
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDefect engineering within clusters to enhance cluster–support interaction boosts catalytic performance
Jiankang Chen†
a,
Yangping Wang†a,
Yu Zhang†a,
Huan Yanb,
Qinzhen Li a,
Jinsong Chaia,
Guiqi Gao*a,
Chunyan Liu*b,
Sha Yang*a and
Manzhou Zhua
a,
Jinsong Chaia,
Guiqi Gao*a,
Chunyan Liu*b,
Sha Yang*a and
Manzhou Zhua
aInstitutes of Physical Science and Information Technology, Department of Chemistry and Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, China. E-mail: gaoguiqi1@126.com; yangshac@ahu.edu.cn
bCollege of Chemistry and Chemical Engineering, Hunan University, Changsha, Hunan 410082, China. E-mail: cyliu@hnu.edu.cn
First published on 19th May 2026
Abstract
Supported cluster catalysts effectively optimize mass transfer and electron transfer efficiency, synergistically boosting catalytic performance. However, the regular morphology of clusters hinders adsorption on the support. Therefore, strengthening the metal–support surface interaction remains a critical challenge requiring urgent solutions. In this study, we constructed the Au40 cluster by modifying the Au44 cluster with phosphine, and the resulting Au40 cluster exhibits features similar to those of the parent Au44 cluster, albeit with three gold atoms missing from its core as defects. Furthermore, we loaded the two clusters onto various support materials to evaluate the interaction strength between the clusters and their supports. The results of experiments as well as DFT calculations showed that the defects enhanced the interaction between the Au40 clusters and their supports. Using the hydrogenation of 4-nitrophenol as a model, the catalytic activity of supported catalysts was investigated. The catalytic activity of the Au40 system was much higher than that of the Au44 system, with a catalytic time ratio of approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4. This work reveals that constructing surface defects on clusters serves as an effective method to enhance the interaction between clusters and supports, providing a novel approach for developing cluster-based heterogeneous catalysts.
:4. This work reveals that constructing surface defects on clusters serves as an effective method to enhance the interaction between clusters and supports, providing a novel approach for developing cluster-based heterogeneous catalysts.
Introduction
The advent of atomically precise metal nanoclusters in catalysis represents a pivotal shift in heterogeneous catalysis, marking the transition from the “nanoscale era” towards an “atomic era”.1,2 This paradigm enables the exquisite regulation of catalytic activity and selectivity through the atom-level engineering of active sites, offering tremendous potential for developing a new generation of industrial catalysts that are highly efficient, selective, and cost-effective.3–5 The integration of these nanoclusters with supports is a critical strategy, serving not only to immobilize the clusters via physicochemical interactions, thereby preventing their migration and sintering and ensuring high dispersion and stability, but also potentially creating novel, highly active sites at the interface.6–10 This can transform the reaction mechanism from operating at a “single active site” to a “composite active centre”.11,12 Consequently, a central research focus is now on precisely designing the cluster-support couple to modulate, or even create, new catalytic centers that are both stable and exhibit performance surpassing the intrinsic activity of the clusters.13–17Given that the composite structure and interaction mechanism between the support and the cluster directly dictate the final catalyst's performance, stability, and activity, their interplay has become a primary research frontier.18–22 Compared to weak physical adsorption via van der Waals or electrostatic forces, the formation of covalent or coordination bonds through strong electronic interactions between the metal atoms of the cluster and defect sites or functional groups on the support surface is generally considered a more desirable composite form.23,24 Current prevalent strategies focus on enhancing this interaction by engineering oxygen vacancies on oxide supports (e.g., CeO2, Fe3O4), generating defects, edges, and step sites via acid treatment or calcination, or incorporating heterometals to create new anchoring sites (Scheme 1).25–27 Employing an “inverse” defect engineering strategy, the deliberate design and synthesis of defect clusters (defined as a cluster obtained by modifying the parent cluster through varying reaction conditions, e.g. altering ligand types or doping heterometal, that retains a structural framework similar to that of the parent yet features localized atomic deficiencies). These atomic vacancies typically result in reduced cluster symmetry but improved catalytic performance.28,29 However, their subsequent hybridization with the support together establishes deeper and stronger interactions that have never been explored.
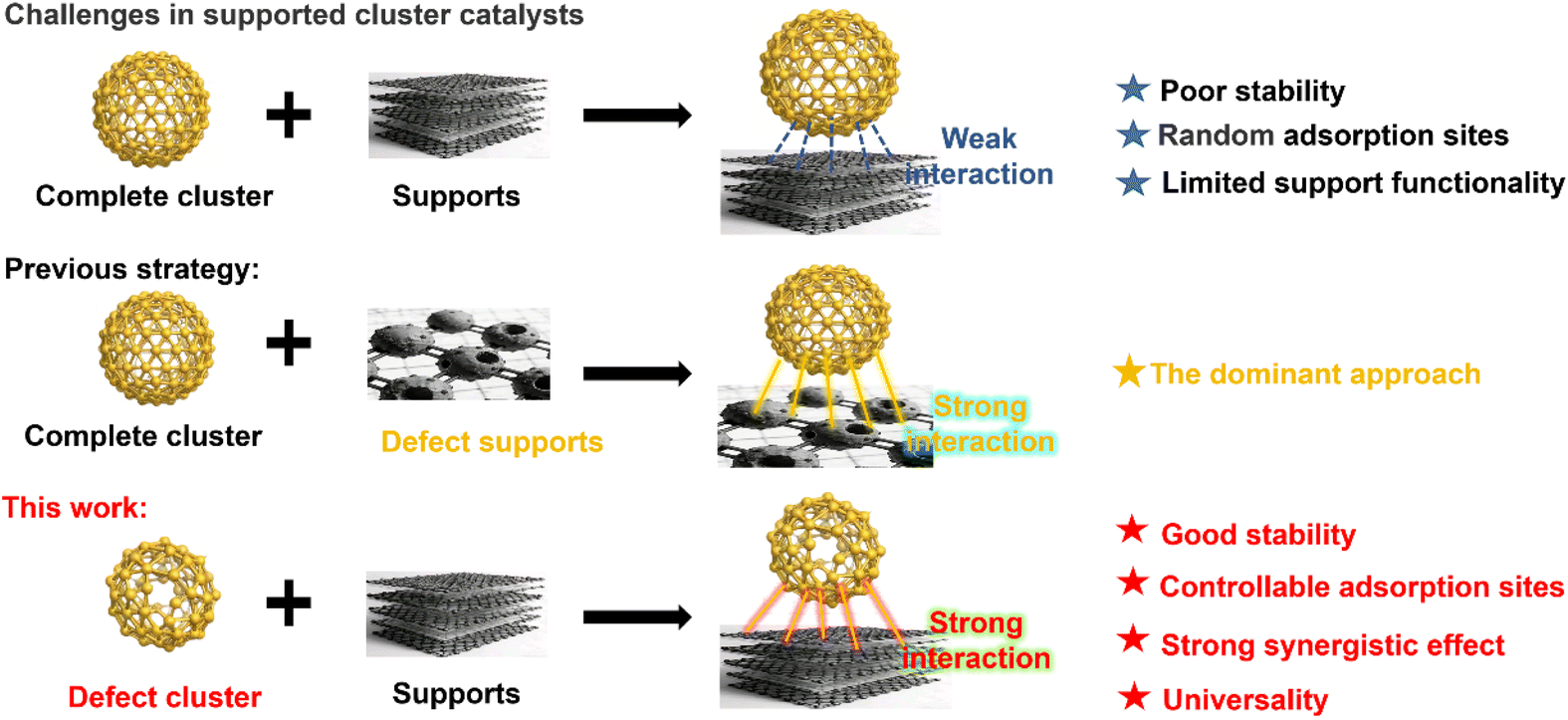 | ||
| Scheme 1 Comparison of the strategies for enhancing the interaction between clusters and supports. | ||
Herein, we constructed a defect-induced Au40(TBBT)24(TPMP) (Au40 for short, TPMP = tri(m-tolyl)phosphine) cluster on the surface of Au44(TBBT)28 (ref. 30) (Au44 for short) cluster through phosphine modification. Structural analysis revealed that both Au40 and Au44 clusters share similar structures, primarily due to the absence of three gold atoms on the core surface, which induces local motif rearrangement and defect formation. Taking these clusters as a research platform, the influence of the defects on the interaction between the cluster and the carrier is investigated. By analyzing changes in Au 4f binding energy via XPS before and after loading gold clusters onto an activated carbon (AC) support, the strength of cluster–support interactions was assessed. Results showed no shift in binding energy for Au44 (ΔE = 0 eV), whereas a 0.3 eV shift occurred for Au40, indicating stronger interactions between Au40 and AC. Density functional theory (DFT) calculations also showed that the interaction between Au40 and the carrier is stronger than that of Au44 systems, by analyzing their adsorption energies and Bader charge. Using the reduction of 4-nitrophenol as a model, we evaluated the catalytic activity of the two catalysts. As expected, the Au40/AC catalyst exhibited higher catalytic activity, reducing the catalytic time to 1/5 of that of the Au44/AC catalyst. We also expanded various carriers, including Graphene (GR), Carbon Nanotubes (CNT), and Silica (SiO2). The results showed that Au40 had a higher binding energy with all carriers than Au44, and its catalytic effect was far superior to that of Au44.
Results and discussion
In this study, Au44 was selected as the parent cluster due to its ease of synthesis, high yield, and particularly its well-defined structure.30 This structural regularity facilitates the construction of surface defects in subsequent cluster formation and enables intuitive observation of defect locations, which is crucial for investigating its effects in heterogeneous catalysis. Based on our preliminary findings, phosphine ligands tend to modify the local structure of cluster surfaces, demonstrating potential to create defects on the cluster surface.31 Therefore, we selected Au44 as the parent compound and employed phosphine ligands to modify its surface, aiming to obtain defect products (Fig. 1a). As shown in Fig. S1, when Au44 clusters were reacted with various monophosphine ligands, the resulting products exhibited distinct UV-visible absorption spectra compared to the parent clusters, indicating that phosphine incorporation induced the transformation of Au44 clusters into novel structures.32 Furthermore, we observed that the UV-visible absorption spectra of Au44 clusters modified with different phosphine ligands were highly similar (Fig. S1), indicating that the R-group has minimal structural influence on the transformation products. | ||
| Fig. 1 (a) Schematic diagram of the cleavage of Au44 surface by phosphorus ligand (atom colors: Au = lavender/turquoise; S = yellow; P = pink); (b) UV-vis spectra of Au44 and Au40; (c) XPS peaks of Au44 and Au40. | ||
Consequently, the product obtained by adding tri(m-tolyl)phosphine (TPMP), which exhibited high yield and excellent crystallinity, was selected for further structural modification studies. The specific preparation process is also illustrated using this example. The conversion process was first observed and investigated through UV-visible absorption spectroscopy and visually observable thin-layer chromatography plates (TLC). Based on time-dependent UV-vis spectroscopy, the conversion of Au44 was confirmed and can be considered rapid and efficient. At the initiation of the reaction, absorption peaks at about 380 nm and 750 nm were observed, attributed to the intrinsic absorption of Au44.29 As the reaction progressed, the characteristic absorption peaks of Au44 gradually weakened, while a new set of characteristic absorption peaks emerged at 410 nm and 600 nm, which gradually became noticeable (Fig. S2). This demonstrates that 44 indeed underwent alteration under the influence of the phosphine ligand. Further, TLC was used for visual monitoring of the transformation kinetics of Au44 clusters.33 As shown in Fig. S3, the TLC plate revealed that Au44 concentration progressively decreased with reaction time, while a new nanocluster concentration gradually increased, ultimately resulting in complete conversion of Au44 clusters. The new TLC band was collected for subsequent testing and characterization.
To characterize the molecular formula of the new cluster, we conducted ESI-MS analysis in cation mode with cesium acetate as the ionization assistant.34 Fig. S4 reveals a strong signal peak at 6207.56 Da in the ESI-MS spectrum. Analysis of the amplified signal region indicates an isotopic nuclear ratio of 0.5 Da (interpolated in Fig. S4), confirming that the cluster carries two charges. This corresponds to the [Au40(TBBT)24TPMP+2Cs]2+ ion (theoretical value: 6207.93 Da), with excellent agreement between experimental and theoretical spectra. Based on the above findings, we propose that the stable product after Au44 cluster modification has the molecular formula Au40 (TBBT)24TPMP. Similar to the Au44 cluster, the modified cluster exhibits electrical neutrality and contains 16 free electrons. As shown in Fig. 1b, the two characteristic absorption peaks of the new cluster are concentrated at 430 and 610 nm. These differ from the peaks of the parent cluster Au44, whose main peak is located at 750 nm and exhibits an absorption peak at 380 nm. This discrepancy also suggests that Au40 exhibits greater structural differences relative to Au44 or structural defects within its core.35 The structural differences also require consideration of their crystallographic variations. Then, we introduced XPS analysis to examine its charge distribution. As shown in Fig. 1c, the Au 4f for Au40 exhibits a negative shift relative to Au44 cluster, indicating that defects cause the valence state of gold to shift toward Au(0). The results of peak splitting also demonstrate an increase in the proportion of Au(0):Au(I) from 0.34 to 0.52. This implies an increase in the number of gold atoms in the outer shell or an increase in the number of unsaturated coordinating atoms around gold atoms. This is beneficial for complexation interactions with carriers.
Given that previous results confirm structural changes in Au40 under the influence of phosphine ligands, verification of the crystal structure is required to determine whether atomic-level defects are present. Therefore, the Au40 clusters were cultured into single crystals via methanol–toluene solution diffusion. The high-quality Au40 single crystals were obtained and subjected to single-crystal X-ray diffraction (SC-XRD) analysis to determine their precise structure. The results revealed that the Au40 crystals belong to the monoclinic P21/c space group (Table S1). Furthermore, we identified Au40 clusters as a racemic mixture within the unit cell (Fig. S5), with one cluster selected for structural analysis. As shown in Fig. 2, the Au40 core is formed by three Au13 units interlocked to create an Au31 core (Fig. 2a and b). This core is further protected by nine bridging thiol ligands (Fig. 2c). Additionally, three AuSR2 ligands and three Au2SR3 ligands are arranged around the Au31 core, enhancing its stability (Fig. 2d and e). Finally, a phosphine ligand is attached to an Au atom in the upper left corner of the Au31 core, forming the Au40SR24P skeletal structure (Fig. 2f). The structural analysis reveals that Au40 clusters exhibit structural similarities to their parent Au44 cluster, as anticipated.
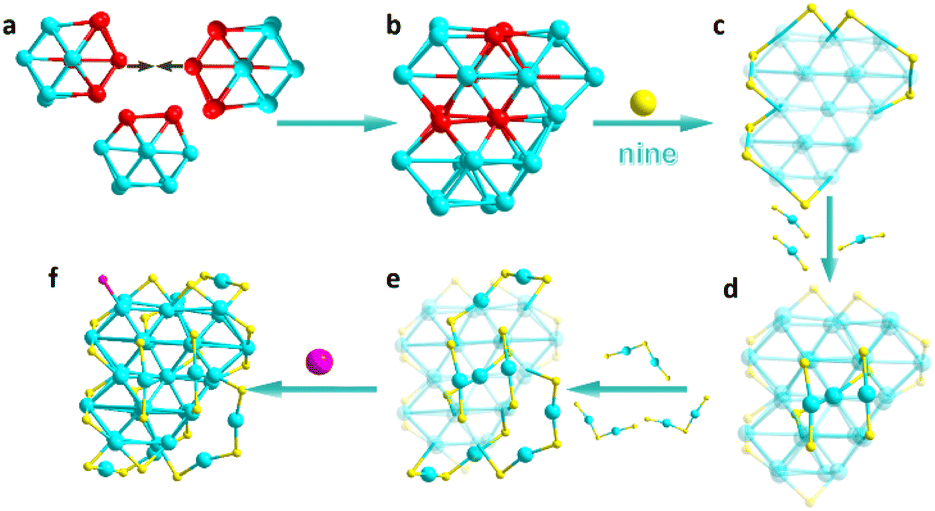 | ||
| Fig. 2 Structural analysis of the Au40 cluster. (a) Three Au13 octahedral units; (b) Au31 core; (c) Au31 core with nine bridging thiol ligands; (d) Au34SR15 unit composed of other AuSR2 ligands; (e) Au40(TBBT)24 formed by other Au2SR3 ligands; (f) Au40(TBBT)24(TPMP) with phosphine ligands (atom colors: Au = red, turquoise; S = yellow; P = pink). | ||
To better demonstrate the structural differences and similarities between Au40 and Au44 clusters, we conducted a step-by-step analysis based on the experimentally determined single-crystal structures (a manual structural comparison). The analysis proceeds from the core to the periphery as follows. First, we compared the core structures. As shown in Fig. 3a, the Au31 core of Au40 exhibits significant similarity with the face-centred cubic (fcc) Au34 core of Au44. By removing a layer of Au3 units from the lower-right corner of the Au34 core, the Au31 core of Au40 is obtained. Additionally, to investigate the effects of phosphine ligands and structural modifications on the peripheral framework, we performed an in-depth analysis of the Au40 surface motif. As shown in Fig. S6, the core changes induce significant alterations in the ligands attached to the core surface. Specifically, the AuSR2 motif at the core center is cleaved due to these structural modifications, generating two AuSR2 motifs that connect to the new Au31 core. The opposing AuSR2 motif undergoes distortion, while the AuSR2 and Au2SR3 motifs linked to the cleaved Au3 unit undergo cleavage and recombination to form a novel Au2SR3 motif, which then attaches to the new nucleus (Fig. 3b and S6). Thus, modifying the Au44 cluster with phosphine ligands enables core trimming, where the motif reconstruction at the trimming site further amplifies the defect.
 | ||
| Fig. 3 Defect structures of the Au40 cluster. (a) Core defect; (b) structural comparison between Au44 and Au40 clusters (atom colors: Au = lavender, turquoise; S = yellow; P = pink). | ||
In current single-atom or nanoparticle heterogeneous catalysis, researchers typically construct defect-stabilized metal atoms on support materials and enhance the interaction between the support and metal to improve catalytic performance.36,37 Having obtained nanoclusters with atomic-level defects and distinct electronic structures, we can now explore the differences in interactions between these cluster defects and the support, as well as their impact on catalytic activity. Therefore, the defective Au40 cluster was loaded onto an activated carbon (AC) support, with the Au44 cluster similarly loaded for comparison. A transmission electron microscope (TEM) was employed to examine the interaction between the clusters and the support. From TEM, the clusters were uniformly loaded onto the support with the size remaining around 1 nanometer (Fig. S7a–d). To directly visualize ligand spatial distribution, high-resolution TEM Mapping on AC-supported clusters revealed colocalized Au, S (thiolate), and P (phosphine) signals (Fig. S8 and S9), confirming dual-ligand preservation post-deposition. UV-vis spectroscopy, serving as an electronic fingerprint for atomically precise gold clusters, detected no structural alterations (e.g., ligand loss, core distortion).38 After loading clusters onto SiO2 to avoid carbon interference (Au40/SiO2, Au44/SiO2, Au36/SiO2), solid-state spectra matched solution profiles (Fig. S10), verifying intact ligand environments and metal cores.
To investigate the relationship between structure and performance, the reaction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) was employed. The reaction was monitored via time-resolved UV-visible absorption spectroscopy, where 4-NP exhibits a distinct absorption peak at 400 nm, while the reduced product, 4-AP, shows a peak at about 295 nm. The results were confirmed by 1H NMR and 13C NMR of the product (Fig. S11). As shown in Fig. 4a and b, the addition of the catalyst caused the absorption peak of NP to gradually weaken, while simultaneously the peak of AP gradually intensified. However, a detailed comparison revealed differences between catalysts Au40/AC and Au44/AC; under catalyst Au40/AC, the reaction time was shorter. Fig. 4c illustrates the plot of the conversion of 4-NP (−Ct) against time; the 4-NP hydrogenation by NaBH4 on Au40/AC or Au44/AC obeys zero-order reaction kinetics, which corresponds with the Eley–Rideal mechanism.39 The calculated apparent rate constant k for Au40/AC is 3.77 × 10−3 M−1 min−1, which is 5 times as much as that of Au44/AC (7.81 × 10−4 M−1 min−1). The calculated TON values for Au40/AC reached 3601, with a TOF of 119 min−1. For Au44/AC, the TON value was 1739, and the TOF was 49 min−1. To evaluate the stability of the defect cluster, we performed solid-state UV-vis spectroscopy on Au40 and Au44 before and after loading on the carbon support (Fig. S10). The absorption features remained essentially unchanged, indicating that both clusters are stable under the loading conditions. Furthermore, catalytic recycling tests (Fig. 4d) showed no significant loss of activity over five cycles for Au40/AC, confirming its good stability in the reaction environment.
 | ||
| Fig. 4 UV-vis spectra of reaction solution under catalysts of (a) Au40/AC and (b) Au44/AC; (c) plot of −Ct versus time for gradual reduction of 4-nitrophenol with Au40 and Au44 clusters; (d) complete degradation time of recycling catalytic experiments catalyzed with Au44 and Au40 clusters; XPS spectra of Au40 (e) and Au44 (f) clusters on activated carbon before and after catalytic reaction; (g) adsorption energy plots of Au44 and Au40; (h) differential charge density map of Au40 clusters; (i) catalytic apparent rate constants (Kapp) of Au44 and Au40 clusters on different supports. | ||
To clarify the impact of interactions between the cluster and the support on its catalytic activity, XPS was again employed to investigate the electronic interactions between the clusters and the support in detail. As shown in Fig. 4e, the Au40 nanocluster exhibits a significant positive shift with the ΔE = 0.3 eV in its Au 4f peak after complexation with the carrier (note: the positive shift in binding energy indicates electron transfer from the cluster to the support, while a negative shift indicates the reverse, and a larger absolute value reflects stronger electronic interaction between the cluster and the support). In contrast, the 4f peak in the parent Au44 nanocluster remains unchanged before and after it is loaded on the AC support (Fig. 4f). Additionally, the binding energy of Au in both the Au40 and Au44 nanoclusters remains unchanged before and after the reaction (Fig. 4e and f). Control experiments with Au36/AC confirm identical XPS profiles before and after loading (Fig. S12), while catalytic performance (Fig. S13) and 5-cycle retention (Fig. S14) further exclude size effects. These results demonstrate that the binding energy shift originates specifically from Au40–AC interactions, enhancing interfacial electron transfer.
Furthermore, we employed DFT calculations to analyze the interactions between the cluster and the support. As illustrated in Fig. S15–S17 and Table S2, the cluster maintains its structural integrity on the support, and there exist interactive forces between them. As shown in Fig. 4g, the adsorption energies (defined as Eads = Etotal − Ecluster − Esubstrate, where a more negative value indicates a stronger and more stable adsorption) of Au40 and Au44 are −0.526 eV and −0.427 eV, respectively. This indicates that Au40 has a thermodynamically more favourable interaction with the support. It is worth mentioning that the total adsorption energy of −0.526 eV for Au40 is a global thermodynamic quantity, and a simple per-atom average would give only about −0.013 eV, which might seem too small to cause an observable XPS shift. However, the XPS binding energy shift is not determined by such an average. Instead, it reflects local changes in the initial-state potential and final-state screening at individual atomic sites. As shown in Table S2, the internal deformation energy of the Au40 cluster upon adsorption is only +0.013 eV, indicating that the cluster geometry remains almost intact. Hence, the adsorption energy mainly originates from interfacial electronic coupling rather than structural distortion. Thus, combining the DFT adsorption energy (a global thermodynamic quantity) with the XPS binding energy shift (a local core-level excitation) provides complementary evidence of stronger interaction for Au40, from thermodynamic and electronic structure perspectives, respectively. Moreover, a further Bader charge analysis was conducted (Fig. 4h). In the Au40 system, all Au atoms combined lose a total of 0.025 electrons after adsorption. However, in the Au44 system, Au gains a total of 0.002 electrons after adsorption (Fig. S18). In comparison, there is a relatively larger charge transfer between Au atoms and the substrate in the Au40 system, also reflecting a relatively stronger interaction between Au40 and the substrate.
To verify the universal enhancement of carrier interaction by engineered surface defects on clusters, we tested Au36,40 Au40, and Au44 clusters on other carriers such as CNT, SiO2, and GR. In the reaction of 4-NP to 4-AP, Au40 required significantly shorter reaction times compared to Au44 (Fig. 4i, S19a–f and S20). In addition, Au40 clusters were employed as catalysts, but no reaction was observed. When Au44 clusters were used as catalysts, the complete reaction required 4 hours (Fig. S19g–i). Additionally, XPS also demonstrates the distinction between the three. After interacting with multiple carriers, the binding energy of Au energy levels in Au40 increased (Fig. S21), while those of Au36 and Au44 remained unchanged (Fig. S22 and S23). The above results demonstrate that the presence of defects in Au40 enhances interactions between deacetylases, thereby optimizing its catalytic activity. Indicating that surface defects on the cluster enhanced its interaction with the supports.
Conclusions
In this study, we modified the Au44 structure by introducing a monofluorophosphine ligand to form an Au40 cluster. Loaded Au44 and Au40 clusters onto various substrates, TEM analysis revealed a uniform distribution of the clusters without aggregation. XPS measurements of Au 4f binding energy before and after loading demonstrated stronger binding forces between the Au40 clusters and substrates, indicating that surface defects enhance intercluster–substrate interactions. Density functional theory (DFT) calculations show that the interaction between Au40 and the carrier is stronger than that of Au44 systems, with the adsorption energies being −0.526 and −0.427 eV, respectively. To further evaluate the catalytic performance of supported catalysts, we used the reduction of p-nitrophenol as a model system. The results showed that the Au40 cluster series catalysts demonstrated significantly higher catalytic activity compared to the Au44 series, with a catalytic time ratio of approximately 1:4.
Author contributions
All authors have approved the final version of the manuscript. J. C., Y. W., and Y. Z. executed the experimental synthesis, characterization, and data analysis. C. L. and H. Y. carried out the theoretical calculations. Q. L. carried out single-crystal X-ray diffraction data collection and structure refinement; J. C., J. C. and S. Y. prepared the first draft of the manuscript, and all authors contributed to the preparation of the manuscript; G. G., S. Y., and M. Z. supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2496950 contains the supplementary crystallographic data for this paper.41The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: SI figures and tables. See DOI: https://doi.org/10.1039/d6sc00569a.
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22371003, 22301001, 22575001, 22003015, and U24A20480), the Anhui Provincial Natural Science Foundation (2508085MB032).References
- R. Jin, G. Li, S. Sharma, Y. Li and X. Du, Chem. Rev., 2021, 121, 567–648 CrossRef CAS PubMed.
- Z. Yang, Y. Wang, R. Zhang, T. Chen and J. Xie, Adv. Mater., 2025, 37, e08578 CrossRef CAS PubMed.
- S. M. Han, M. Park, J. Kim and D. Lee, Angew. Chem., Int. Ed., 2024, 63, e202404387 CrossRef CAS PubMed.
- Q. Li, T. Jiang, S. Yang, J. Chai, H. Yu and M. Zhu, Chem. Sci., 2025, 16, 19669–19676 RSC.
- H. Sakai, S. Hiramatsu, A. Akiyama, Y. Negishi and T. Hasobe, J. Am. Chem. Soc., 2025, 147, 13483–13490 CrossRef CAS PubMed.
- P. Yin, X. Luo, Y. Ma, S.-Q. Chu, S. Chen, X. Zheng, J. Lu, X.-J. Wu and H.-W. Liang, Nat. Commun., 2021, 12, 3135 CrossRef CAS PubMed.
- K. Liu, H. Shen, Z. Sun, Q. Zhou, G. Liu, Z. Sun, W. Chen, X. Gao and P. Chen, Nat. Commun., 2025, 16, 1203 CrossRef CAS PubMed.
- Y. Tan, G. Sun, T. Jiang, D. Liu, Q. Li, S. Yang, J. Chai, S. Gao, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317471 CrossRef CAS PubMed.
- S. Wang, C. Han, X. Chen, Y. Xiang, Q. Li, J. Chai, S. Yang, Y. Du, Q. Luo and M. Zhu, ACS Nano, 2025, 19, 25334–25341 CrossRef CAS PubMed.
- X. Cai, Y. Liu, G. Li, W. Hu, X. Liu, M. Chen, W. Ding and Y. Zhu, Adv. Mater., 2023, 35, 2301466 CrossRef CAS PubMed.
- X. Tang, S. Ge, Y. Lv, G. Sun, Z. Wang, J. Xie, M. Peng, Y. Xu, J. Zhang, B. Yao, Q. He, Y. Guo, W. Zhan, L. Wang, L. Zhou, B. Xu, S. Dai, Y. Guo and D. Ma, Angew. Chem., Int. Ed., 2025, 64, e202505507 CrossRef CAS PubMed.
- T. Zhou, X. Li, J. Zhao, L. Luo, Y. Wang, Z. Xiao, S. Hu, R. Wang, Z. Zhao, C. Liu, W. Wu, H. Li, Z. Zhang, L. Zhao, H. Yan and J. Zeng, Nat. Mater., 2025, 24, 891–899 CrossRef CAS PubMed.
- J. Yu, X. Qin, Y. Yang, M. Lv, P. Yin, L. Wang, Z. Ren, B. Song, Q. Li, L. Zheng, S. Hong, X. Xing, D. Ma, M. Wei and X. Duan, J. Am. Chem. Soc., 2024, 146, 1071–1080 CrossRef CAS PubMed.
- Q. Chu, Y. Niu, H. Tao, H. Liu, Q. Li, C. Lian and J. Li, ACS Catal., 2025, 15, 1942–1951 CrossRef CAS.
- H. Wang, Y. Jiao, G. Zhang, W. Ma, W. Fan, X. Liu, Y. Zhao, H. Xie, W. Ma and X. Zong, Adv. Funct. Mater., 2025, 35, 2418617 CrossRef CAS.
- Z. Qin, A. Lakra, R. R. Somni, W. Peng, G. Li, G. Hu and Z. Tong, ACS Catal., 2025, 15, 20270–20283 CrossRef CAS PubMed.
- X. Guan, X. Wang, X. Zhang, C. Zhang, S. S. C. Chuang and Z. Li, Angew. Chem., Int. Ed., 2025, 64, e202423958 CrossRef CAS PubMed.
- J. Chai, S. Yang, Y. Lv, H. Chong, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2019, 58, 15671–15674 CrossRef CAS PubMed.
- S. Masuda, K. Sakamoto and T. Tsukuda, Nanoscale, 2024, 16, 4514–4528 RSC.
- M. Zhang, S. Ji, Z. Zhang, M. Zhu, Q. Yao and J. Xie, CCS Chem., 2026, 8, 37–59 CrossRef.
- S. Li, Q. Wu, X. You, X. Ren, P. Du, F. Li, N. Zheng and H. Shen, J. Am. Chem. Soc., 2024, 146, 27852–27860 CrossRef CAS PubMed.
- M. F. Alotaibi, A. Sagadevan, P. Yuan, M. Bodiuzzaman, K. Murugesan, R. Zhou, J. Yin, S. Thomas, R.-W. Huang, N. M. Halappa, C. Dong, M. Abulikemu, H. N. Alshareef, O. F. Mohammed, M. Rueping and O. M. Bakr, J. Am. Chem. Soc., 2025, 147, 28932–28942 CrossRef CAS PubMed.
- S. Yang, S. Wu, F. Qian, H. Yu, J. Chai and M. Zhu, Coord. Chem. Rev., 2026, 556, 217698 CrossRef CAS.
- D. Leybo, U. J. Etim, M. Monai, S. R. Bare, Z. Zhong and C. Vogt, Chem. Soc. Rev., 2024, 53, 10450–10490 RSC.
- J. Yang, L. J. Falling, S. Yan, B. Zhang, P. Verma, L. Daemen, Y. Cheng, X. Zhao, S. Zhang, J.-L. Chen, B. Yao, S. Tan, S. Chae, Q. He, S. Nemsak, Z. Wu, D. Prendergast, Y. Guo, J. Liu, M. Salmeron and J. Su, Science, 2025, 388, 514–519 CrossRef CAS PubMed.
- V.-D. Quach, A. Harsan, M. C. Spadaro, M. Botifoll, J. Arbiol, M. Knezevic, C. Colbeau-Justin, F. Dumeignil, H. Vezin, R. Wojcieszak, T. L. Bahers, C. Michel and M. N. Ghazzal, Adv. Sci., 2025, 12, e01835 CrossRef CAS PubMed.
- J. Jeon, H.-S. Bang, Y.-J. Ko, E. Huh, J. Kang, X. Zhang, S. Ka, Y. Kim, W. H. Lee, K. Kim, H. K. Yu, H.-S. Oh and J.-Y. Choi, Chem. Eng. J., 2025, 523, 168350 CrossRef CAS.
- S. Nematulloev, A. Sagadevan, B. Alamer, A. Shkurenko, R. Huang, J. Yin, C. Dong, P. Yuan, K. E. Yorov, A. A. Karluk, W. J. Mir, B. E. Hasanov, M. N. Hedhili, N. M. Halappa, M. Eddaoudi, O. F. Mohammed, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202303572 CrossRef CAS PubMed.
- C. Dong, R.-W. Huang, A. Sagadevan, P. Yuan, L. Gutiérrez-Arzaluz, A. Ghosh, S. Nematulloev, B. Alamer, O. F. Mohammed, I. Hussain, M. Rueping and O. M. Bakr, Angew. Chem., Int. Ed., 2023, 62, e202307140 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Iida, K. Nobusada, K. Kirschbaum, K. J. Lambright and R. Jin, J. Am. Chem. Soc., 2016, 138, 3950–3953 CrossRef CAS PubMed.
- Q. Li, B. Huang, S. Yang, H. Zhang, J. Chai, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2021, 143, 15224–15232 CrossRef CAS PubMed.
- J. Chai, S. Yang, Y. Lv, T. Chen, S. Wang, H. Yu and M. Zhu, J. Am. Chem. Soc., 2018, 140, 15582–15585 CrossRef CAS PubMed.
- Q. Li, Y. Tan, B. Huang, S. Yang, J. Chai, X. Wang, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2023, 145, 15859–15868 CrossRef CAS PubMed.
- W. Liu, H. Wang, X. Zhang, Q. Li, J. Chai, S. Yang and M. Zhu, Small Methods, 2025, 9, 2500044 CrossRef CAS PubMed.
- J. Zhou, X. Yang, P. Zheng, Q. Li, X. Li, J. Chai, B. Huang, S. Yang and M. Zhu, Chem. Sci., 2024, 15, 4853–4859 RSC.
- L. Tian, F. Xu, G. Sastre, A. Primo, J. Yu and H. García, Chem. Soc. Rev., 2026, 55, 4244–4302 RSC.
- S.-H. Ryu, G. Kim, S. Gupta, S. Bhattacharjee, S.-C. Lee, H. Lee, J.-H. Choi and H. Jeong, Chem. Eng. J., 2024, 485, 149487 CrossRef CAS.
- Y. Tian, C. Shen, M. Han, Q. Zhai, G. Wu, W. Ding and Y. Zhu, J. Am. Chem. Soc., 2026, 148, 16204–16210 CrossRef CAS PubMed.
- S.-F. Yuan, Z.-J. Guan and Q.-M. Wang, J. Am. Chem. Soc., 2022, 144, 11405–11412 CrossRef CAS PubMed.
- C. Zeng, H. Qian, T. Li, G. Li, N. L. Rosi, B. Yoon, R. N. Barnett, R. L. Whetten, U. Landman and R. Jin, Angew. Chem., Int. Ed., 2012, 51, 13114–13118 CrossRef CAS PubMed.
- CCDC 2496950: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2pt8sl.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |