
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePicolinate-based acyclic ligand for rare earth element extraction and separation
Yangyang
Gao
 ab,
Sean
Medin
c,
Alexa M.
Schmitz
c and
Justin J.
Wilson
*ab
ab,
Sean
Medin
c,
Alexa M.
Schmitz
c and
Justin J.
Wilson
*ab
aDepartment of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, California 93106, USA. E-mail: justinjwilson@ucsb.edu
bDepartment of Chemistry and Chemical Biology, Cornell University, Ithaca, New York 14853, USA
cREEgen Inc., Ithaca, New York 14853, USA
First published on 13th April 2026
Abstract
The rare earth elements (REEs) play an important role in many modern technologies, particularly those relevant to clean energy. Despite their increasing importance, obtaining them in elementally pure forms suitable for downstream applications is challenging due to their similar chemical properties. This problem has impeded efforts to efficiently and selectively extract them from end-of-life materials and electronic waste. Here, we report a cost-efficient acyclic picolinate-based chelator H4aapa. The REE stability constants (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) KML) of this chelator were measured via pH potentiometric and UV-Visible spectrophotometric titrations, revealing it to preferably bind light over heavy REEs like many recently reported 18-membered macrocycles. Its REE complexes were characterized by X-ray crystallography and NMR spectroscopy, demonstrating that this chelator can attain different conformations. The unique properties of aapa were subsequently used to separate REEs via the dissolution of insoluble REE oxalate mixtures. This dissolution-based separation led to large separation factors, the most significant being that for the Ce3+/Lu3+ pair (38.6) at pH 4. Leveraging the strong REE binding affinity of aapa, we further demonstrated this chelator can leach REEs from authentic end-of-life materials in the form of magnet waste and autocatalyst smelting (autocat) slag. With this approach, exposure of these materials to a 20 mM solution of aapa at neutral pH generates a metal-containing solution enriched in Nd3+ and Dy3+ by 56.9 wt% and 3.0 wt%, marking a 4-fold improvement over the use of 4 M HNO3.
KML) of this chelator were measured via pH potentiometric and UV-Visible spectrophotometric titrations, revealing it to preferably bind light over heavy REEs like many recently reported 18-membered macrocycles. Its REE complexes were characterized by X-ray crystallography and NMR spectroscopy, demonstrating that this chelator can attain different conformations. The unique properties of aapa were subsequently used to separate REEs via the dissolution of insoluble REE oxalate mixtures. This dissolution-based separation led to large separation factors, the most significant being that for the Ce3+/Lu3+ pair (38.6) at pH 4. Leveraging the strong REE binding affinity of aapa, we further demonstrated this chelator can leach REEs from authentic end-of-life materials in the form of magnet waste and autocatalyst smelting (autocat) slag. With this approach, exposure of these materials to a 20 mM solution of aapa at neutral pH generates a metal-containing solution enriched in Nd3+ and Dy3+ by 56.9 wt% and 3.0 wt%, marking a 4-fold improvement over the use of 4 M HNO3.
Introduction
The unique electronic, magnetic, and physical properties of rare earth elements (REEs) make them essential components in over 200 commercial products, many of which are crucial for clean technologies.1–6 With these REE-based products on the market across different industrial sectors, the demand for these critical minerals is projected to climb from 171300 tons in 2022 to nearly 240000 tons by 2030.7 For example, the global need for NdFeB magnets is expected to triple by 2035, while global production for neodymium (Nd) will only double over this same timeframe.8,9 Likewise, other light REEs like lanthanum (La), cerium (Ce), and praseodymium (Pr) are also subject to shortages because of their use in battery electrodes, catalysts, and polishing.10–12 To meet these growing demands, increasing mining and refining efforts is one option, but these processes come at significant environmental and health costs.13–17 Alternatively, the implementation of recycling strategies to obtain these critical minerals from their end-of-life materials can contribute to a robust and sustainable supply chain.18–20
Currently, hydrometallurgy is the state-of-the-art method to recover REEs from magnet and electronic waste (e-waste) compared to pyrometallurgy because of its lower energy demands, better efficacy at low temperatures, and scalability.8,21 In this approach, REE-bearing e-waste are dissolved using strong acid lixiviants, like HNO3, HCl, and H2SO4, to transform the insoluble REE-containing matrix into more soluble forms,22,23 or by employing a concentrated (NH4)2SO4 solution to form water soluble (NH4)RE(SO4)2 salts.24,25 These leachates are then subjected to chemical separations with either solvent extraction or ion exchange chromatography methods to obtain individual REEs. Despite their sufficient efficacy and cost-efficiency, these REE-leaching solutions suffer from some drawbacks. First, these strategies require large quantities of concentrated acid or aqueous (NH4)2SO4 for the initial e-waste dissolution step, followed by the requirement of volatile organic solvents for large-scale solvent extraction separation. For example, 4 M HNO3 or HCl and 0.5 M (NH4)2SO4 are typically required, and their use generates acidic and ammonium-containing wastewater,26–29 which are difficult and costly to remediate. Furthermore, these agents are not selective and leach other metals co-occurring in the matrix as well, reducing the purity of the leached REEs. Therefore, more efficient and environmentally friendly leaching and separation approaches are needed.
Greener and less energy-intensive bio-metallurgical strategies employ heterotrophic microorganisms to leach REEs into solution by biosynthesizing unique mixtures of organic acids, like gluconic, citric, and oxalic, from sugar-based carbon sources. Some engineered microorganisms like Gluconobacter oxydans, Acidithiobacillus thiooxidans, and Aspergillus niger have been successfully used to bioleach REEs from discarded LED lamps, spent automobile catalyst, and fluid cracking catalysts.30–36 The success of these efforts has highlighted the growing interest in biologically inspired solutions for sustainable REE processing and have stimulated both academic and industrial efforts aimed at developing scalable bio-based REE recovery technologies. Although these bioleaching strategies provide a promising path forward for the clean dissolution of REEs from e-waste, they also often extract other metal ions at higher concentrations, requiring a subsequent separation process.
To increase the efficiency of e-waste leaching, organic chelators have been designed to strip REEs from their ores and secondary materials. For instance, the addition of the commonly used chelator ethylenediamine acetic acid (EDTA, 0.005 M) to a 0.2% (NH4)2SO4 solution enabled the extraction of 90% of La from weathered crusts, achieving the same efficiency as a 2.0% (NH4)2SO4 solution alone.37 Similarly, 0.1 M ascorbic acid and acetic acid were able to leach 86% and 45% of REEs, respectively, from colloidal sediment phases.38 Furthermore, gluconate and its metabolites (2-ketogluconate and 5-ketogluconate), which are the effective constituents in Gluconobacter oxydans for REE leaching, were also investigated as biolixiviants for extracting REEs from nickel–metal hydride (NiMH) batteries,39 leading to a leaching yield of 56.1% at pH 3. These leaching approaches highlight how the use of chelators can accomplish efficient REE extraction from solid waste. An ideal chelator for this application should exhibit both strong affinity and excellent selectivity for REEs, compared to other metal ions present in such materials.
Chelators bearing picolinic groups have been intensively studied and applied for REE separations. For example, the large macrocyclic chelator macropa (Scheme 1), which has been extensively used in the nuclear medicine realm for actinium-225 targeted alpha therapy,40,41 is also promising for REE chelation because of its good selectivity for large over small metal ions. For example, this chelator and its variants have been used for solvent extraction separations of REEs and actinides,42–44 as well as within precipitation-based and resin-based separations to extract REEs in the presence of other small metal ions.45,46 Additionally, macropa can rapidly dissolve barite due to its strong affinity for Ba2+, suggesting its use for leaching metal ions from solid materials.47 Despite these promising applications, the macrocyclic nature of this chelator makes its large-scale synthesis challenging and costly, providing a potential limitation for its industrial use. Acyclic chelators, which tend to have more facile and high-yielding syntheses, have been investigated as alternatives by designing them to exhibit selectivity for large metal ions, like macropa. Most notably, the linear analogues of macropa, acyclopa and its amphiphilic version octadecyl acyclopa (ODA, Scheme 1), exhibit the same unique reverse-size selectivity properties of the parent chelator. Crystallographic and extended X-ray absorption spectroscopy (EXAFS) studies of the La3+ complexes of these two chelators show that they attain pseudo-cyclic structures that are nearly isostructural with those of macropa.48,49 Another analogue H2ampa (Scheme 1), which has two carboxamide pendent groups, may also be promising for REE extraction because of its ability to bind large metal ions like Pb2+, Bi3+, and La3+.50 To optimize acyclic chelators like acyclopa, ODA and H2ampa for REE leaching from end-of-life materials, more hydrophilic and stronger-donor end-cap groups may improve their properties for this application. This hypothesis is somewhat supported by related acyclic chelators that bear anionic picolinate groups. Octapa, for example, can form extremely stable complexes with lanthanides under mild condition.51,52 The expansion of this acyclic chelator via the addition of a neutral oxygen donor into its backbone generates a ligand named oxyaapa or H4noneunpa (Scheme 1).53,54 which makes stable lanthanide complexes across the entire series.
 | ||
| Scheme 1 The chelators discussed in this study. | ||
Inspired by the success of acyclopa for REE dissolution and leaching,48 in this study we investigated a close structural analogue bearing anionic carboxylates in place of the neutral alcohol donors. This new chelator, called aapa, is also similar to related acyclic chelators octapa and oxyaapa, but contains an additional ether oxygen donor atom to afford a donor atom set similar to that of macropa. As shown here, the structural similarity of aapa to macropa confers it with a similar reverse-size selectivity for lanthanides, akin to the trends observed for acylcopa and ODA. Leveraging these properties, H4aapa was investigated for its ability to separate REEs and extract them from relevant waste sources. This study demonstrates how acyclic chelators can be designed to mimic macrocycles and be leveraged effectively for green REE separations and leaching applications.
Results and discussion
Synthesis of H4aapa
The chelator H4aapa was targeted because it provides a similar donor atom set as macropa, while comprising an acyclic scaffold. Likewise, the investigation of H4aapa was also motivated by the success of H2ampa, which was shown to be an effective chelator for many large metal ions like Pb2+, Bi3+, and La3+,50 and acyclopa, which can directly dissolve and separate REEs from REE(OH)3 materials.48 To access H4aapa, it was synthesized from 2,2′-(ethane-1,2-diylbis(oxy))bis(ethan-1-amine) via a reductive amination of methyl 6-formylpicolinate to install the two picolinate methyl ester arms. The remaining secondary amines were subsequently functionalized with methyl 2-bromoacetate through an SN2 reaction.55–57 Finally, the esters on both the carboxylate and picolinate groups were removed under acidic conditions to afford H4aapa with an overall yield of 47.0% (Scheme 2). This new chelator, as well as all of its synthetic intermediates, were characterized by NMR spectroscopy, elemental analysis (EA), and liquid chromatography-mass spectrometry (LC-MS, Fig. S1–S9, SI). This synthetic route is streamlined in comparison to those used to access related bis(picolinate)-bearing acyclic chelators, which used a less efficient sequential protection and deprotection of the amines to prevent overalkylation.48–50 Additionally, H4aapa is not a derivative of the expensive diaza-18-crown-6 macrocyclic precursor like analogues of macropa, and its synthesis is therefore more cost-effective. For example, we estimate the cost for 1 g macropa, based solely on chemical reagents and average yields for each reaction in our lab, to be $42.70, of which diaza-18-crown-6 contributes $35. By contrast, the reagent cost of aapa via this synthetic route is only $11.4 per g (Tables S1 and S2).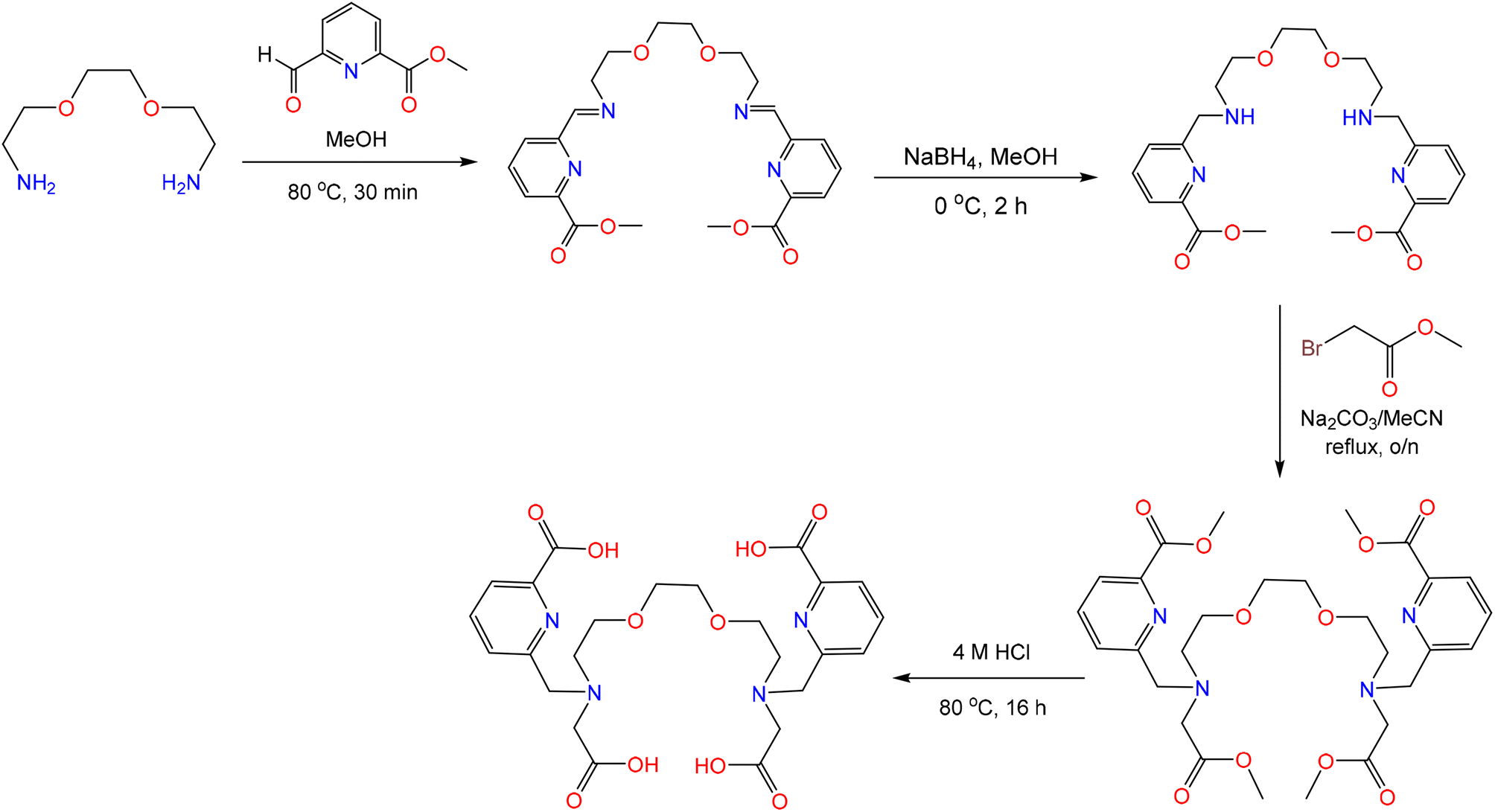 | ||
| Scheme 2 Synthetic scheme of H4aapa. | ||
Solution characterization of the Ln3+ complexes of aapa
NMR spectroscopy (1H, 13C{1H}, 2D HSQC and HMBC), HPLC, LC-MS, and UV-Vis spectroscopy were carried out to investigate the coordination chemistry of aapa with REEs in solution. These studies focused on the largest and smallest lanthanide ions, La3+ and Lu3+, because they are diamagnetic, making them amenable for characterization by NMR spectroscopy and providing an opportunity to understand how aapa can differentially bind small and large REEs. The 1H and 13C{1H} NMR spectra of a 1:1 mixture of La3+ and aapa show the presence of two conformers in aqueous solution at pD 8 (Fig. S10 and S11, SI), and all resonances were assigned with 2D NMR spectroscopy using the numbering scheme shown in Fig. 1 and S10–S15. The integration of 1H resonances of each conformation shows that they are present in a 1:3 ratio, with the major species lacking symmetry. This lack of symmetry is revealed by the appearance of two distinct sets of diastereotopically split CH2 resonances of the picolinate arms (H-7 and H-8 in blue) and CH2 resonances of the backbone carbon adjacent to the tertiary nitrogen (H-1 and H-6 in blue). By contrast, the minor species is symmetric, as reflected by the appearance of only one set of diastereotopic CH2 resonances for each type of hydrogen (yellow labels). HPLC analysis of this mixture also reveals the presence of two species, characterized by peaks at retention times of 15.11 and 16.44 min, also at a ratio of 1:3 (Fig. S16, SI). The presence of conformers of different symmetries for La3+ complexes of related picolinate or picolinamide-based acyclic chelators has previously been observed.50,54,58 Notably, the 1H NMR spectrum of the La3+–aapa complex does not vary over the pD range of 4–10, indicating that the conformer equilibrium is not dependent on H+ concentrations (Fig. S17, SI). The mass spectrum of La3+–aapa only reveals one peak with an m/z = 670.9 (Fig. S18, SI), which matches the expected m/z value for [La(H2aapa)]+. The observation of a single peak with the expected mass for the 1:1 ligand:metal complex by mass spectrometry supports our interpretation that the two species detected by NMR spectroscopy are conformers of the same complex.
 | ||
| Fig. 1
1H NMR spectrum of the aapa complex formed with La3+ at a 1:1 ratio (500 MHz, D2O, pD = 8, 25 °C). Resonances are assigned via the numbering scheme shown in the inset. The major conformer, labeled with blue numbers, is asymmetric, whereas the minor conformer, labeled with yellow numbers is symmetric. The peak at 4.70 ppm is from D2O. Acetoitrile, apparent as a singlet at 2.00 ppm, was added as an internal reference. | ||
The formation of the Lu3+ complex of aapa was afforded from a 1:1 mixture of the metal ion and ligand at pD 8. In contrast to that of the La3+–aapa complex, the 1H NMR spectrum of the Lu3+ complex at room temperature revealed very broad resonances, possibly signifying fluxionality or conformational dynamics (Fig. S19, SI). The formation of the Lu3+–aapa complex was further confirmed by mass spectrometry, revealing a peak at m/z = 706.9 that matches the [Lu(H2aapa)]+ ion (Fig. S20, SI), and by analytical HPLC, which showed a single peak with a retention time at 14.83 min, distinct from the free ligand (Fig. S21, SI). Under more acidic conditions, however, aapa cannot bind Lu3+ appreciably. For example, the 1H NMR spectra of 1:1 solutions of Lu3+ and aapa at pD 2 reveal that no Lu3+–aapa complex forms (Fig. S19, SI), contrasting the 1H NMR spectra for the 1:1: solution of La3+ and aapa, where partial complexation occurs at this pD value (Fig. S17, SI). These results suggest that aapa binds more strongly and stably to the light large La3+ over the heavy small Lu3+ ion, matching the properties observed for diaza 18-crown-6 macrocyclic chelators like macropa and its analogues. Lastly, the UV-Vis spectrum of the Lu3+ complex is red-shifted (λmax = 269 nm) compared to the free ligand (λmax = 265 nm). This shift is identical to that seen for the La3+ complex (Fig. S22, SI). Collectively, these data indicate that the acyclic aapa can form complexes with both large and small Ln3+ under mild conditions, while also retaining a reverse-size selectivity for light over heavy lanthanides like macropa.
X-ray crystallographic studies
Despite extensive efforts to obtain X-ray quality crystals of multiple different Ln3+ ions, we were only successful in accessing those of the La3+–aapa complex, which were grown from a mixture of La3+ and aapa in methanol via slow evaporation. The crystal structure is shown in Fig. 2, and its crystallographic data collection and refinement parameters are listed in Tables S3 and S4, SI. Within the asymmetric unit, two La3+–aapa complexes are present, and both complexes reside on crystallographic C2 axes, conferring them with this symmetry. An analysis of the C–O distances on the picolinate groups, intermolecular hydrogen-bonding interactions, and residual electron density on the difference Fourier map led us to assign these two complexes in the asymmetric unit to be the doubly protonated cationic [La(H2aapa)]+ and the fully deprotonated anionic [La(aapa)]−. Their existence in a 1:1 ratio leads to charge balance within the crystal. Within both the cation [La(H2aapa)]+ and the anion [La(aapa)]−, the La3+ centers attain a 10-coordinate geometry, afforded by close interactions with all of the donor atoms available on the aapa ligand. Furthermore, the conformations attained by both ions are identical, with the two picolinate arms binding on opposite faces of the La3+ coordination sphere, as enforced by the crystallographic C2 axes. The respective symmetries of these solid-state complexes are consistent with the symmetric minor conformation observed by 1H NMR spectroscopy. Thus, even though there is a slight preference for the asymmetric conformer in solution, as revealed by NMR spectroscopy, the energy difference between these conformers is small enough to be influenced by solid-state packing interactions, which lead to crystallization of the less favored conformation. The La–N distances from the picolinate groups range from 2.737 to 2.739 Å, whereas the La–N distances from the backbone are longer, falling between 2.780 to 2.798 Å (Table S4). These distances do not vary substantially between the anion and cation within the structure. For the La–O picolinate distances, however, these values are larger (2.619 Å) for the protonated cationic species, compared to those within the anion (2.534 Å). This result is expected given the decrease electrostatic interaction afforded once the picolinate groups are protonated.
 | ||
| Fig. 2 Structures of [La(H2aapa)]+ (left) and [La(aapa)]− (right) with thermal ellipsoids drawn at the 50% probability level. Counteranions, nonacidic hydrogen atoms, and outer-sphere solvent molecules are omitted for clarity. Grey = C, blue = N, red = O, teal = La, and white = H. | ||
Overall, the La3+–aapa complex geometries are similar to those observed in the crystal structure of the Bi3+ complex of the carboxamide analogue ampa.50 However, the conformation is substantially different from those of the La3+–ODAme, and La3+–acyclopa complexes, where two picolinate arms binding to the same face of La3+ like macropa,48,49 and the DFT-optimized structure of the La3+–ampa, which reveal a 9-coordinate geometry that arises from a lack of coordination of one of the carboxamide.50 We note, however, that the crystal structure of La3+–aapa does not represent the major species in solution, which is asymmetric. The major conformer in solution may more closely match those of the macropa and acyclopa complexes of La3+, where the picolinate arms bind with a syn orientation and lose their symmetry equivalence. Thus, like macropa, acylcopa and aapa may be able to attain a conformation that exhibits reverse-size selectivity.
Solution thermodynamics
To determine the quantitative binding affinity of H4aapa for the lanthanide (Ln) series, the protonation constants (Ki) and stability constants of aapa with the Ln series (KLnL) were measured by potentiometric titrations over a pH range of 2.2–11.3 (Fig. S23–S36). The stability constants of the light lanthanides (La3+–Nd3+) were also verified with UV-Vis titrations over the more acidic pH range of 1.28–2.22 (Fig. S37–S40). The constants Ki and KLnL, which are defined in eqn (S1) and (S2), are summarized in Fig. 3 and Table 1. The logKi and logKLnL of the related chelators oxyaapa, octapa, and macropa are also provided for comparison. Similar to the acyclic oxyaapa and octapa, five protonation constants could be determined for aapa. These constants most likely correspond to the successive protonation of two tertiary nitrogen atoms (logKa1 = 8.53 and logKa2 = 7.48), two picolinate nitrogen atoms (logKa3 = 3.70 and logKa4 = 2.78), and one acetate donor (logKa5 = 2.20). The sum of these five logKi values, which corresponds to the overall basicity of the chelator, is 24.69, comparable to those for oxyaapa (24.52) and octapa (22.2). This result indicates that the additional ether oxygen atom within the linear backbone of this chelator does not appreciably affect their basicity.
 | ||
| Fig. 3 The Ln3+ stability constants (logKLnL) of aapa, oxyaapa, octapa, and macropa, versus ionic radii (CN = 6). | ||
 | ||
| Fig. 4 The effect of the additional neutral oxygen donor atom on Ln3+ complex stability and selectivity. | ||
| Aapa | Oxyaapac | Octapa | Macropae | ||||
|---|---|---|---|---|---|---|---|
| a 0.1 M KCl, results obtained from potentiometric titration (pH 2.2–11.3), this work. b 0.1 M KCl, results obtained from UV-vis titration (pH 1.28–2.22), this work. The values in the parentheses are one standard deviation of the last significant figure. c 0.1 M KCl, ref. 54. d 0.15 M NaCl, ref. 52. e 0.16 M KCl, ref. 60 f 0.1 M KCl, ref. 61. g pLn = −log[Ln3+]free. Calculated for 10 µM total ligand and 1 µM total metal at pH 7.4 and 25 °C. | |||||||
| pKa1 | 8.53(5)a | 8.47 | 8.52d, 8.58e | 7.41 | |||
| pKa2 | 7.48(5)a | 7.63 | 5.40d, 5.43e | 6.85 | |||
| pKa3 | 3.70(8)a | 3.66 | 3.65d, 3.75e | 3.32 | |||
| pKa4 | 2.78(1)a | 2.99 | 2.97d, 3.05e | 2.36 | |||
| pKa5 | 2.20(3)a | 1.77 | 1.66d, 2.21e | 1.69 | |||
|
|||||||
| Ln 3+ | pLng |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) K
LnL K
LnL
|
logK
LnHL
|
logK
LnL
|
logK
LnL
|
logK
LnL
|
logK
LnHL
|
| La3+ | 19.2 | 18.21(7)a | 2.63(5)a | 19.06 | 20.13d | 14.99 | 2.28 |
| 18.69(4)b | 2.13(5)b | ||||||
| Ce3+ | 19.1 | 18.50(8)a | 2.27(6)a | 15.11 | 2.07 | ||
| 18.63(6)b | 2.10(3)b | ||||||
| Pr3+ | 19.9 | 18.90(8)a | 2.56(5)a | 14.70 | 2.96 | ||
| 19.55(4)b | 2.04(5)b | ||||||
| Nd3+ | 19.7 | 18.68(6)a | 2.32(3)a | 19.77 | 14.36 | 2.08 | |
| 18.98(2)b | 1.97(4)b | ||||||
| Sm3+ | 19.8 | 18.75(10)a | 3.00(8)a | 20.20e | 13.80 | 2.70 | |
| Eu3+ | 19.8 | 18.76(6)a | 2.49(3)a | 13.01 | 1.97 | ||
| Gd3+ | 19.5 | 18.50(9)a | 2.23(5)a | 20.50 | 20.23d | 13.02 | 2.48 |
| Tb3+ | 19.4 | 18.38(5)a | 2.36(6)a | 11.79 | 2.91 | ||
| Dy3+ | 19.2 | 18.21(5)a | 2.45(3)a | 20.35e | 11.72 | 2.42 | |
| Ho3+ | 18.6 | 17.60(3)a | 2.58(2)a | 10.59 | |||
| Er3+ | 18.3 | 17.31(3)a | 2.67(5)a | 21.33 | 10.10 | ||
| Tm3+ | 17.4 | 16.44(5)a | 2.95(2)a | 9.59 | |||
| Yb3+ | 16.9 | 15.86(2)a | 2.98(2)a | 20.10e | 8.89 | ||
| Lu3+ | 16.3 | 15.28(4)a | 3.47(2)a | 21.49 | 20.49d | 8.25 | |
The logKLnL values for aapa shows that this chelator has a preference for binding large over small REEs. This result is consistent with the 1H NMR data described above, which showed the failure of Lu3+ complex formation at pH values that led to La3+ complexation. Notably, the size preference of aapa is distinct from those of oxyaapa and octapa. The greater number of oxygen donor atoms on aapa can more stably accommodate the larger light and middle REEs, as indicated by the greater stability constants (logKLnL > 18) for the La3+–Dy3+ complexes compared to those of Ho3+–Lu3+ (logKLnL = 17.60–15.28). In particular, the high stability of the La3+–Nd3+ complexes required the use of low pH UV-Vis titrations to confirm the logKLnL values (Table 1). The titration data was best fit by including an equilibrium for the protonated MHL species (Table 1). This type of species is consistent with the X-ray crystal structure, which shows that [La(aapa)]− can exist in a protonated form. In comparing the Ln3+ stability constant trend between these three acyclic chelators, the reverse-size selectivity of aapa is driven by a decrease in affinity for heavy REEs compared to light REEs. For example, the ΔlogKLaL between oxyaapa and aapa is −0.85, whereas the ΔlogKLuL between oxyaapa and aapa is −6.21 (Fig. 4). By contrast, the ΔlogKLnL between octapa and oxyaapa are generally much smaller, as reflected by values of −1.07 for La3+, 0.27 for Gd3+, and 1.00 for Lu3+. Moreover, a 3.22 log unit difference between the largest logKPrL (18.90) and smallest logKLuL (15.28) for aapa is more significant than those for oxyaapa (2.43) and octapa (only 0.36). The larger difference in logKLnL values for aapa indicates that it is a more promising candidate for REE separations, compared to oxyaapa and octapa. In addition, the increased selectivity of aapa for large metal ions is consistent with established chelator design principles that suggest the inclusion of additional neutral oxygen donor atoms leads to this type of metal-binding preference (Fig. 4).59
 | ||
| Fig. 5 REE oxalate dissolution using aapa. (a) La2(C2O4)3 was fully dissolved by aapa in 2 minutes at pH 5. (b) Photograph of dissolution of single REE2(C2O4)3 and (c) dissolution efficiencies of REE2(C2O4)3 in the presence of aapa at different pH values. Conditions: [REE3+] = 5 mM, [C2O42−] = 15 mM, [aapa] = 5 mM; buffer: 100 mM ammonium acetate; t = 1.5 h. | ||
In comparison to macropa and acyclopa, aapa exhibits systematically larger Ln3+-binding affinities (logKLnL = 18.90–15.23 for aapa, logKLnL = 14.99–8.25 for macropa, and logKLnL are in a range of 14–9 for acyclopa respectively).48,61 Additionally, aapa provides uniformly strong binding for early Ln3+ (La3+–Dy3+), whereas macropa has stability constants for these metal ions that decrease steadily across the series by ∼3 logK units. For the small Ln3+ ions (Dy3+–Lu3+), the stability constants of the aapa complexes decrease gradually by 2.93logK units, which is comparable to the 3.47 and ∼3logK unit decrease observed for the macropa and acyclopa complexes over the same range. Following the trends based on the higher affinity of aapa for larger REEs, this chelator is also expected to only weakly bind smaller non-REE metal ions like Fe3+, and Ca2+, which are present within common end-of-life REE materials. This type of selectivity was observed for BZmacropa, which enabled its use for enriching REEs from complex mixtures containing multiple metal ion types.45,62 To further evaluate the selectivity of aapa for REEs over non-REE metals, we investigated its coordination chemistry Fe3+ and Cu2+, two of the most common contaminants in end-of-life materials. Mass spectrometry showed that aapa forms primarily a 2:1 M:L complex with Cu2+, whereas Fe3+ forms a 1:1 complex (Fig. S41 and S42). Potentiometric titrations were carried out, and the equilibria were modeled based on the stoichiometry observations from mass spectrometry. From these data, the pM values of these two metal ions and Ln3+ under standard conditions (10 µM total ligand and 1 µM total metal at pH 7.4 and 25 °C) were calculated to provide a direct comparison of conditional affinity of these ligands (Tables 1 and S5). The results show that pM values for the Ln3+ range from 16.3 (Lu3+) to 19.9 (Pr3+), whereas the non-REE metals exhibit substantially lower pM values (Fe3+: 12.75; Cu2+: 6.3), indicating that the ligand preferentially sequesters REEs over these competing ions. Overall, the difference between and overall larger stability constants for the Ln3+ ions makes aapa advantageous for applications in both REE extraction and separation.
Selective dissolution of REE oxalates
In a typical hydrometallurgical approach, REEs are recovered in the form of insoluble oxalate salts from the strongly acidic leachate solutions. This method is specifically efficient for REE extraction from Fe-containing matrix because Fe(C2O4)33− is water soluble.45,63,64 Although this precipitation strategy can remove non-REE ions reasonably well, the oxalate salts are obtained a mixture of the different REEs. Given both the binding affinity and selectivity of aapa for large REEs, we investigated whether this chelator could be used to simultaneously extract and separate REEs from solid REE oxalate mixtures. Because light REE oxalates have a larger thermodynamic stability (logK values of Ln2(C2O4)3 for La3+–Lu3+ increase from 4.5 to 5.3) but poorer solubility than heavy REE oxalates (Ksp values decrease from 10−29 to 10−32),64,65 we hypothesized that aapa, which more strongly binds to more soluble light REE oxalates, could achieve effective dissolution-based separations of these elements.
First, the ability of aapa to dissolve pure individual REE oxalates at different pH values was investigated. Nine different solid REE2(C2O4)3 salts were prepared in ammonium acetate buffer by combining solutions of LnCl3 (final concentration 5 mM) and excess ammonium oxalate (final concentration 15 mM) for 30 min. The precipitation efficiencies (E%), calculated based on eqn (S3), from this process were determined by measuring the remaining Ln3+ concentration in solution by inductively coupled plasma optical emission spectroscopy (ICP-OES) after removal of the solid REE2(C2O4)3 by filtration, as summarized in Tables S6, S7, Fig. 5, and S45. The REE concentration remaining in solution was below the detection limit, reflecting near-quantitative precipitation of the metal ions with oxalate at pH 5. Once these solids were formed, they were subsequently treated with 1 equiv. of aapa (5 mM) for 1.5 h at pH 5 to determine its dissolution efficacy. Under these conditions, aapa could dissolve nearly 100% of La2(C2O4)3 and Ce2(C2O4)3 (Fig. 5b, c and S41), highlighting its strong binding affinity for light REEs. Notably, La2(C2O4)3 can be fully dissolved by aapa in 2 minutes with stirring (Fig. 5a), underscoring the rapid dissolution kinetics achieved by aapa. In comparison, only 58.8% Yb2(C2O4)3 and 52% Lu2(C2O4)3 were solubilized by aapa under identical conditions (Fig. 5c). The large differences in dissolution efficiency of aapa for the light and heavy REEs is not entirely expected based on the relatively small differences in stability constants between these metal ions. However, the large dissolution efficiency differences are also a result of the poorer affinity of the light REEs for oxalate, which enables aapa to more easily bring these species back into solution via chelation.65–67 Notably, the difference in dissolution efficiencies between light and heavy REEs can be enhanced significantly at lower pH values (Fig. 5c). At pH 4.5, aapa can dissolve 89.7% Ce2(C2O4)3, but only 57.7% Dy2(C2O4)3 and 37.0% Lu2(C2O4)3. At pH 4, the differences in dissolution efficiency between the light Ce3+ and the heavy Dy3+ and Lu3+ reached 66.0% and 71.6%, respectively.
 | ||
| Fig. 6 The selective dissolution of REE oxalates using aapa within 96-well plate. (a) A photograph of REE2(C2O4)3 binary mixtures in the 96-well plate. (b) A photograph of the REE2(C2O4)3 binary mixtures after being mixed with aapa for 1.5 h. (c) The SF values of selective dissolution separation using aapa. Condition: [REE1] = [REE2] = 2.5 mM, [C2O42−] = 15 mM; [aapa] = 5 mM. pH 4. Buffer: 100 mM ammonium acetate; t = 1.5 h. | ||
Because aapa afforded the largest differences within the dissolution of the Ln2(C2O4)3 solids at pH 4, this pH was selected for subsequent separation experiments. A total of 36 binary Ln2(C2O4)3 mixtures were prepared and then contacted with aapa (Fig. 6a and b), following the procedures used for the single-element oxalate salts. The metal concentrations remaining the solution were measured by ICP-OES, and based on these data the separation factors (SFs), which quantify the extent of REE separation, were calculated with eqn (S4) and are summarized in Tables S8 and S9. The SF values increase as the difference in ionic radii between the REEs gets larger. The highest SF of 38.7 was achieved for the Ce3+/Lu3+ pair, followed by 33.1 for La3+/Lu3+ and 26.6 for Ce3+/Yb3+ (Fig. 6c). These binary REE separation data are consistent with the single Ln2(C2O4)3 dissolution experiments and provide a promising strategy for direct dissolution-based REE separations. Although these SFs are somewhat smaller than might be expected based on relative logK value, the experimentally observed SFs reflects the combined effects of metal–ligand complexation, precipitation–dissolution equilibria, and the specific experimental conditions employed. Consequently, further optimization of separation conditions could potentially improve the observed SFs. The chelator acyclopa was also used for dissolution-based separations of La3+ and Y3+ from insoluble REE(OH)3. With acyclopa, SFs for these two metal ions up to 295 could be obtained, albeit with higher loading concentrations of insoluble REE(OH)3 mixtures at pH 9.48
 | ||
| Fig. 7 REE leaching from waste magnet using different chelators. (a) Metal concentration in the filtrate of magnet waste leaching; (b) ligand efficiency of REEs for magnet waste; (c) weight ratio of REE to non-REEs in the filtrate of magnet waste leaching. Conditions: 20 mg magnet waste. 1 mL chelator solution (20 mM); buffer: 100 mM MOPS solution (pH 7.4); t = 24 h, rotated at 40 rpm. | ||
REE-selective extraction from magnet waste and autocatalyst smelting slag
Building upon the ability of aapa to dissolve REE2(C2O4)3 solids, we investigated its use for selectively leaching REEs from Nd2Fe14B magnet waste and autocatalyst smelting slag (autocat slag). The Nd2Fe14B magnet waste was acquired from a computer hard drive that was ground and separated into 30-mesh (0.595 mm) particle size. These leaching experiments were performed at pH 7.4, buffered by 100 mM 3-(morpholin-4-yl)propane-1-sulfonic acid (MOPS), and studied in comparison to diethylenetriaminepentaacetic acid (DTPA) and macropa. Buffered water was used as a leaching control. After contacting the solid waste materials for 24 h with continuous mixing via end-over-end rotation at 40 rpm, the solid was removed by filtration, and the metal concentration in the filtrate was determined by ICP-OES. From these data, dissolution efficacy was calculated via the ligand efficiency parameter shown in eqn (1): | (1) |
:1 M:L binding model. For the Nd2Fe14B waste, a pulp density of 20 g L−1 was employed. As shown in Fig. 7a, Tables S10 and S11, 1 mL of buffered water does not extract any metals from the Nd2Fe14B magnet waste at neutral pH. By contrast, the addition of 20 µmol (20 mM × 1 mL) aapa or macropa to the magnet waste leads to efficient dissolution of the REEs. This effect is most pronounced for Nd3+, the REE of highest abundance in these materials. Under this condition, the use of aapa and macropa gives leachates containing 828 ppm and 819 ppm Nd3+, corresponding to ligand efficiencies of 28.9% and 28.6% (Fig. 7a), respectively. Both of these chelators are more effective than DTPA, which gives rise to a leachate containing 521 ppm Nd3+ corresponding to a ligand efficiency of 18.1%. Moreover, both aapa and macropa achieved superior leaching of Dy3+ and Pr3+ compared to DTPA, even though DTPA complexes of these REEs have larger logK values than those of aapa and macropa,68 as reflected by overall ligand efficiencies for REEs of 30.5%, 29.8%, and 19.1% for aapa, macropa, and DTPA, respectively (Fig. 7b). The poorer performance of DTPA can potentially be explained by the less selective metal-binding properties of this chelator. For example, DTPA also extracts large quantities of transition metals (342 ppm Fe3+ and 363 ppm Cu2+) into the solution. These values are 2–6 times larger than those extracted by aapa (179 ppm Fe3+ and 120 ppm Cu2+) and macropa (58 ppm Fe3+ and 265 ppm Cu2+). The difference in metal composition of the leachate obtained from these different chelators could be easily recognized from their colors (insert of Fig. 7a), as complexes of Cu2+ and Fe3+ give characteristic blue and yellow solutions. The selectivity of these ligands for REEs over non-REEs was calculated by determining the weight concentration ratio between the total quantity of REEs (∑REEs) and total quantity of non-REEs (∑non-REEs) in the filtrates (Fig. 7c). The chelator aapa shows the highest selectivity for REEs with a ∑REEs/∑non-REEs ratio of 1.70, somewhat larger than the 1.57 value obtained for macropa. The larger transition metal-leaching ability of DTPA is characterized by a poorer selectivity ratio of only 0.66. Next, the rate of dissolution of Nd3+ and Dy3+ from magnet waste by these chelators were investigated over a 0.5–24 h timescale (Fig. S46, SI). Faster Nd3+-leaching kinetics are observed for aapa and macropa compared to DTPA. Furthermore, even though aapa and macropa have a similar Nd3+-dissolution performance after 24 h, aapa dissolves Nd3+ much faster, achieving soluble concentrations of this metal ion of 730 ppm after only 4 h (Fig. S46a, SI). By contrast, macropa requires 16 h to dissolve Nd3+ to the same extent (774 ppm). The faster leaching properties of aapa are also apparent for Dy3+, as this chelator exhibits the fastest rates for dissolution of this metal ion from magnet waste throughout the entire leaching process (Fig. S46b, SI).
 | ||
| Fig. 8 Metal weight percentages of different magnet waste leachates using different ligands. | ||
To evaluate the REE-selective dissolution of aapa and macropa, they were compared to the most commonly used lixiviant 4 M HNO3. As shown in Tables S10 and S11 (SI), 4 M HNO3 extracts both Nd3+ (2132.4 ppm) and Dy3+ (107.5 ppm), 2.6 times more effectively than aapa. However, this increased dissolution efficacy also leads to poorer REE selectively, leaching 89-, 35-, 5-, 23-, 53-, and 3-fold higher levels of Al3+, Fe3+, Ca2+, Cu2+, Ni2+ and Zn2+, respectively, than that of 20 mM aapa. Consequently, the REE weight percentage of the resulting HNO3 leachate is very low. As shown in Fig. 8, this HNO3 leachate contains only 15.6% Nd3+, 0.1% Pr3+, and 0.8% Dy3+, with larger weight percentages of Fe3+ (44.8%) and Cu2+ (20.3%). By contrast, the Nd3+ and Dy3+ content in the aapa and macropa leachate range between 58.4–59.4% and 2.1–3%, respectively, and the Fe3+ and Cu2+ weight percentages are only 19.9–4.1% and 8.7–19.0%, respectively. For comparison, 20 mM DTPA exhibits moderate selectivity for REEs, leading to a leachate that contains 37.6% Nd3+ and 1.7% Dy3+, but also with 26.2% Cu2+. Collectively, these leaching results demonstrate that aapa enables the most efficient and selective recovery of Nd3+, Dy3+, and Pr3+ from Nd2Fe14B magnet waste under neutral aqueous conditions, performing better than the macrocycle macropa, the conventional chelator DTPA, and 4 M HNO3.
Similar leaching experiments were conducted with autocat slag. Compared to the Nd2Fe14B magnet waste, the relative amounts of main group and transition metals are hundreds of times larger than those of REEs within the autocat slag,45 making selective REE leaching a more challenging process. First, the effects of the pulp density of the autocat slag on its chelator-assisted dissolution was studied (Tables S12, S13 and Fig. S47, SI). At larger pulp densities, both aapa and macropa were more effective at dissolving the REEs. For example, at a pulp density of 10 g L−1, aapa and macropa led to soluble Ce3+ concentrations of 12.4 and 2.36 ppm, respectively, whereas at a pulp density of 50 g L−1, these soluble concentrations increased to 28.4 and 10.2 ppm, respectively. Although higher pulp densities appear to lead to more efficient dissolution, the maximum practical pulp density used in these studies was 50 g L−1, due to challenges arising from the mechanical mixture of higher pulp density suspensions. For direct comparison, all subsequent studies with aapa, macropa, DTPA, and buffered water used a pulp density of 50 g L−1 (Tables S14 and S15, SI). As expected, pH 7.4 buffered water is unable to leach any metals from the autocat slag (Fig. S48a, SI). Remarkably, aapa solubilizes more REEs from autocat slag than macropa, yielding a leachate containing 3.76 ppm La3+, 28.4 ppm Ce3+, 0.92 ppm Pr3+, and 2.25 ppm Nd3+ that corresponds to an overall ligand efficiency of 1.26% (Fig. S48b, SI). In comparison, the concentration of these REEs solubilized by macropa were 1.62, 10.2, 0.26, and 0.83 ppm, respectively, which corresponds to only ∼30% of the REE content that can be dissolved by aapa (Fig. S48b, SI). These results indicate that aapa is more effective than macropa in leaching REEs from autocat slag, presumably due to its higher binding affinities for these elements. Under the same condition, DTPA extracted more Ce3+ (33.0 ppm vs. 28.4 ppm) and La3+ (4.8 ppm vs. 3.8 ppm) than aapa and gave a higher ligand efficiency of 1.67% (Fig. S48b, SI). However, this chelator also dissolved nearly two times as much Ca2+ (125 vs. 78 ppm) and more than twice as much Fe3+ (50 vs. 21.2 ppm) than aapa, which can be recognized from the color of chelate in insert of Fig. S48a, resulting in a slightly lower ∑REEs/∑non-REEs (0.23%) than that of aapa (0.25%, Fig. S48c, SI). Moreover, aapa and DTPA have similar kinetics for autocat slag leaching, as both reach equilibrium at 4 h. By comparison, macropa leaching is slower; it doesn't reach equilibrium until 24 h (Fig. S49, SI). However, the leaching efficiencies of those three chelators for autocat slag (2–3% of 4 M HNO3 REEs leaching) is much lower than that for magnet waste (∼40% of 4 M HNO3 REEs leaching), demonstrating that the composition of the secondary REEs materials is also an important factor for REEs leaching employing chelators.
Conclusion
Inspired by recent efforts to make acyclic versions of macropa, like acyclopa and ODA, we developed and evaluated a cost-efficient acyclic ligand, H4aapa, for its abilities to chelate REEs from solid waste. This ligand has a high binding affinity for REEs, as marked by logKLnL values that range from 18.95–15.28. Furthermore, it retains a preference for light over heavy REEs. These properties enable aapa to achieve a direct dissolution-based REE separation via its action on REE oxalate mixtures. The subsequent application of aapa for use in authentic magnet waste, as well as autocat slag, also reveals its potential for enhanced REE solubilization and enrichment, especially compared to a conventional chelator like DTPA and a strong acid, like HNO3, which exhibit poor selectivity. Collectively, these studies highlight how chelator design efforts can be applied for the direct leaching of REEs from end-of-life materials. However, further efforts are needed to optimize and improve the overall selectivity of these chelators to achieve both higher leaching efficiency as well as REE selectivity.
Author contributions
Y. Gao designed and conducted experimental data and wrote the paper. A. M. Schmitz and S. Medin provided research materials and designed and aided in interpretation of the autocat slag experiments. J. J. Wilson conceived and supervised the project and wrote the paper.Conflicts of interest
Alexa M. Schmitz and Sean Medin declare a financial interest in the company REEgen, Inc. which is exploring biological-based REE extraction technologies partially related to this research.Data availability
CCDC 2528345 ([La(aapa)][La(H2aapa)]) contains the supplementary crystallographic data for this paper.69Data to support the conclusions in this paper are available in the main text or the supplementary information (SI). Supplementary information: syntheses, potentiometric titrations, UV-vis titrations, NMR spectroscopy studies, X-ray crystallography studies, REE oxalate separation and REE waste leaching studies. See DOI: https://doi.org/10.1039/d6sc00975a.
Acknowledgements
This material is based upon work supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under award number DE-SC0025792 and work supported by the U.S. National Science Foundation, Directorate of Technology, Innovation and Partnerships under Award No. 2527909. The hard drive magnet waste powder was provided by Dr Erik Spiller and Dr Brandon Ott at Colorado School of Mines by way of Dr David Reed at Idaho National Lab, and the autocat slag was provided by REEgen, Inc.References
- V. Balaram, Rare earth elements: A review of applications, occurrence, exploration, analysis, recycling, and environmental impact, Geosci. Front., 2019, 10, 1285–1303 CrossRef CAS.
- A. M. Brown, Reclaiming rare earths, Nat. Rev. Chem., 2025, 9, 654 CrossRef PubMed.
- B. Ghosh, H. Vapnik, H. E. Kim, Y. Kim, R. Birawat, Y. Lu, X. Su and H. Yang, Electrochemical Separation and Clean Energy Applications of Rare Earth Elements, Chem. Rev., 2025, 125, 7965–8023 CrossRef CAS PubMed.
- Z.-Q. Bian and C.-H. Huang, Rare Earth Coordination Chemistry: Fundamentals and Applications, Wiley-VCH Verlag GmbH & Co. KGaA, 2008 Search PubMed.
- T. Cheisson and E. J. Schelter, Rare earth elements: Mendeleev's bane, modern marvels, Science, 2019, 363, 489–493 CrossRef CAS PubMed.
- M. K. Hossain, G. A. Raihan, M. A. Akbar, M. H. Kabir Rubel, M. H. Ahmed, M. I. Khan, S. Hossain, S. K. Sen, M. I. E. Jalal and A. El-Denglawey, Current Applications and Future Potential of Rare Earth Oxides in Sustainable Nuclear, Radiation, and Energy Devices: A Review, ACS Appl. Electron. Mater., 2022, 4, 3327–3353 CrossRef CAS.
- Y. Khan, The U.S. Wants a Rare-Earths Supply Chain. Here's Why It Won't Come Easily, Wall St. J., 2023 Search PubMed , Available from: https://www.wsj.com/articles/the-u-s-wants-a-rare-earths-supply-chain-heres-why-it-wont-come-easily-dfc3b632?st=vA4w77&reflink=desktopwebshare_permalink.
- B. J. Smith, R. E. Matthew, M. R. Earlam, C. Iloeje and D. Diamond, Rare Earth Permanent Magnets: Supply Chain Deep Dive Assessment, U.S. Department of Energy, Washington, DC. United States. 2022, DOI:10.2172/1871577.
- Adamas Intelligence, Rare Earth Magnet Market Outlook to 2035, Toronto, Canada: Adamas Inside, 2022 April 20, Available from: https://www.adamasintel.com/rare-earth-magnet-market-outlook-to-2035/.
- L. Omodara, S. Pitkäaho, E. M. Turpeinen, P. Saavalainen, K. Oravisjärvi and R. L. Keiski, Recycling and substitution of light rare earth elements, cerium, lanthanum, neodymium, and praseodymium from end-of-life applications - A review, J. Cleaner Prod., 2019, 236, 11573 CrossRef.
- S. B. Jadhav, D. B. Malavekar, R. A. Mohite, S. B. Shaikh, K. V. Kadam, P. N. Pawaskar, J. H. Kim and N. E. Lee, A critical review of lanthanum and lanthanum-based materials: synthesis, applications, and challenges, Rare Met., 2025, 44, 5201–5232 CrossRef CAS.
- X. Song, M. H. Chang and M. Pecht, Rare-earth elements in lighting and optical applications and their recycling, JOM, 2013, 65, 1276–1282 CrossRef.
- J. Nayar, Not So “Green” Tec hnology: The Complicated Legacy of Rare Earth Mining, https://hir.harvard.edu/not-so-green-technology-the-complicated-legacy-of-rare-earth-mining/, accessed 12 August 2021.
- E. O. Opare, E. Struhs and A. Mirkouei, A comparative state-of-technology review and future directions for rare earth element separation, Renewable Sustainable Energy Rev., 2021, 143, 110917 CrossRef CAS.
- U. Epa, L. Remediation and P. C. Division, Rare Earth Elements: A Review of Production, Processing, Recycling, and Associated Environmental Issues, Cincinnati, U.S. Environment protection Agency, United States, 2012, Report No. EPA 600/R-12/572, Available from: https://nepis.epa.gov/Exe/ZyPURL.cgi?Dockey=P100EUBC.txt Search PubMed.
- K. S. Patel, S. Sharma, J. P. Maity, P. Martín-Ramos, Ž. Fiket, P. Bhattacharya and Y. Zhu, Occurrence of uranium, thorium and rare earth elements in the environment: A review, Front. Environ. Sci., 2023, 10, 1058053 CrossRef.
- A. Romero-Freire, L. Minguez, M. Pelletier, A. Cayer, C. Caillet, S. Devin, E. M. Gross, F. Guérold, S. Pain-Devin, D. A. L. Vignati and L. Giamberini, Assessment of baseline ecotoxicity of sediments from a prospective mining area enriched in light rare earth elements, Sci. Total Environ., 2018, 612, 831–839 CrossRef CAS PubMed.
- M. Evans, C. Brooks and M. K. Johnson, United States Energy Association: Critical Material Recovery from E-waste, United States Energy Association, Columbus, United States, 2023, Report No. 633-2023-004-01, Available from: https://usea.org/publication/critical-material-recovery-e-waste Search PubMed.
- G. Gaustad, E. Williams and A. Leader, Rare earth metals from secondary sources: Review of potential supply from waste and byproducts, Resour. Conserv. Recycl., 2021, 167, 105213 CrossRef CAS.
- Y. Fujita, S. K. McCall and D. Ginosar, Recycling rare earths: Perspectives and recent advances, MRS Bull., 2022, 47, 283–288 CrossRef CAS.
- M. K. Jha, A. Kumari, R. Panda, J. Rajesh Kumar, K. Yoo and J. Y. Lee, Review on hydrometallurgical recovery of rare earth metals, Hydrometallurgy, 2016, 161, 77–101 CrossRef CAS.
- M. Walawalkar, C. K. Nichol and G. Azimi, Process investigation of the acid leaching of rare earth elements from phosphogypsum using HCl, HNO3, and H2SO4, Hydrometallurgy, 2016, 166, 195–204 CrossRef CAS.
- S. Peelman, Z. H. I. Sun, J. Sietsma and Y. Yang, Leaching of Rare Earth Elements: Past and Present Technologies, ed. I. B. De Lima and W. Leal Filho, Rare Earths Industry, Delft, The Netherlands. 2014, pp. 446–456 Search PubMed.
- A. K. Saim, Ammoniaca l Leaching for the Extraction of Valuable Metals from Secondary Resources: A Review, Miner. Process. Extr. Metall. Rev., 2025, 46, 284–305 CrossRef CAS.
- A. Shoppert, D. Valeev, J. Napol'skikh, I. Loginova, J. Pan, H. Chen and L. Zhang, Materials, Rare-Earth Elements Extraction from Low-Alkali Desilicated Coal Fly Ash by (NH4)2SO4 + H2SO4, Materials, 2023, 16, 6 CrossRef CAS PubMed.
- Q. Zhang, F. Ren, F. Li, G. Chen, G. Yang, J. Wang, K. Du, S. Liu and Z. Li, Ammonia nitrogen sources and pollution along soil profiles in an in-situ leaching rare earth ore, Environ. Pollut., 2020, 267, 115449 CrossRef CAS PubMed.
- E. Vahidi and F. Zhao, Environmental Life Cycle Assessment on the Separation of Rare Earth Oxides Through Solvent Extraction, J. Environ. Manage., 2017, 203, 255–263 CrossRef CAS PubMed.
- A. Pan, S. Feng, X. Hu and Y. Li, How environmental regulation affects China's rare earth export?, PLoS One, 2021, 16, 1–20 Search PubMed.
- P. Zapp, A. Schreiber, J. Marx and W. Kuckshinrichs, Recycling rare earths: Perspectives and recent advances, MRS Bull., 2022, 47, 267–275 CrossRef CAS PubMed.
- A. M. Schmitz, B. Pian, S. Marecos, M. Wu, M. Holycross, E. Gazel, M. C. Reid and B. Barstow, High efficiency rare earth element bioleaching with systems biology guided engineering of Gluconobacter oxydans, Commun. Biol., 2025, 8, 815 CrossRef CAS PubMed.
- F. Hosseinzadeh, S. O. Rastegar and M. Ashengroph, Bioleaching of rare earth elements from spent automobile catalyst as pretreatment method to improve Pt and Pd recovery: Process optimization and kinetic study, Process Biochem., 2021, 105, 1–7 CrossRef CAS.
- S. Nadi, F. Pourhossein and S. M. Mousavi, Sustainable bio-extraction of rare earth elements from discarded LED lamps, Sci. Rep., 2025, 15, 21655 CrossRef PubMed.
- D. W. Reed, Y. Fujita, D. L. Daubaras, Y. Jiao and V. S. Thompson, Bioleaching of Rare Earth Elements from Waste Phosphors and Cracking Catalysts, Hydrometallurgy, 2016, 166, 34–40 CrossRef CAS.
- K. Joshi, S. Magdouli and S. K. Brar, Bioleaching for the Recovery of Rare Earth Elements from Industrial Waste: A Sustainable Approach, Resour. Conserv. Recycl., 2025, 215, 108129 CrossRef CAS.
- P. Rasoulnia, R. Barthen and A. M. Lakaniemi, A Critical Review of Bioleaching of Rare Earth Elements: The Mechanisms and Effect of Process Parameters, Crit. Rev. Environ. Sci. Technol., 2021, 51, 378–427 CrossRef CAS.
- V. S. Thompson, M. Gupta, H. Jin, E. Vahidi, M. Yim, M. A. Jindra, V. Nguyen, Y. Fujita, J. W. Sutherland, Y. Jiao and D. W. Reed, Techno-Economic and Life Cycle Analysis for Bioleaching Rare-Earth Elements from Waste Materials, ACS Sustain. Chem. Eng., 2018, 6, 1602–1609 CrossRef CAS.
- J. Tang, J. Qiao, Q. Xue, F. Liu, H. Chen and G. Zhang, Leach of the weathering crust elution-deposited rare earth ore for low environmental pollution with a combination of (NH4)2SO4 and EDTA, Chemosphere, 2018, 199, 160–167 CrossRef CAS PubMed.
- M. Zhao, X. Hou, H. Zhang, H. Han, Y. Yang and T. Li, Selection of Organic Acids for Leaching Rare Earth Elements from the Colloidal Sediment Phase: Leaching Characteristics and Mechanism, ACS Sustainable Resour. Manage., 2025, 2, 1869–1878 CrossRef CAS.
- P. Rasoulnia, R. Barthen, J. A. Puhakka and A. M. Lakaniemi, Leaching of rare earth elements and base metals from spent NiMH batteries using gluconate and its potential bio-oxidation products, J. Hazard. Mater., 2021, 414, 125564 CrossRef CAS PubMed.
- N. A. Thiele, V. Brown, J. M. Kelly, A. Amor-Coarasa, U. Jermilova, S. N. MacMillan, A. Nikolopoulou, S. Ponnala, C. F. Ramogida, A. K. H. Robertson, C. Rodríguez-Rodríguez, P. Schaffer, C. Williams, J. W. Babich, V. Radchenko and J. J. Wilson, An Eighteen-Membered Macrocyclic Ligand for Actinium-225 Targeted Alpha Therapy, Angew. Chem., Int. Ed., 2017, 129, 14904–14909 CrossRef.
- A. P. King, N. T. Gutsche, N. Raju, S. Fayn, K. E. Baidoo, M. M. Bell, C. S. Olkowski, R. E. Swenson, F. I. Lin, S. M. Sadowski, S. S. Adler, N. A. Thiele, J. J. Wilson, P. L. Choyke and F. E. Escorcia, 225Ac-MACROPATATE: A Novel a-Particle Peptide Receptor Radionuclide Therapy for Neuroendocrine Tumors, J. Nucl. Med., 2023, 64, 549–554 CrossRef CAS PubMed.
- M. P. Jensen, R. Chiarizia, J. S. Ulicki, B. D. Spindler, D. J. Murphy, M. M. Hossain, A. Roca-Sabio, A. de Blas and T. Rodríguez-Blas, Solvent Extraction Separation of Trivalent Americium from Curium and the Lanthanides, Solvent Extr. Ion Exch., 2015, 33, 329–345 CrossRef CAS.
- M. P. Jensen, R. Chiarizia, I. A. Shkrob, J. S. Ulicki, B. D. Spindler, D. J. Murphy, M. Hossain, A. Roca-Sabio, C. Platas-Iglesias, A. De Blas and T. Rodríguez-Blas, Aqueous complexes for efficient size-based separation of americium from curium, Inorg. Chem., 2014, 53, 6003–6012 CrossRef CAS PubMed.
- N. A. Thiele, D. Fiszbein, J. J. Woods and J. J. Wilson, Tuning the Separation of Light Lanthanides Using a Reverse-Size Selective Aqueous Complexant, Inorg. Chem., 2020, 59, 16522–16530 CrossRef CAS PubMed.
- Y. Gao, K. Ivanovich, S. Medin, B. Pian, S. N. MacMillan, A. M. Schmitz and J. J. Wilson, 18-membered macrocycle appended on resin for selective rare earth element extraction and separation, Commun. Chem., 2025, 8, 176 CrossRef CAS PubMed.
- Y. Gao, G. L. Licup, N. P. Bigham, D. C. Cantu and J. J. Wilson, Chelator-Assisted Precipitation-Based Separation of the Rare Earth Elements Neodymium and Dysprosium from Aqueous Solutions, Angew. Chem., Int. Ed., 2024, 63, e202410233 CAS.
- N. A. Thiele, S. N. Macmillan and J. J. Wilson, Rapid Dissolution of BaSO4 by Macropa, an 18-Membered Macrocycle with High Affinity for Ba2+, J. Am. Chem. Soc., 2018, 140, 17071–17078 CrossRef CAS PubMed.
- N. A. Thiele, B. A. Moyer and J. Dempsey, Size-selective acyclic chelators and their use for the recovery of rare earth elements, US Pat., US18/443585, Oak Ridge National Laboratory, Filing date February 16, 2024 Search PubMed.
- M. F. Islam, L. Lin, D. Ray, U. I. Premadasa, Y. Z. Ma, R. L. Sacci, V. Kertesz, R. Custelcean, V. S. Bryantsev, B. Doughty and N. A. Thiele, Conformationally Adaptable Extractant Flexes Strong Lanthanide Reverse-Size Selectivity, J. Am. Chem. Soc., 2025, 147, 5080–5088 CrossRef CAS PubMed.
- A. Ingham, L. Wharton, T. El Sayed, L. Southcott, B. L. McNeil, M. B. Ezhova, B. O. Patrick, M. D. G. Jaraquemada-Peláez and C. Orvig, H2ampa? Versatile Chelator for [203Pb]Pb2+, [213Bi]Bi3+, and [225Ac]Ac3+, Inorg. Chem., 2022, 61, 9119–9137 CrossRef CAS PubMed.
- E. W. Price, B. M. Zeglis, J. F. Cawthray, C. F. Ramogida, N. Ramos, J. S. Lewis, M. J. Adam and C. Orvig, H4octapa-trastuzumab: Versatile acyclic chelate system for 111In and 177Lu imaging and therapy, J. Am. Chem. Soc., 2013, 135, 12707–12721 CrossRef CAS PubMed.
- F. K. Kálmán, A. Végh, M. Regueiro-Figueroa, É. Tóth, C. Platas-Iglesias and G. Tircsó, H4octapa: Highly stable complexation of lanthanide(III) ions and copper(II), Inorg. Chem., 2015, 54, 2345–2356 CrossRef PubMed.
- L. Wharton, E. Kurakina, V. Radchenko, P. Schaffer and C. Orvig, Chemical Promiscuity of Non-Macrocyclic Multidentate Chelating Ligands for Radiometal Ions: H4neunpa-NH2 vs H4noneunpa, Inorg. Chem., 2021, 60, 4076–4092 CrossRef CAS PubMed.
- A. Hu, I. Keresztes, S. N. MacMillan, Y. Yang, E. Ding, W. R. Zipfel, R. A. Distasio Jr, J. W. Babich and J. J. Wilson, Oxyaapa: A Picolinate-Based Ligand with Five Oxygen Donors that Strongly Chelates Lanthanides, Inorg. Chem., 2020, 59, 5116–5132 CrossRef CAS PubMed.
- K. K. Lee, M. Chakraborty, A. Hu, T. Kanagasundaram, D. L. J. Thorek and J. J. Wilson, Chelation of [111In]In3+ with the dual-size-selective macrocycles py-macrodipa and py2-macrodipa, Dalton Trans., 2024, 53, 14634 RSC.
- A. Hu, E. Aluicio-sarduy, V. Brown, S. N. Macmillan, K. V Becker, T. E. Barnhart, V. Radchenko, C. F. Ramogida, J. W. Engle and J. J. Wilson, Py-Macrodipa: A Janus Chelator Capable of Binding Medicinally Relevant Rare-Earth Radiometals of Disparate Sizes, J. Am. Chem. Soc., 2021, 143, 10429–10440 CrossRef CAS PubMed.
- J. J. Woods, R. Unnerstall, A. Hasson, D. S. Abou, V. Radchenko, D. L. J. Thorek and J. J. Wilson, Stable Chelation of the Uranyl Ion by Acyclic Hexadentate Ligands: Potential Applications for 230U Targeted α-Therapy, Inorg. Chem., 2022, 61, 3337–3350 CrossRef CAS PubMed.
- L. Li, M. D. G. Jaraquemada-Peláez, H. T. Kuo, H. Merkens, N. Choudhary, K. Gitschtaler, U. Jermilova, N. Colpo, C. Uribe-Munoz, V. Radchenko, P. Schaffer, K. S. Lin, F. Bénard and C. Orvig, Functionally Versatile and Highly Stable Chelator for 111In and 177Lu: Proof-of-Principle Prostate-Specific Membrane Antigen Targeting, Bioconjugate Chem., 2019, 30, 1539–1553 CrossRef CAS PubMed.
- R. D. Hancock and A. E. Martell, Metal Complexes in Aqueous Solutions, Plenum Publishing Corporation, New York, 1996 Search PubMed.
- M. D. G. Jaraquemada-Peláez, X. Wang, T. J. Clough, Y. Cao, N. Choudhary, K. Emler, B. O. Patrick and C. Orvig, H4octapa: Synthesis, Solution Equilibria and Complexes with Useful Radiopharmaceutical Metal Ions, Dalton Trans., 2017, 46, 14647–14658 RSC.
- A. Roca-Sabio, M. Mato-Iglesias, D. Esteban-Gómez, É. Toth, A. De Bias, C. Platas-Iglesias and T. Rodríguez-Blas, Macrocyclic Receptor Exhibiting Unprecedented Selectivity for Light Lanthanides, J. Am. Chem. Soc., 2009, 131, 3331–3341 CrossRef CAS PubMed.
- P. A. Panchenko, A. D. Zubenko, E. Y. Chernikova, Y. V. Fedorov, A. V. Pashanova, V. A. Karnoukhova, I. V. Fedyanin and O. A. Fedorova, Synthesis, structure and metal ion coordination of novel benzodiazamacrocyclic ligands bearing pyridyl and picolinate pendant side-arms, New J. Chem., 2019, 43, 15072–15086 RSC.
- T. V. Hoogerstraete, B. Blanpain, T. Van Gerven and K. Binnemans, From NdFeB magnets towards the rare-earth oxides: A recycling process consuming only oxalic acid, RSC Adv., 2014, 4, 64099–64111 RSC.
- D. Prodius, M. Klocke, V. Smetana, T. Alammar, M. Perez Garcia, T. L. Windus, I. C. Nlebedim and A. V. Mudring, Rationally designed rare earth separation by selective oxalate solubilization, Chem. Commun., 2020, 56, 11386–11389 RSC.
- K. J. Cantrell and R. H. Byrne, Rare earth element complexation by carbonate and oxalate ions, Geochim. Cosmochim. Acta, 1987, 51, 591–605 CrossRef.
- L. A. Sarver and P. H. M.-P. Brinton, The solubilities of some rare-earth oxalates, J. Am. Chem. Soc., 1927, 49, 943–958 CrossRef CAS.
- B. Weaver, Fractional Separation of Rare Earths by Oxalate Precipitation from Homogeneous. Solution, Anal. Chem., 1954, 26, 479–480 CrossRef CAS.
- T. S. Grimes and K. L. Nash, Acid dissociation constants and rare earth stability constants for DTPA, J. Solution Chem., 2014, 43, 298–313 CrossRef CAS.
- CCDC 2528345: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qvyj2..
| This journal is © The Royal Society of Chemistry 2026 |