
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanochemically assembled organometallic complexes: a mechanistic study
Lama Hamdounaab,
Gianmarco Pisanò a,
Andrew G. M. Rankinc,
Julien Tréboscc,
Laura Falivened,
Ida Ritaccod,
Olivier Devose,
Laurent Delevoyeb,
Pascal Rousselb,
Lucia Caporasod,
Luigi Cavallof,
Steven P. Nolana,
Olivier Lafon*b and
Catherine S. J. Cazin*a
a,
Andrew G. M. Rankinc,
Julien Tréboscc,
Laura Falivened,
Ida Ritaccod,
Olivier Devose,
Laurent Delevoyeb,
Pascal Rousselb,
Lucia Caporasod,
Luigi Cavallof,
Steven P. Nolana,
Olivier Lafon*b and
Catherine S. J. Cazin*a
aDepartment of Chemistry and Centre for Sustainable Chemistry, Ghent University, Krijgslaan 289 - S3, 9000 Ghent, Belgium. E-mail: catherine.cazin@ugent.be
bUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181-UCCS-Unité de Catalyse et Chimie du Solide, 59000 Lille, France
cUniv. Lille, CNRS, INRAE, Centrale Lille, Univ. Artois, FR2638 - IMEC – Fédération Chevreul, 59000 Lille, France
dDepartment of Chemistry and Biology, University of Salerno, Via Papa Giovanni Paolo II 132, 84084 Fisciano (SA), Italy
eUniversité Lille, CNRS, UMR 8516, Laboratoire Avancé de Spectroscopie pour les Intéractions la Réactivité et l’Environnement (LASIRE), Lille, France
fKAUST Catalysis Center, Physical Sciences and Engineering Division, King Abdullah University of Science and Technology, 23955-6900 Thuwal, Saudi Arabia
First published on 22nd May 2026
Abstract
A combination of mechanical synthesis, solid-state analytical techniques (ssNMR, powder XRD, ATR-FTIR) and DFT calculations sheds light on the mechanisms operating in two organometallic solid-state syntheses and highlight the importance of workup protocols in mechanochemical syntheses. The data clearly indicate that product formation can occur during or post grinding.
Introduction
The use of mechanochemical methodologies to carry out chemical transformations of solid reactants is experiencing a period of great activity,1,2 with numerous applications being reported in a variety of areas of chemical science: organic,3,4 organometallic,5,6 main group,7 and supramolecular chemistry,8 as well as their uses in metal-mediated,9 and enzymatic-mediated10 transformations. The ability to provide a reaction environment devoid of solvents has offered the opportunity to tackle a major problem in the environmental efficiency of synthetic routes: the preponderant issue of solvent waste management.11 Mechanochemical conditions are generally characterized by higher yields and shorter reaction times when compared to their solution phase incarnations,12 and have resulted in opportunities to access products otherwise inaccessible by normal solvent-based methodologies13 (e.g., by eliminating issues associated with solubility of starting materials).14 Despite its simplicity, the outcome of a mechanochemical reaction involving the treatment of only solid reactants is still difficult to predict a priori and there is no established consensus as to what really is a reaction occurring through mechanochemical means alone.15,16 Hanusa and co-workers have recently reported on how the interaction with the solvent during the workup phase of the synthesis by ball-milling of an allyl calciate species altered the structural arrangement of the final product to its more thermodynamically stable form, and underlined how compounds produced under “dry” milling conditions “undergo a type of selection by solution”.17 Therefore, the understanding of mechanochemical reaction mechanism requires the characterization of the structure of chemical species in the solid state, prior to workup. The most employed characterization techniques for that purpose are powder X-ray diffraction (XRD) and vibrational spectroscopy.18 Nevertheless, a limited number of studies have recently demonstrated the potential of solid-state nuclear magnetic resonance spectroscopy (ssNMR) to monitor mechanochemical reactions.19–23 This technique is advantageous since it is quantitative and permits the observation of disordered or amorphous solids.Herein, we investigate the mechanochemical synthesis of organometallic complexes of Cu(I) and Rh(I) bearing N-heterocyclic carbene (NHC) ligands by combining results from various analytical techniques, including attenuated total reflectance infrared (ATR-FTIR) and ssNMR spectroscopies as well as powder XRD, with density functional theory (DFT) calculations. This project is part of recent efforts by our groups to investigate and exploit the so-called weak base route for the assembly of well-defined transition metal complexes bearing N-heterocyclic carbene (NHC) ligands, using both solution-based and mechanochemical methodologies.24 The goal of this study is to probe the reaction mechanism leading to the formation of NHC complexes in the solid state, and to understand the sequence of events leading to product formation.
As an initial system to probe mechanochemical reaction mechanism, we have targeted a classically studied copper-NHC based system. Among the various synthetic strategies leading to these complexes, the mechanochemical weak base approach to transition metal-NHC complexes reported concomitantly in 2020 by the Cazin and Udvardy groups stands out not only as it employs readily available starting materials in an operationally simple way and makes use of an inexpensive base (e.g., K2CO3, Cs2CO3) but also because of its wide applicability to different metal centers (i.e., Cu(I), Ag(I), Au(I), Rh(I), Pd(II)),25–27 in combination with a variety of NHC ligands [benzimidazolium, imidazol(in)ium,25–27 as well as cyclic (alkyl)(amino)carbene (CAAC)] (Scheme 1).28 A second system we recently studied also uses the weak base route to produce [Rh(acac)(CO)(NHC)]. In all known cases, however, mechanical treatment of the solid reactants was followed by the subsequent addition of solvents for the workup and purification of the products, and/or the processed samples were dissolved in solvents for characterization. Consequently, little evidence is available to confirm that the observed reactions occur during the mechanical event or because of any follow-up treatment. We now provide evidence for two possible reaction mechanisms involved in mechanochemical reactions: one that is in-line with the reaction occurring because of mechanochemical grinding and a second, that proceeds to product post-grinding after the workup.
 | ||
| Scheme 1 General weak base approach toward the mechanochemical synthesis of transition metal-NHC complexes.25 | ||
Results and discussion
The goal of the initial phase of the present study was to elucidate the mechanism operating in the weak base approach applied to the mechanical synthesis of [Cu(Cl)(NHC)] complexes (Scheme 2), as previously investigated.26 We hypothesized that the [Cu(Cl)(NHC)] complex is generated through the formation of a cuprate intermediate, drawing upon prior mechanistic studies on the solution-based weak base approach and the isolation of such species.29 We therefore investigated whether (1) these complexes and cuprate intermediates are formed in the solid state during the ball milling process or during the subsequent workup and (2) whether the final neutral complex formed purely based on the mechanochemical treatment (Scheme 3).30 For this purpose, we employed ssNMR analysis to monitor the reaction. The imidazolium salt IPr·HCl 1 (IPr = N,N′-bis-[2,6-(di-iso-propyl)phenyl]imidazol-2-ylidene), was selected as a model substrate and the corresponding [Cu(Cl)(IPr)] complex was obtained in two different ways: (i) one-step one-pot (OP) synthesis and (ii) stepwise (SW) synthesis (Scheme 3).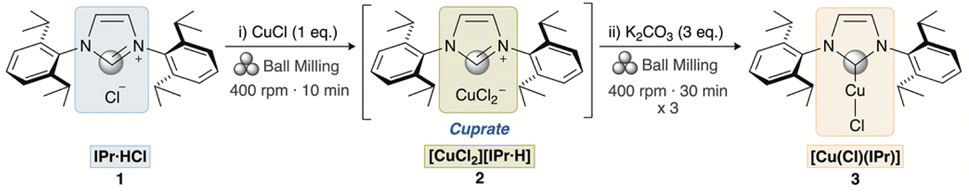 | ||
| Scheme 2 Weak base approach towards the mechanochemical synthesis of [Cu(Cl)(IPr)].29 The gray circle denotes the 13C2 carbon of the imidazolylidene moiety. | ||
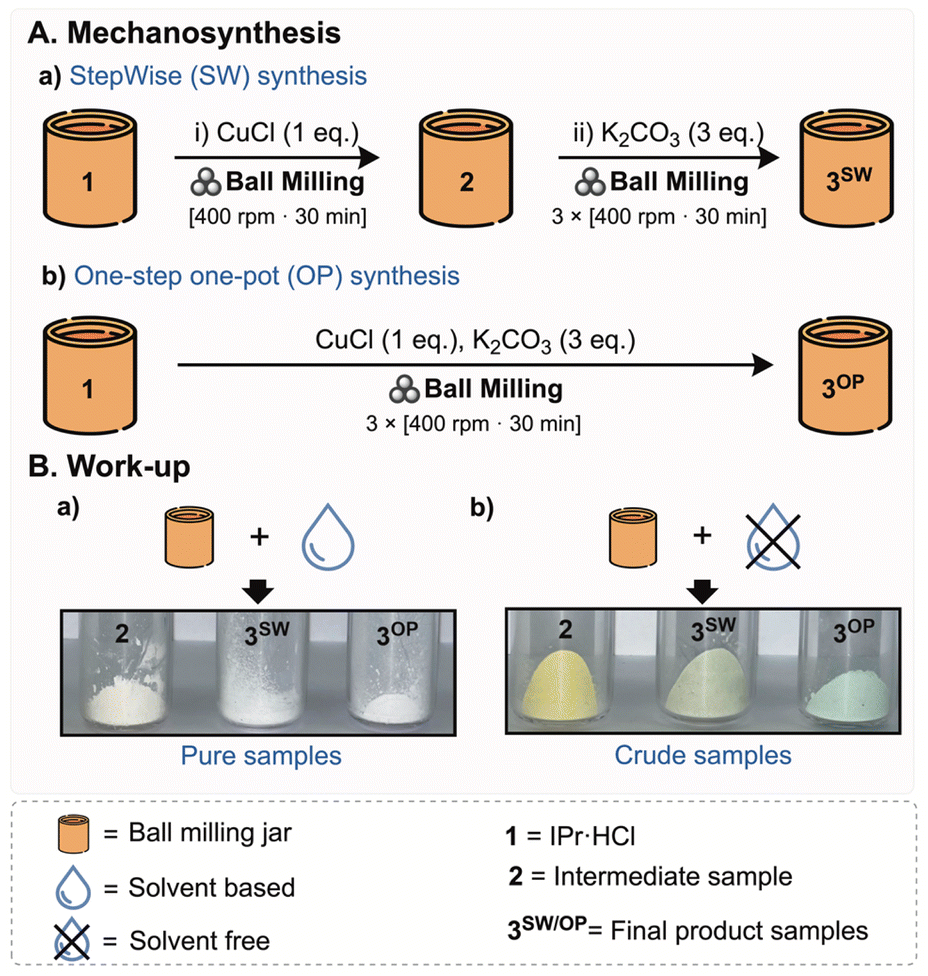 | ||
| Scheme 3 (A) Schematic outline of the mechanosynthesis: (a) step-wise (SW) synthesis and (b) one-step one-pot (OP) synthesis. (B) Purified and crude samples collected after (a) solvent-based and (b) solvent-free workup procedures. The noticeable colour difference between the crude and purified compounds results from variations in the quantities of unreacted reactants present in the crude mixtures, as verified by the absorbance and luminescence spectra (see Fig. S8). | ||
Proton and 13C solid-state NMR experiments at B0 = 9.4, 18.8, and 28.2 T, i.e., 1H Larmor frequencies of 400, 800, and 1200 MHz, respectively, were conducted to monitor the disappearance of the reactants and the formation of the intermediate and final product for the different synthetic routes and workup protocols (Scheme 2).
Solid-state NMR spectroscopy
 | ||
| Fig. 1 1D 1H MAS NMR spectra of (a) purified and (b) crude 2 (green), 3OP (blue), and 3SW (orange) acquired at B0 = 18.8 T with a MAS frequency, νR = 60 kHz. The spectrum of 1 compound is also shown in black in panel (a) for the sake of comparison. The shaded area denotes the region where the H2 signal of the 1 and 2 compounds resonates. The 1H signal denoted by the black box in panel b corresponds to KHCO3. The signal resonating near 5 ppm in 1D 1H NMR spectrum of crude 3OP and 3SW is assigned to water. | ||
After 10 min ball milling of [IPr·HCl] in the presence of CuCl, the H2 signal of [CuCl2][IPr·H] cuprate is detected in the purified sample but not in the crude (Fig. S3). However, by increasing the ball-milling time to 30 min, the cuprate H2 signal is also detected in the crude sample 2 (Fig. 1b), which confirms the formation of the cuprate in the solid-state during ball milling. The 1H spectra of crude 2 and 3OP/SW, depicted in Fig. 1b, exhibit a H2 signal resonating at 12.5 ppm, indicating the presence of unreacted IPr·HCl in the crude samples. In addition, a 1H signal at 13.5 ppm attributed to KHCO3 is present in the 1H spectra of crude 3OP/SW.31
 | ||
| Fig. 2 1D 1H → 13C CP-MAS NMR spectra of (a) purified and (b) crude 2 (green), 3OP (blue), and 3SW (orange) samples acquired at B0 = 9.4 T with νR = 12.5 kHz. The spectrum of the purified 1 is also shown in black in panel (a) for the sake of comparison. The multiplet assigned to the C2 site for samples 3OP and 3SW is shown as an inset in panels (a) and (b). The isotropic chemical shifts of the 13C2 nucleus are indicated on the spectra. Furthermore, the vertical dashed line in panel (b) denotes the isotropic chemical shift of the 13C2 nucleus in compound 1. | ||
The 1D 1H → 13C NMR spectra of compounds 3OP and 3SW also exhibit a multiplet around 180 ppm assigned to 13C2 nuclei in [Cu(Cl)(IPr)]. This multiplet arises from J-coupling and second-order quadrupolar-dipolar cross-terms with 63,65Cu isotopes with spin I = 3/2.36,37 This interpretation is confirmed by smaller splitting at a higher magnetic field, as seen in Fig. 3, since the second-order quadrupolar-dipolar cross-terms are inversely proportional to the B0 field strength. Assuming that the shortest C–Cu distance is equal to that measured by X-ray diffraction, 1.953 Å,38 the 13C–63,65Cu dipolar coupling constants are equal to b(13C–63Cu)/(2π) = 0.93 b(13C–65Cu)/(2π) = 1.078 kHz since the ratio of 63,65Cu gyromagnetic ratios is γ(63Cu)/γ(65Cu) = 0.93. Using these dipolar coupling constants, these spectra can be simulated with δiso = 180 ppm, the isotropic value of the indirect 13C–63,65Cu coupling tensor, 1Jiso(13C–63Cu) = 0.93 1Jiso(13C–65Cu) = 682 ± 8 Hz, its anisotropy, Δ1J(13C–63Cu) = 0.93 Δ1J(13C–65Cu) = −160 ± 260 Hz and quadrupolar coupling constants, CQ(63Cu) = 1.08 CQ(65Cu) = 80 ± 5 MHz since the ratio of electric quadrupolar moments of 63,65Cu isotopes is Q(63Cu)/Q(65Cu) = 1.08. These parameters are close to those recently measured for [Cu(Cl)(NHC)] complex, in which the NHC ligand was N,N′-bis-[2,4,6-trimethylphenyl]imidazole-2-ylidene.37,38 The best-fit CQ(63Cu) value agrees well with that calculated using DFT from the periodic crystal structure of [Cu(Cl)(IPr)] (67 MHz). Its large value stems from a large electric field gradient owing to the highly asymmetric environment of the Cu atom in this compound. The measured 1Jiso(13C–63Cu) coupling constant agrees with that calculated using DFT for the isolated [Cu(Cl)(IPr)] complex (660 Hz). Furthermore, the calculated Δ1J(13C–63Cu) value of 112 Hz is consistent with experimental one given the high standard deviation in the measured Δ1J(13C–63Cu) value.
 | ||
| Fig. 3 Expansions of experimental (blue) and simulated (black) 1D13 C2 signals in 1D 1H → 13C CP-MAS NMR spectra of 3OP recorded at (a) B0 = 9.4 T with νR = 12 kHz and (b) B0 = 18.8 T with νR = 18 kHz. | ||
As seen in Fig. 2b, the 1D 1H → 13C CPMAS spectrum of crude 2 sample exhibits a peak at 138 ppm showing the formation of [CuCl2][IPr·H] cuprate (2) in the solid state during the ball milling in agreement with 1H NMR spectrum. The 1H → 13C CPMAS spectra of crude 3OP/SW samples do not exhibit any signal at 138 ppm but a broad signal near 180 ppm. Furthermore, no cross peak between 1H and 13C signals at 10 and 138 ppm, respectively, is detected in their 2D 1H{13C} HETCOR spectra shown in Fig. S7b and c. This observation shows that during ball milling, the [Cu(Cl)(IPr)] complex is formed in the solid state, whereas the cuprate is totally transformed into this complex. The spectra of crude 2 and 3OP/SW samples also exhibit an additional signal at 143 ppm indicating the presence of unreacted IPr·HCl. Moreover, the signals at 162 and 169 ppm detected in the 13C spectra of crude 3OP/SW indicate the presence of KHCO3 and K2CO3, respectively.31 The spectra of crude samples exhibit broader signals than those of purified samples owing to the disorder created by ball milling. The 1H, 13C and 15N NMR resonances of pure 3SW compound are narrower than those of pure 3OP because the former is better crystallized than the latter. This line narrowing improves spectral resolution and allows the observation of additional peaks in the 1H and 1H → 13C spectra of pure 3SW shown in Fig. 1a and 2a, respectively.
This broadening is especially large for the C2 multiplet of [Cu(Cl)(IPr)] complex since the disorder results in a distribution of CQ(63,65Cu) values, and hence, splitting due to second order quadrupolar-dipolar cross-terms.
For this initial part of the study, a clear mechanistic picture of the formation of [Cu(Cl)(NHC)] complexes was obtained. The ssNMR spectroscopic data along with DFT calculations of NMR parameters show unequivocally that the cuprate and the desired Cu–NHC complex are formed in the solid state during the ball milling event and not during workup when the crude reaction solids are in contact with solvent. Additionally, the cuprate intermediate is not detected in the crude samples of the final product, indicating the simultaneous conversion of cuprate into [Cu(Cl)(NHC)]. We have also shown that solid-state NMR can be used to assess the presence of unreacted reagents, such as NHC·HCl and K2CO3, as well as the formation of other reaction products, such as KHCO3.
While the formation of cuprates has now been confirmed in the mechanochemical solid-state synthesis (as well as in solution, from our previous work on Cu, Au and Pd), our effort in identifying metalates in solution for rhodium systems have met with little success. A detailed examination of the solid-state transformation was initiated to understand the mechanism at play in the formation of Rh(I)–NHC complexes of the type [Rh(acac)(CO)(NHC)] (5), obtained from [Rh(acac)(CO)2] (4) (with acac = acetylacetonato) and an imidazolium salt (Scheme 4) with the goal of possibly observing rhodate intermediates.
 | ||
| Scheme 4 (A). Synthetic approach to [Rh(acac)(CO)(L)] (6) complexes. (B) Mechanochemical synthesis of [Rh(acac)(CO)(NHC)] (5) complexes. | ||
The [Rh(acac)(CO)(L)] (L = two-electron-donor) complexes are obtained in a one-step ligand displacement reaction leading to the elimination of one CO ligand (Scheme 4A). Nolan39 and more recently Carrow and co-workers40 have exploited this class of complexes to assess the Tolman electronic parameter (TEP) of ligands through linear correlation analysis involving the carbonyl stretching frequency found in [Rh(acac)(CO)(PR3)] complexes and the TEP values (from Ni(CO)3L) for this same set of phosphine ligands.39,40 Using the same linear correlation analysis, Gessner and co-workers have assessed the TEP value for a di(amino)-substituted carbodiphosphorane (CDP) ligand by recording the IR stretching frequency in the corresponding [Rh(acac)(CO)(L)] (6).41 Recently, Cazin has used such complexes as electrochemical probes to assess ligand electronic properties, enabling direct correlation of νCO and E1/2.42 This class of complexes has undoubtedly become an interesting platform to assess ligand electronic properties.
All [Rh(acac)(CO)(NHC)] (5) complexes reported to date42–48 have been obtained through synthetic routes that require strictly anhydrous conditions. While we have recently disclosed a solvent-based synthetic protocol that allows for the assembly of these complexes using operationally simple conditions and readily available starting materials,42,48 this solvent-based method is marked by relatively long reaction times (ranging from 3 to 18 hours, depending on the nature of the NHC·HBF4 salt employed) at high temperatures. This is in striking contrast with the reaction time needed when reactions are carried out in solid-state. Indeed, when we investigated the feasibility of the reaction under mechanochemical conditions, we found that 30 minutes of grinding were sufficient for the assembly of a series of N,N′-diaryl NHC–Rh(I) complexes of the type [Rh(acac)(CO)(NHC)] (5). Initial experiments were carried out using N,N′-bis-[(2,6-di(isopropylphenyl)]imidazolium, (IPr·HBF4), an excess of K2CO3, and [Rh(acac)(CO)2] (4) (Scheme 4B). Note that the BF4 salt was used instead of the imidazolium chloride in view of the hygroscopic nature of the latter and the water resistance and high stability, crystallinity of the former. The reaction (as were subsequent others) was carried out in a planetary ball mill, using a ZrO2 reactor charged with 28 ZrO2 balls (10.3 g, ∅ = 5 mm, corresponding to a filling degree for the milling bodies of ΦMB = 0.15) on a 100 mg scale of the NHC·HBF4 salt. Gratifyingly, these conditions resulted in the formation of the desired complex.49 An intriguing feature of this reaction was the colour change of the solid reagents throughout the process up to the final desired product (Scheme 5). When [Rh(acac)(CO)2] (4), which displays a pronounced dichroic character (appearing green by diffraction and red in transmission),50 was milled with IPr·HBF4 and an excess of K2CO3, the solids were converted to a red amaranth powder (5a*) which affords the desired [Rh(acac)(CO)(IPr)] complex (5a) as a yellow powder after workup. This last colour change occurs spontaneously upon extraction of 5a* from the reactor using a solvent, this with concomitant gas evolution (Scheme 5). To confirm the identity of the gas, an experiment was carried out in a Landolt tube (Scheme S1), with the first tube charged with the milled solids mixture 5a* and the second chamber loaded with a solution of [Ir(Cl)(COD)(IPr)] 7.
 | ||
| Scheme 5 Mechanochemical synthesis of [Rh(acac)(CO)(IPr)] (5a). Reaction conditions: (i) IPr·HBF4 (100 mg, 0.21 mmol), [Rh(acac)(CO)2] (4) (54 mg, 0.21 mmol), K2CO3 (87 mg, 0.63 mmol), planetary ball mill, 400 rpm, 30 min. (ii) Solvent workup. a Isolated yield. | ||
Addition of solvent to the first chamber resulted in vigorous frothing/gas evolution which led to reaction with 7 in the second chamber and to formation of [Ir(Cl)(CO)2(IPr)] (8).51 This unequivocally confirms that CO(g) is the gas released upon solvent addition in the first chamber (see SI for details). This observation suggests that the workup itself is key to product formation.
Considering these data, the necessity for the mechanical treatment prior to reaction was questioned and was determined by recording the kinetic profiles (in situ IR monitoring) of reactions carried out in solution using the pre-ground solids (5a*) and the unground solids. These experiments show the exceptional effect of the pre-grinding, leading to reaction completion within a few minutes, while the same reaction carried out with non-pre-ground material requires several hours at room temperature.52 Fig. 4 shows the profile obtained with the IPr derivative. This effect of the pre-grinding on reaction rate is even more striking when the bulkier congener IPr* is used (SI Fig. S40), with which the reaction using pre-ground starting materials is essentially instantaneous upon solvent addition, while the same reaction carried with non-pre-ground starting material is very sluggish. In addition, we observed that pre-grinding the base alone also increases reaction rate, but not as drastically as when all reactants are pre-ground together. We therefore note that particle size but also mixing of the solids have an important influence on the reaction kinetics. We next examined whether such results could also be achieved with manual grinding using simple mortar and pestle. As shown in Scheme 6, this is indeed the case, as after manually pre-grinding the components of the reaction, followed by solvent addition/workup, a 72% yield of the Rh–NHC complex was isolated.
 | ||
| Fig. 4 Kinetic profiles of reactions with starting material pre-ground (red); not ground (blue). In situ IR monitoring of νCO product (1961 cm−1). | ||
 | ||
| Scheme 6 Synthesis of [Rh(acac)(CO)(IPr)] (5a) using manual grinding. Conditions: IPr·HBF4 (100 mg, 0.21 mmol), [Rh(acac)(CO)2] (4) (54 mg, 0.21 mmol), and K2CO3 (87 mg, 0.63 mmol), in a mortar and pestle with grinding time of 10 min. a Isolated yield after workup involving solvent. | ||
To obtain experimental evidence as to the exact identity of the solids obtained after milling and to offer insights into how this reaction does indeed proceed, we investigated the nature of the crude reaction mixture without resorting to analytical methods that require prior dissolution of solids in a liquid medium.
ATR-FTIR (Attenuated total reflectance Fourier transform infrared) spectroscopic analyses were performed directly on the solids of the crude reaction mixture 5a*, the [Rh(acac)(CO)2] 4, and the [Rh(acac)(CO)(IPr)] final product 5a. The fingerprint infrared carbonyl region unambiguously establishes that no significant differences exist between the spectrum of [Rh(acac)(CO)2] 4 and the one of the resulting solid 5* after milling. The CO stretching frequencies related to the A1 and B1 vibrational modes are present in both spectra but are slightly shifted. Upon workup of the milled solids using solvents, clear conversion to [Rh(acac)(CO)(IPr)] 5a is observed, with a single CO band appearing at 1958 cm−1 (Fig. 5).
 | ||
| Fig. 5 ATR-FTIR spectra of solids [carbonyl stretching frequencies region] for: (A) [Rh(acac)(CO)2] 4; (B) the crude reaction mixture 5a*; (C) [Rh(acac)(CO)(IPr)] 5a. | ||
These pre-ground solids were then studied using solid-state NMR spectroscopy (Fig. 6). The 1D 1H NMR spectra of these solids were recorded under MAS with the homonuclear dipolar decoupling scheme wDUMBO53 to enhance spectral resolution (Fig. 6B). The spectral assignments are given in Fig. S11 (see SI). The 1H spectrum of reaction mixture 5a* contains signals of the reactants, IPr·HBF4 and 4, but does not display those of the final product 5a. Similar conclusions were reached from 13C NMR spectra acquired using 1H → 13C cross-polarization under MAS (CPMAS) sequence (see Fig. 6A and assignment given in Fig. S12).
 | ||
| Fig. 6 (A) 1D 1H → 13C CPMAS, (B) 1H wDUMBO-decoupled, (C) 1H → 15N CPMAS, (D) 11B and (E) 19F MAS NMR spectra of IPr·HBF4 (black), [Rh(acac)(CO)2] (4) (green), the crude reaction mixture (5a*) (red) and [Rh(acac)(CO)(IPr)] (5a) complex (olive) acquired at 9.4 T with a MAS frequency of 12.5 kHz. An expansion of the 1H → 13C CPMAS spectrum of (5a) showed as an inset in panel A displays the doublet assigned to the 13C2 nucleus of the imidazolylidene ring split by the J-coupling constant 1J(13C–103Rh) = 73 Hz. | ||
Specifically, the 13C spectrum of 5a* does not exhibit a doublet at 192 ppm, corresponding to the 13C2 signal of the imidazolylidene moiety in the final product 5a. Conversely, the spectrum of this compound contains a singlet at 140 ppm assigned to the 13C2 signal of IPr·HBF4. Furthermore, the carbonyl 13C signals are similar for solids 4 and 5a* and differ from those of 5a. In particular, the 13C signal of the CO ligand of 5a at 178 ppm is not observed in the spectrum of 5a*. The signal at 170 ppm in the 13C spectrum of 5a* also indicates the presence of K2CO3.21 Hence, the crude mixture contains unreacted 4, IPr·HBF4, and K2CO3 but no final product 5a.
The 1D 1H → 15N CPMAS, 11B, and 19F MAS spectra, shown in Fig. 6C–E, respectively, further confirm the presence of IPr·HBF4 in the crude mixture and the absence of 5a. The 19F signal of IPr·HBF4 is broadened in the 5a* mixture with respect to the pure reagent. This broadening indicates a distribution of the local environment of 19F nuclei in 5a* mixture arising from the creation of crystallographic defects or reactant mixing by ball milling. In addition, the 15N signal at 184 ppm is identical to that of IPr·HBF4. This result is consistent with the 1H and 13C data and supports our initial suspicion that 5a is not formed during the ball milling event but is formed only after the workup of the milled solids upon solvent addition.
The powder XRD pattern of 5a* (see SI Fig. S13) shows that this mixture contains 4, IPr·HBF4 and K2CO3 crystallites. Furthermore, reflections from 5a crystals were not detected in the XRD pattern of 5a*. This observation confirms that the final complex is not formed before the addition of the solvent. The reflections of 5a* powder pattern are broader than those of the reagents since ball milling reduces the size of the crystallites and results in the formation of crystallographic defects (Fig. S14). Therefore, all experimental data collected show that 5a* is a physical mixture of the reagents resulting from mechanical treatment. When the solids are treated with various solvents, product 5a is smoothly formed with concomitant gas evolution (Fig. S15).
Computational studies (DFT) were conducted to gain further insights into the thermodynamics of this process, intermediates, and transition states (TSs) involved in the reaction of IPr·HBF4 with 4 in the presence of K2CO3 to form 5a. As 5a corresponds experimentally to a heterogeneous physical mixture of solid components and is therefore not directly amenable to molecular DFT treatment, calculations were performed starting from the product of the addition of [IPr·H]+ and CO32−, namely [IPr·HCO3]− (8′), and the Rh complex (4) at infinite distance, which was set as zero energy.
Fig. 7A presents the free energy changes resulting from calculations in the gas phase (values in black) and in CH2Cl2 as the solvent (values in red). From (8′) the hydrogen of [IPr·H]+ is transferred to CO32− forming HCO3− species through a transition state (TS) at +14.2 and +16.6 kcal mol−1, in the gas phase and in CH2Cl2, respectively. In this TS, the rhodium complex interacts with the oxygen of the CO32− through the hydrogen of the methyl group of the acac ligand (see Fig. 8A), resulting in a small stabilization effect (see Fig. S16).
 | ||
| Fig. 7 (A). Computed reaction pathways from 8′ and 4 to 5a. Values reported are ΔG in the gas phase (black values) and in CH2Cl2 (red values) in kcal mol−1.54,55 (B). Computed reaction pathway from 1 to 2 and finally to 3 in solution (red values) and in the gas phase (black values) in kcal mol−1. | ||
 | ||
| Fig. 8 Geometries of: (A) transition state (TS) and (B) the kinetic product of the pentacoordinate intermediate (8″·HCO3−) with HCO3− anion interaction with acac ligand. | ||
Moving forward, deprotonation of IPr in the TS results in its spontaneous coordination to the rhodium complex, generating a kinetic product (5″·HCO3−), at −3.3 kcal mol−1 in the gas phase and at −2.0 kcal mol−1 in CH2Cl2 in which a pentacoordinate species (5″) is stabilized by the interaction of one acac oxygen atom with the proton of the non-coordinating HCO3− anion (see Fig. 8B). Finally, the pentacoordinate intermediate (5″·HCO3−) evolves by HCO3− and CO elimination to the product (5a), which is energetically favoured only in a solvent (+2.1 kcal mol−1 in gas phase and −16.6 kcal mol−1 in CH2Cl2). This difference is attributed to solvent stabilization of the leaving HCO3− anion, while CO dissociation is essentially thermoneutral.
Comparison of the vibrational frequencies of the CO groups in our intermediates with the experimental data (Fig. S17) clearly shows that the CO stretching frequencies found in our intermediate spectra are comparable with the experimentally found frequencies.
To address the energetics of the mechanochemical reaction in the copper system, a similar profile was calculated (see Fig. 7B). Calculations clearly show that the initial interaction between IPr·HCl and CuCl is remarkably exergonic. This favourable interaction drives the reaction to overall negative free energies along the entire reaction pathway, both in gas-phase and solvent simulations. These computational results align well with the experimental formation of 3, observed in both the gas-phase and in solution.
In grinding experiments conducted with both a mixer mill and a planetary orbital shaker under liquid assisted grinding (LAG), product 5a was formed after grinding, without the need for any workup. For details and IR spectra, see SI Fig. S48 and S49. Temperature (external or during grinding) may also contribute to product formation. Using either milling instruments identical results were obtained where 5a is not formed. However, heating ground samples permits some product formation at temperatures of 120 °C as evidenced by FT-IR ATR spectroscopy in the solid state (Fig. S50 and S51).
Conclusion
The mechanisms leading to the formation of [Cu(NHC)Cl] and [Rh(acac)(CO)(NHC)] complexes were examined through a combination of spectroscopic and computational means. These methods permit to confirm that for the Cu system, the chemical transformation is achieved during milling/grinding. However, for the rhodium system, the need for solvent is evident to bring the ligand binding/substitution event to completion and to product formation for the Rh system, but milling/grinding assists the reaction but the drive towards product is brought about by events occurring upon or after solvent addition for the Rh system. Particle size and mixing affect solution reaction kinetics. The results for the Rh system are somewhat unexpected, but the described method and observations should prove convenient to the community for synthetic access to organometallic complexes and might be applicable to other systems obtained through mechanochemical means. The pre-grinding/milling effect can be of significant practical use to accelerate reaction kinetics.As a closing remark, the two systems studied clearly show that there is no single reaction mechanism involved in mechanosynthesis. This may not come as a surprise to the practitioners of physical organometallic chemistry but here, we show that reaction or conversion can occur during or post mechanochemical grinding. The evolution of a gas during the reaction favours product formation and this entropic factor may be crucial in dictating the mechanistic route by which mechanochemical reactions proceed. This study may also serve as a caveat to those making common use of mechanochemical synthesis in highlighting that each system can be unique in possessing given thermodynamic driving forces (entropic and enthalpic) and that these should be considered before proposing (or assuming) mechanochemical reaction mechanisms.
Author contributions
GP, LH, JT, OD, AGMR and PR carried out experimental work and contributed to the writing of the manuscript. IR, LF, LD and LC conducted computational work and contributed to the writing of the manuscript. OL, LC, SPN and CSJC conceived and supervised the work, secured funding, and contributed to the writing and editing of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary Information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6sc01038e.Acknowledgements
We gratefully acknowledge the Special Research Found (BOF) of Ghent University. The Chevreul Institute (FR 2638), Ministère de l’Enseignement Supérieur de la Recherche et de l’Innovation, Hauts-de-France Région, IR Infranalytics (FR 2054) and FEDER are acknowledged. This project has received funding from the European Union Horizon 2020 research and innovation program under the Skłodowska-Curie agreement No. 847568. Umicore AG and Johnson Matthey are gratefully acknowledged for gifts of materials. Gianluca Carí is thanked for conducting the LAG and high temperature experiments. We thank the reviewers for helpful suggestions.Notes and references
- J. L. Do and T. Friščić, ACS Central Sci., 2017, 3, 13–19 CrossRef CAS PubMed.
- T. Friščić and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef PubMed.
- A. Stolle, T. Szuppa, S. E. S. Leonhardt and B. Ondruschka, Chem. Soc. Rev., 2011, 40, 2317–2329 RSC.
- D. Tan and T. Friščić, Eur. J. Org Chem., 2018, 2018, 18–33 CrossRef CAS.
- N. R. Rightmire and T. P. Hanusa, Dalton Trans., 2016, 45, 2352–2362 RSC.
- A. Beillard, X. Bantreil, T. X. Métro, J. Martinez and F. Lamaty, Chem. Rev., 2019, 119, 7529–7609 CrossRef CAS PubMed.
- D. Tan and F. Garcia, Chem. Soc. Rev., 2019, 48, 2274–2292 RSC.
- T. Friščić, Chem. Soc. Rev., 2012, 41, 3493–3510 RSC.
- A. Porcheddu, E. Colacino, L. De Luca and F. Delogu, ACS Catal., 2020, 10, 8344–8394 CrossRef CAS.
- C. Bolm and J. G. Hernandez, ChemSusChem, 2018, 11, 1410–1420 CrossRef CAS PubMed.
- C. J. Clarke, W.-C. Tu, O. Levers, A. Bröhl and J. P. Hallett, Chem. Rev., 2018, 118, 747–800 CrossRef CAS PubMed.
- J. L. Howard, Q. Cao and D. L. Browne, Chem. Sci., 2018, 9, 3080–3094 RSC.
- J. G. Hernandez and C. Bolm, J. Org. Chem., 2017, 82, 4007–4019 CrossRef CAS PubMed.
- For a recent example, see: T. Seo, N. Toyoshima, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 6165–6175 CrossRef CAS PubMed.
- G. Kaupp, CrystEngComm, 2009, 11, 388–403 RSC.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719–7738 RSC.
- R. F. Koby, A. M. Doerr, N. R. Rightmire, N. D. Schley, W. W. Brennessel, B. K. Long and T. P. Hanusa, Chem. Eur. J., 2021, 27, 8195–8202 CrossRef CAS PubMed.
- S. Lukin, L. S. Germann, T. Friščić and I. Halasz, Acc. Chem. Res., 2022, 55, 1262–1277 CrossRef CAS PubMed.
- J. G. Schiffmann, F. Emmerling, I. C. B. Martins and L. Van Wüllen, Solid State Nucl. Magn. Res., 2020, 109, 101687 CrossRef CAS PubMed.
- S. Gupta, T. Kobayashi, J. F. Goldston and V. K. Pecharsky, Green Chem., 2014, 16, 4378–4388 RSC.
- Y. Xu, L. Champion, B. Gabidullin and D. L. Bryce, Chem. Commun., 2017, 53, 9930–9933 RSC.
- S. Kaabel, R. S. Stein, M. Fomitšenko, I. Järving, T. Friščić and R. Aav, Angew. Chem., Int. Ed., 2019, 58, 6230–6234 CrossRef CAS PubMed.
- I. d'Anciães Almeida Silva, E. Bartalucci, C. Bolm and T. Wiegand, Adv. Mat., 2023, 35, 2304092 CrossRef PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- S. De, F. Joó, H. Horváth, A. Udvardy and C. E. Czégéni, J. Organomet. Chem., 2020, 918, 121308 CrossRef CAS.
- G. Pisanò and C. S. J. Cazin, Green Chem., 2020, 22, 5253–5256 RSC.
- G. Pisanò and C. S. J. Cazin, ACS Sustainable Chem. Eng., 2021, 9, 9625–9631 CrossRef.
- Q. Liao, K. Su, H. Cai, T. Zhao and F. Liu, J. CO2 Util., 2022, 59, 101963 CrossRef CAS.
- N. V. Tzouras, F. Nahra, L. Falivene, L. Cavallo, M. Saab, K. Van Hecke, A. Collado, C. J. Collett, A. D. Smith, C. S. J. Cazin and S. P. Nolan, Chem. Eur. J., 2020, 26, 4515–4519 CrossRef CAS PubMed.
- K. J. Ardila-Fiero and J. G. Hernández, Angew. Chem., Int. Ed., 2024, 63, e202317638 CrossRef PubMed.
- D. Stueber, D. Patterson, C. L. Mayne, A. M. Orendt, D. M. Grant and R. W. Parry, Inorg. Chem., 2001, 40, 1902–1911 CrossRef CAS PubMed.
- O. Santoro, A. Collado, A. M. Z. Slawin, S. P. Nolan and C. S. J. Cazin, Chem. Commun., 2013, 49, 10483–10485 RSC.
- A. Beillard, T.-X. Métro, X. Bantreil, J. Martinez and F. Lamaty, Chem. Sci., 2017, 8, 1086–1089 RSC.
- Y. Ishii, J. P. Yesinowski and R. Tycko, J. Am. Chem. Soc., 2001, 123, 2921–2922 CrossRef CAS PubMed.
- J. W. Wiench, C. E. Bronnimann, V. S.-Y. Lin and M. Pruski, J. Am. Chem. Soc., 2007, 129, 12076–12077 CrossRef CAS PubMed.
- R. K. Harris and A. C. Olivieri, Prog. Nucl. Magn. Reson. Spectrosc., 1992, 24, 435–456 CrossRef CAS.
- N. Kaeffer, D. Mance and C. Copéret, Angew. Chem., Int. Ed., 2020, 132, 20174–20182 CrossRef.
- H. Kaur, F. K. Zinn, E. D. Stevens and S. P. Nolan, Organometallics, 2004, 23, 1157–1160 CrossRef CAS.
- S. A. Serron, J. Huang and S. P. Nolan, Organometallics, 1998, 17, 534–539 CrossRef CAS.
- L. Chen, P. Ren and B. P. Carrow, J. Am. Chem. Soc., 2016, 138, 6392–6395 CrossRef CAS PubMed.
- A. Kroll, H. Steinert, L. T. Scharf, T. Scherpf, B. Mallick and V. H. Gessner, Chem. Commun., 2020, 56, 8051–8054 RSC.
- F. Bru, R. S. C. Charman, L. Bourda, K. Van Hecke, L. Grimaud, D. J. Liptrot and C. S. J. Cazin, Dalton Trans., 2024, 53, 16030–16037 RSC.
- M. S. Datt, J. J. Nair and S. Otto, J. Organomet. Chem., 2005, 690, 3422–3426 CrossRef CAS.
- G. Berthon-Gelloz, M. A. Siegler, A. L. Spek, B. Tinant, J. N. Reek and I. E. Marko, Dalton Trans., 2010, 39, 1444–1446 RSC.
- M. Iglesias, D. J. Beetstra, K. J. Cavell, A. Deryisi, I. A. Fallis, B. Kariuki, R. W. Harrington, W. Clegg, P. N. Horton, S. J. Coles and M. B. Hursthouse, Eur. J. Inorg. Chem., 2010, 2010, 1604–1607 CrossRef.
- W. Y. Lu, K. J. Cavell, J. S. Wixey and B. Kariuki, Organometallics, 2011, 30, 5649–5655 CrossRef CAS.
- Q. Zhao, G. Meng, G. Li, R. Mendelsohn, R. Lalancette, R. Szostak and M. Szostak, Chem. Sci., 2021, 12, 10583–10589 RSC.
- S. G. Guillet, G. Pisanò, S. Chakrabortty, B. H. Müller, J. G. de Vries, P. C. J. Kamer, C. S. J. Cazin and S. P. Nolan, Eur. J. Inorg. Chem., 2021, 43, 3506–3511 CrossRef.
- Milling of other NHC salts used as reactants in this transformation for 30 min at 400 rpm afforded the desired metal complexes (5a–5e) in good to excellent isolated yields (see SI for details)..
- F. Bonati and G. Wilkinson, J. Chem. Soc., 1964, 3156–3160 RSC.
- R. A. Kelly, III, H. Clavier, S. Guidice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef.
- For grinding effects in mechanochemical reactions, see: J. Templ and L. Borchardt, Angew. Chem., Int. Ed., 2026, 65, e23191 CrossRef CAS PubMed.
- S. Paul, R. S. Thakur, M. Goswami, A. C. Sauerweim, S. Mamone, M. Concistré, H. Förster, M. H. Levitt and P. K. Madhu, J. Mag. Reson., 2009, 197, 14–19 CrossRef CAS PubMed.
- Geometries were optimized with Gaussian09 package at the PBE0-D3 level of theory. The valence triple-zeta with two sets of polarization functions (Def2TZVPP) was used for Rh, Cu, H, C, N, Cl and O atoms. In gas phase, the reported free energies have been obtained considering the thermal free energies. Instead, the solvent energy profile has been computed adding the thermal corrections in gas-phase to the electronic energy in solvent (PCM model) calculated via single point energy calculations in CH2Cl2 with Def2TZVPP basis set for metal and main group atoms and the triple-z with two sets of polarization functions and a set of diffuse functions (Def2TZVPPD) on one of the three oxygen atoms of the weak base and on Cl atoms..
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |