossRef CAS PubMed.
- We studied the potential for fluorine-retentive addition to (4,4-difluorobut-3-en-1-yl)benzene and 2-ethylhexan-1-ol (2); use of strong organic (e.g., guanidines and phosphazenes) and inorganic bases (e.g., KO-t-Bu, KHMDS) under various conditions do not yield fluorine retentive addition and results in either no reaction or formation of vinyl ether products. These results indicate that β-fluoride elimination after nucleophilic addition is very facile and, in a similar vein, all attempts to achieve fluorine-retentive addition as part of a dehydrofluorination sequence from (trifluoromethyl)alkanes (e.g., 1) did not lead to α,α-difluoroether products.
- We note that similar elimination-type products are known to form for polyhalogenated (trifluoromethyl)alkanes under basic conditions; for examples, see: (a) Y. Karuo, A. Tarui, K. Sato, K. Kawai and M. Omote, Beilstein J. Org. Chem., 2022, 18, 1567–1574 CrossRef CAS PubMed; (b) T. Kubota, K. Yamamoto and T. Tanaka, Chem. Lett., 1983, 12, 167–168 CrossRef.
- For reviews on hydrofluorination reactions, see: (a) X. Bertrand, L. Chabaud and J.-F. Paquin, Chem.–Asian J., 2021, 16, 563–574 CrossRef CAS PubMed; (b) R. Gauthier and J.-F. Paquin, Chem.–Eur. J., 2023, 29, e202301896 CrossRef CAS PubMed.
- For reviews on the value of α-fluoro(thio)ethers, see: (a) F. Leroux, P. Jeschke and M. Schlosser, Chem. Rev., 2005, 105, 827–856 CrossRef CAS PubMed; (b) B. Manteau, S. Pazenok, J.-P. Vors and F. R. Leroux, J. Fluorine Chem., 2010, 131, 140–158 CrossRef CAS; (c) P. Jeschke, E. Baston and F. R. Leroux, Mini-Rev. Med. Chem., 2007, 7, 1027–1034 CrossRef CAS PubMed; (d) H.-Y. Xiong, X. Pannecoucke and T. Besset, Chem.–Eur. J., 2016, 22, 16734–16749 CrossRef CAS PubMed.
- For discussions of specific α,α-difluoroether molecules studied in these applications, see: (a) A. K. Podichetty, S. Wagner, A. Faust, M. Schäfers, O. Schober, K. Kopka and G. Haufe, Future Med. Chem., 2009, 1, 969–989 CrossRef CAS PubMed; (b) D. Bianchi, P. Cesti, S. Spezia, C. Garavaglia and L. Mirenna, J. Agric. Food Chem., 1991, 39, 197–201 CrossRef CAS; (c) P. Kirsch, M. Bremer, A. Taugerbeck and T. Wallmichrath, Angew. Chem., Int. Ed., 2001, 40, 1480–1484 CrossRef CAS; (d) P. T. Meinke, J. Med. Chem., 2001, 44, 641–659 CrossRef CAS PubMed; (e) C. V. Amanchukwu, Z. Yu, X. Kong, J. Qin, Y. Cui and Z. Bao, J. Am. Chem. Soc., 2020, 142, 7393–7403 CrossRef CAS PubMed; (f) A. Casimiro-Garcia, J. I. Trujillo, F. Vajdos, B. Juba, M. E. Banker, A. Aulabaugh, P. Balbo, J. Bauman, J. Chrencik, J. W. Coe, R. Czerwinski, M. Dowty, J. D. Knafels, S. Kwon, L. Leung, S. Liang, R. P. Robinson, J.-B. Telliez, R. Unwalla, X. Yang and A. Thorarensen, J. Med. Chem., 2018, 61, 10665–10699 CrossRef CAS PubMed.
- (a) P. Kirsch and M. Bremer, ChemPhysChem, 2010, 11, 357–360 CrossRef CAS PubMed; (b) M. Hird, Chem. Soc. Rev., 2007, 36, 2070–2095 RSC.
- For examples and discussions of difluoromethylene and α,α-difluoro(thio)ethers in medicinal chemistry, see ref. 26 and 27, and: B. M. Johnson, Y.-Z. Shu and N. A. Meanwell, J. Med. Chem., 2020, 63, 6315–6386 CrossRef CAS PubMed.
- For discussions and examples of traditional routes to α,α-difluoroethers, see ref. 15 and: (a) W. H. Bunnelle, B. R. McKinnis and B. A. Narayanan, J. Org. Chem., 1990, 55, 768–770 CrossRef CAS; (b) M. Kuroboshi and T. Hiyama, Synlett, 1994, 251–252 CrossRef CAS.
- For reviews on HMDS bases, see: (a) R. E. Mulvey and S. D. Robertson, Angew. Chem., Int. Ed., 2013, 52, 11470–11487 CrossRef CAS PubMed; (b) B. T. Watson, H. Lebel, H. C. Malinakova and J. C. Hershberger, Potassium Hexamethyldisilazide, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2007 Search PubMed; (c) B. T. Watson and H. Lebel, Sodium Hexamethyldisilazide, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2005 Search PubMed.
- For discussions of common HF reagents, see: (a) S. Caron, Org. Process Res. Dev., 2020, 24, 470–480 CrossRef CAS; (b) S. G. Sweeting and A. J. J. Lennox, J. Am. Chem. Soc., 2025, 147, 19329–19341 CrossRef CAS PubMed; (c) G. Haufe, Chem. Rec., 2023, 23, e202300140 CrossRef CAS PubMed; (d) S. Liang, G. B. Hammond and B. Xu, Chem.–Eur. J., 2017, 23, 17850–17861 CrossRef CAS PubMed.
- For discussions of Olah's reagent and DMPU·HF, as well as their safety considerations, see ref. 31 and: (a) G. A. Olah, J. T. Welch, Y. D. Vankar, M. Nojima, I. Kerekes and J. A. Olah, J. Org. Chem., 1979, 44, 3872–3881 CrossRef CAS; (b) S. P. Kotun, G. K. S. Prakash and J. Hu, Pyridinium Poly(Hydrogen Fluoride), in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2007 Search PubMed; (c) O. E. Okoromoba, J. Han, G. B. Hammond and B. Xu, J. Am. Chem. Soc., 2014, 136, 14381–14384 CrossRef CAS PubMed; (d) P. A. Champagne, J. Desroches, J.-D. Hamel, M. Vandamme and J.-F. Paquin, Chem. Rev., 2015, 115, 9073–9174 CrossRef CAS PubMed.
- For discussions and safety considerations of Et3N·3HF, ref. 32 and: (a) S. Hara, Triethylamine Trihydrofluoride, in Encyclopedia of Reagents for Organic Synthesis, John Wiley & Sons, Ltd, 2005 Search PubMed; (b) R. Franz, J. Fluorine Chem., 1980, 15, 423–434 CrossRef CAS.
- X. Bertrand and J.-F. Paquin, Org. Lett., 2019, 21, 9759–9762 CrossRef CAS PubMed.
- F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS.
- For a safety discussion on the use of strong bases in DMF, see: Q. Yang, M. Sheng and Y. Huang, Org. Process Res. Dev., 2020, 24, 1586–1601 CrossRef CAS.
- Et3N·3HF does not etch glass and can be transferred using typical glass syringes. However, we conducted step 2 in PTFE vessels to avoid any potential glass etching caused by the dual acid system. We note that the entire procedure (steps 1 and 2) can be conducted in a one-pot manner using a PTFE round bottom flask; this was done for substrate 5 and is described in the SI.
- For example, the use of KO-t-Bu for the substrates in Chart 1a typically leads to 15–20% remaining (trifluoromethyl)alkane 1 that is difficult to separate from the α,α-difluoroether product. In contrast, KHMDS generally led to full consumption of 1. For similar reasons, step 1 was monitored by TLC and the reaction time for each substrate (generally 4 h or 16 h) is described in the SI.
- (a) J. A. Bull, R. A. Croft, O. A. Davis, R. Doran and K. F. Morgan, Chem. Rev., 2016, 116, 12150–12233 CrossRef CAS PubMed; (b) H. Mughal and M. Szostak, Org. Biomol. Chem., 2021, 19, 3274–3286 RSC.
- In these cases, we do not observe competing deprotection of the acid-sensitive groups. For examples of related acidic deprotections, see: (a) S. E. López and J. Salazar, J. Fluorine Chem., 2013, 156, 73–100 CrossRef; (b) I. W. Ashworth, B. G. Cox and B. Meyrick, J. Org. Chem., 2010, 75, 8117–8125 CrossRef CAS PubMed; (c) W. Li, J. Li, Y. Wu, N. Fuller and M. A. Markus, J. Org. Chem., 2010, 75, 1077–1086 CrossRef CAS PubMed; (d) J. Tian and L. Zhou, Chem. Sci., 2023, 14, 6045–6051 RSC.
- Highly base-sensitive groups (e.g., alkyl halides) are not tolerated in this protocol. For a discussion of nucleophilic substitution reactions of electron-deficient aryl ethers, see: M. Shigeno, K. Hayashi, K. Nozawa-Kumada and Y. Kondo, Org. Lett., 2019, 21, 5505–5508 CrossRef CAS PubMed.
- 18-Crown-6 is likely needed to enhance the nucleophilicity of the phenoxide nucleophile for addition to the gem-difluoroalkene intermediate, while use of the stronger acid MsOH is likely need for protonation of the less basic fluorovinyl aryl ether intermediate.
- We did not observe hydrofluorination of vinyl thioethers using Brønsted acid systems alone. We hypothesize that CuCl either disrupts H-bonding in Olah's reagent (see ref. 32) or may act as an acid to activate the vinylthioether towards fluoride addition; see: Y. Yamamoto, J. Org. Chem., 2007, 72, 7817–7831 CrossRef CAS PubMed.
- For radical-based routes for phenol/thiol addition to gem-difluoroalkenes, see ref. 22. For a nucleophilic phenol addition process to 2,2-difluorostyrenes, see: D. L. Orsi, M. R. Yadav and R. A. Altman, Tetrahedron, 2019, 75, 4325–4336 CrossRef CAS PubMed.
- (a) J. Irurre, J. Casas and A. Messeguer, Bioorg. Med. Chem. Lett., 1993, 3, 179–182 CrossRef CAS; (b) R. Szpera, P. G. Isenegger, M. Ghosez, N. J. W. Straathof, R. Cookson, D. C. Blakemore, P. Richardson and V. Gouverneur, Org. Lett., 2020, 22, 6573–6577 CrossRef CAS PubMed.
- For the synthesis of ArOCH2CF3 compounds, see: B. Pethő and Z. Novák, Asian J. Org. Chem., 2019, 8, 568–575 CrossRef.
- We note that addition reactions to structurally related trifluorovinyl aryl ethers have been reported. (a) S. T. Iacono, N. J. Weeks and D. W. Smith Jr, J. Fluorine Chem., 2023, 271, 110187 CrossRef CAS; (b) J. D. Moody, D. VanDerveer, D. W. Smith Jr and S. T. Iacono, Org. Biomol. Chem., 2011, 9, 4842–4849 RSC.
- For references to the drug substructures used in Fig. 5c, see: (a) J. Rybniker, A. Vocat, C. Sala, P. Busso, F. Pojer, A. Benjak and S. T. Cole, Nat. Commun., 2015, 6, 7659 CrossRef PubMed; (b) T. S. Reger, Z.-Q. Yang, K.-A. S. Schlegel, Y. Shu, C. Mattern, R. Cube, K. E. Rittle, G. B. McGaughey, G. D. Hartman, C. Tang, J. Ballard, Y. Kuo, T. Prueksaritanont, C. E. Nuss, S. M. Doran, S. V. Fox, S. L. Garson, Y. Li, R. L. Kraus, V. N. Uebele, J. J. Renger and J. C. Barrow, Bioorg. Med. Chem. Lett., 2011, 21, 1692–1696 CrossRef CAS PubMed; (c) K. Shibata, R. Foglar, K. Horie, K. Obika, A. Sakamoto, S. Ogawa and G. Tsujimoto, Mol. Pharmacol., 1995, 48, 250–258 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |