
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotoinduced deacylative epoxidation of ketone derivatives with allylic peroxides
Jiangtao
Lin
a,
Mingqian
Zhou
a,
Ziyang
Li
a,
Jiang
Duan
a,
Xiaoyu
Liu
a,
Yue
Wang
a and
Chao
Shu
 *ab
*ab
aState Key Laboratory of Green Pesticide, Engineering Research Center of Photoenergy Utilization for Pollution Control and Carbon Reduction, CCNU-uOttawa Joint Research Centre, College of Chemistry, Central China Normal University, 152 Luoyu Road, Wuhan, Hubei 430079, China. E-mail: chaoshu@ccnu.edu.cn
bWuhan Institute of Photochemistry and Technology, 7 North Bingang Road, Wuhan, Hubei 430083, China
First published on 17th April 2026
Abstract
Epoxide compounds represent a privileged scaffold with diverse applications in organic synthesis, pharmaceuticals, agrochemicals, and materials science. However, the available routes to obtain such high-value structures from feedstocks, such as ketones, remain less developed. In this work, we report an alternative deacylative epoxidation of ketones using allylic peroxides to produce fused epoxides via the photoinduced radical cyclization of ketone-derived pro-aromatic dihydroquinazolinones. This system operates under very mild conditions and showcases a broad substrate scope with excellent functional group tolerance (over 50 examples). Its gram-scale preparation and late-stage transformations emphasize its potential in drug discovery. A possible mechanism has been proposed based on mechanistic studies.
Introduction
Strained three-membered oxygen heterocycles, known as epoxides or oxiranes, are highly valuable motifs in the drug industry with remarkable biological and pharmaceutical activities and are commonly observed in naturally occurring substances, pharmaceutical compounds, materials and synthetic molecules, including 14 FDA-approved drugs (Scheme 1a).1 In addition, spring-loaded oxiranes have inherent angle strain and two polar bonds, making them highly reactive and rendering them excellent reactive intermediates for constructing new bonds in larger and more complex molecules through nucleophilic ring-opening reactions in the presence of different nucleophiles, such as organolithium, amine, cyanides, thiols, halides and azides.2,3 The global market for oxiranes is expected to reach $78 billion by 2025, fueled by various applications, such as the production of epoxy resins, surfactants, adhesives, coatings, agrochemicals, and pharmaceuticals.4 | ||
| Scheme 1 Substrate scope for ketones. Reaction conditions: ketone derivatives (0.2 mmol, 1.0 equiv.), 1b (0.4 mmol, 2.0 equiv.), 4CzIPN (5 mol%), DBU (0.1 mmol, 0.5 equiv.), Cs2CO3 (0.1 mmol, 0.5 equiv.), chlorobenzene (0.05 M), room temperature, under 20 W blue LEDs (λmax = 445 nm), 24 h, in a vial. Isolated yield. The R′ group of the ketone used as the raw material is a phenyl (R′ = Ph) unless otherwise noted. The specific reaction raw materials are referenced in the Supporting Information. aThe R′ group of the ketone used as the raw material is a methyl group (R′ = Me). | ||
Therefore, the efficient incorporation of oxirane units into organic molecules is an important and challenging objective, particularly when utilizing abundant feedstock chemicals under simple conditions. However, the majority of state-of-the-art methodologies for synthesizing oxiranes are limited to the epoxidation of specific single alkenes using strong oxidizing agents, such as peroxy acids or hydrogen peroxide.1,5 To explore more readily available substrates and facilitate operational processes, as well as to achieve greater structural diversity in oxiranes, it would be immensely beneficial to develop a straightforward method for converting libraries of cheap feedstocks into their corresponding oxiranes, addressing growing demands in chemical biology and drug discovery.
Among them, ketones (carbonyl compounds) are one of the most common and widely used fundamental skeletons found in many industrial chemicals and pharmaceutical architectures.6 For example, Darzens condensation involves the reaction of aldehydes or ketones with α-mono- or dihalocarbonyl compounds in the presence of bases to form epoxides (Fig. 1b, left),7 and the Johnson–Corey–Chaykovsky method8 is based on the reaction of carbonyl compounds with sulfonium and sulfoxonium ylides, which are often derived in situ from sulfonium or sulfoxonium halides containing the mono- or disubstituted methyl group (Fig. 1b, right). However, these well-known reactions still suffer from limitations, such as the requirement for strong bases, aldehyde self-condensation, and the failure to achieve tri-alkyl sulfur ylides. The further elaboration of complex functionalities associated with ketone-containing molecules, such as amino acids, peptides and sensitive drug candidates, poses a synthetic challenge for epoxidation reactions, primarily due to the limited number of reliable methods currently available and their poor compatibility under harsh conditions.
 | ||
| Fig. 1 Background for the synthesis of epoxides and our design. (a) Selected bioactive examples containing oxirane unit. (b) Typical well-known epoxidation of ketone and aldehyde. (c) Photoinduced radical decarbonylation transformations of ketones. (d) This work: the first photoinduced deacylative radical epoxidation of ketones. | ||
Despite this outstanding work, the exploration of conceptually distinct strategies—especially those avoiding harsh reaction conditions, such as high temperature, excessive additives, long reaction times and limited substrate scope—remains an urgent issue. Developing alternative methods for the direct activation and functionalization of these carbonyl compounds to oxiranes is crucial for improving the quality of chemical raw materials and for the late-stage modification of complex natural products and pharmaceuticals. However, due to the strong bond dissociation energy (BDE) of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond, deoxygenation transformations of these functional groups present considerable challenges, especially for unstrained common ketones. Based on Dong's pioneering work on the deconstructive transformations of ketones,9 alkyl radicals can be generated via deacylation through pre-aromatization intermediates like dihydropyrazoles and dihydrotriazoles. Recently, innovative methods have emerged for the deconstructive reactions of ketones, involving their transformation into pro-aromatic compounds known as dihydroquinazolinones (DHQZs), with related studies being reported consecutively (Fig. 1c).10,11 Seeking a complementary route that would allow the direct transformation of unstrained ketones to oxiranes, we questioned whether deacylative epoxidation could occur through the aromatization-driven deconstructive reaction of ketones.
O double bond, deoxygenation transformations of these functional groups present considerable challenges, especially for unstrained common ketones. Based on Dong's pioneering work on the deconstructive transformations of ketones,9 alkyl radicals can be generated via deacylation through pre-aromatization intermediates like dihydropyrazoles and dihydrotriazoles. Recently, innovative methods have emerged for the deconstructive reactions of ketones, involving their transformation into pro-aromatic compounds known as dihydroquinazolinones (DHQZs), with related studies being reported consecutively (Fig. 1c).10,11 Seeking a complementary route that would allow the direct transformation of unstrained ketones to oxiranes, we questioned whether deacylative epoxidation could occur through the aromatization-driven deconstructive reaction of ketones.
Based on our continuing interest in environmentally friendly visible-light catalytic cyclization,12 herein, we report our efforts to develop a new method for the direct deacylative epoxidation of unstrained ketones under exceptionally mild and highly compatible conditions, providing an alternative avenue to access diverse epoxides (Fig. 1D). This versatile protocol requires no additional oxidants or organometallic reagents and features considerable substrate scope, including not only primary, secondary, and tertiary ketones, but also pharmaceutical molecules. Furthermore, the synthetic utility of this protocol is highlighted by its ability to convert epoxide into various valuable functional groups (thiirane, aldehyde, alcohol and other bulky hindered skeletons with quaternary carbon), highlighting its potential applications.
Results and discussion
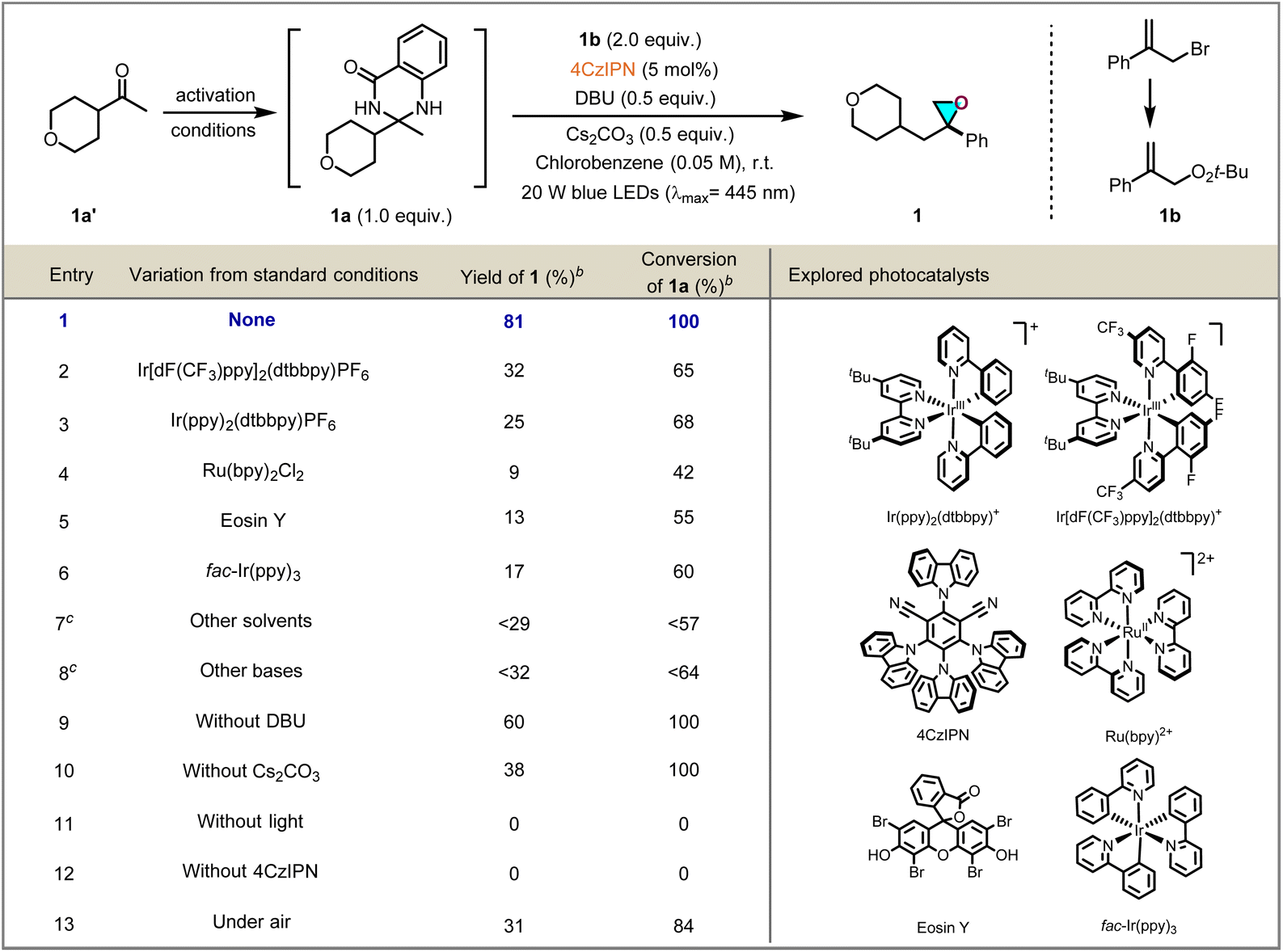
At the outset of the reaction, DHQZ substrate 1a, synthesized from a ketone feedstock through a single condensation step, and easily accessible 1b were chosen as the model substrates to explore epoxidation conditions, and selected results are displayed in Table 1. Delightfully, after a survey of reaction conditions, it was found that when a solution of 1a and 1b with 0.5 equivalents of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) and cesium carbonate (Cs2CO3) in the presence of 5 mol% of 4,5,6-tetra(9H-carbazol-9-yl)isophthalonitrile (4CzIPN) in chlorobenzene was stirred for 24 h under irradiation by 20 W blue LEDs (λmax = 445 nm) at ambient temperature under N2 atmosphere, the desired oxirane 1 was obtained in 81% yield (Table 1, entry 1). Other common photocatalysts, such as Ir[(dF(CF3)ppy)]2(dtbpy)PF6, Ir[(ppy)2(dtbpy)]PF6, Ru(bpy)2Cl2, Eosin Y or fac-Ir(ppy)3 were screened, but provided no improvement over 4CzIPN (Table 1, entries 2–6). The screening of solvents, such as DMSO, DMC, DMA, and NMP, showed that MeCN was more suitable than others (Table 1, entry 7; for more details please see the SI). In addition, different organic or inorganic bases were examined, but no further improvement in yield was obtained (Table 1, entry 8). The developed approach still worked without the involvement of bases DBU or Cs2CO3, however, both leading to significantly lower efficiency (Table 1, entries 9 and 10), control experiments revealed that visible light and a photocatalyst are indispensable to the success of the reaction (Table 1, entries 11 and 12). When the reaction was carried out under air conditions, it led to 4a in lower yield (Table 1, entry 13).| a Reaction conditions: 1a (0.1 mmol, 1.0 equiv.), 1b (0.2 mmol, 2.0 equiv.), photocatalyst (5 mol%), DBU (0.05 mmol, 0.5 equiv.), Cs2CO3 (0.05 mmol, 0.5 equiv.), chlorobenzene (0.05 M), room temperature, under 20 W blue LEDs (λmax = 445 nm), 24 h, in vial. b 1H NMR yield using 1,2-diethyl phthalate as the internal standard. c See SI for details. |
|---|
|
With the optimal conditions in hand for the deacylative epoxidation reaction, we first sought to explore the scope of ketones. As presented in Scheme 1, a range of structurally diverse methyl and phenyl ketones underwent epoxidation site-selectively at the nonmethyl/phenyl side with allylic peroxide to easily afford the corresponding oxiranes in generally moderate to good yields. First, various carbon cyclic and heterocyclic methyl ketones with various ring sizes reacted smoothly to generate secondary alkyl radicals and to form the desired oxiranes 1–5 in 64–79% yields. Benzo-fused cyclic methyl ketone as an indanyl radical precursor and acetal-derived 2,2-diethoxyacetophenone as a masked formyl radical source proved to be competent substrates, to give the corresponding oxiranes 6 and 7 efficiently in 76% and 59% yields, respectively. The protocol successfully accommodated different benzyl methyl ketones, and the substituted benzyl methyl ketone bearing groups, such as Me, OMe, F and Cl, on the aromatic ring were applied, yielding the corresponding oxiranes 8–15 in 56–77% yields, and demonstrating that the reaction efficiency is not affected by the electronic properties of the aromatic ring. In addition, primary methyl ketones with alkene (16), alkyl (17), ether (18), thioether (19), amino (20) or silyl (21) residues were compatible partners under current reaction conditions, to deliver the targeted products in 58–87% yields. Linear alkenyl-substituted ketones undergo partial self-cyclization, leading to decreased product yield. Notably, an array of structurally diverse tertiary alkyl methyl ketones could undergo efficient deacylative epoxidation. For example, adamantyl epoxides 22 were obtained in 82% yield. Cyclopropyl ketone tethered with substituted benzene, pyridine or naphthalene also successfully furnished the desired sterically hindered epoxides 23–28 in 42–78% yields. A substrate-bearing pyridine unit is highly polar and poorly soluble in the reaction system, resulting in a lower product yield (27). Gratifyingly, ketone substrates derived from bioactive molecules dihydro-β-ionone and estrone afford highly functionalized epoxides 29 and 30 in 84% and 64% yields, respectively, further highlighting the high functional group tolerance and the potential for late-stage functionalization in complex systems. These results significantly expand the synthetic utility of ketones and the accessibility of epoxides, facilitating efficient deacylative epoxidation to versatile oxiranes.
Encouraged by the above results, we next investigated the scope of allylic peroxides (Scheme 2). In general, a collection of aryl allylic peroxides were proven to be viable substrates, affording the 1,1-disubstituted epoxides in moderate to good yields. Aryl substituted by different electron-donating groups, including methyl (31, 32), cyclohexyl (33), methoxyl (34) and phenyl (35), reacted smoothly with 1a to provide products in good yields (64–82%). Electron-withdrawing groups on the aromatic ring were tolerated as well, leading to the formation of epoxides 36–44 in 42–74% yields. Apart from various halogens (F, Cl, Br, I) on the phenyl of peroxides, giving the products efficiently, other useful substituted groups (CF3, CN, CO2Me) also generate the corresponding epoxides in good yields. Notably, some of these groups, such as Br, I and CN, could offer a valuable synthetic handle for further transformations. Additionally, disubstituted aryl peroxides were also tolerated to produce oxiranes 45–48 in 47–78% yield. Neither electron-withdrawing nor electron-donating substituents has a significant influence on the reaction. Nevertheless, ortho-substituted styrene peroxides markedly reduce the reaction yield, presumably owing to steric hindrance impeding the reaction pathway, as evidenced by products 40 and 47. Heteroaryl peroxide could also be involved to form oxirane 49 in satisfactory yield (68%) under the current conditions. However, alkyl allylic peroxide resulted in a significantly reduced yield of 50, likely due to the relatively weak reactivity of alkyl peroxides.
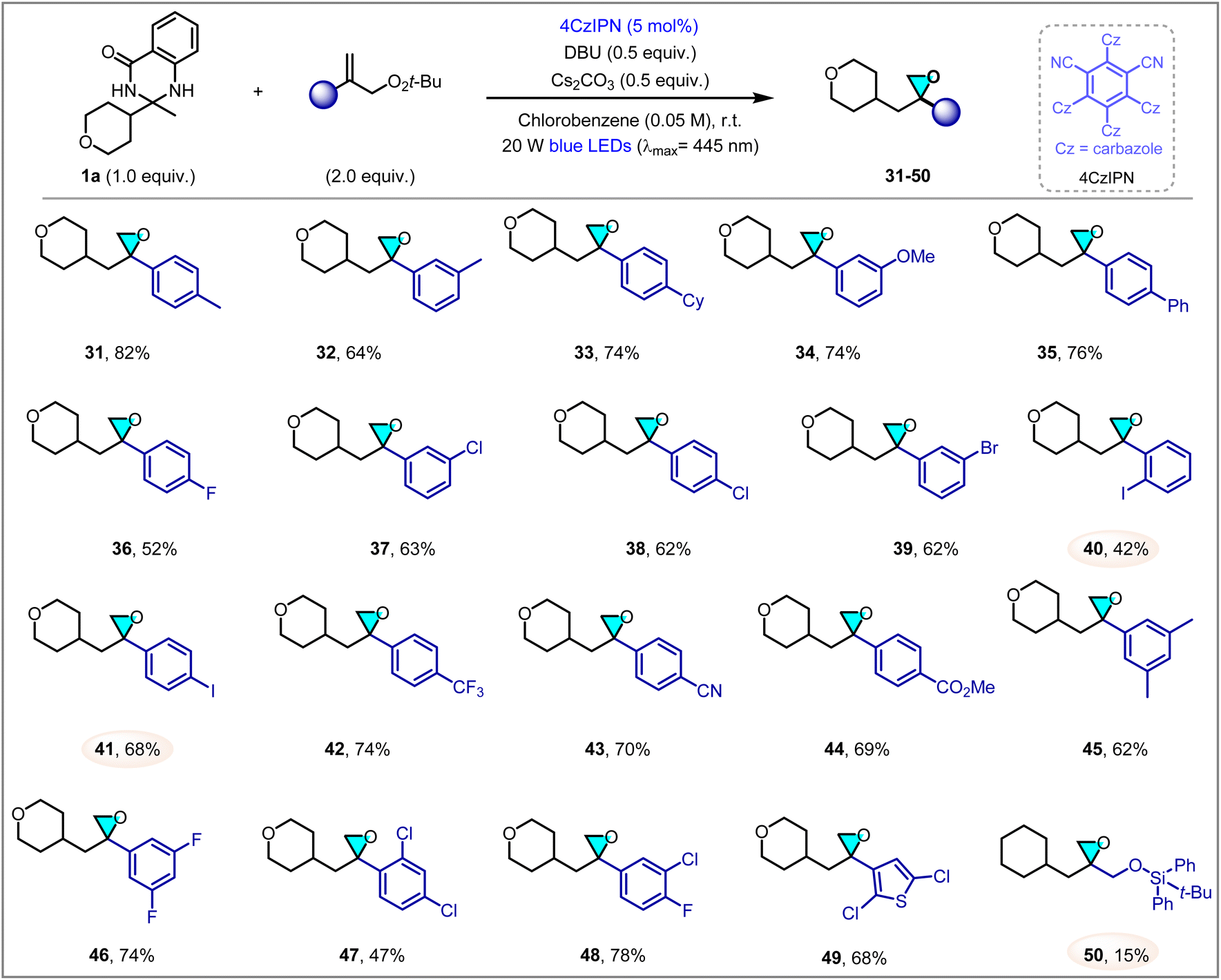 | ||
| Scheme 2 The scope of allylic peroxides. Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), allylic peroxide (0.4 mmol, 2.0 equiv.), 4CzIPN (5 mol%), DBU (0.1 mmol, 0.5 equiv.), Cs2CO3 (0.1 mmol, 0.5 equiv.), chlorobenzene (0.05 M), room temperature, under 20 W blue LEDs (λmax = 445 nm), 24 h, in vial. Isolated yields. | ||
Homologation is a general and valuable synthetic approach used for the elongation of a carbon chain.13 As demonstrated in Scheme 3A, one-carbon-inserted ketones 51 and 53 were obtained in 53% and 48% yields, respectively, suggesting an alternative way to achieve the one-carbon homologation of carbonyl derivatives without modification of the backbone. Moreover, to demonstrate the scalability and robustness of the method, epoxide 25 was synthesized at gram scale (5.0 mmol) with comparable efficiency (68%) (Scheme 3B). In particular, strained oxiranes, as privileged heterocyclic building blocks, are very valuable motifs in organic synthesis and drug development. By the addition of thiourea, thiirane 54 was obtained in 67% yield. Selective oxidation by ZnCl2 and reduction by Et2SiH gave primary aldehyde 55 and alcohol 56 in 88% and 72% yields, respectively. Treatment with PPh3 led directly to the generation of terminal alkene 57 in 74% yield. Ring-opening by Et3N·3HF formed fluorinated product 58 in 73% yield. Other nucleophiles could open the ring to give compounds 59 and 60 under basic and acidic conditions. Sterically hindered tertiary alcohol 61 was delivered with 71% yield in the presence of LiAlH4.
 | ||
Scheme 3 Gram-scale synthesis and synthetic derivatizations of epoxide. Reaction conditions: (A) 51![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 (1.0 equiv.), DABCO (4.0 equiv.), mesitylene, 165 °C. 53:52 (1.0 equiv.), DABCO (4.0 equiv.), mesitylene, 165 °C. (B) (a) Thiourea (2.0 equiv.), 25 (0.20 mmol, 1.0 equiv.), MeOH (1.0 mL), r.t., 18 h. (b) ZnCl2 (0.5 equiv.), 25 (0.20 mmol, 1.0 equiv.), toluene (2.0 mL), 110 °C, 4 h. (c) Et2SiH (2.0 equiv.), TfOH (0.05 equiv.), 25 (0.20 mmol, 1.0 equiv.), HFIP (1.0 mL), r.t., 1 h. (d) PPh3 (1.2 equiv.), 25 (0.20 mmol, 1.0 equiv.), i-PrOH (1.0 mL), 120 °C, 36 h. (e) Et3N·3HF (2.0 equiv.), BF3·Et2O (0.2 equiv.), 25 (0.20 mmol, 1.0 equiv.), DCM (2.0 mL), r.t., 12 h. (f) C6H5SNa (1.5 equiv.), 25 (0.20 mmol, 1.0 equiv.), DMF (4.0 mL), 40 °C, 12 h. (g) HCl (37% in H2O, 0.005 equiv.), 25 (0.20 mmol, 1.0 equiv.), MeOH (2.0 mL), r.t., 2 h. (h) LiAlH4 (2.0 equiv.), 25 (0.20 mmol, 1.0 equiv.), THF (2.0 mL), r.t., 4 h. :2 (1.0 equiv.), DABCO (4.0 equiv.), mesitylene, 165 °C. 53:52 (1.0 equiv.), DABCO (4.0 equiv.), mesitylene, 165 °C. (B) (a) Thiourea (2.0 equiv.), 25 (0.20 mmol, 1.0 equiv.), MeOH (1.0 mL), r.t., 18 h. (b) ZnCl2 (0.5 equiv.), 25 (0.20 mmol, 1.0 equiv.), toluene (2.0 mL), 110 °C, 4 h. (c) Et2SiH (2.0 equiv.), TfOH (0.05 equiv.), 25 (0.20 mmol, 1.0 equiv.), HFIP (1.0 mL), r.t., 1 h. (d) PPh3 (1.2 equiv.), 25 (0.20 mmol, 1.0 equiv.), i-PrOH (1.0 mL), 120 °C, 36 h. (e) Et3N·3HF (2.0 equiv.), BF3·Et2O (0.2 equiv.), 25 (0.20 mmol, 1.0 equiv.), DCM (2.0 mL), r.t., 12 h. (f) C6H5SNa (1.5 equiv.), 25 (0.20 mmol, 1.0 equiv.), DMF (4.0 mL), 40 °C, 12 h. (g) HCl (37% in H2O, 0.005 equiv.), 25 (0.20 mmol, 1.0 equiv.), MeOH (2.0 mL), r.t., 2 h. (h) LiAlH4 (2.0 equiv.), 25 (0.20 mmol, 1.0 equiv.), THF (2.0 mL), r.t., 4 h. | ||
To probe the mechanism of this deacylative epoxidation, the following control experiments were conducted, as shown in Scheme 4. First, radical scavengers 2,2,6,6-tetramethylpiperidinooxy (TEMPO) and galvinoxyl fully inhibited the reaction, and cyclohexyl radical adduct 62 was detected by high-resolution mass spectrometry (Scheme 4a). Subjecting cyclopropane-containing dihydroquinazolinone 63 to a radical clock experiment, ring-opening product 16 was isolated in 52% yield (Scheme 4b). Using alkene 64a as a radical acceptor, no radical addition product 64 was observed, and only a trace of cyclopropanation product 65 was observed, implying that perhaps no direct radical-polar crossover process may be involved (Scheme 4c). Direct irradiation of the reaction with UV light in the absence of a photocatalyst failed to work (Scheme 4d). In addition, short-term photo-irradiation experiments suggested that a radical chain process is unlikely (Scheme 4e). Stern–Volmer fluorescence quenching experiments revealed that dihydroquinazolinone 1a is an effective quencher for excited-state 4CzIPN* (Scheme 4f). Cyclic voltammetry (CV) studies provided compelling evidence for the ability of the photocatalyst to activate the substrate, leading to the generation of radical species (Scheme 4g).
 | ||
| Scheme 4 Mechanistic studies and proposed mechanism. | ||
Based on the above results and previous reports,11 a probable catalytic cycle is proposed (Scheme 4h). Initially, photocatalyst 4CzIPN reached excited-state 4CzIPN* by irradiation with visible light. Then, single-electron transfer (SET) between 1a (E1a˙+/1a = +1.29 V vs. SCE) and 4CzIPN* (E4CzIPN*/4CzIPN˙− = +1.35 V vs. SCE)14 generated 4CzIPN˙− and radical cation I, which underwent rapid homolytic cleavage into carbon radical II and quinazolinone III under basic conditions. Carbon radical II was subsequently captured by allylic peroxide to form open-shell intermediate IV. An intramolecular radical homolytic substitution (SH2) on the oxygen atom of IV furnished target oxirane 1, alongside the release of t-BuO˙ radical intermediate V. Finally, single-electron reduction of the t-BuO˙ radical to t-BuO− by 4CzIPN˙− completed the catalytic cycle and regenerated the ground state of the photocatalyst. Meanwhile, a t-BuO˙ radical could also abstract a hydrogen from 1a to form carbon radical intermediate II.
Conclusions
In conclusion, we introduced a unique concept of epoxidation and developed an efficient deacylative epoxidation strategy for synthesizing diverse oxirane-containing scaffolds. This method uses easily available unstrained ketones and allylic peroxides under exceptionally mild and highly compatible conditions. This transformation proceeds via a photoinduced aromatization-driven deconstructive reaction of ketones. This platform is particularly well-suited to the late-stage functionalization of complex natural products and pharmaceuticals with unstrained ketones, owing to its broad substrate scope and high functional group compatibility. Additionally, the ease of subsequent transformations of oxiranes, providing an appealing alternative avenue to polar Corey–Chaykovsky epoxidation for accessing diverse epoxides and their derivatives. Mechanistic investigations suggested that a deacylative radical substitution and cyclization pathway might be involved in the process.Author contributions
C. Shu designed and directed the investigations and composed the manuscript with revisions provided by the other authors. J. Lin and M. Zhou developed the catalytic method. Z. Li, J. Duan, X. Liu and Y. Wang studied the substrate scope. All the authors were involved the analysis of results and discussions of the project.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterization data, and nuclear magnetic resonance spectra. See DOI: https://doi.org/10.1039/d6sc00898d.Acknowledgements
We are grateful for financial support from the National Natural Science Foundation of China (22571109, 22301093), National Key R&D Program of China (2023YFD1700500), Department of Science and Technology of Hubei Province (2025DJB052) and financial support from self-determined research funds of CCNU from the college's basic research and operation of MOE.References
- (a) D. M. Hodgson, M. A. H. Stent, M. Reilly and E. Gras, in Comprehensive Heterocyclic Chemistry III, Elsevier, Oxford, U.K, 2008, vol. 1, pp. 235–298 Search PubMed; (b) C. J. Thibodeaux, W. C. Chang and H. W. Liu, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS PubMed; (c) J. Herzberger, K. Niederer, H. Pohlit, J. Seiwert, M. Worm, F. R. Wurm and H. Frey, Chem. Rev., 2016, 116, 2170–2243 CrossRef CAS PubMed; (d) S. S. Uthumange, A. J. H. Liew, X. W. Chee and K. Y. Yeong, Bioorg. Med. Chem., 2024, 116, 117980 CrossRef CAS PubMed.
- (a) T. B. Hughes, G. P. Miller and S. J. Swamidass, ACS Cent. Sci., 2015, 1, 168–180 CrossRef CAS PubMed; (b) A. R. Gomes, C. L. Varela, E. J. Tavares-da-Silva and F. M. F. Roleira, Eur. J. Med. Chem., 2020, 201, 112327 CrossRef CAS PubMed; (c) B. Kaur and P. Singh, Bioorg. Chem., 2022, 125, 105862 CrossRef CAS PubMed; (d) V. M. Dembitsky, Biomedicines, 2023, 11, 2237 CrossRef CAS PubMed.
- (a) J. He, J. Ling and P. Chiu, Chem. Rev., 2014, 114, 8037–8128 CrossRef CAS PubMed; (b) C. Y. Huang and A. G. Doyle, Chem. Rev., 2014, 114, 8153–8198 CrossRef CAS PubMed; (c) F. Moschona, I. Savvopoulou, M. Tsitopoulou, D. Tataraki and G. Rassias, Catalysts, 2020, 10, 1117 CrossRef CAS; (d) M. Thirumalaikumar, Org. Prep. Proced. Int., 2022, 54, 1–39 CrossRef CAS; (e) R. R. Rodríguez-Berríos, S. R. Isbel and A. Bugarin, Int. J. Mol. Sci., 2023, 24, 6195 CrossRef PubMed; (f) Y. Zhang, B. Hu, Y. Chen and Z. Wang, Chem. Eur. J., 2024, 30, e202402469 CrossRef CAS PubMed; (g) M. Hanif, A. Mansha, K. G. Ali, M. A. Saeed, S. Mahmood, A. R. Chaudhry, A. Irfan, A. Mushtaq and A. F. Zahoor, Mol. Divers., 2025, 29, 4919–4952 CrossRef CAS PubMed; (h) N. Orbach, Z. P. Sercel, R. Suresh and I. Marek, Nat. Rev. Chem., 2025, 10, 31–49 CrossRef PubMed.
- V. Juyal, Epoxide Global Market Report 2025, The Business Research Company, 2025 Search PubMed.
- (a) C. J. Thibodeaux, W. C. Chang and H. W. Liu, Chem. Rev., 2012, 112, 1681–1709 CrossRef CAS PubMed; (b) Y. Zhu, Q. Wang, R. G. Cornwall and Y. Shi, Chem. Rev., 2014, 114, 8199–8256 CrossRef CAS PubMed; (c) Y. Tanaka, in Epoxy Resins, Routledge, London, 2018, pp. 9–283 Search PubMed; (d) I. Triandallidi, D. I. Tzaras and C. G. Kokotos, ChemCatChem, 2018, 10, 2521–2535 CrossRef; (e) V. L. Mamedova, G. Z. Khikmatova, D. E. Korshin, S. V. Mamedova, E. L. Gavrilova and V. A. Mamedov, Russ. Chem. Rev., 2022, 91, RCR5049 CrossRef; (f) D. Tang, K. Dang, J. Wang, C. Chen, J. Zhao and Y. Zhang, Sci. China Chem., 2023, 66, 3415–3425 CrossRef CAS; (g) X. Han, N. Zhang, Q. Li, Y. Zhang and S. Das, Chem. Sci., 2024, 15, 13576–13604 RSC; (h) Q.-Z. Wang, Y. Zheng, W.-T. Wu and H.-M. Huang, J. Am. Chem. Soc., 2025, 147, 16248–16254 CrossRef CAS PubMed; (i) D.-N. Chen, L.-L. Qin, D.-J. Luo, Y. Lv, H.-Y. Yang, D.-D. Ye and P.-J. Xia, Org. Lett., 2025, 27, 5744–5749 CrossRef CAS PubMed.
- (a) F. A. Carey and R. J. Sundberg, Advanced Organic Chemistry, Part B: Reactions and Synthesis, Springer, New York, NY, 2007 Search PubMed; (b) P. Ertl and T. A. Schuhmann, J. Nat. Prod., 2019, 82, 1258–1263 CrossRef CAS PubMed; (c) D. J. Foley and H. Waldmann, Chem. Soc. Rev., 2022, 51, 4094–4120 RSC; (d) A. R. Varma, B. S. Shrirame, S. Gadkari, K. R. Vanapalli, V. Kumar and S. K. Maity, Chem. Eng. J., 2024, 489, 151297 CrossRef CAS.
- (a) Z. Wang, Comprehensive Organic Name Reactions and Reagents, Wiley, Hoboken, 2009, vol. 178, pp. 841–845 Search PubMed; (b) M. S. Newman and B. J. Magerlein, in Organic Reactions, Wiley, New York, 2011, pp. 413–440 Search PubMed; (c) L. J. Qi, B. Zhou and L. W. Ye, Chem.–Eur. J., 2024, 30, e202401389 CrossRef CAS PubMed.
- (a) A. H. Li, L. X. Dai and V. K. Aggarwal, Chem. Rev., 1997, 97, 2341–2372 CrossRef CAS PubMed; (b) V. K. Aggarwal and J. Richardson, Chem. Commun., 2003, 2644–2651 RSC; (c) E. M. McGarrigle, E. L. Myers, O. Illa, M. A. Shaw, S. L. Riches and V. K. Aggarwal, Chem. Rev., 2007, 107, 5841–5883 CrossRef CAS PubMed; (d) M. M. Heravi, S. Asadi, N. Nazari and B. M. Lashkariani, Curr. Org. Synth., 2016, 13, 308–333 CrossRef CAS.
- (a) Y. Xu, X. Qi, P. Zheng, C. C. Berti, P. Liu and G. Dong, Nature, 2019, 567, 373–378 CrossRef CAS PubMed; (b) X. Zhou, Y. Xu and G. Dong, Nat. Catal., 2021, 4, 703–710 CrossRef CAS PubMed.
- (a) Z. Li, Z. Hao, S. Yang, W. Yang and T. Jin, Chin. J. Org. Chem., 2025, 45, 3644–3654 CrossRef CAS; (b) X. Yang, X. Ma and C. Shu, J. Org. Chem., 2025, 90, 12029–12049 CrossRef CAS PubMed.
- (a) L. Li, L. Fang, W. Wu and J. Zhu, Org. Lett., 2020, 22, 5401–5406 CrossRef CAS PubMed; (b) X. Y. Lv, R. Abrams and P. R. Martin, Angew. Chem., Int. Ed., 2023, 62, e202217386 CrossRef CAS PubMed; (c) P. P. Mondal, S. Das, S. Venugopalan, M. Krishnan and B. Sahoo, Org. Lett., 2023, 25, 1441–1446 CrossRef CAS PubMed; (d) J. Li, D. Zhang, L. Tan and C. J. Li, Angew. Chem., Int. Ed., 2024, 63, e20241036 Search PubMed; (e) H. J. Miao, J. H. Zhang, W. Li, W. Yang, H. Xin, P. Gao, X. H. Duan and L. N. Guo, Chem. Sci., 2024, 15, 8993–8999 RSC; (f) Q. Z. Li, M. H. He, R. Zeng, Y. Y. Lei, Z. Y. Yu, M. Jiang, X. Zhang and J. L. Li, J. Am. Chem. Soc., 2024, 146, 22829–22839 CrossRef CAS PubMed; (g) K. H. He, N. Jin, J. C. Chen, Y. F. Zheng and F. Pan, Org. Lett., 2024, 26, 9503–9507 CrossRef CAS PubMed; (h) R. Liu and H. Zeng, Green Chem., 2025, 27, 7329–7335 RSC; (i) K. Wei, Y. L. Tang, X. P. Chen, B. B. Zhan and X. H. Zhang, Angew. Chem., Int. Ed., 2025, e202518599 Search PubMed.
- (a) H. Li, Y. Zhang, X. Yang, Z. Deng, Z. Zhu, P. Zhou, X. Ouyang, Y. Yuan, X. Chen, L. Yang, M. Liu and C. Shu, Angew. Chem., Int. Ed., 2023, 62, e202300159 CrossRef CAS PubMed; (b) M. Liu, X. Ouyang, C. Xuan and C. Shu, Org. Chem. Front., 2024, 11, 895–915 RSC; (c) H. Li, Y. X. Zhang, X. Y. Zou, X. X. Yang, P. Zhou, X. Y. Ma, S. Q. Lu, Q. Sun and C. Shu, ACS Catal., 2024, 14, 3664–3674 CrossRef CAS; (d) Z. M. Zhu, Y. X. Zhang, Z. Y. Li and C. Shu, Chem Catal., 2024, 4, 100945 CAS; (e) Y.-X. Zhang, P. Zhou, X.-Y. Ma, X.-X. Yang, X. Fang, Y.-X. Wang and C. Shu, Sci. China Chem., 2025, 68, 622–630 CrossRef CAS; (f) C.-L. Xuan, Z.-M. Zhu, Z.-Y. Li and C. Shu, Adv. Agrochem., 2025, 4, 13–29 CrossRef CAS; (g) Y.-Y. Zhao, Z.-M. Zhu, L. Li, B.-Y. Shi, Z.-Y. Li, Y.-Y. Huang, L.-J. Jiang and C. Shu, Chin. Chem. Lett., 2025, 36, 110900 CrossRef CAS; (h) B. Y. Shi, X. Y. Li, Z. M. Zhu, Z. Y. Li, Y. Y. Zhao, M. J. Wang, J. Zeng and C. Shu, CCS Chem., 2025, 7, 2987–2995 CrossRef CAS; (i) Y. X. Zhang, H. Xu, L. G. Huang, Z. M. Zhu, Z. Y. Li, J. Duan and C. Shu, ACS Catal., 2025, 15, 13145–13156 CrossRef.
- (a) M. Adamczyk, E. K. Dolence, D. S. Watt, M. R. Christy, J. H. Reibenspies and O. P. Anderson, J. Org. Chem., 1984, 49, 1378–1382 CrossRef CAS; (b) N. R. Candeias, R. Paterna and P. M. P. Gois, Chem. Rev., 2016, 116, 2937–2981 CrossRef CAS PubMed.
- T. Y. Shang, L. H. Lu, Z. Cao, Y. Liu, W. M. He and B. Yu, Chem. Commun., 2019, 55, 5408–5419 RSC.
| This journal is © The Royal Society of Chemistry 2026 |