
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAcridinium amidate as a hydrogen-bonding photocatalyst for direct decarboxylative alkylation of native carboxylic acids
Duc An
Truong†
a,
Soichiro
Mori†
a,
Taiki
Ninomiya
b,
Ryoya
Niwa
a,
Bumpei
Maeda
 b,
Haruka
Fujino
c,
Masayuki
Inoue
c,
Kohsuke
Ohmatsu
*b and
Takashi
Ooi
*a
b,
Haruka
Fujino
c,
Masayuki
Inoue
c,
Kohsuke
Ohmatsu
*b and
Takashi
Ooi
*a
aInstitute of Transformative Bio-Molecules (WPI-ITbM), and Department of Molecular and Macromolecular Chemistry, Graduate School of Engineering, Nagoya University, Nagoya, 464-8601, Japan
bDepartment of Chemistry, Faculty of Science and Technology, Keio University, Yokohama 223-8522, Japan
cGraduate School of Pharmaceutical Sciences, The University of Tokyo, Tokyo 113-0033, Japan
First published on 21st April 2026
Abstract
The direct generation of carbon radicals from carboxylic acids under visible-light photocatalysis offers an appealing strategy for molecular diversification. Here, we report a decarboxylative Giese-type addition using a zwitterionic acridinium amidate as a uniquely effective photocatalyst, which enables the efficient and selective activation and functionalization of a broad range of carboxylic acids without an external base or oxidant. The catalytic process relies on the formation of a hydrogen-bonded complex between the carboxylic acid and amidate, which triggers proton-coupled electron transfer upon photoexcitation to generate the corresponding carboxyl radical. This method featuring neutral conditions demonstrates excellent functional group tolerance and is applicable to structurally demanding substrates, including complex natural products and pharmaceutically relevant compounds.
Introduction
Carboxy groups are fundamental functional groups that are prevalent in a range of organic molecules, including biomolecules, natural products, and pharmaceuticals. Because of its large dipole moment and ability to act as a hydrogen-bond donor and acceptor, the carboxy group plays a cardinal role in biochemical processes as well as in drug design as a constituent of pharmacophores.1 Given that replacement of this functional group with other fragments can profoundly alter the physicochemical properties, it offers an effective strategy for modulating molecular behaviour for specific purposes, such as understanding biological mechanisms, upgrading natural products, and improving the efficacy of drug candidates. Accordingly, direct decarboxylative functionalization has garnered particular attention as an ideal means of fulfilling the synthetic demand in this pursuit.2,3Photocatalysis has emerged as an enabling platform for decarboxylative transformations under mild conditions through the generation of carbon radicals from carboxylic acids. The methodologies developed for this purpose rely on one of the three distinct modes of activation: single-electron transfer (SET),4–15 ligand-to-metal charge transfer (LMCT),16–20 and proton-coupled electron transfer (PCET) (Fig. 1A).21–41 The SET-based approach typically involves oxidation of carboxylate ions, but the relatively high oxidation potential of these species (ca. +1.3 V vs. SCE) often results in poor chemoselectivity, especially in the presence of electron-rich functional groups. While LMCT allows for the direct generation of carboxyl radicals via the homolytic cleavage of photoexcited metal carboxylates, it generally requires a stoichiometric amount of base and is sensitive to polar or Lewis basic functionalities, which compromise both the efficiency and substrate scope of the reaction. In this regard, methods exploiting PCET obviate the use of an external base, and recent development of azaanthraquinone catalysts significantly expanded the scope of this approach.42 In parallel with this progress, the effectiveness of photoinduced hydrogen atom transfer (HAT) catalysis with ketone derivatives has been explored for the direct radical generation from carboxylic acids.43 Despite these significant advances in catalytic activation of carboxy groups, efficient and chemoselective decarboxylative functionalization in the presence of multiple polar and reactive functional groups, which are common features of natural products and therapeutic agents, remains a considerable challenge. Therefore, further development of photocatalysts that operate with high reactivity and chemoselectivity in such demanding settings is a highly desirable goal.
 | ||
| Fig. 1 Background and summary. (A) Representative strategy for photocatalytic activation of carboxylic acids. (B) Acridinium amidate 1 as a hydrogen-bonding photocatalyst. | ||
Recently, we introduced zwitterionic acridinium amidate 1a as a novel organic molecular photocatalyst capable of facilitating HAT for the direct functionalization of structurally unbiased aliphatic C–H bonds (Fig. 1B).44 The salient feature of this catalyst is that attractive noncovalent interaction between the pertinent hydrogen-bond donor and the amidate moiety is crucial for exerting productive photocatalytic reactivity. Considering the basicity of 1a (pKa of 1a·H = 17.0 in acetonitrile (MeCN)), we reasoned that carboxylic acids could serve as hydrogen-bond donors, which would not only ensure the precise recognition of the carboxy group but also set a reactivity basis for the generation of the corresponding carbon radicals via PCET with 1 upon photoexcitation followed by decarboxylation, thereby allowing precisely chemoselective transformation. Herein, we report the decarboxylative Giese-type addition of carboxylic acids to electron-deficient olefins in the presence of acridinium amidate 1 as the sole catalyst under visible-light irradiation. This protocol enables the direct decarboxylative alkylation of a broad range of carboxylic acids, including those with polar and/or redox-sensitive functional groups, using an equimolar amount of olefins with high efficiency, demonstrating the vast synthetic utility of hydrogen-bond-induced photocatalysis of 1 as a practical tool for unlocking the long-standing limitations in carboxy group activation.
Results and discussion
To assess the feasibility of carboxy group-targeted photocatalytic functionalization by harnessing the characteristic attributes of 1, we chose gibberellic acid A3 (2a), an important plant hormone with an ent-gibberellane skeleton decorated with a carboxy group, two hydroxy groups, internal and terminal alkenes, and a lactone as a model substrate. With N-phthaloyl dehydroalanine ester 3a as the radical acceptor, our investigation was initiated by subjecting a mixture of 2a, 3a, and N-benzoyl acridinium amidate 1a (3 mol%) in 1,2-dichloroethane (DCE) to blue-light irradiation for 6 h, affording the desired product 4a in 13% nuclear magnetic resonance (NMR) yield as a mixture of diastereomers (Table 1, entry 1). This low efficiency can be ascribed to the poor solubility of 2a, which is rich in polar functional groups, in DCE. In fact, changing the solvent to MeCN significantly improved the product yield (entry 2), and the use of an MeCN/H2O (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) co-solvent system, which completely dissolved 2a, led to the formation of 4a in 62% yield (entry 3). Additional screening of polar solvents revealed that acetone was optimal for eliciting the full performance of 1a in this transformation (entries 4–6). We then examined the effect of the catalyst structure, specifically the substituent of the amidate carbonyl, on the reactivity profile, as its steric and electronic properties would affect the hydrogen-bonding ability of the amidate moiety (entries 7–9). While introduction of a substituent derived from an α-amino acid, such as L-valine (1c) or L-tert-leucine (1d), was not beneficial, pivaloyl-substituted 1b delivered a slight improvement in the reaction efficiency, giving rise to 4a in 72% isolated yield (diastereomeric ratio = 7:3.5:1:1) (entry 7). Control experiments under the influence of TiO2 or 9-(2-chlorophenyl)acridine (5a) with violet-light irradiation resulted in markedly diminished yields (entries 10–13). To further clarify the relationship between acridinium amidates and conventional acridine photocatalysts, we evaluated the performance of 3,6-di-tert-butyl-9-mesitylacridine (5b). Under 390 nm light irradiation, the reaction with 10 mol% of 5b afforded 4a in 67% yield, demonstrating the ability of the acridine-type catalysts to promote decarboxylative alkylation of 2a under high-energy shorter-wavelength light irradiation (entries 14 and 15). UV-vis absorption spectroscopic analysis showed that 1b exhibits a pronounced absorption band centered at 420 nm, extending well into the visible-light region, whereas 5b displays negligible absorption above 400 nm, even in the presence of carboxylic acid (Fig. S4). Consistent with these absorption profiles, comparison of the catalytic activity of 5b with that of 1b in the reaction of 2a with 3a under carefully controlled blue-light irradiation conditions revealed that the product formation was not detected with 5b as the catalyst, while 1b facilitated the reaction in a time-dependent manner (Fig. S3). These results demonstrate that acridinium amidates operate in a distinct photophysical and catalytic regime, enabling efficient PCET-driven decarboxylation under longer-wavelength visible-light irradiation.
:1) co-solvent system, which completely dissolved 2a, led to the formation of 4a in 62% yield (entry 3). Additional screening of polar solvents revealed that acetone was optimal for eliciting the full performance of 1a in this transformation (entries 4–6). We then examined the effect of the catalyst structure, specifically the substituent of the amidate carbonyl, on the reactivity profile, as its steric and electronic properties would affect the hydrogen-bonding ability of the amidate moiety (entries 7–9). While introduction of a substituent derived from an α-amino acid, such as L-valine (1c) or L-tert-leucine (1d), was not beneficial, pivaloyl-substituted 1b delivered a slight improvement in the reaction efficiency, giving rise to 4a in 72% isolated yield (diastereomeric ratio = 7:3.5:1:1) (entry 7). Control experiments under the influence of TiO2 or 9-(2-chlorophenyl)acridine (5a) with violet-light irradiation resulted in markedly diminished yields (entries 10–13). To further clarify the relationship between acridinium amidates and conventional acridine photocatalysts, we evaluated the performance of 3,6-di-tert-butyl-9-mesitylacridine (5b). Under 390 nm light irradiation, the reaction with 10 mol% of 5b afforded 4a in 67% yield, demonstrating the ability of the acridine-type catalysts to promote decarboxylative alkylation of 2a under high-energy shorter-wavelength light irradiation (entries 14 and 15). UV-vis absorption spectroscopic analysis showed that 1b exhibits a pronounced absorption band centered at 420 nm, extending well into the visible-light region, whereas 5b displays negligible absorption above 400 nm, even in the presence of carboxylic acid (Fig. S4). Consistent with these absorption profiles, comparison of the catalytic activity of 5b with that of 1b in the reaction of 2a with 3a under carefully controlled blue-light irradiation conditions revealed that the product formation was not detected with 5b as the catalyst, while 1b facilitated the reaction in a time-dependent manner (Fig. S3). These results demonstrate that acridinium amidates operate in a distinct photophysical and catalytic regime, enabling efficient PCET-driven decarboxylation under longer-wavelength visible-light irradiation.
|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent | Yieldb (%) |
| a Unless otherwise noted, the reactions were conducted with 0.10 mmol of 2a and 0.11 mmol of 3a in the presence of catalyst (3 mol%) in solvent (0.2 mL) for 6 h under an argon atmosphere with 450 nm light-emitting diode (LED) irradiation and a fan to maintain the temperature. b NMR yield using trimethoxybenzene as an internal standard. Values in parentheses are isolated yields. c 0.20 mmol scale. d With 10 mg of anatase TiO2 instead of 1 with 390 nm LED irradiation. e With 10 mol% of 5a/b instead of 1 with 390 nm LED irradiation. f With 3 mol% of 5b instead of 1 with 390 nm LED irradiation. Abbreviations: DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; DCM, dichloromethane. | |||
| 1 | 1a | DCE | 13 |
| 2 | 1a | MeCN | 52 |
| 3 | 1a | MeCN/H2O (4:1) |
62 |
| 4 | 1a | DMF | 49 |
| 5 | 1a | DMSO | 28 |
| 6 | 1a | Acetone | 65 |
| 7c | 1b | Acetone | 77 (72) |
| 8 | 1c | Acetone | 52 |
| 9 | 1d | Acetone | 62 |
| 10d | TiO2 | Acetone | Trace |
| 11d | TiO2 | MeCN | Trace |
| 12e | 5a (10 mol%) | Acetone | 12 |
| 13e | 5a (10 mol%) | DCM | 8 |
| 14e | 5b (10 mol%) | Acetone | 67 |
| 15f | 5b (3 mol%) | Acetone | 40 |
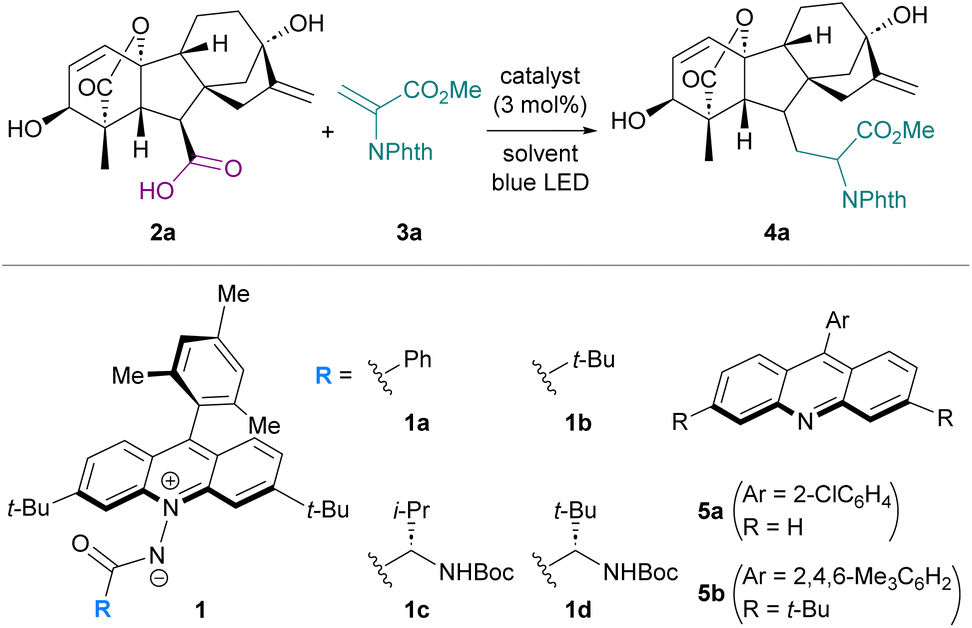
Next, we explored the general applicability of this photocatalytic protocol for the decarboxylative alkylation of various carboxylic acids bearing diverse functional groups using benzalmalononitrile (3b) as the radical acceptor (Fig. 2). When simple aliphatic carboxylic acids were employed, the Giese-type addition proceeded efficiently with 1 mol% of 1a as a catalyst in MeCN to give the corresponding alkylated products 4 in high yields, irrespective of the substitution pattern at the α-position (see the SI for optimization details). Notably, even with a redox-active indole component in the side chain, whose oxidation potential is lower than that of the carboxylate, N-Boc-tryptophan underwent smooth transformation to afford 4b in nearly quantitative yield. Furthermore, decreasing the loading of 1a to 0.1 mol% still provided 4b in good yield (Table S1).45 Likewise, a carboxylic acid with a more electron-rich 5-hydroxyindole unit appeared to be a viable substrate, furnishing 4c in good yield. In addition, substrates with electron-rich aromatic substituents, such as poly-methoxylated benzenes and methoxyphenols, were well tolerated, leading to the formation of 4d–4g without any observable inhibition. The broad functional-group compatibility of this method was also demonstrated in the reactions with carboxylic acids incorporating sulfide, hydroxy, bromo, and carbamate functionalities, which produced 4h–4k in uniformly good yields.
 | ||
| Fig. 2 Decarboxylative Giese-type addition of functionalized carboxylic acids to benzalmalononitrile 3b. Isolated yields are shown. Diastereomeric ratio (d.r.) was determined by 1H NMR analysis of the crude reaction mixture. aUsing 3 mol% of 1a. bIsolated yield of the corresponding cyclized product 4i′ is shown. See the SI for details. | ||
Moreover, this catalytic system was reliably applicable to more complex substrate settings by adjusting the reaction conditions, as exemplified by the selective functionalization of cholic acid, a bile acid featuring a fused polycyclic framework with multiple hydroxy groups (Fig. 3). In the presence of 1d (3 mol%) as a catalyst, decarboxylative radical addition to 3b occurred in MeCN/DMF (1:1), affording the desired product 4l. As radical acceptors for this decarboxylative alkylation, not only olefins with two electron-withdrawing groups but also simple vinyl ketones, α-phenyl acrylate, and dehydroalanine derivatives were all employable, and the corresponding alkylated products 4m–4q were obtained in moderate to good yields. In contrast, the reactions with less activated acceptors, such as simple acylates and acrylamides, gave only trace amounts of the desired products under the standard conditions (see SI, Section “Unsuccessful examples of radical acceptors”). The possibility of the chemoselective transformation of functionalized carboxylic acids was also showcased by the effective conversion of valsartan into 4r. Base-labile 2-cyano-2-cyclohexenone was applicable to the coupling reaction to yield 4s, accentuating the neutral nature of the present conditions.20 The versatility of this protocol was further demonstrated by its application to complex molecules bearing diverse reactive functionalities. Specifically, substrates containing acetal, cyclopropane, alkene, and alkyne groups were irradiated with 1a to realize the intermolecular formation of the sterically hindered linkages of 4t and 4u, which were previously utilized as pivotal intermediates for the total synthesis of taxol and euphorbialoid A.46,47
 | ||
| Fig. 3 Decarboxylative Giese-type addition of complex carboxylic acids. Isolated yields are shown. aDiastereomeric ratios (d.r.) were determined by 1H NMR analysis of the crude reaction mixture. bIsolated yields after the acetylation of enol moiety are shown; d.r. values were determined after acetylation and isolation. cObtained as a 1:4.7:1.4 mixture of the enol and diastereomeric keto nitriles. | ||
With the broad applicability in mind, our investigation focused on elucidating the activation mechanism of the carboxy group in this photocatalytic transformation. The excited-state reduction potential of acridinium amidate 1a was estimated to be +1.63 V vs. SCE,44 which exceeds the oxidation potential of typical carboxylate ions (ca. +1.3 V vs. SCE). Nevertheless, when the reaction was conducted in the presence of hydroxide ions to ensure the in situ generation of carboxylate ions, little to no product formation was observed (Fig. 4A). This outcome indicates that direct oxidation of free carboxylate ions is unlikely to be a dominant pathway under the standard reaction conditions. 1H NMR titration study in CD3CN revealed a significant association between 1a and the carboxylic acid, consistent with the formation of a ground-state complex under catalytically relevant conditions (Fig. S7). Since the NMR data alone is insufficient to distinguish between a hydrogen-bonded assembly (1/RCO2H) and an ion-pairing interaction (1·H+/RCO2−), cyclic voltammetry (CV) measurements were performed to clarify the nature of this interaction (Fig. 4B). The CV of a solution containing 1a and butyric acid showed the reduction wave corresponding to 1a without additional signals attributable to the protonated form of the acridinium amidate, amide acridinium 1a·H+/BF4− (Fig. 4C). This result suggests that 1 and carboxylic acid predominantly form a hydrogen-bonded assembly rather than an ion pair.
 | ||
| Fig. 4 Mechanistic investigations. (A) Control experiments with Brønsted base additives. (B) Structures of hydrogen-bonded complex and amide-acridinium/carboxylate ion pair. (C) Cyclic voltammetry of 1a (blue line), the mixture of 1a and HBF4 (1a·H+/BF4−, red line), and the mixture of 1a and butyric acid (1a/n-PrCO2H, green line). | ||
Based on these experimental findings, a plausible mech-anism for the acridinium amidate 1-catalyzed decarboxylative Giese-type addition is illustrated in Fig. 5. Initially, 1 engages in a hydrogen-bonding interaction with carboxylic acid to form ground-state complex A. Upon blue-light excitation, A undergoes PCET to generate carboxyl radical B, accompanied by the formation of 1-derived acridinyl radical C. Decarboxylation of the resultant B readily occurs to generate carbon radical D, which adds to the electron-deficient olefin to afford radical intermediate E. Subsequent SET between E and C, followed by protonation, gives the desired Giese-type adduct and concurrently regenerates ground-state catalyst 1.
 | ||
| Fig. 5 Proposed catalytic cycle. | ||
Conclusions
In summary, we developed a chemoselective decarboxylative Giese-type addition by exploiting the unique behaviour of a zwitterionic acridinium amidate as a photocatalyst. This transformation proceeds with remarkable efficiency under essentially neutral, visible-light-driven conditions and exhibits a broad substrate scope, tolerating a wide range of polar and redox-sensitive functional groups. The advantage of this distinct feature is underscored by its application to the direct remodelling of natural product scaffolds and reactions of multiply oxygenated carboxylic acids with highly electron-deficient olefins, which constitute relevant bond-forming processes in the total synthesis of natural products. Mechanistic investigations indicated that the key to this prominent selectivity lies in the formation of a hydrogen-bonded complex between the catalyst and carboxylic acid, which triggers PCET to generate the requisite carboxyl radical without requiring pre-activation or external additives. This study demonstrates the immense potential of rationally designed zwitterionic photocatalysts for expanding the versatility of decarboxylative radical transformations, particularly for addressing the reactivity and selectivity challenges in high-complexity structural settings.Author contributions
The manuscript was prepared through the contributions of all authors. All the authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting the findings of this study are available within the supplementary information (SI). Supplementary information: the exploratory investigation, experimental procedures, and characterization data. See DOI: https://doi.org/10.1039/d6sc00657d.Acknowledgements
This work was financially supported by JSPS KAKENHI Grants JP23H04901, JP23H04907, JP24H01838 (Green Catalysis Science), JP25H02059 (Integrated Science of Synthesis by Chemical Structure Reprogramming (SReP)), JP23H00296, JP22K21346, and JP24K01481, and JST FOREST Grant JPMJFR221L.Notes and references
- C. Ballatore, D. M. Huryn and A. B. Smith III, ChemMedChem, 2013, 8, 385–395 CrossRef CAS PubMed.
- A. Hall, M. Chatzopoulou and J. Frost, Bioorg. Med. Chem., 2024, 104, 117653 CrossRef CAS PubMed.
- N. A. Nguyen, J. H. Forstater and J. A. McIntosh, JACS Au, 2024, 4, 2715–2745 CrossRef CAS PubMed.
- L. Chu, C. Ohta, Z. Zuo and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 10886–10889 CrossRef CAS PubMed.
- A. Noble, R. S. Mega, D. Pflästerer, E. L. Myers and V. K. Aggarwal, Angew. Chem., Int. Ed., 2018, 57, 2155–2159 CrossRef CAS PubMed.
- K. C. Cartwright and J. A. Tunge, ACS Catal., 2018, 8, 11801–11806 CrossRef CAS.
- K. C. Cartwright, S. B. Lang and J. A. Tunge, J. Org. Chem., 2019, 84, 2933–2940 CrossRef CAS PubMed.
- F. El-Hage, C. Schöll and J. Pospech, J. Org. Chem., 2020, 85, 13853–13867 CrossRef CAS PubMed.
- M. Shao, H. Liang, Y.-L. Liu, W. Qin and Z. Li, Asian J. Org. Chem., 2020, 9, 782–787 CrossRef CAS.
- Q. Wei, Y. Lee, W. Liang, X. Chen, B.-S. Mu, X.-Y. Cui, W. Wu, S. Bai and Z. Liu, Nat. Commun., 2022, 13, 7112 CrossRef CAS PubMed.
- T. S. Mayer, T. Taeufer, S. Brandt, J. Rabeah and J. Pospech, J. Org. Chem., 2023, 88, 6347–6353 CrossRef CAS PubMed.
- D. M. Kitcatt, K. A. Scott, E. Rongione, S. Nicolle and A.-L. Lee, Chem. Sci., 2023, 14, 9806–9813 RSC.
- C.-H. Hu, Y. Sang, Y.-W. Yang, W.-W. Li, H.-L. Wang, Z. Zhang, C. Ye, L.-Z. Wu, X.-S. Xue and Y. Li, Chem, 2023, 9, 2997–3012 CAS.
- D. M. Kitcatt, S. Nicolle and A.-L. Lee, Chem. Soc. Rev., 2022, 51, 1415–1453 RSC.
- S. Mondal, S. Mandal, S. Mondal, S. P. Midya and P. Ghosh, Chem. Commun., 2024, 60, 9645–9658 RSC.
- Q. Y. Li, S. N. Gockel, G. A. Lutovsky, K. S. DeGlopper, N. J. Baldwin, M. W. Bundesmann, J. W. Tucker, S. W. Bagley and T. P. Yoon, Nat. Chem., 2022, 14, 94–99 CrossRef CAS PubMed.
- S.-C. Kao, K.-J. Bian, X.-W. Chen, Y. Chen, A. A. Martí and J. G. West, Chem Catal., 2023, 3, 100603 CAS.
- S. Tamaki, R. Kuwata, S. Wakita, Y. Osakada, M. Fujitsuka, T. Kusamoto, K. Mashima and H. Tsurugi, Angew. Chem., Int. Ed., 2025, 64, e202505639 CrossRef CAS PubMed.
- Q. Zhu and D. G. Nocera, J. Am. Chem. Soc., 2020, 142, 17913–17918 CrossRef CAS PubMed.
- D. Kuwana, Y. Komori, M. Nagatomo and M. Inoue, J. Org. Chem., 2022, 87, 730–736 CrossRef CAS PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, H. T. Dang, S. Jin, Z. Li, C. Flores-Hansen, B. S. Benavides, H. D. Arman and O. V. Larionov, ACS Catal., 2019, 9, 9485–9498 CrossRef CAS PubMed.
- V. T. Nguyen, V. D. Nguyen, G. C. Haug, N. T. H. Vuong, H. T. Dang, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2020, 59, 7921–7927 CrossRef CAS PubMed.
- H. T. Dang, G. C. Haug, V. T. Nguyen, N. T. H. Vuong, V. D. Nguyen, H. D. Arman and O. V. Larionov, ACS Catal., 2020, 10, 11448–11457 CrossRef CAS PubMed.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, H. D. Arman and O. V. Larionov, Chem. Sci., 2021, 12, 6429–6436 RSC.
- I. A. Dmitriev, V. V. Levin and A. D. Dilman, Org. Lett., 2021, 23, 8973–8977 CrossRef CAS PubMed.
- V. D. Nguyen, G. C. Haug, S. G. Greco, R. Trevino, G. B. Karki, H. D. Arman and O. V. Larionov, Angew. Chem., Int. Ed., 2022, 61, e202210525 CrossRef CAS PubMed.
- K. A. Zhilyaev, D. L. Lipilin, M. D. Kosobokov, A. I. Samigullina and A. D. Dilman, Adv. Synth. Catal., 2022, 364, 3295–3301 CrossRef CAS.
- A. Adili, A. B. Korpusik, D. Seidel and B. S. Sumerlin, Angew. Chem., Int. Ed., 2022, 61, e202209085 CrossRef CAS PubMed.
- J.-T. Yi, X. Zhou, Q.-L. Chen, Z.-D. Chen, G. Lu and J. Weng, Chem. Commun., 2022, 58, 9409–9412 RSC.
- V. T. Nguyen, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, G. B. Karki, H. D. Arman and O. V. Larionov, Chem. Sci., 2022, 13, 4170–4179 RSC.
- Q. Chen, Y. Wang and G. Luo, Chem. Eng. J., 2023, 461, 141767 CrossRef CAS.
- J. A. Andrews, J. Kalepu, C. F. Palmer, D. L. Poole, K. E. Christensen and M. C. Willis, J. Am. Chem. Soc., 2023, 145, 21623–21629 CrossRef CAS PubMed.
- J. G. L. de Araujo, M. d. S. B. da Silva, J. C. C. V. Bento, A. M. de Azevêdo, A. M. de M. Araújo, A. S. D. dos Anjos, C. A. Martínez-Huitle, E. V. dos Santos, A. D. Gondim and L. N. Cavalcanti, Chem.–Eur. J., 2023, 29, e202302330 CrossRef CAS PubMed.
- H. T. Dang, G. C. Haug, V. D. Nguyen, N. T. H. Vuong, H. D. Arman and O. V. Larionov, JACS Au, 2023, 3, 813–822 CrossRef CAS PubMed.
- H. T. Dang, A. Porey, S. Nand, R. Trevino, P. Manning-Lorino, W. B. Hughes, S. O. Fremin, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2023, 14, 13384–13391 RSC.
- J. A. Andrews, R. G. Woodger, C. F. Palmer, D. L. Poole and M. C. Willis, Angew. Chem., Int. Ed., 2024, 63, e202407970 CrossRef CAS PubMed.
- K. Bhatt, A. Adili, A. H. Tran, K. M. Elmallah, I. Ghiviriga and D. Seidel, J. Am. Chem. Soc., 2024, 146, 26331–26339 CrossRef CAS PubMed.
- X. Sui, H. T. Dang, A. Porey, R. Trevino, A. Das, S. O. Fremin, W. B. Hughes, W. T. Thompson, S. K. Dhakal, H. D. Arman and O. V. Larionov, Chem. Sci., 2024, 15, 9582–9590 RSC.
- D. L. Lipilin, M. O. Zubkov, M. D. Kosobokov and A. D. Dilman, Chem. Sci., 2024, 15, 644–650 RSC.
- E. Wheatley, H. Melnychenko and M. Silvi, J. Am. Chem. Soc., 2024, 146, 34285–34291 CrossRef CAS PubMed.
- Z. M. Rubanov, V. V. Levin and A. D. Dilman, Org. Chem. Front., 2025, 12, 2255–2259 RSC.
- T. Inoue, D. Tomiya, M. Fuki, Y. Kobori, M. Higashi, K. Uesaka, A. Yamakata, S. A. Kawashima, K. Yamatsugu, H. Mitsunuma and M. Kanai, J. Am. Chem. Soc., 2025, 147, 40272–40281 CrossRef CAS PubMed.
- K. Yamashita, H. Sano, Y. Goto, H. Hayashi and Y. Hamashima, J. Am. Chem. Soc., 2025, 147, 29711–29721 CrossRef CAS PubMed.
- L.-M. Entgelmeier, S. Mori, S. Sendo, R. Yamaguchi, R. Suzuki, T. Yanai, O. García Mancheño, K. Ohmatsu and T. Ooi, Angew. Chem., Int. Ed., 2024, 63, e202404890 CAS.
- Attempted reaction by employing 1 mol% of acridine-derived 5b under 390 nm light irradiation led to a substantial decrease in reaction efficiency. See the SI for details (Table S1)..
- Y. Imamura, K. Takaoka, Y. Komori, M. Nagatomo and M. Inoue, Angew. Chem., Int. Ed., 2023, 62, e202219114 CrossRef CAS PubMed.
- J. Taguchi, S. Fukaya, H. Fujino and M. Inoue, J. Am. Chem. Soc., 2024, 146, 34221–34230 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |