
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElectrocatalytic methane oxidation via integrated design of mechanism, microenvironment, and mass transfer
Hui-Jian Zhangab,
Xiao-Tong Wangb,
Luxiao Zhoubd,
Jiguang Deng *a,
Zhong-Wen Liu*d and
Zhao-Qing Liu*bc
*a,
Zhong-Wen Liu*d and
Zhao-Qing Liu*bc
aSchool of Materials Science and Engineering, Beijing University of Technology, Beijing, 100124, P. R. China. E-mail: jgdeng@bjut.edu.cn
bDepartment School of Chemistry and Chemical Engineering, Guangzhou University, Guangzhou 510006, China. E-mail: lzqgzu@gzhu.edu.cn
cSchool of Chemistry, South China Normal University, Guangzhou 510006, China
dSchool of Chemistry and Chemical Engineering, Shaanxi Normal University, Xi'an 710119, China. E-mail: zwliu@snnu.edu.cn
First published on 7th May 2026
Abstract
Electrocatalytic methane oxidation (EMO) offers a promising approach by utilizing renewable electricity to drive electron-transfer reactions, enabling C–H bond activation and transformation into value-added chemicals at ambient temperature and pressure while offering advantages in sustainability and process control. However, challenges such as the inherent inertness of methane, its low solubility in conventional electrolytes, and tendencies toward over-oxidation severely limit catalytic efficiency. This review summarizes recent advances in EMO across three key scales. At atomic and molecular scales, mechanisms of C–H bond activation through direct electron transfer and reactive oxygen species are discussed. At the catalyst and microenvironment level, practical strategies to enhance catalysis are reviewed, including structural modification, heterogeneous interfaces, single-atom or dual-atom catalysis, and electrolyte/reactor designs. At the macroscopic transport level, methods to optimize methane transport via pressurization, electrolyte engineering, transport systems, and porous nanoarchitectures are examined. Integrating insights from reaction kinetics, catalytic microenvironments, and mass transport offers a theoretical foundation for the rational design of highly efficient, stable, and scalable electrocatalytic methane conversion.
1. Introduction
Methane (CH4) is the most abundant hydrocarbon resource on Earth. According to 2024 data, global natural gas reserves have reached approximately 2.09 × 105 billion cubic meters.1,2 However, it is also the second largest greenhouse gas after CO2, with a 100-year global warming potential 28 times that of CO2, contributing about 30% to the warming effect.3–5 Against the backdrop of global efforts to mitigate climate change and achieve carbon neutrality, the selective conversion of methane to liquid oxygenated compounds (such as methanol, ethanol, formaldehyde, or acetic acid) not only effectively curbs fugitive emissions but also allows convenient storage, transportation, and high-value utilization of this gaseous resource, becoming a key link in the circular carbon economy.6–10 However, the extremely high chemical inertness of the methane molecule—symmetric tetrahedral structure, low polarizability (2.6 × 10−40 C2 m2 J−1), high ionization energy (12.6 eV), and a first C–H bond dissociation energy of 439 kJ mol−1—has long made its direct activation known as the “Holy Grail” in the field of catalysis.11,12 Traditional industrial routes (such as steam reforming coupled with Fischer–Tropsch synthesis) require operation at >700 °C and high pressure, with high energy consumption, heavy carbon emissions, and generally using syngas as an intermediate, making it difficult to achieve high selectivity for oxygenated compounds.13–17 Even with the introduction of external oxidants like H2O2 or O3, it is difficult to balance activity and selectivity below 500 °C.18–20Electrocatalytic methane oxidation (EMO) provides a green solution to this challenge, utilizing renewable electricity to drive C–H activation under mild conditions, thereby achieving precise control of reaction pathways through electrode potential regulation, opening sustainable new paths for the resource utilization of methane.21–23 In EMO, methane is oxidized at the anode, potentially producing value-added products with high faradaic efficiency (FE), while coupling with cathode reactions such as hydrogen evolution or CO2 reduction to form integrated energy systems.24,25 Recent advances in tailored catalysts (such as spherical IrO2, Cu–Pd/anatase, or Co-based heterojunctions) have demonstrated FE values exceeding 80% for methanol production, highlighting the application potential of this technology.26–28 Additionally, EMO promotes methane activation by electro-generating reactive oxygen species (ROS), including hydroxyl radicals (–OH), atomic oxygen (O*), or superoxide (–O2−), without the use of chemical oxidants.26,29 However, the weak adsorption of methane on the electrode surface hinders initial activation, and the generated intermediates (such as *CH3, *CH2OH) are prone to further oxidation at anodic potentials, leading to complete overoxidation to CO2.30 Moreover, the solubility of methane in aqueous phases is only 1.3 mM (25 °C, 1 atm), resulting in local reactant depletion at the electrode surface, with current densities lingering at 10–50 mA cm−2, far below the industrial threshold (>200 mA cm−2).31,32 Such challenges are also made worse by the propensity of catalysts to be poisoned under reaction conditions due to carbon accumulation and/or changes of structure.33,34 Although a lot of progress has been made in catalyst design based on the efforts of preceding research, we are still in need of a comprehensive approach that bridges knowledge ranging from atomic level mechanisms to reactor design. In order to overcome the long-standing challenges, the focus of recent research has moved away from the usual method of merely testing individual catalysts. Instead, there has been great enthusiasm for an integrated design approach at various scales. One such area that has recently been enabled for real-time study is in situ and operando spectroscopic analysis of lattice oxygen confinement and reactive oxygen species evolution paths.35–38 Single-atom catalysts, heterojunction composites, and alloyed structures have shown tremendous potential in maximizing overpotential reduction. However, it is the synergy between cleverly designed structural properties, such as the coordination of the d-band center with strategic oxygen vacancies, that makes them more effective.39–41 In addition to the development of more efficient catalysts, tremendous progress is being made in reactor engineering to counter transport-related limitations. The adoption of gas diffusion electrodes, hierarchically porous platforms, and microfluidic reactors has profoundly improved methane mass transfer rates by up to 1000-fold.42,43 On the theoretical front, DFT calculations have contributed greatly to supporting empirical findings. The underlying adsorption energy-catalyst activity correlation is being unscrambled through DFT calculations, providing the blueprints for more efficient catalyst development in the future.44
This review aims to respond to these challenges by connecting microscopic principles with macroscopic reactor design. Differing from other reviews that, to a great extent, emphasize screening new catalytic materials, this review will give a holistic panorama by connecting electronic structure modulation, micro-environmental control, and mass transfer enhancement. Initially, this review will deconstruct the basic process of C–H cleavage. In addition, it will explore ways of promoting kinetic rates via optimized catalysts and system design. Finally, it will also try to connect engineering strategies that could effectively counteract solubility limitations at the inter-phase of multiphase interfaces (Fig. 1). Through in-depth discussions of these three dimensions, we aim to provide forward-looking theoretical references and technical guidance for constructing efficient, stable, and industrially promising methane electrocatalytic conversion systems.
 | ||
| Fig. 1 Schematic diagram of synergistic strategy for EMO, analysis of atomic reaction mechanism, micro-environment regulation engineering and macro-mass transfer enhancement. | ||
2. Origins of activation and selectivity: a mechanistic analysis of methane C–H bond cleavage
As a highly stable molecule, methane presents a significant challenge in its initial activation due to the high dissociation energy of its C–H bond (439 kJ mol−1), making its cleavage the core step in the EMO process. Concurrently, once the C–H bond is broken, the generated intermediates are prone to further oxidation under anodic potentials, leading to low product selectivity and a tendency for complete oxidation to CO2.33,34 This section dissects the pathways of C–H bond cleavage from a microscopic mechanistic perspective, focusing on mechanisms such as DET, ROS mediation, and interface/defect regulation. Based on recent in situ analysis studies, these mechanisms reveal how to achieve highly efficient activation and selective conversion through precise modulation of active sites and the reaction microenvironment.352.1. Direct C–H activation via surface electronic and structural regulation
In EMO processes, C–H bond cleavage is often achieved via DET, where methane molecules adsorb directly onto the electrode surface and lose electrons without the need for ROS mediation. This mechanism relies on the electronic structure and surface adsorption energy of the catalyst, typically observed with transition metal oxides or interfacial catalysts.36,37A study based on a Sm-doped CeO2 (SDC) thin-film model cell combined electrochemical impedance spectroscopy, in situ ambient-pressure X-ray photoelectron spectroscopy (AP-XPS), and DFT calculation to reveal the direct electrooxidation pathway of CH4 on the SDC surface at the molecular scale (Fig. 2a).37 The reaction follows the Mars–van Krevelen mechanism. CH4 first chemisorbs on the SDC surface with an adsorption energy of −3.90 eV, which is about 80% stronger than on pure CeO2. Stepwise dehydrogenation then occurs (Fig. 2b). According to DFT, the first C–H cleavage (CH4 → *CH3 + *H) has a barrier of 0.71 eV and is exothermic by −0.12 eV. The subsequent three dehydrogenation steps (*CH3 → *CH2 → *CH → *C) have barriers of 0.41, 0.68, and 0.82 eV, respectively. Impedance analysis exhibits a reaction order of only −0.25 with respect to CH4 partial pressure, indicating that C–H activation is not rate-limiting. Under anodic overpotential, AP-XPS detects no carbon-containing intermediates. Instead, a clear accumulation of surface hydroxyl groups is observed, and their concentration decreases as the overpotential increases. DFT calculation further reveals that the barrier for H2O formation reaches 2.09 eV, far exceeding all C–H cleavage steps. Thus, the formation and desorption of H2O govern the overall reaction kinetics (Fig. 2c). This finding challenges the conventional view that C–H bond activation is the rate-limiting step and highlights the central role of surface hydrogen recombination and removal in controlling the reaction rate.
 | ||
| Fig. 2 (a) Schematic illustration of the SDC thin-film model cell for studying direct electrochemical oxidation of methane at the ceria/gas interface. (b) Schematic of the direct electro-oxidation pathway of CH4 on an SDC electrode surface following the Mars–van Krevelen mechanism. (c) DFT calculations indicating that H2O formation is the rate-determining step in CH4 electro-oxidation. Reproduced from ref. 37 with permission from Wiley-VCH GmbH, copyright 2024. (d) Schematic illustration of the electrocatalytic oxidation of methane to ethanol and methanol at the NiO/Ni interface. Reproduced from ref. 36 with permission from Elsevier, copyright 2020. (e) Proposed mechanism for the electrocatalytic generation of methyl formate from methane on a CN-defective Ni–Co PBA-VCN catalyst. Reproduced from ref. 38 with permission from Elsevier, copyright 2025. (f) Reaction mechanism for the radical pathway of methane electro-oxidation to formic acid on a CuOx/V2O5 catalyst. Reproduced from ref. 39 with permission from Elsevier, copyright 2024. | ||
In the NiO/Ni interface catalyst, the synergy between Ni3+ and metallic Ni0 significantly enhances CH4 adsorption. CH4 temperature-programmed desorption experiments show that the optimized NiO/Ni interface exhibits the highest CH4 desorption amount compared to pure Ni foam (Fig. 2d).36 DFT calculation reveals the molecular origin of the interface-enhanced C–H activation and C–C coupling. At the NiO(200)/Ni(111) interface, the first dehydrogenation step  has a barrier of only 0.30 eV and is exothermic by −0.99 eV, much lower than on pure Ni(111). In contrast, the pure NiO(200) surface binds *CH3 and *CH2OH too weakly to facilitate CH4 conversion. Starting from *CH3, hydroxylation to methanol requires a barrier of 0.66 eV and is endothermic by 0.32 eV. Dehydrogenation to *CH2 has a barrier of 0.62 eV and is exothermic by −0.22 eV. The *CH2 species then undergoes hydroxylation to *CH2OH (barrier 0.38 eV, exothermic −0.77 eV), followed by C–C coupling with *CH3 to form ethanol. This coupling step has a barrier of only 0.06 eV and is exothermic by −1.64 eV. The difference between endothermic and exothermic steps explains thermodynamically why the interface favors ethanol over methanol. With this interface catalyst, an ethanol faradaic efficiency of 89% and production rate of 25 µmol gNiO−1 h−1 are achieved at 1.40 V vs. RHE. Similarly, in Mg-MOF-74, Mg-oxo-Mg nodes serve as active sites, creating interfaces. These nodes not only possess good methane adsorption capacity but also promote C–H bond polarization and activation under an electric field, enabling the selective conversion of methane to formate and acetate (FE 10.9%).40 This illustrates the decisive role of interface structures in guiding reaction path selectivity.
has a barrier of only 0.30 eV and is exothermic by −0.99 eV, much lower than on pure Ni(111). In contrast, the pure NiO(200) surface binds *CH3 and *CH2OH too weakly to facilitate CH4 conversion. Starting from *CH3, hydroxylation to methanol requires a barrier of 0.66 eV and is endothermic by 0.32 eV. Dehydrogenation to *CH2 has a barrier of 0.62 eV and is exothermic by −0.22 eV. The *CH2 species then undergoes hydroxylation to *CH2OH (barrier 0.38 eV, exothermic −0.77 eV), followed by C–C coupling with *CH3 to form ethanol. This coupling step has a barrier of only 0.06 eV and is exothermic by −1.64 eV. The difference between endothermic and exothermic steps explains thermodynamically why the interface favors ethanol over methanol. With this interface catalyst, an ethanol faradaic efficiency of 89% and production rate of 25 µmol gNiO−1 h−1 are achieved at 1.40 V vs. RHE. Similarly, in Mg-MOF-74, Mg-oxo-Mg nodes serve as active sites, creating interfaces. These nodes not only possess good methane adsorption capacity but also promote C–H bond polarization and activation under an electric field, enabling the selective conversion of methane to formate and acetate (FE 10.9%).40 This illustrates the decisive role of interface structures in guiding reaction path selectivity.
Defect engineering, as a powerful tool for electronic and structural regulation, can create sites with both high activity and unique selectivity. Ge He et al. constructed a Ni–Co Prussian blue analogue (PBA) tandem catalyst rich in CN vacancies (VCN). The introduction of CN vacancies shifts the d-band center of Ni atoms upward, enhancing chemical adsorption of CH4 and lowering the C–N bond cleavage barrier. Upon activation and stepwise dehydrogenation at Ni sites to generate *CH3 and *CH2, the *CH2 intermediate can “spill over” to adjacent Co atoms within the same CN vacancy, subsequently being oxidized to HCOOH.38 Meanwhile, the Ni site generates *CH3OH, and the two ultimately undergo esterification at room temperature to achieve direct electro-oxidation of CH4 to methyl formate (Fig. 2e). In atomically dispersed CuOx catalysts supported on V2O5, the Cu–O–V bridging structure activates CH4 via direct electron transfer. The Lewis acid sites (oxygen vacancies) induced by CuOx not only enhance CH4 adsorption but also promote C–H bond polarization under an electric field, lowering its cleavage barrier and facilitating the generation of *OH for C–H bond cleavage, ultimately enabling selective oxidation of methane to formic acid (FE up to 92%) (Fig. 2f).39
These examples demonstrate that the core of the direct activation mechanism lies in the precise design of the surface microenvironment through doping, interfaces, and defects to lower energy barriers, control pathways, and avoid over-oxidation.
2.2. ROS-mediated C–H bond oxidation
In most EMO systems, C–H bond cleavage is mediated by ROS (e.g., *OH, *O, *Cl). These reactive oxygen species serve as key intermediates, directly participating in the activation and oxidation processes of the C–H bond, significantly influencing reaction selectivity and efficiency. Unlike gaseous oxidants such as O2 or N2O common in thermocatalysis, ROS in electrocatalytic systems often originate from oxyanions in the electrolyte (e.g., CO32−, OH−), which are generated in situ on the catalyst surface under an applied electric field, enabling efficient and selective methane oxidation.26 However, their concentration needs to be regulated to suppress over-oxidation.Study on IrO2 catalysts shows that the dissociation of CO32− on the IrO2(110) surface to form terminal oxygen species 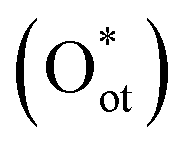 is highly favorable under electrochemical conditions (Fig. 3a).26 DFT calculations indicate that CO32− adsorbs on surface Ircus sites to form CO3 and then dissociates to release Oot. At an anodic bias of 1.5 V vs. RHE, this reaction is exothermic by nearly 3 eV. On a surface pre-adsorbed with three
is highly favorable under electrochemical conditions (Fig. 3a).26 DFT calculations indicate that CO32− adsorbs on surface Ircus sites to form CO3 and then dissociates to release Oot. At an anodic bias of 1.5 V vs. RHE, this reaction is exothermic by nearly 3 eV. On a surface pre-adsorbed with three  species (75% coverage), the dissociation barrier is only 0.05 eV, about an order of magnitude lower than on the bare surface. Such high coverage of
species (75% coverage), the dissociation barrier is only 0.05 eV, about an order of magnitude lower than on the bare surface. Such high coverage of  species (>0.8 ML) activates CH4 via a methoxy pathway. Two adjacent
species (>0.8 ML) activates CH4 via a methoxy pathway. Two adjacent  species cooperatively abstract a hydrogen atom to form
species cooperatively abstract a hydrogen atom to form  with a barrier of 0.87 eV, which is thermally accessible at room temperature. The
with a barrier of 0.87 eV, which is thermally accessible at room temperature. The  intermediate then combines with H+ from
intermediate then combines with H+ from  to yield methanol with a barrier of only 0.35 eV. 13C NMR confirms that the carbon originates from CH4 and the oxygen from CO32−. In situ Raman spectroscopy captures a peak at ∼1445 cm−1 assigned to the δ-CH vibration of the CH3O* intermediate at 1.50 V, directly verifying the methoxy pathway. This system achieves a methanol selectivity of 95%, a production rate of 11.1 mmol gcat−1 h−1 in a flow electrolyzer, and only 8% decay after 100 hours of operation at 1.50 V vs. RHE.
to yield methanol with a barrier of only 0.35 eV. 13C NMR confirms that the carbon originates from CH4 and the oxygen from CO32−. In situ Raman spectroscopy captures a peak at ∼1445 cm−1 assigned to the δ-CH vibration of the CH3O* intermediate at 1.50 V, directly verifying the methoxy pathway. This system achieves a methanol selectivity of 95%, a production rate of 11.1 mmol gcat−1 h−1 in a flow electrolyzer, and only 8% decay after 100 hours of operation at 1.50 V vs. RHE.
 | ||
Fig. 3 (a) Schematic of CO32− electrochemical dissociation on an IrO2(110) surface, generating a high coverage of terminal oxygen species that promotes the selective conversion of methane to methanol via a methoxy intermediate pathway. Reproduced from ref. 26 with permission from Springer Nature, copyright 2025. (b) Schematic of C–H bond activation in methane via hydrogen atom abstraction by Fe(IV)![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species generated from water oxidation on an α-Fe2O3 catalyst surface, forming *OCH3 intermediates that undergo oxidation and C–C coupling to ultimately yield C2+ products such as acetone. Reproduced from ref. 41 with permission from the American Chemical Society, copyright 2024. (c) Proposed mechanism for methane electro-oxidation based on a peroxide-assisted pathway on a Cu–Fe–Ni multimetal catalyst. Reproduced from ref. 42 with permission from Elsevier, copyright 2023. (d) Schematic illustrating the CH activation pathways via *O or *Cl, and the *Cl-intermediate-promoted methane electro-oxidation mechanism on a Co–Ni mixed spinel catalyst. Reproduced from ref. 43 with permission from Wiley-VCH GmbH, copyright 2021. (e) Schematic of high-pressure electro-Fenton CH4 conversion at room temperature. Ag cathode reduces O2 to H2O2, and Fe2+ generates *OH to oxidize CH4 to HCOOH. Reproduced from ref. 44 with permission from the American Chemical Society, copyright 2024. O species generated from water oxidation on an α-Fe2O3 catalyst surface, forming *OCH3 intermediates that undergo oxidation and C–C coupling to ultimately yield C2+ products such as acetone. Reproduced from ref. 41 with permission from the American Chemical Society, copyright 2024. (c) Proposed mechanism for methane electro-oxidation based on a peroxide-assisted pathway on a Cu–Fe–Ni multimetal catalyst. Reproduced from ref. 42 with permission from Elsevier, copyright 2023. (d) Schematic illustrating the CH activation pathways via *O or *Cl, and the *Cl-intermediate-promoted methane electro-oxidation mechanism on a Co–Ni mixed spinel catalyst. Reproduced from ref. 43 with permission from Wiley-VCH GmbH, copyright 2021. (e) Schematic of high-pressure electro-Fenton CH4 conversion at room temperature. Ag cathode reduces O2 to H2O2, and Fe2+ generates *OH to oxidize CH4 to HCOOH. Reproduced from ref. 44 with permission from the American Chemical Society, copyright 2024. | ||
In electrocatalytic methane oxidation, high-valent metal-oxo species (MO) can also be employed for the selective activation of C–H bonds, representing an effective strategy to avoid over-oxidation to CO2. This mechanism drives the reaction via electrochemically generated, highly reactive MO species on the catalyst surface.
Taking Md Golam Kibria et al.'s study on α-Fe2O3 catalysts as an example, water oxidation on α-Fe2O3 generates Fe(IV)O species, which cleave the C–H bond via H-atom abstraction, generating *OCH3 that subsequently couples to form C2+ products (e.g., acetone, FE 6.3%) (Fig. 3b). On the α-Fe2O3 catalyst, water oxidation generates Fe(IV)O species in situ at potentials above 1.8 V vs. RHE. In situ ATR-SEIRAS spectroscopy captures a characteristic absorption peak at 880 cm−1. This peak disappears completely when KI is added as an oxygen species scavenger, directly confirming the involvement of Fe(IV)O in CH4 activation.41 Electrochemical impedance spectroscopy shows that this high-valent metal-oxo species activates CH4 via a hydrogen atom abstraction mechanism. In the low-potential region below 2.0 V vs. RHE, CH4 undergoes non-faradaic adsorption. DFT calculations indicate that Fe(IV)O lowers the CH4 dehydrogenation barrier by 0.16 eV compared to Fe(III), corresponding to a kinetic rate enhancement of approximately 450-fold. The resulting *OCH3 intermediate, identified by asymmetric and symmetric C–H stretching peaks at 2935 and 2822 cm−1 in infrared spectra, is subsequently oxidized to formic acid or undergoes C–C coupling with formate to yield acetic acid. Further ketonization with *CH3 leads to acetone formation. In a Cu–Fe–Ni polymetallic catalyst system for selective electro-oxidation of CH4 to formate (HCOO−), H2O2 oxidation on the Ni surface generates ROS, triggering the formation of Fe(IV)O at low voltage. This promotes CH4 activation via a peroxymethyl (CH3OOH) intermediate, while Cu inhibits over-oxidation to prevent CO2 formation (Fig. 3c).42
Beyond traditional oxygen-containing ROS like *OH or O, chlorine intermediates (Cl), as strongly electrophilic non-oxygen radical species, exhibit unique C–H bond activation capabilities in electrocatalytic methane oxidation. Gengfeng Zheng et al. reported that in saturated NaCl electrolyte, a Co–Ni spinel catalyst can electrochemically generate *Cl intermediates in situ, efficiently converting CH4 to CH3Cl (yield 364 mmol g−1 h−1, CH3Cl/CO2 selectivity >400). As CH3Cl is volatile, this pathway effectively suppresses over-oxidation to CO2. DET analysis showed that Ni2+ sites are instrumental in stabilizing *Cl species, thereby facilitating electrophilic activation of the C–H bond and boosting both selectivity and efficiency of CH4 activation (Fig. 3d).43 This work uncovers a novel mechanism for C–H bond activation, which proceeds via *Cl-mediated electrophilic attack. However, chloride-containing electrolytes can cause equipment corrosion. Industrial application would require higher-cost materials such as titanium or fluoropolymer linings. Moreover, CH3Cl itself is a toxic gas. If collected as a product, the entire setup must meet hazardous chemical standards. We also note that Gengfeng Zheng et al.24 used CH3Cl as an intermediate and coupled it with formate from cathodic CO2 reduction to produce methyl formate. Chlorine was cycled within the system without significant release. This integrated design concept merits attention. Additionally, bromine and iodine are less toxic than chlorine. Replacing Cl with Br or I is a direction worth pursuing in the future.
Besides direct generation of reactive oxygen species at the anode, in situ cathodic H2O2 production provides another effective route for ROS supply. At ambient temperature and pressure, an acid-treated carbon catalyst generates H2O2 in situ via two-electron oxygen reduction, along with *OH and *OOH radicals. These reactive oxygen species activate CH4 in acidic electrolyte, yielding methanol, methyl hydroperoxide, and formic acid. The total liquid product formation rate reaches 18.9 µmol h−1, and HCOOH selectivity is 80.7%. The metal-free catalyst remains stable over six hours of continuous operation.29 Building on this approach, a high-pressure electro-Fenton system further couples mass transport enhancement with H2O2 generation (Fig. 3e). An Ag foil cathode efficiently reduces O2 to H2O2 at 4.0 MPa. Fe2+ then catalyzes H2O2 decomposition to produce *OH radicals, which activate CH4. This system achieves a HCOOH faradaic efficiency of 81.4% at an overpotential of only 0.38 V, with productivity enhanced 220-fold over ambient pressure.44 The above studies show that cathodic in situ H2O2 strategies can decouple oxidant generation from C–H activation, with or without elevated pressure, opening a low-energy pathway for electrocatalytic methane conversion.
For ROS-mediated pathways, achieving high product selectivity critically depends on generating active species in situ and controlling their concentration with precision. The real difficulty is maintaining this balance consistently enough to minimize unwanted side reactions. In general, the mechanisms proposed for initial methane C–H activation tend to fall into two main categories: direct electron transfer and those mediated by reactive oxygen species. With the direct activation route, researchers mainly focus on intentionally tuning the catalyst's electronic structure and its immediate surface environment. The goal here is to lower the inherent energy barrier for breaking the C–H bond and to guide the reaction's course right from the initial steps. In contrast, the ROS-mediated pathway depends on in situ generated active species (e.g., *OH, *O, or *Cl) under an electric field, which activate C–H bonds through electrophilic attack; meanwhile, over-oxidation is suppressed by regulating their surface coverage. Essentially, both mechanisms share a core goal: realizing efficient C–H bond activation alongside highly selective oxidation. Future efforts should combine in situ characterization and theoretical calculations to elucidate the dynamic processes at active sites, thereby guiding the design of highly efficient catalytic systems.
3. Enabling reaction acceleration: synergistic design of catalysts and reaction systems
The core challenge in EMO lies in achieving efficient activation of the C–H bond and the highly selective synthesis of target products under mild conditions. Single-dimensional optimization of either material or system often struggles to balance activity, selectivity, and stability. Therefore, the synergistic design of catalysts and reaction systems emerges as a key pathway to break through existing performance bottlenecks and permit reaction acceleration. In this section, we examine how coupling electronic structure tuning with multi-dimensional system engineering can accelerate reaction kinetics. We specifically focus on the integration of material design with external fields to achieve controllable methane conversion, demonstrating that synergistic optimization is key to breaking current efficiency bottlenecks.3.1. Tuning electronic structure and active sites in catalyst design
In EMO processes, modulating the electronic structure and active sites of catalyst materials is a core strategy for achieving efficient activation and selective conversion. Through electronic structure optimization and active site engineering, it is possible to lower activation barriers, enhance ROS generation, and regulate the stability and desorption of intermediates, thereby accelerating reaction kinetics and improving selectivity.45The work by Ning Yan et al.46 utilized phosphotungstic acid (PTA) as a redox mediator. By electrochemically regulating the valence state of its tungsten centers, they constructed catalytic sites with high activity and stability. The reduced PTA (containing W5+) could activate oxygen in solution to generate hydroxyl radicals, which then efficiently cleaved the C–H bonds of methane, enabling the high-selective conversion of methane to methanol (73%) at room temperature. This strategy not only enhanced the migration and activation capabilities of oxygen species but also ensured the stable regeneration of active sites during cycling due to the excellent reversibility of the PTA structure (Fig. 4a). Zhenmeng Peng et al.47 constructed a Fe–Ni–OH catalyst with a well-defined layered structure. Their study found that the introduction of Fe promoted the Ni(II) → Ni(III) oxidation transition and modulated the local electronic structure, making Ni3+OOH the key active center for the electro-oxidation of methane to ethanol (Fig. 4b). This system achieved an ethanol production rate of 9.09 mmol g−1 h−1 (FE up to 87%) and exhibited excellent C–C coupling selectivity. The study indicated that Fe doping significantly improved oxygen migration and C–H bond activation efficiency. Steve S.-F. Yu et al.48 achieved efficient and highly selective electrocatalytic oxidation of methane by immobilizing a tricopper cluster catalyst on the cathode surface under a threshold cathodic potential. This catalyst could be electrochemically activated in air. A voltage-gating mechanism was used to regulate the electron density of the tricopper cluster, enhancing oxygen activation and site stability, promoting its combination with O2 to form active intermediates, and thereby facilitating the conversion of methane to methanol (Fig. 4c and d), with a turnover frequency as high as >40 min−1.
 | ||
| Fig. 4 (a) Schematic of the electro-thermal synergistic strategy for methane-to-methanol conversion mediated by phosphotungstic acid. Reproduced from ref. 46 with permission from Wiley-VCH GmbH, copyright 2024. (b) Schematic showing Fe-doping modulating the electronic structure of Ni(OH)2 to form highly active Ni(III)OOH species, enabling efficient electrocatalytic oxidation of methane to ethanol on Fe3Ni7(OH)x catalyst. Reproduced from ref. 47 with permission from Elsevier, copyright 2022. (c) Catalytic cycle of a tricopper cluster catalyst, illustrating the voltage-gating mechanism for modulating electron density to drive oxygen activation and achieve highly selective oxidation of methane to methanol under mild conditions. (d) Performance of the electrocatalytic system based on a cathode-supported tricopper cluster catalyst, demonstrating high efficiency and selectivity for methane oxidation to methanol upon applying a threshold potential, with superior turnover frequency and product flux. Reproduced from ref. 48 with permission from the American Chemical Society, copyright 2022. (e) Schematic of the reversible adsorption process of CH4 on a transition metal oxide surface. Reproduced from ref. 49 with permission from PNAS, copyright 2021. (f) Correlation between the C–H bond activation energy barrier for methane and the proton affinity on MXenes. Reproduced from ref. 50 with permission from Elsevier, copyright 2021. | ||
Transition metal oxides (TMOs) have attracted significant attention due to their potential in catalytic activity and stability. Meenesh R. Singh et al.49 investigated the behavior of 12 TMOs during methane electro-oxidation. They found that active catalysts generally possessed higher CH4 binding energy (>0.23 eV) and lower Madelung potential (<−40 V). In the Cu2O3–TiO2 system, the introduction of Cu effectively promoted methanol desorption, thereby significantly increasing the faradaic efficiency for methanol (Fig. 4e). Zhen Zhao et al.,50 through large-scale DFT calculations on 66 MXenes, discovered that the p-band center of active oxygen could serve as an effective descriptor for balancing its stability (preventing excessive conversion to OOH* participating in OER) and reactivity (enabling efficient C–H bond activation) (Fig. 4f). Furthermore, due to the greater filling of metal–oxygen bonding orbitals, MXene surfaces tend to favor the formation of oxygenated derivatives, which is beneficial for the formation of target products like methanol and helps avoid coking. Similarly, PdZn/C catalysts, through Pd–Zn alloying to tune the electronic structure, activated methane at low temperatures in an acidic electrolyte. The study confirmed that Zn doping optimized the Pd d-band, induced oxygen vacancies, enhanced oxygen migration, and strengthened CH4 adsorption.51
Oxide composite catalysts can also improve oxygen vacancy concentration and active site stability, and enhance oxygen migration to suppress carbon deposition and promote partial oxidation, through doping or impregnation to optimize the electronic structure. For example, the introduction of Co3O4 into NiO-YSZ significantly increased the exchange current density for methane electro-oxidation, reaching 87.9 mA cm−2. The Ni3+ sites generated in this process served as active centers for methane C–H bond dissociation, effectively lowering the activation barrier.57
In CuO/CeO2 heterojunction catalysts, the high oxygen storage capacity of CeO2 is utilized to regulate the electronic structure, efficiently promoting lattice oxygen migration.52 This system achieves a methanol production rate of 752.9 µmol gcat−1 h−1 with 79% selectivity under ambient temperature and pressure (Fig. 5a). The Cu2O/CuO interface enhances CH4 adsorption. The –OH generated via water oxidation promote CH4 activation to methanol. In the Cu2O/CuO heterogeneous interface catalyst system, the strong electronic interaction induced by the heterointerface not only optimizes active sites and enhances methane adsorption and activation, but also promotes the in situ generation of hydroxyl radicals (*OH)—a key oxidizing mediator—during the anodic water oxidation reaction (Fig. 5b). This leads to an ethanol production rate of 441.3 µmol gcat−1 h−1.53
 | ||
| Fig. 5 (a) Schematic of the room-temperature electrocatalytic methane-to-methanol system using a CuO/CeO2 catalyst driven by a solar cell. Reproduced from ref. 52 with permission from the American Chemical Society, copyright 2021. (b) Proposed mechanism for the electrocatalytic conversion of methane to ethanol on a Cu2O/CuO heterointerface catalyst. Reproduced from ref. 53 with permission from the American Chemical Society, copyright 2024. (c) Schematic of the core–shell structure and charge transfer mechanism in a NiO/ZnO heterojunction nanorod catalyst. Reproduced from ref. 54 with permission from Elsevier, copyright 2023. (d) Structure of the Fe–N–C single-atom catalyst and schematic of the EMO mechanism. (e) Microkinetic analysis showing the coverage of OER intermediates on the Fe–N–C catalyst surface as a function of electrode potential. Reproduced from ref. 55 with permission from the Royal Society of Chemistry, copyright 2023. (f) Schematic illustrating current-modulated dynamic electronic transfer at the Pd–Ce dual-atom site. (g) Comparison of the current-assisted methane oxidation turnover frequency for the Pd1–Ce1/ATO catalyst with reported catalysts in the literature. Reproduced from ref. 56 with permission from Wiley-VCH GmbH, copyright 2025. All the above are reproduced with permission. | ||
NiO/ZnO heterojunction nanorod catalysts regulate the electronic structure through a core/shell architecture (Fig. 5c).54 The built-in potential and one-dimensional morphology facilitate rapid charge transfer, resulting in an ethanol production rate of 1084.2 µmol gNiO−1 h−1 and 81% selectivity (for 600 nm long nanorods). The NiO/ZnO heterointerface optimizes the d-band center and enhances ROS generation efficiency. In NiO/Ni interface catalysts, the synergy between oxidized Ni3+ in NiO and metallic Ni optimizes the d-band center position, induces oxygen vacancy formation, and increases the –OH generation rate.36 This significantly lowers the C–H bond cleavage barrier, leading to the formation of *CH3 and *CH2 intermediates, which subsequently couple to generate ethanol. This system achieves an ethanol FE of up to 89% and a production rate of 25 µmol gNiO−1 h−1.
For example, constructing Fe–N–C SACs allowed the efficient electrocatalytic conversion of methane to ethanol under mild conditions, achieving a production rate of 4668.3 µmol g−1 h−1 and an FE of 68% (Fig. 5d). This catalyst precisely tunes the electronic structure of the active site via the Fe–Nx coordination structure, shifting the rate-determining step of the OER to *OOH formation. Consequently, it stabilizes the crucial *O intermediate within the potential range of 1.14–1.79 V, effectively suppressing competitive OER and promoting methane activation and stepwise oxidation (Fig. 5e).55 Pd–Ce dual-atom catalysts utilize dynamic current regulation (Fig. 5f) to induce electron transfer from Ce to Pd, enhancing the electron density on Pd and weakening the Pd–O and Ce–O bonds. This synergistic effect promotes methane C–H bond cleavage and lattice oxygen migration, forming an H3Cδ−–Pd–O–Hδ+ four-center transition state that significantly lowers the reaction energy barrier. This allows efficient methane conversion at low temperatures with a TOF of 4.25 × 10−2 s−1 (Fig. 5g).56 These site engineering strategies, through precise electronic structure optimization, enhance the reaction rate and selectivity of EMO and are suitable for scenarios with low noble metal loading. Compared to traditional catalysts, SACs/DACs reduce cluster formation, inhibit coking, and improve stability.
The key to advancing electrocatalytic methane oxidation lies in precisely tuning the catalyst's electronic structure and active sites. This can be achieved through approaches such as oxide compositing or doping, heterojunction and interface engineering, and single- or dual-atom site design. By tailoring the electronic environment of active centers, promoting interfacial charge transfer, and creating adaptable, cooperative active centers, these methods directly enhance the production and mobility of reactive oxygen species, lower the energy required to activate C–H bonds, and guide the transformation of key reaction intermediates along desired pathways. Thus, they collaboratively promote the efficient and highly selective conversion of methane at the atomic/molecular level.
3.2. Engineering optimization of reaction systems through electrolyte, reactor, and operating conditions
In EMO processes, the engineering optimization of the reaction system is a key component for enhancing overall performance, encompassing electrolyte selection, reactor design, and the regulation of operating parameters. These optimizations aim to address challenges such as the low solubility of methane, mass transfer limitations, and over-oxidation, thereby improving current density, faradaic efficiency, and product selectivity.11 This subsection explores the mechanisms and applications of these strategies.The selection and regulation of the electrolyte directly influence the reaction pathway, intermediate stability, and product distribution. Yogesh Surendranath et al.59 employed concentrated sulfuric acid as the electrolyte. Its strong oxidizing nature and acidic environment not only promoted the electrochemical oxidation of Pd2+ but also stabilized highly active PdIII,II2 dimers, thereby enabling efficient methane activation while suppressing over-oxidation and enhancing the selectivity for methanol precursors (Fig. 6a). Yun Jeong Hwang et al.29 demonstrated that a weakly acidic electrolyte (0.05 M H2SO4) favors the in situ generation of H2O2 and its derived reactive oxygen species via a two-electron oxygen reduction reaction (ORR) to activate CH4. Under identical conditions, no oxidation products were generated in an alkaline electrolyte (Fig. 6b). These examples underscore the crucial role of the electrolyte in modulating the reaction pathway and the interfacial microenvironment.
 | ||
| Fig. 6 (a) Schematic of the electrochemical oxidation of Pd(II) in concentrated sulfuric acid to generate highly active PdIII,II2 dimers, which subsequently catalyze the selective conversion of methane to a methanol precursor. Reproduced from ref. 59 with permission from the American Chemical Society, copyright 2017. (b) Schematic showing the in situ generation of H2O2 and reactive oxygen species via the oxygen reduction reaction at the cathode in an acidic electrolyte, driving the electrocatalytic oxidation of methane to liquid-phase products like formic acid with high selectivity under ambient conditions. Reproduced from ref. 29 with permission from Springer Nature, copyright 2023. (c) Schematic of a solid oxide electrolysis cell where electrochemically driven oxygen ion migration to an SFMO-based porous anode enables efficient methane oxidation to ethylene at the in situ constructed metal-oxide interface. Reproduced from ref. 60 with permission from Springer Nature, copyright 2019. (d) Schematic of a flow electrolyzer system employing a gas diffusion electrode. Reproduced from ref. 55 with permission from the Royal Society of Chemistry, copyright 2023. (e) Schematic of a pH-asymmetric electrolyzer cell. Reproduced from ref. 61 with permission from Elsevier, copyright 2025. (f) Plot of current density as a function of methane pressure. Reproduced from ref. 62 with permission from Springer Nature, copyright 2020. (g) Predicted faradaic efficiency for the Ag(II)-mediated electrocatalytic methane functionalization reaction under different methane pressures and temperatures. Reproduced from ref. 63 with permission from Wiley-VCH GmbH, copyright 2021. (h) Potential-dependent reaction pathways for methane electro-oxidation to formic acid versus the OER on a transition metal oxide surface. Reproduced from ref. 49 with permission from PNAS, copyright 2021. | ||
Traditional H-type electrolysis cells are widely used in fundamental research due to their simple structure, but their low mass transfer efficiency severely limits the reaction rate. Kui Xie et al.60 designed a conduction-type solid oxide electrolysis cell (SOEC), which possesses excellent high-temperature oxygen ion conductivity, ensuring efficient transport of oxygen ions during the reaction. Pure methane was fed to the anode side, while air or CO2 was supplied to the cathode side. An applied voltage drove oxygen ions to migrate from the cathode to the anode, participating in the activation and conversion of methane (Fig. 6c). This system significantly increased the current density and selectivity for C2 products (81.2%). Jun Hyuk Moon et al.55 innovatively introduced a gas diffusion electrode (GDE) and a flow cell reactor (Fig. 6d). This design allows methane gas to diffuse directly through the hydrophobic GDE to the catalyst–electrolyte–gas triple-phase boundary, greatly shortening the mass transfer distance and significantly enhancing the local methane concentration. In the flow cell, the ethanol production rate reached a remarkable 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 480.6 µmol g−1 h−1. Weng et al.61 designed a pH-asymmetric electrolysis cell (Fig. 6e). A bipolar membrane (BPM) separates an acidic cathode chamber from an alkaline anode chamber, ingeniously leveraging electrochemical neutralization energy to couple sustainable and efficient electrocatalytic methane oxidation with low-energy-consumption hydrogen production.
480.6 µmol g−1 h−1. Weng et al.61 designed a pH-asymmetric electrolysis cell (Fig. 6e). A bipolar membrane (BPM) separates an acidic cathode chamber from an alkaline anode chamber, ingeniously leveraging electrochemical neutralization energy to couple sustainable and efficient electrocatalytic methane oxidation with low-energy-consumption hydrogen production.
In EMO, operating pressure is a critical engineering parameter for regulating reaction efficiency and selectivity.62 For traditional Shilov-type Pt catalyst systems, high methane pressures (e.g., 500–675 psi) are required to drive the reaction, ensuring effective mass transfer of reactants in the liquid phase to achieve continuous catalysis.64 Early oxidative coupling of methane (OCM) studies also indicated that increasing pressure promotes *CH3 radical recombination, thereby enhancing C2 hydrocarbon selectivity, with an inert gas atmosphere providing a superior stabilization effect.65 In contrast, systems employing novel molecular catalysts (e.g., vanadium-oxo dimers) achieved a breakthrough by operating efficiently at ambient pressure (1 bar) and room temperature. A moderate increase in pressure (e.g., to 3 bar) to enhance the methane current response (Fig. 6f) could further increase the faradaic efficiency from 63.5% to 84.5%.62
In EMO, temperature is a key operating parameter that significantly influences reaction kinetics, selectivity, and catalyst stability.62,64 Different catalyst systems exhibit markedly different temperature requirements. AgII-based systems operate efficiently at ambient temperature (∼25 °C), demonstrating low activation energy (13.1 kcal mol−1) and high reaction rates. However, elevated temperatures can significantly reduce their selectivity due to accelerated solvent oxidation side reactions (Fig. 6g).63 In contrast, Pd-based systems require moderate to high temperatures (80–140 °C) to improve reaction kinetics, achieving a high turnover frequency of 2000 h−1 at 140 °C, with an activation energy of 25.9 kcal mol−1. Similarly, Pt-based Shilov catalytic systems also rely on elevated temperatures (∼130 °C) to ensure sufficient reaction rates and long-term catalyst stability.59
Operating voltage is a key engineering parameter for regulating the EMO reaction pathway, selectivity, and efficiency.26 Proper selection of voltage can promote the generation of target products. Singh et al. found that the onset potential for methane oxidation on transition metal oxides (e.g., TiO2, IrO2) lies between 1.65 and 2.10 V vs. RHE. Excessively high voltages exacerbate the competing OER, reducing methanol selectivity (Fig. 6h).49 Higher applied voltages intensify competition with OER, leading to a decline in yield. Therefore, optimizing the applied voltage is crucial for achieving efficient conversion, striking a balance between activating methane and suppressing side reactions.
Collectively, the performance of an EMO reaction system is the result of synergistic optimization of electrolyte, reactor, and operating conditions. The electrolyte modulates the reaction microenvironment and active species, generating synergistic effects with the catalyst. The core of reactor engineering innovation lies in overcoming mass transfer limitations and improving energy utilization efficiency. Operating parameters such as temperature, pressure, and potential collectively determine reaction kinetics, pathway selection, and catalytic stability. These three aspects are interlinked, systematically strengthening the reaction process from microscopic activity to macroscopic mass transfer, ultimately achieving comprehensive enhancement of rate, selectivity, and energy efficiency.
3.3. Synergistic design mechanisms through material-system integration and multi-field coupling
In EMO processes, the synergistic design mechanism between materials and systems represents an advanced strategy for achieving efficient and stable conversion, encompassing material integration and multi-field coupling. These mechanisms utilize the interaction between the electronic structure of materials and the dynamics of the system to lower energy barriers, enhance ROS generation, and suppress over-oxidation and coking.45 This subsection explores the principles and applications of these mechanisms.Under mild conditions, Feng Ru Fan et al.66 proposed a methane oxidation strategy based on contact-electrocatalysis. This system utilizes an ultrasonic field (mechanical field) to drive interfacial electron transfer between fluorinated ethylene propylene and water (Fig. 7a), generating ROS such as *OH and *OOH (Fig. 7b). This process converts methane to formaldehyde and methanol under ambient conditions, achieving HCHO and CH3OH production rates of 467.5 and 151.2 µmol g−1 mat, respectively, with an HCHO selectivity of 66.6%. Furthermore, Wenzhong Wang et al.68 employed polydopamine (PDA) as a contact-electrocatalytic material and enhanced its performance via pre-treatment with a light field. Under ultrasonication, the strongly electron-withdrawing CO groups on the PDA surface promote O2 reduction to generate ROS, thereby enabling the partial oxidation of methane, with HCHO and CH3OH production rates reaching 1.5 and 0.9 mmol g−1, respectively. The coupling of light-sound-electric triple fields not only improved product yields but also optimized and stabilized catalytic performance through dynamic regulation of the PDA oxidation state. These examples highlight the pivotal role of multi-field synergistic design.
 | ||
| Fig. 7 (a) Schematic of a contact-electrocatalysis system for methane oxidation driven by an ultrasonic field (mechanical field). (b) Generation of ROS in contact-electrocatalysis and the corresponding reaction pathway for methane conversion. Reproduced from ref. 66 with permission from Wiley-VCH GmbH, copyright 2024. (c) Schematic of the electrocatalytic methane conversion system in the aprotic ionic liquid [BMIM]BF4. Reproduced from ref. 67 with permission from Elsevier, copyright 2025. (d) Schematic of an integrated electrochemical system coupling CO2 reduction with CH4 oxidation. Reproduced from ref. 24 with permission from the Royal Society of Chemistry, copyright 2024. (e) Schematic of a solid oxide steam electrolyzer system for synergistic methane conversion and hydrogen production. Reproduced from ref. 25 with permission from Wiley-VCH GmbH, copyright 2025. | ||
Yongmei Chen et al.67 utilized an aprotic ionic liquid, [BMIM]BF4, as the electrolyte to electrocatalytically convert methane into methanol and ethanol at room temperature. This system employed V3O7·H2O nanorods as the anode catalyst to activate CH4. By controlling the cathode potential, O2 underwent one-electron reduction, stably generating –O2− as the key reactive oxygen species (Fig. 7c). A methanol selectivity of 76.8% and a production rate of 149.5 µmol g−1 were achieved. This study demonstrated controllable reaction pathways through the effective synergy between the ionic liquid electrolyte and the catalyst, enabling a stable supply of active oxygen and efficient methane conversion. Gengfeng Zheng et al.24 designed an integrated electrochemical system coupling CO2 reduction with EMO for the efficient synthesis of methyl formate (Fig. 7d). In this system, a Bi catalyst at the cathode selectively reduces CO2 to formate, while IrO2 nanowires with Cl− at the anode oxidize CH4 to CH3Cl. The formate migrates across an anion exchange membrane driven by the electric field and undergoes a nucleophilic reaction with CH3Cl in the anode compartment to yield the target product, methyl formate. This strategy successfully transforms CO2 and CH4 into high-value-added chemicals through the synergy of cathodic and anodic reactions coupled with product transport.
In a high-temperature electrochemical system, Xinhe Bao et al.25 designed an Ag/YSZ solid oxide electrolysis cell. By coupling electrochemical and thermal fields, it achieved the non-oxidative coupling of methane with co-production of hydrogen (Fig. 7e). This system achieved a CH4 consumption rate of 584.46 mL per g mat per h at a current density of 127 mA cm−2, with a high C2+ hydrocarbon selectivity of 76.86%. Concurrently, the faradaic efficiency for hydrogen production at the cathode was nearly 100%, and the system operated stably for 90 hours without significant carbon deposition. This system exemplifies the synergistic design of electrode materials and the electrolysis cell system under high-potential and high-temperature fields.
The core of synergistic design mechanisms lies in the deep integration of materials and systems and the coupling of multiple fields, enabling the dynamic intensification and global empowerment of the reaction process. It overcomes the limitations of traditional single-factor regulation. By harnessing the synergistic effects of multiple physical fields (e.g., light, sound, electricity, heat), it not only substantially lowers the activation barrier for methane but also precisely regulates the generation of reactive oxygen species and the reaction pathway. Consequently, it achieves significant acceleration of reaction kinetics under mild, and even extreme, conditions.
4. Mass transfer process intensification: overcoming solubility and diffusion limitations
In EMO processes, intensifying mass transfer is a crucial strategy to overcome the challenges of methane's low solubility (∼1.3 mM at 25 °C, 1 atm) and diffusion limitations, which can significantly enhance current density and FE. These intensifications include solubility enhancement, mass transfer optimization, and diffusion improvement. To increase the effective concentration of reactants at catalyst active sites, researchers have substantially addressed the slow mass transfer in EMO through approaches such as high pressure, electrolyte regulation, additives, system optimization, and the use of porous structures and surface modifications.69 This section explores the mechanisms and applications of these strategies.4.1. Enhancing solubility through high pressure, electrolyte, and additive strategies
Solubility enhancement strategies address mass transfer bottlenecks by employing high pressure, tuning electrolyte polarizability, and optimizing the dissolution environment with additives, thereby increasing reaction rates.For example, in a system combining the ionic liquid [bmim][BF4] with a TiO2–CuOx composite interface and a gas-oriented transport structure, efficient conversion of CH4 to ethanol was achieved at ambient temperature and pressure (Fig. 8a).70 The hydrophobic alkyl chains of the [bmim]+ cation reduce the solution's surface tension, while the strongly polar BF4− anion can polarize methane molecules. Their synergistic effect results in methane solubility in this electrolyte being 2–3 times higher than in traditional inorganic salt electrolytes (Fig. 8b). Driven by the anodic potential, [bmim][BF4] can directionally adsorb and arrange on the catalyst surface, forming an “ionic channel” with an outer hydrophobic and inner electrophilic structure. This greatly promotes the directional diffusion and enrichment of methane from the bulk phase to the catalyst active sites. This strategy allowed the system to achieve a high ethanol production rate of 48 mmol gcat−1 h−1 and high selectivity (76.8%). For the NiO/ZnO heterojunction nanorod catalyst, the organic solvent sulfolane was introduced as a co-solvent in an aqueous Na2CO3 electrolyte. Given that methane solubility in sulfolane (∼78.93 mg L−1) is about 3.5 times higher than in water, this strategy directly increases reactant concentration. Experiments showed that adding 10% sulfolane increased the ethanol production rate by 80% (Fig. 8c) while maintaining product selectivity.54 This demonstrates that physically enhancing mass transfer by introducing high-solubility co-solvents is a simple yet effective strategy. Notably, the introduction of organic solvents or ionic liquids often reduces the ionic conductivity of the electrolyte and increases ohmic polarization. In practical system design, a compromise can be struck to balance enhanced solubility and high conductivity. For instance, using a water–ionic liquid mixed electrolyte with a 7:3 volume ratio of [bmim][BF4] to water raises methane solubility nearly twofold compared to pure water while maintaining conductivity an order of magnitude higher than that of the neat ionic liquid.67 In addition, screening low-viscosity, high-conductivity ionic liquids or shortening the ion transport path via thin-layer electrodes and zero-gap reactors can serve as viable compensatory measures.
 | ||
| Fig. 8 (a) Schematic of the synergistic reaction mechanism for the electro-oxidative synthesis of ethanol from methane on an ionic liquid TiO2–CuOx composite catalyst. (b) Molecular structure of the ionic liquid [bmim][BF4] and schematic illustrating its hydrophobic-polar synergistic effect, which significantly enhances methane solubility and forms efficient diffusion channels. Reproduced from ref. 70 with permission from Elsevier, copyright 2025. (c) Schematic illustrating the significant enhancement of ethanol yield by adding sulfolane, a high-methane-solubility organic cosolvent, in the NiO/ZnO heterojunction catalyst system. Reproduced from ref. 54 with permission from Elsevier, copyright 2023. (d) Effect of methane pressure (pCH4) on the electrocatalytic current response in an AgII-mediated methane oxidation system. Reproduced from ref. 63 with permission from Wiley-VCH GmbH, copyright 2021. | ||
Increasing the pressure of the reaction system is one of the most effective methods to directly enhance methane solubility in the liquid phase, significantly boosting its mass transfer driving force and reaction rate. A more systematic kinetic study of the pressure effect was conducted in an AgII-mediated homogeneous catalytic system. By progressively increasing methane pressure from 15 psi (∼1 bar) to 125 psi (∼8.6 bar), the researchers observed a continuous rise in current density (Fig. 8d), along with significant increases in the reaction rate constant and faradaic efficiency.63 This work not only confirmed the yield-enhancing effect of high pressure but also theoretically elucidated its critical role in suppressing solvent oxidation side reactions and achieving ultra-high selectivity, providing clear design goals for high-pressure reactors. In the CuO/CeO2 catalyst system, increasing the reaction pressure from ambient to 10 bar was investigated. Under this condition, methane solubility in water increased approximately 8-fold. After a 12-hour reaction, the cumulative methanol yield surged from 7165.0 µmol gcat−1 at ambient pressure to 21986.6 µmol gcat−1, representing a 236% increase.52 This study successfully coupled a high-pressure reactor with a solar cell-driven system, demonstrating the feasibility of achieving yield multiplication through engineering means under mild conditions.
These findings confirm that low solubility is no longer an insurmountable barrier. Through the combined use of high-pressure reactors, electrolyte engineering, and functional additives, recent studies have successfully amplified the mass transfer driving force, laying a solid foundation for scaling up EMO technologies.
4.2. Optimizing diffusion via flow systems, porous structures, and interfacial fields
The diffusion process is the core link of mass transfer limitation in electrochemical methane conversion. Its efficiency directly depends on the diffusion path length of reactants/products, the mass transfer driving force, and the interfacial transport resistance.71 Systemic synergy through flow system design, porous structure regulation, and interface engineering can overcome bottlenecks in traditional systems, such as slow methane diffusion, long species residence times, and high interfacial resistance, thereby achieving efficient matching between mass transfer and catalysis.11Meenesh R. Singh et al.72 constructed a flow-type three-electrode system by loading patterned Cu–Ti bimetallic oxide onto a gas diffusion layer (GDL) (Fig. 9a). The porous structure of the GDL provided rapid diffusion channels, delivering gaseous methane directly to the catalytic active sites. The total current density for methane oxidation reached 40 mA cm−2, the highest value reported under ambient conditions. This confirmed that the synergy between the flow cell and GDE can effectively address the issues of slow methane diffusion and insufficient contact. Similarly, constructing a flow electrolyzer using a GDE to deliver gaseous methane directly to the interface of an Fe–N–C single-atom catalyst shortened the diffusion distance from the millimeter to the micrometer scale.55 The study found that as the feed flow rate increased from 10 sccm to 50 sccm, the ethanol production rate increased significantly, reaching a maximum of 11480.6 µmol gcat−1 h−1 at 50 sccm (Fig. 9b). This represented a 2.5-fold enhancement compared to batch reactions, significantly intensifying mass transfer during the reaction and enabling continuous, efficient electrochemical methane conversion.
 | ||
| Fig. 9 (a) Schematic of the fabrication of a patterned Cu–Ti bimetallic oxide catalyst and its application on a gas diffusion electrode. Reproduced from ref. 72 with permission from the American Chemical Society, copyright 2022. (b) Effect of methane feed flow rate on ethanol yield for an Fe–N–C single-atom catalyst in a flow cell system. Reproduced from ref. 55 with permission from the Royal Society of Chemistry, copyright 2023. (c) Systematic study of the effect of electrode rotation speed on mass transfer and reaction selectivity during the electrochemical oxidation of methane to methanol using a sealed rotating cylinder electrode reactor. Reproduced from ref. 73 with permission from the American Chemical Society, copyright 2023. (d) Performance data showing limited methane conversion but high C2+ selectivity (86.6%) under high gas hourly space velocity (GHSV) conditions. (e) Performance data showing significantly improved methane conversion but reduced C2+ selectivity (47.2%) under lower gas hourly space velocity conditions. Reproduced from ref. 25 with permission from Wiley-VCH GmbH, copyright 2025. (f) Schematic showing the tunable selectivity distribution for C2 products from methane electro-oxidation via in situ construction of metal-oxide interfaces within a porous SFMO anode. Reproduced from ref. 60 with permission from Springer Nature, copyright 2019. (g) Schematic of electric-field-driven directional cross-membrane migration of formate ions. (h) Schematic illustrating the competitive pathways for the nucleophilic substitution reaction between formate and chloromethane. Reproduced from ref. 24 with permission from the Royal Society of Chemistry, copyright 2024. All the above are reproduced with permission. | ||
Morales-Guio et al.73 employed a sealed rotating cylinder electrode reactor to investigate the effect of electrode rotation speed on mass transfer and reaction selectivity in the methane electro-oxidation to methanol. By adjusting the rotation speed (0–800 rpm) to enhance convective mass transfer, the diffusion rate of methane to the electrode surface was increased by approximately 3.3 times (from 1.9 × 10−9 to 6.3 × 10−9 mol cm−2 s−1). At 800 rpm, the maximum accumulated methanol concentration reached 55 µM (Fig. 9c), representing a ∼65% improvement over the static system. This indicates that optimized hydrodynamic conditions can effectively match mass transfer with catalytic reaction rates, enabling nearly half of the methane delivered to the electrode to be converted to methanol.
Porous structured electrodes are the core carriers for optimizing diffusion pathways. Their pore size distribution, specific surface area, and pore connectivity directly determine reactant diffusion efficiency and active site accessibility. Bao et al.25 designed a porous nano-silver anode in a SOEC, whose pore structure effectively promoted the bulk diffusion of gaseous methane to the active interface. By adjusting the gas hourly space velocity (GHSV), the methane mass transfer process and reaction pathway could be optimized: at a higher GHSV (80000 mL gcat−1 h−1), methane conversion was limited but C2+ selectivity was as high as 86.6%; whereas at a lower GHSV (24000 mL gcat−1 h−1), conversion increased significantly but selectivity dropped to 47.2% (Fig. 9d and e). In another SOEC, constructing a porous Sr2Fe1.5+xMo0.5O6−δ (SFMO) anode with a nano metal-oxide interface allowed efficient electrochemical conversion of methane to ethylene (Fig. 9f). The porosity of this structure reached 65%, forming interconnected three-dimensional diffusion channels, which reduced the diffusion resistance of methane within the electrode by 40%. Furthermore, the high specific surface area of the porous structure also inhibited the agglomeration of Fe nanoparticles, maintaining catalyst stability during 100 h of high-temperature operation.60 This exemplifies the critical role of porous structure and interface engineering in enhancing mass transfer and improving reaction performance.
Electric field-driven directional migration is a key means to regulate ion transport paths and match reaction spaces. Its migration efficiency, directionality, and matching degree with the reaction zone directly determine the utilization efficiency of ionic intermediates and the selectivity of coupling reactions. In an integrated electrochemical system, the synergistic effect of an anion exchange membrane and an applied electric field allowed the directional migration of HCOO− (produced from cathodic CO2 reduction) to the anode compartment (Fig. 9g). Subsequently, it underwent a nucleophilic substitution reaction with CH3Cl generated from anodic CH4 oxidation, achieving a methyl formate production rate of 1660 µmol h−1 cm−2 and an overall cell energy efficiency of approximately 15.2% (Fig. 9h).24
Collectively, the systematic optimization of diffusion relies on the integrated application of flow system engineering, tailored porous architectures, and electric field manipulation. These strategies work in concert to drastically shorten transport distances, enhance convective/diffusive fluxes, and guide the directional delivery of key species.
5. Conclusion and prospect
5.1. Core findings and research progress
EMO as a green technology for efficient methane conversion under mild conditions, represents a critical pathway toward achieving the carbon-cycle economy. Its core challenge lies in overcoming the three major bottlenecks: the inertness of methane molecules, low solubility, and tendency for over-oxidation. From an integrated design perspective, we organically summarize recent advances in the EMO field (Fig. 10a). At the atomic/molecular scale, the mechanisms of C–H bond activation via direct electron transfer and ROS mediation are elucidated, highlighting the role of electronic structure modulation and defect engineering in steering reaction pathways. At the catalyst and reaction-system scale, strategies such as precise electronic structure tuning, hetero-interface construction, and site-specific design have been employed to optimize catalytic active centers. Coupled with electrolyte composition regulation, reactor innovations (e.g., flow cells, gas-diffusion electrodes), and operational parameter optimization, synergistic enhancements in activity, selectivity, and stability have been achieved. At the macroscopic mass-transfer scale, techniques including high-pressure operation, electrolyte engineering, porous structure design, and flow-system construction have significantly improved methane solubility and diffusion efficiency, thereby alleviating interfacial mass-transfer limitations. The deep integration of these three dimensions forms the core development framework of EMO technology: microscopic mechanisms provide theoretical guidance for catalytic system design, catalyst innovation and system optimization reinforce reaction kinetics, and macroscopic mass-transfer enhancement breaks throughput bottlenecks for industrial application. Together, they form a complete chain from fundamental research to technological translation, offering systematic solutions to key issues such as low reaction efficiency, poor selectivity, and insufficient current density in EMO. | ||
| Fig. 10 (a) Synergistic optimization of mechanisms, materials, and mass transport for EMO. (b) Framework of future full-chain solutions to address EMO challenges. | ||
5.2. Current challenges and technical bottlenecks
However, we should acknowledge that EMO remains in the early phases of transitioning from fundamental discovery to practical application, with its core scientific and technological bottlenecks still largely unresolved. The formidable C–H bond dissociation energy (439 kJ mol−1) continues to pose a significant activation barrier, meaning most catalytic systems still demand substantial overpotentials to proceed. Adding to the challenge, the identity of the true active species and the detailed reaction pathways are still hotly debated. There is simply not enough in situ or operando evidence available yet, and noticeable gaps often persist between what theoretical models predict and what experimental results actually show. Furthermore, the product spectrum remains rather limited. Current research is largely confined to a narrow set of C1–C2 oxygenated products—methanol, formic acid, or ethanol—while routes to more complex, higher value chemicals are still largely uncharted territory. Improving product yields is constrained jointly by methane mass transport and oxygen evolution competition. Engineering approaches such as gas diffusion electrodes, superaerophilic interfaces, and ionic liquid solubilization can mitigate mass transport bottlenecks. At the same time, developing non noble metal catalysts with high intrinsic activity remains essential for lowering overpotential and raising current density. The synthesis of higher carbon products (C2+) poses more fundamental difficulties. C–C coupling requires a reductive step to proceed within an oxidative environment. This demands both adequate coverage and spatial proximity of intermediates on the catalyst surface. Three strategies appear feasible. First, construct interfacial synergistic sites to stabilize *CH3 and lower the coupling barrier. Second, design spatially separated tandem catalytic systems. C–H activation and C–C coupling are assigned to distinct active components, preventing mutual interference at the same site. Third, employ non oxygen mediated pathways such as those involving *Cl. Methane is first converted to a platform molecule like CH3Cl. Subsequent nucleophilic substitution with a cathode derived product (e.g., formate) yields esters, which bypasses the kinetic hurdle of direct C–C coupling.Beyond the strategies outlined above for expanding the product scope, a more fundamental understanding of selectivity control is essential. Product selectivity in EMO ultimately depends on the coordinated interplay of three factors: intermediate adsorption strength, reactive oxygen species coverage, and the surface coordination environment. In terms of coordination environment, single-atom catalysts achieve atomic-level selectivity control through precisely designed coordination structures. High-entropy alloys could exploit the abundant unsaturated metal sites in a multi-component coordination environment. They simultaneously promote *CH3 dehydrogenation and the adsorption of *CH2OH and *CH3 intermediates, thereby driving C–C coupling toward ethanol. In solid oxide electrolyzers, stabilizing active oxygen species during the oxygen evolution reaction promotes C–H activation and C–C coupling, thereby enhancing C2 selectivity. Taken together, the key to selectivity control lies in the multi-objective co-optimization of electronic structure, coordination environment, and reactive oxygen species engineering, guided by the targeted product.
Beyond this selectivity challenge, today's best-performing systems continue to rely heavily on precious metals like Ir, Pd, and Ru. Catalysts based on non-precious alternatives still struggle to match them, showing significant gaps in both activity and long-term durability. Perhaps the most persistent hurdle, however, stems from methane's notoriously low solubility in water. This creates severe mass-transfer bottlenecks at the gas–liquid–solid interface, and even with advanced gas diffusion electrodes and increased pressure, achieving the industrial benchmark of >500 mA cm−2 remains out of reach.
5.3. Future outlook and development strategies
To move beyond the current bottlenecks, future work needs to tackle several interconnected issues at once. First, we still lack a clear picture of how active sites evolve and how reactive oxygen species behave under working conditions. Better in situ and operando characterization, combined with DFT and machine learning, should help close this gap. Second, most high-performing catalysts still rely on precious metals. We need to design non-noble alternatives—high-entropy alloys, single-atom catalysts, defect-rich oxides—without sacrificing activity or stability. Third, methane's poor solubility in water remains a hard physical limit. Even with flow cells and gas-diffusion electrodes, the current densities are far from industrial levels. New electrolyte formulations, better electrode architectures, and perhaps non-aqueous reaction media deserve more attention. Fourth, the field lacks standardized stability tests. Comparing catalyst lifetimes across different reports is nearly impossible right now. Establishing clear protocols for long-term operation and deactivation analysis would help separate real progress from incremental noise. The following sections discuss these directions in more detail.Elucidating catalytic mechanisms and confirming reaction pathways rely heavily on advanced characterization techniques. In EMO research, commonly employed methods fall into three main categories: spectroscopy, diffraction and scattering, and mass spectrometry coupling. In terms of spectroscopy, in situ Raman tracks potential-dependent vibrational fingerprints of surface species to identify key intermediates. Operando infrared absorption spectroscopy directly captures the vibrational signatures of low-coverage transient species after C–H cleavage. Regarding diffraction and scattering methods, the combination of X-ray diffraction and X-ray photoelectron spectroscopy depth profiling provides complementary information on bulk crystal structure and surface elemental valence evolution during reaction. As for mass spectrometry coupling, differential electrochemical mass spectrometry (DEMS) enables real-time online analysis of volatile products and intermediates, offering direct insight into product distribution as a function of potential. Meanwhile, 18O and 13C isotope labeling experiments respectively trace the origin of oxygen atoms and confirm the carbon backbone of products, serving as critical tools to distinguish homogeneous radical pathways from surface-catalyzed routes. Each technique has its own strengths, and the combined use of multiple methods is becoming standard practice for systematically unraveling the complex mechanisms of EMO.
Furthermore, the role of defect engineering in transition-metal oxides and carbides should be thoroughly explored to enhance intrinsic activity and suppress over-oxidation. Catalyst design should evolve from single-function toward integrated “activation–stabilization-coupling” multi-functionality, while simultaneously pioneering new C–C coupling pathways to allow the directed synthesis of C3+ high-value oxygenated compounds. Exploring electro-thermochemical coupling processes may open routes from methane to esters, aldehydes, and even aromatic compounds. Inspired by *Cl-mediated activation strategies, investigating the electrocatalytic co-conversion of methane with Br−, I−, or NO3−/NO2− could lead to the direct synthesis of high-value platform molecules such as halogenated methanes and nitromethane.
Ultimately, by deeply integrating high-performance non-precious-metal catalysts, clarified reaction mechanisms, and advanced reactor/electrolyte engineering, a new paradigm of integrated design and synergistic optimization spanning materials, interfaces, and systems can be established. Looking forward, the maturation and industrialization of EMO technology will rely not only on breakthroughs in traditional disciplines such as electrochemistry, materials science, and engineering, but also on the convergence of artificial intelligence, high-throughput computation, and automated experimentation to allow form the intelligent design and dynamic control of catalyst–electrolyte–reactor systems. Through tight coupling with renewable-energy power systems (e.g., photovoltaic, wind), EMO is poised to become a pivotal hub technology for realizing the “carbon cycle” and “hydrogen economy”, thereby providing substantive support for building a green, low-carbon, and sustainable energy and chemical industry.
Author contributions
Z. L., J. D. and X. W. conceive and supervise research, funding, writing – criticism and editing. H. Z. conceived the framework of the review, analyzed and sorted out the logical context between the papers, drew the main picture of the review, and finally wrote the manuscript. L. Z. optimized the picture and reviewed the manuscript. All the authors contributed to the interpretation of the results.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by National Key Research and Development Program Project (No. 2025YFF0516504, and 2022YFA1504500), National Natural Science Foundation of China (No. 22278094, U24A20541 and W2421038), Basic and Applied Basic Research Program of Guangzhou (No. 2024A03J0236).Notes and references
- B. Yarlagadda, G. Iyer, M. Binsted, P. Patel, M. Wise and J. McLeod, iScience, 2024, 27, 108902 CrossRef CAS PubMed.
- L. Zhang, Energy, 2025, 320, 135253 CrossRef.
- E. G. Nisbet, R. E. Fisher, D. Lowry, J. L. France, G. Allen, S. Bakkaloglu, T. J. Broderick, M. Cain, M. Coleman, J. Fernandez, G. Forster, P. T. Griffiths, C. P. Iverach, B. F. J. Kelly, M. R. Manning, P. B. R. Nisbet-Jones, J. A. Pyle, A. Townsend-Small, A. al-Shalaan, N. Warwick and G. Zazzeri, Rev. Geophys., 2020, 58, e2019RG000675 CrossRef.
- I. Pérez-Domínguez, A. del Prado, K. Mittenzwei, J. Hristov, S. Frank, A. Tabeau, P. Witzke, P. Havlik, H. van Meijl, J. Lynch, E. Stehfest, G. Pardo, J. Barreiro-Hurle, J. F. L. Koopman and M. J. Sanz-Sánchez, Nat. Food, 2021, 2, 970–980 CrossRef PubMed.
- J. C. Ku-Vera, R. Jiménez-Ocampo, S. S. Valencia-Salazar, M. D. Montoya-Flores, I. C. Molina-Botero, J. Arango, C. A. Gómez-Bravo, C. F. Aguilar-Pérez and F. J. Solorio-Sánchez, Front. Vet. Sci., 2020, 7, 584 CrossRef PubMed.
- P. G. Levi and J. M. Cullen, Environ. Sci. Technol., 2018, 52, 1725–1734 CrossRef CAS PubMed.
- A. Caballero and P. J. Pérez, Chem. Soc. Rev., 2013, 42, 8809–8820 RSC.
- N. J. Gunsalus, A. Koppaka, S. H. Park, S. M. Bischof, B. G. Hashiguchi and R. A. Periana, Chem. Rev., 2017, 117, 8521–8573 CrossRef CAS PubMed.
- H. T. Zhang, Z. X. Sun and Y. H. Hu, Renewable Sustainable Energy Rev., 2021, 149, 111330 CrossRef CAS.
- P. Schwach, X. L. Pan and X. H. Bao, Chem. Rev., 2017, 117, 8497–8520 CrossRef CAS PubMed.
- L. Yan, L. Jiang, C. Qian and S. Zhou, Energy Rev., 2024, 3, 100065 CrossRef CAS.
- J. C. Fornaciari, D. Primc, K. Kawashima, B. R. Wygant, S. Verma, L. Spanu, C. B. Mullins, A. T. Bell and A. Z. Weber, ACS Energy Lett., 2020, 5, 2954–2963 CrossRef CAS.
- B. Wang, S. Albarracín-Suazo, Y. Pagán-Torres and E. Nikolla, Catal. Today, 2017, 285, 147–158 CrossRef CAS.
- W. J. Jang, J. O. Shim, H. M. Kim, S. Y. Yoo and H. S. Roh, Catal. Today, 2019, 324, 15–26 CrossRef CAS.
- Y. C. Xing, Z. Yao, W. Y. Li, W. T. Wu, X. Q. Lu, J. Tian, Z. T. Li, H. Hu and M. B. Wu, Angew. Chem., Int. Ed., 2021, 60, 8889–8895 CrossRef CAS PubMed.
- F. B. Gu, X. T. Qin, M. W. Li, Y. Xu, S. Hong, M. Y. Ouyang, G. Giannakakis, S. F. Cao, M. Peng, J. L. Xie, M. Wang, D. M. Han, D. Q. Xiao, X. Y. Wang, Z. H. Wang and D. Ma, Angew. Chem., Int. Ed., 2022, 61, e202201540 Search PubMed.
- X. J. Cui, H. B. Li, Y. Wang, Y. L. Hu, L. Hua, H. Y. Li, X. W. Han, Q. F. Liu, F. Yang, L. M. He, X. Q. Chen, Q. Y. Li, J. P. Xiao, D. H. Deng and X. H. Bao, Chem, 2018, 4, 1902–1910 CAS.
- K. Tabata, Y. Teng, T. Takemoto, E. Suzuki, M. A. Bañares, M. A. Peña and J. L. G. Fierro, Catal. Rev.-Sci. Eng., 2002, 44, 1–58 CrossRef CAS.
- H. D. Gesser, N. R. Hunter and P. A. Das, Catal. Lett., 1992, 16, 217–221 CrossRef CAS.
- T. Li, S. J. Wang, C. S. Yu, Y. C. Ma, K. L. Li and L. W. Lin, Appl. Catal., A, 2011, 398, 150–154 CrossRef CAS.
- S. K. Wang, V. Fung, M. J. Hülsey, X. C. Liang, Z. Y. Yu, J. Q. Chang, A. Folli, R. J. Lewis, G. J. Hutchings, Q. He and N. Yan, Nat. Catal., 2023, 6, 895–905 CrossRef CAS.
- H. Fujisaki, T. Ishizuka, H. Kotani, Y. Shiota, K. Yoshizawa and T. Kojima, Nature, 2023, 616, 476–481 CrossRef CAS PubMed.
- W. Y. Wang, W. Zhou, Y. C. Tang, W. C. Cao, S. R. Docherty, F. W. Wu, K. Cheng, Q. H. Zhang, C. Copéret and Y. Wang, J. Am. Chem. Soc., 2023, 145, 12928–12934 CrossRef CAS PubMed.
- Q. Zhang, Y. Chen, S. Yan, X. Lv, C. Yang, M. Kuang and G. Zheng, Energy Environ. Sci., 2024, 17, 2309–2314 Search PubMed.
- Y. Guo, T. Liu, M. Xu, S. Zhang, X. Zhang, B. Zhao, L. Zhang, L. Zhao, Y. Pan, Y. Song, G. Wang and X. Bao, Angew. Chem., Int. Ed., 2025, 64, e202512935 CrossRef CAS PubMed.
- C. Kim, J. Lee, S. Lee, W. Jung, H. Min, J. Choi, S. Kim, Y. T. Kim, J. Lee, J. S. Yoo and J. H. Moon, Nat. Catal., 2025, 8, 688–696 CrossRef CAS.
- H. M. Jiang, L. Zhang, Z. W. Han, Y. Tang, Y. Z. Sun, P. Y. Wan, Y. M. Chen, M. D. Argyle and M. Fan, Energy Environ. Sci., 2022, 7, 1132–1142 CAS.
- L. Q. Wang, J. T. Jin, W. Z. Li, C. S. Li, L. Y. Zhu, Z. Zhou, L. L. Zhang, X. Zhang and L. Yuan, Energy Environ. Sci., 2024, 17, 9122–9133 Search PubMed.
- J. Kim, J. H. Kim, C. Oh, H. Yun, E. Lee, H.-S. Oh, J. H. Park and Y. J. Hwang, Nat. Commun., 2023, 14, 4704 CrossRef CAS PubMed.
- J. A. Arminio-Ravelo and M. Escudero-Escribano, Curr. Opin. Green Sustainable Chem., 2021, 30, 100489 Search PubMed.
- D. Richard, Y. C. Huang and C. G. Morales-Guio, Curr. Opin. Electrochem., 2021, 30, 100793 CrossRef CAS.
- S. Yuan, Y. D. Li, J. Y. Peng, Y. M. Questell-Santiago, K. Akkiraju, L. Giordano, D. J. Zheng, S. Bagi, Y. Román-Leshkov and Y. Shao-Horn, Adv. Energy Mater., 2020, 10, 2002154 CrossRef CAS.
- Q. H. Wang, M. Kan, Q. Han and G. F. Zheng, Small Struct., 2021, 2, 2100037 CrossRef CAS.
- A. H. B. Mostaghimi, T. A. Al-Attas, M. G. Kibria and S. Siahrostami, J. Mater. Chem. A, 2020, 8, 15575–15590 RSC.
- B. Yu, W. Li, X. Tang, L. Cheng, J. Zhang, S. Yang, F. Liu, J. Xu, Y. Zhang, C. Pan, X.-M. Cao, Y. Zhu and Y. Lou, Nat. Commun., 2025, 16, 9471 CrossRef CAS PubMed.
- Y. Song, Y. Zhao, G. Nan, W. Chen, Z. Guo, S. Li, Z. Tang, W. Wei and Y. Sun, Appl. Catal., B, 2020, 270, 118888 CrossRef CAS.
- Y. Choi, H. Ha, J. Kim, H. G. Seo, H. Choi, B. Jeong, J. Yoo, E. J. Crumlin, G. Henkelman, H. Y. Kim and W. Jung, Adv. Mater., 2024, 36, 2403626 CrossRef CAS PubMed.
- Q. Zhang, J. Peng, H. Xiong, S. Jiang, W. Li, X. Fu, S. Shang, J. Xu and G. He, Appl. Catal., B, 2025, 362, 124759 CrossRef CAS.
- H. Tian, Z.-Y. Zhang, H. Fang, H. Jiao, T.-T. Gao, J.-T. Yang, L. Bian and Z.-L. Wang, Appl. Catal., B, 2024, 351, 124001 CrossRef CAS.
- M. Chen, X. Lv, A. Guan, C. Peng, L. Qian and G. Zheng, J. Colloid Interface Sci., 2022, 623, 348–353 CrossRef CAS PubMed.
- T. Al-Attas, K. Kannimuthu, M. A. Khan and M. G. Kibria, ACS Catal., 2024, 14, 10614–10623 CrossRef CAS.
- T. Al-Attas, M. A. Khan, T. J. Goncalves, N. G. Yasri, S. Roy, A. S. Zeraati, P. Kumar, K. A. Miller, P. M. Ajayan, I. D. Gates, J. Hu, V. Thangadurai, S. Siahrostami and M. G. Kibria, Chem. Eng. J., 2023, 474, 145827 CrossRef CAS.
- Q. Wang, T. Li, C. Yang, M. Chen, A. Guan, L. Yang, S. Li, X. Lv, Y. Wang and G. Zheng, Angew. Chem., Int. Ed., 2021, 60, 17398–17403 CrossRef CAS PubMed.
- Y. Song, X. Yang, H. Liu, S. Liang, Y. Cai, W. Yang, K. Zhu, L. Yu, X. Cui and D. Deng, J. Am. Chem. Soc., 2024, 146, 5834–5842 CrossRef CAS PubMed.
- Y. Huang, L. Zou, Y.-B. Huang and R. Cao, Chin. J. Catal., 2025, 70, 207–229 CrossRef.
- J. Chang, S. Wang, M. J. Hülsey, S. Zhang, S. Nee Lou, X. Ma and N. Yan, Angew. Chem., Int. Ed., 2024, 64, e202417251 Search PubMed.
- J. Li, L. Yao, D. Wu, J. King, S. S. C. Chuang, B. Liu and Z. Peng, Appl. Catal., B, 2022, 316, 121657 CrossRef CAS.
- Y. F. Tsai, T. Natarajan, Z. H. Lin, I. K. Tsai, D. Janmanchi, S. I. Chan and S. S. Yu, J. Am. Chem. Soc., 2022, 144, 9695–9706 CrossRef CAS PubMed.
- A. Prajapati, B. A. Collins, J. D. Goodpaster and M. R. Singh, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2023233118 CrossRef CAS PubMed.
- Y. Kang, Z. Li, X. Lv, W. Song, Y. Wei, X. Zhang, J. Liu and Z. Zhao, J. Catal., 2021, 393, 20–29 CrossRef CAS.
- J. Nandenha, I. H. F. Nagahama, J. Y. Yamashita, E. H. Fontes, J. M. S. Ayoub, R. F. B. de Souza, F. C. Fonseca and A. O. Neto, Int. J. Electrochem. Sci., 2019, 14, 10819–10834 CrossRef CAS.
- J. Lee, J. Yang and J. H. Moon, ACS Energy Lett., 2021, 6, 893–899 CrossRef CAS.
- A. Li, H. Qiu, Z. Wang, Y. Sun, Y. Tang, P. Wan, H. Jiang and Y. Chen, ACS Sustainable Chem. Eng., 2024, 12, 9558–9567 CrossRef CAS.
- C. Kim, H. Min, J. Kim, J. Sul, J. Yang and J. H. Moon, Appl. Catal., B, 2023, 323, 122129 CrossRef CAS.
- C. Kim, H. Min, J. Kim and J. H. Moon, Energy Environ. Sci., 2023, 16, 3158–3165 RSC.
- S. Tao, X. Wang, C. Rao, K. Liu, Y. Li, Z. Yuan, G. Peng, Y. Lou, J. Ye, H. Liu, Z. Zhang, X. Yang, Y. Zhang and S. Song, Adv. Funct. Mater., 2025, 36, e19202 CrossRef.
- M. Shahid, C. He, S. Sankarasubramanian, V. K. Ramani and S. Basu, ACS Appl. Mater. Interfaces, 2020, 12, 32578–32590 CrossRef CAS PubMed.
- B. Yu, L. Cheng, J. Wu, B. Yang, H. Li, J. Xu, Y. Zhang, C. Pan, X.-M. Cao, Y. Zhu and Y. Lou, Energy Environ. Sci., 2024, 17, 8127–8139 Search PubMed.
- M. E. O'Reilly, R. S. Kim, S. Oh and Y. Surendranath, ACS Cent. Sci., 2017, 3, 1174–1179 CrossRef PubMed.
- C. Zhu, S. Hou, X. Hu, J. Lu, F. Chen and K. Xie, Nat. Commun., 2019, 10, 1173 CrossRef PubMed.
- R. Chen, F. Xie, Z. Wu, X. Li, C. Deng, Z. Yao and G.-M. Weng, Sci. Bull., 2025, 70, 2629–2640 CrossRef CAS PubMed.
- J. Deng, S. C. Lin, J. Fuller III, J. A. Iniguez, D. Xiang, D. Yang, G. Chan, H. M. Chen, A. N. Alexandrova and C. Liu, Nat. Commun., 2020, 11, 3686 Search PubMed.
- D. Xiang, J. A. Iniguez, J. Deng, X. Guan, A. Martinez and C. Liu, Angew. Chem., Int. Ed., 2021, 60, 18152–18161 Search PubMed.
- R. S. Kim and Y. Surendranath, ACS Cent. Sci., 2019, 5, 1179–1186 CrossRef CAS PubMed.
- M. Y. Sinev, Y. P. Tulenin, O. V. Kalashnikova, V. Y. Bychkov and V. N. Korchak, Catal. Today, 1996, 32, 157–162 CrossRef CAS.
- W. Li, J. Sun, M. Wang, J. Xu, Y. Wang, L. Yang, R. Yan, H. He, S. Wang, W. Q. Deng, Z. Q. Tian and F. R. Fan, Angew. Chem., Int. Ed., 2024, 63, e202403114 CrossRef CAS PubMed.
- H. Qiu, A. Li, Z. Wang, Q. Shangguan, Y. Sun, Y. Tang, P. Wan, H. Jiang and Y. Chen, J. Colloid Interface Sci., 2025, 684, 449–456 CrossRef CAS PubMed.
- T. Jia, W. Wang, C. Zhang, L. Zhang and W. Wang, Angew. Chem., Int. Ed., 2024, 64, e202413343 CrossRef PubMed.
- L. K. Dhandole, S. H. Kim and G.-h. Moon, J. Mater. Chem. A, 2022, 10, 19107–19128 RSC.
- H. Tian, J.-T. Yang, X. Wang, H. Jiao, Z.-F. Gao, K.-Y. Zhu, Q. He and Z.-L. Wang, Appl. Catal., B, 2025, 375, 125411 Search PubMed.
- W. Shen, C. Yao, B. Yu, S. Ge, X. Tang, J. Xu, Y. Zhang, C. Pan, Y. Zhu and Y. Lou, Chem. Eng. J., 2026, 528, 14321–14329 Search PubMed.
- A. Prajapati, R. Sartape, N. C. Kani, J. A. Gauthier and M. R. Singh, ACS Catal., 2022, 12, 14321–14329 Search PubMed.
- K. Shen, S. Kumari, Y.-C. Huang, J. Jang, P. Sautet and C. G. Morales-Guio, J. Am. Chem. Soc., 2023, 145, 6927–6943 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |