
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA general strategy for high-efficiency live bacteria imaging and targeted phototherapy
Xinci Wang†
a,
Lihao Ding† a,
Jiang Liua,
Jiahui Lva,
Junjie Liua,
Yu Yana,
Xutong Lib,
Huan Shia,
Hui Wangad,
Jiajia Wanga,
Senquan Liu*ae,
Duanhua Cao*c,
Xinxing Yang*ad and
Lin Xue*ad
a,
Jiang Liua,
Jiahui Lva,
Junjie Liua,
Yu Yana,
Xutong Lib,
Huan Shia,
Hui Wangad,
Jiajia Wanga,
Senquan Liu*ae,
Duanhua Cao*c,
Xinxing Yang*ad and
Lin Xue*ad
aMOE Key Laboratory for Cellular Dynamics, Center for Advanced Interdisciplinary Science and Biomedicine of IHM, Hefei National Research Center for Interdisciplinary Sciences at the Microscale, Division of Life Sciences and Medicine, University of Science and Technology of China, Hefei, 230026, China. E-mail: linxue@ustc.edu.cn; xinxingyang@ustc.edu.cn; liusenquan1988@ustc.edu.cn
bDrug Discovery and Design Center, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, 555 Zuchongzhi Road, Shanghai 201203, China
cMedical Artificial Intelligence Innovation Center, Shanghai East Hospital, School of Medicine, School of Life Sciences and Technology, Tongji University, Shanghai, 200092, China. E-mail: caodh@tongji.edu.cn
dAnhui Basic Discipline Research Center of Artificial Intelligence Biotechnology and Synthetic Biology, Hefei, 230026, China
eClinical and Experimental Research Center, The First Affiliated Hospital of Guilin Medical University, Guilin, 541001, China
First published on 14th May 2026
Abstract
Bacterial peptidoglycan (PG) serves as a unique bacterial signature for imaging and therapeutic targeting, yet existing chemical probes require high concentrations and involve disruptive washing steps. Herein, we report a general strategy for designing high-performance PG probes by incorporating a fluorine-substituted benzenesulfonamide moiety. The structural modification promotes the binding of probes to PBPs and significantly improves their PG-incorporation efficiency. Using this approach, we developed a full-color probe palette spanning the visible spectrum, enabling wash-free imaging of live bacteria at low concentrations with outstanding signal-to-background ratios. Beyond imaging, this strategy further led to the creation of FluoEosinY, a PG-targeted photosensitizer that effectively inactivates antibiotic-resistant bacteria without inducing resistance. In a mouse model of methicillin-resistant Staphylococcus aureus (MRSA)-infected skin wounds, FluoEosinY-mediated antimicrobial photodynamic therapy not only achieved marked reductions in bacterial burden but also accelerated wound healing, with negligible toxicity to host tissues. This versatile design strategy advances both the mechanistic investigation of bacterial cell wall physiology and the development of precision-targeted therapeutics to combat antibiotic-resistant infections.
Introduction
Bacterial infections, particularly those driven by antibiotic-resistant pathogens, remain a pressing global public health threat, with rising mortality and socioeconomic burdens.1 To address this crisis, two interconnected priorities demand immediate attention: first, gaining comprehensive insights into bacterial physiology to identify actionable targets; second, developing targeted therapeutic strategies that selectively eliminate pathogens while sparing host cells. Critically, these two goals share a foundational requirement: bacterial-specific molecular signatures for key cellular processes.2–4 These signatures not only enable in-depth studies of bacterial physiology, but also facilitate the design of targeted interventions by distinguishing pathogens from host tissues.5–7Among such bacterial-specific signatures, the peptidoglycan (PG) cell wall, stands out as a pivotal target for both advanced imaging and antimicrobial interventions.8–11 PG biogenesis is not only essential for bacterial viability and morphogenesis but also a conserved, bacteria-exclusive feature, making it an ideal marker to differentiate bacterial cells from host tissues.12,13 However, traditional methods for labeling and visualizing PG suffer from notable limitations that undermine their utility in advancing the above two goals.14,15 Current PG-specific probes are inherently limited by their relatively low labeling efficiency and hence require high working concentrations. This requirement markedly elevates the risk of probe aggregation, non-specific binding to off-target cellular components, and cytotoxicity toward both bacteria and host cells. Non-specific interactions with off-target biomolecules in both bacterial and host cells not only generate high background noise that obscures visualization but may also disrupt normal cellular processes. Such interference can lead to misinterpretation of PG biogenesis mechanisms and compromise the validity of experimental findings. The problem is further exacerbated by cumbersome washing steps required to reduce non-specific binding, which are poorly compatible with live-cell imaging. Therefore, those probes are limited in use of monitoring PG synthesis dynamics, particularly in studies of changes in cell morphology and pathogen–host interactions.16–18
Recently, photodynamic therapy (PDT) has emerged as a promising antimicrobial strategy. It utilizes photosensitizers (PSs) that generate reactive oxygen species (ROS) upon light irradiation to induce microbial death.19,20 However, conventional PSs often lack specificity for bacterial cells, leading to off-target damage to host tissues.21–23 Moreover, their integration with imaging modalities remains fragmented, requiring separate probes for visualization and therapy, complicating experimental workflows and reducing spatial–temporal correlation between imaging and treatment effects.
To address these challenges, we established a new PG labeling strategy based on the rational design of a fluorine-substituted benzenesulfonamide-D-amino amide conjugate. This approach enables the direct conversion of common fluorophores into wash-free probes, achieving high-specificity PG labeling at low concentrations without requiring tedious washing steps, thus facilitating the development of a versatile probe palette for visualizing cell wall dynamics across diverse bacterial species. Beyond imaging, this labeling strategy also permits targeted delivery of photosensitizers to bacteria. Upon light activation, the localized photosensitizer generates ROS, driving potent light-based ablation of antibiotic-resistant pathogens. This capability is demonstrated by >99% killing efficiency in bacterial cultures and significant attenuation of methicillin-resistant Staphylococcus aureus (MRSA)-induced murine skin infections, while exerting minimal toxicity to the host.
Results and discussion
Design strategy of the chemical probes for PG-labeling
One of the most effective strategies for labeling bacterial cell walls hinges on the stereoselective activity of transpeptidases, including penicillin-binding proteins (PBPs) and L,D-transpeptidases (LDTs), that are crucial in PG biogenesis. Conventional D-amino acid (FDAA) probes, as mimics of PBPs or LDTs substrates, are specifically incorporated into nascent PG by PBPs and LDTs, thereby enabling highly precise, in situ visualization of bacterial cell wall biosynthesis (Fig. 1a).9,24 To address limitations such as high background fluorescence and cumbersome washing steps in FDAA labeling, advanced fluorophores, including fluorescent rotors, aggregation-induced emission fluorogens, and fluorogenic rhodamines, have been integrated into probe design to minimize non-specific signals and enable wash-free imaging.16,18,25,26 Our recently developed rhodamine-based D-amino acid probes (FADAs) incorporate precisely tuned hydrophobicity to confer fluorogenic properties, enabling wash-free visualization of live bacterial cell walls with exceptional signal intensity and minimal background noise.18 However, excessive aggregation of the probes potentially compromises labeling efficiency and leads to non-specific adsorption to mammalian cell membranes, which restricts their use in host-cell or tissue imaging applications. | ||
| Fig. 1 A general strategy for fast PG labeling with fluorine substituted benzenesulfonamide. (a) Schematic illustration of the metabolic labeling of traditional FDAA probes. (b) Cartoon representation of the TPase domain of SaPBP1 (PDB ID: 7O4B), SaPBP2 (PDB ID: 2OLU), SaPBP3 (PDB ID: 3VSL) and SaPBP4 (PDB ID: 5TW8) with aromatic residues colored cyan and the ligand molecule colored yellow. The reactive residue (in SaPBP2) was colored in orange. The mutant residues in SaPBP3 were colored pink. (c) Schematic illustration of the metabolic labeling of the probes developed in this work, where benzenesulfonamides serve as the linking group between the dye and the D-amino amide. (d) The different charge distribution of π–π stacking and polar-π stacking. (e) Chemical structures and space-filling models showing the partial charges and electrostatic potentials of three aromatic residues and benzenesulfonamide derivatives. Red and blue indicate negative and positive electrostatic potentials, respectively. | ||
PBPs serve as the most critical PG transpeptidase (TPase) in most cell wall containing bacteria. We noticed that aromatic residues are distributed around the TPase active site, where they form a hydrophobic catalytic channel, among different bacteria27,28 (four Staphylococcus aureus PBPs illustrated in Fig. 1b). In some studies, these aromatic residues have been shown to form an aromatic wall-like structure and are critical for the enzyme activity.29–31 We therefore reasoned that introducing additional functional groups (e.g., a benzene ring), capable of interacting with these aromatic rings via π-stacking, into the probe would enhance the probe's binding affinity and accelerate the reaction rate between the probe and acyl-PBPs (Fig. 1c).
Notably, incorporating fluorine atoms onto benzene rings modulates the electron density at the ring center: shifting it from a negative state (corresponding to the red region in the benzene structure) to a positive state (Fig. 1d).32,33 This electron density redistribution polarizes their π-conjugated systems, thereby enabling enhanced polar–π interactions via electrostatic attraction with non-fluorinated aromatic rings.34 Additionally, fluorine atoms are well-documented to mediate hydrophobic interactions,35 while sulfonamide moieties can form multiple hydrogen bonds between the small-molecule probes and target proteins (Fig. 1e).36
Based on these considerations, we designed three probes, each integrating a D-amino amide as the reactive group for specific PG targeting and 6-carboxytetramethylrhodamine (TMR) as the fluorophore for fluorescent signal readout. A critical structural element in these probes is the fluorobenzenesulfonamide moiety, which serves as the key bridge between the D-amino amide tail and the TMR fluorophore, with its structure modulated by 0, 2, or 4 fluorine substitutions. The resulting probes are designated TMR-0F (1), TMR-2F (2), and TMR-4F (3) (Fig. 2a). This design aimed to systematically assess whether the benzene ring and fluorine substitution enhance the probes' PG-labeling efficiency.
 | ||
Fig. 2 Functional evaluation of fluorinated TMR Probes against SaPBPs. (a) Chemical structures of probes TMR-0F/2F/4F. The benzene ring with fluorine substitution is highlighted in blue. (b–e) Fluorescence polarization (FP) responses of 50 nM different probes as a function of SaPBPs concentration. Data presented as mean ± s.d., n = 3 independent replicates. (f–i) FP responses of 50 nM TMR-4F as a function of the concentration of SaPBPs variants. Data presented as mean ± s.d., n = 3 independent replicates. (j) Schematic of the in vitro transpeptidation assay: probes and the synthetic substrate (PC) were mixed with SaPBP4, the absorption signals of the probes and reaction products were measured via HPLC. (k) Representative HPLC analysis of the transpeptidation products. A shifted retention time was observed when SaPBP4 was added to the probe–substrate mixture. (l) Calculation of the probe conversion rates based on the HPLC results. Data presented as mean ± s.d., n = 3 independent replicates. (m) Fluorescence microscopy images of live S. aureus cells labeled with TADA and TMR-0F/2F/4F. Consistent contrast settings (0–40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) were maintained across all images. Scale bars: 2 µm. (n) Quantification of the SBR for the fluorescence labeling results shown in panel (m). Data presented as mean ± s.d., the number of cells (n) is indicated in the figure. 000) were maintained across all images. Scale bars: 2 µm. (n) Quantification of the SBR for the fluorescence labeling results shown in panel (m). Data presented as mean ± s.d., the number of cells (n) is indicated in the figure. | ||
Fluorobenzenesulfonamide moiety dramatically enhances the binding of probes to PBPs
To characterize the binding properties of the probes with target PBPs, we selected the four PBPs from Staphylococcus aureus (S. aureus) (NCTC 8325),37,38 which represent three distinct classes: SaPBP1 and SaPBP3 (class B), SaPBP2 (class A), and SaPBP4 (class C).As indicated in Fig. 2b, c, e and Table S1, TMR-4F exhibited the highest binding affinity (Kd = 2.2–4.7 µM) for SaPBP1, SaPBP2 and SaPBP4 in fluorescence polarization (FP) assays, whereas TMR-2F had slightly elevated Kd of 5.1 and 7.1 µM for SaPBP1 and SaPBP4, respectively. No detectable binding was observed for TMR-0F or the control probe TADA39 for all PBPs under the tested conditions. Molecular docking analyses revealed offset-stacked polar–π interaction between the fluorobenzenesulfonamide moiety of TMR-4F and the aromatic ring of Phe241 in the protein's binding pocket with centroid distance of ∼4.4 Å. By comparison, the corresponding benzenesulfonamide moiety of TMR-0F exhibited only very weak stacking with Phe241, at a larger centroid distance of ∼5.5 Å (Fig. S1). These results demonstrated that fluorobenzenesulfonamide moiety significantly enhances the binding affinity of the probes to PBPs.
To further validate this special binding, we performed additional experiments: first, removing the aromatic residues from SaPBP1 and SaPBP2 obviously reduced their binding affinity to TMR-4F (Fig. 2f, g, i, and Table S1). Specifically, the F241A mutation in SaPBP4 disrupts the key polar–π interaction with the probe, as demonstrated by molecular docking studies, resulting in a dramatic loss of the protein's binding affinity; second, introducing mutations that add aromatic residues to SaPBP3 (Fig. 1b), whose catalytic channel contains fewer aromatic residues, markedly enhanced its binding to TMR-4F in vitro (Fig. 2d and h). Based on these findings, we conclude that the enhanced binding affinity of the probes toward PBPs mainly arises from the interaction between the fluorobenzenesulfonamide moiety of the probes and the aromatic residues in the TPase domain of the PBPs.
Next, isothermal titration calorimetry (ITC) was conducted to characterize the binding behaviors and thermodynamic profiles of three probes toward SaPBP4. As expected, TMR-4F exhibited the strongest binding affinity with a Kd of 3.4 ± 0.2 µM, accompanied by endothermic ΔH (21.2 ± 2.1 kcal mol−1) and favorable entropic contribution (TΔS = 28.7 ± 2.1 kcal mol−1). TMR-2F showed moderate binding (Kd = 10.9 ± 0.6 µM), with a slightly positive ΔH (13.6 ± 1.3 kcal mol−1) and TΔS of 20.3 ± 1.4 kcal mol−1 (Fig. S2). No detectable binding was observed for TMR-0F. Thermodynamically, the interactions of TMR-2F and TMR-4F with SaPBP4 were predominantly entropy-driven, a typical feature of hydrophobic interactions driven by the release of ordered interfacial water molecules. This is in consistent with that fluorination enhances both hydrophobicity and polar–π interactions and promotes insertion into the SaPBP4 hydrophobic pocket. These synergistic noncovalent forces stabilize the probe–protein complex and enhance molecular recognition, increased with higher fluorination degree. In contrast, the non-fluorinated analog TMR-0F lacks these key interactions and fails to bind effectively. Thus, fluorination finely tunes hydrophobicity and polar–π interaction capacity, determining the binding affinity.
We characterized the photophysical properties of the three probes in HEPES buffer. As shown in Fig. S3a–f and Table 1, all probes exhibited identical maximum absorption (λabs = 552 nm) and emission (λem = 577 nm) wavelengths, and they existed primarily in a soluble form in the buffer (Fig. S3g–i). For TMR-2F and TMR-4F, fluorine substitution slightly increased the probes' aggregation fraction, which in turn resulted in less than 2-fold fluorescence quenching. Treatment with SDS, known to disrupt rhodamine dye aggregates, restored the fluorescence quantum yields (φ ≈ 0.71, Table 1) to levels comparable to those of TMR-0F. These findings indicate that fluorine substitution has minimal impact on the photophysical properties of the fluorophore.
| Probe | λabs/λabs, SDS (nm) | ε/εSDS (104 M−1 cm−1) | λem/λem,SDS (nm) | Φ | F/F0 | SBR, cell count | ||
|---|---|---|---|---|---|---|---|---|
| S. aureus | B. subtilis | C. glutamicum | ||||||
| a FluoDA648 was previously reported as FADA648 in our earlier work.18 | ||||||||
| TMR-0F | 552/552 | 4.3/10.8 | 577/578 | 0.71 | 1.1 | 2.7, 145 | 1.4, 66 | 16.3, 71 |
| TMR-2F | 552/552 | 2.6/10.8 | 577/578 | 0.72 | 1.7 | 5.2, 83 | 2.4, 84 | 15.4, 28 |
| TMR-4F | 552/552 | 3.7/10.8 | 577/578 | 0.70 | 1.8 | 8.0, 130 | 6.1, 70 | 29.4, 72 |
| FluoDA400 | 400/356 | 3.7/3.7 | 447/444 | 0.58 | 0.4 | 15.1, 89 | 8.1, 63 | 45.0, 77 |
| FluoDA480 | 480/482 | 1.0/2.1 | 542/542 | 0.11 | 0.9 | 20.7, 137 | 15.2, 68 | 52.2.100 |
| FluoDA500 | 500/508 | 4.6/8.0 | 526/530 | 0.87 | 0.8 | 46.3, 91 | 10.6, 65 | 45.5, 51 |
| FluoDA504 | 504/510 | 2.2/8.0 | 513/517 | 0.83 | 11.3 | 45.6, 64 | 26.3, 97 | 88.7, 48 |
| FluoDA530 | 530/531 | 3.8/10.2 | 552/552 | 0.98 | 2.6 | 14.3, 82 | 10.0, 69 | 38.9, 52 |
| FluoDA560 | 560/559 | 5.7/12.9 | 580/580 | 0.73 | 3.0 | 30.6, 69 | 37.6, 84 | 113.8, 55 |
| FluoDA580 | 580/580 | 4.0/11.9 | 601/601 | 0.91 | 3.1 | 33.5, 84 | 52.0, 83 | 121.0, 39 |
| FluoDA648a | 648/651 | 17.6/14.9 | 665/668 | 0.68 | 4.5 | 17.6, 83 | 121.0, 39 | 44.0, 48 |
Fluorobenzenesulfonamide moiety significantly accelerates transpeptidation
We next examined the in vitro transpeptidation activity of SaPBP4.16 As a typical initial step, PBPs form an acyl-PBP complex with their PG substrate. To recapitulate this natural process, we prepared a coumarin-conjugated short peptide (Ala-Ala-Lys), designated as PC (Fig. 2j). The probes were then incubated with purified SaPBP4 and PC at room temperature, and the reaction progress was monitored via reverse-phase high-performance liquid chromatography (RP-HPLC) and high-resolution mass spectrometry (HRMS).Consistent with the FP assay, TMR-4F generated the highest yield of cross-linked product (∼50%), followed by TMR-2F (∼35%), while TMR-0F produced minimal adducts (∼20%, Fig. 2k and l). We subsequently extended the in vitro assays to other three PBPs. Similar to the SaPBP4 results, incubation with SaPBP1 yielded conversion efficiencies: 52.4% for TMR-4F, 39.7% for TMR-2F, and 14.3% for TMR-0F (Fig. S4a and S4e). However, no catalytic products were detected with SaPBP2 or SaPBP3, probably due to the lower activity of these two PBPs with PC in vitro. (Fig. S4b and S4c). Nevertheless, these findings of SaPBP1 and SaPBP4 establish a clear structure–activity relationship: increased fluorine substitution proportionally enhances transpeptidation efficiency in vitro.
Fluorobenzenesulfonamide moiety significantly enhances PG labeling efficiency
We then evaluated the probes' labeling performance in live S. aureus cells. To minimize interference from probe aggregation during labeling, cells were incubated with 1 µM of either TADA or the TMR probes for 60 minutes (Fig. 2m, n and S5a). Wash-free imaging revealed comparable background fluorescence among all probes, enabling direct comparison of their signal performance. Specifically, TADA failed to generate distinct cellular envelope signals, with an extremely low signal-to-background ratio (SBR) of 0.8 (Fig. 2n). TMR-0F, lacking fluorine substitution, exhibited moderately enhanced envelope fluorescence, achieving an SBR of 2.7. In sharp contrast, the introduction of fluorine substitution on the benzenesulfonamide moiety of TMR-2F and TMR-4F yielded substantially stronger fluorescence, with correspondingly higher SBR values of 5.2 and 8.0, respectively (Fig. 2n and Table 1). Given the limited fluorescence enhancement of TMR-2F and TMR-4F tested in buffer with SDS (Table 1), the high SBR is likely attributed to enhanced probe incorporation efficiency into PG, mediated by the fluorobenzenesulfonamide moiety.We then optimized the probe working concentration by measuring fluorescence intensity and SBR across probe concentrations ranging from 1 to 100 µM (Fig. S5b–d). The fluorescence signal increased with probe concentration, while the SBR decreased. Notably, the optimal working concentration of 1 µM is far lower than the hundreds of micromolar to millimolar concentrations required for other common FDAA probes.39 This lower concentration is expected to mitigate the potential aggregation of the probe and reduce non-specific labeling.40 Furthermore, TMR-4F-labeled cells showed no obvious fluorescence reduction after ethanol fixation and washing, which principally removes membrane-bound probes, demonstrating the probe's specific targeting of the PG cell wall rather than cell membranes (Fig. S5e and S5f).
Considering the high fluorescence intensity and SBR of TMR-2F and TMR-4F in cellular imaging, their enhanced fluorescent signals could, in principle, be attributed to slower removal by PG hydrolases, such as D,D-carboxypeptidases,41 which cleave the C-terminal peptide bond of PG pentapeptides and thereby eliminate the terminal fluorescent amino acid. To verify this hypothesis, we monitored cellular fluorescence for 1 h after probe labeling and washing. PG hydrolysis did induce a gradual decrease in cellular fluorescence, but the decay rates were comparable across all four probes (Fig. S5g and S5h). These results confirm that the enhanced signals of TMR-2F and TMR-4F stem from a higher incorporation level during PG synthesis, rather than reduced removal by PG hydrolases. Using the PG-labeled cells, we evaluated the photostability of probe TMR-4F and the commercial probe TADA. Cells labeled with the two probes were continuously illuminated at 561 nm, and quantitative analysis showed that both probes exhibited comparable photostability (Fig. S6). Therefore, introduction of the fluorobenzenesulfonamide moiety did not compromise the photostability of the fluorophore.
To assess the broad applicability of our probes, we extended live-cell labeling to Bacillus subtilis (B. subtilis), Corynebacterium glutamicum (C. glutamicum), Enterococcus faecium (E. faecium), and Deinococcus radiodurans (D. radiodurans), representative strains from distinct phyla of Gram-positive bacteria. The fluorine mediated enhancement of labeling efficiency was consistently observed across all bacteria (Fig. S7). Among the probes, TMR-4F performed optimally, exhibiting an SBR comparable to or even higher than that in S. aureus (e.g., ∼32 in C. glutamicum, Table 1). These findings confirm that our strategy, incorporating the fluorobenzenesulfonamide moiety into probes, significantly enhances the labeling efficiency of probes in PG. This advancement enables wash-free imaging in a broad spectrum of bacterial species.
Extending the strategy to a full-color palette for PG labeling in live bacteria
In contrast to previously reported strategies for PG labeling in live bacteria, most of which rely on fluorophore chemical engineering,16,18,25,26 our strategy reported herein does not improve the performance of the fluorophore. Instead, our strategy accelerates the metabolic incorporation of probes into PG, thereby facilitating the development of a new series of fluorescent probes spanning the visible spectrum (Fig. 3). Commercial fluorophores, including coumarin, NBD, BODIPY, and rhodamines, were incorporated into the tetrafluorobenzenesulfonamide moiety to generate probes, termed “FluoDA probes”, referring to fluorescent fluorinated D-amino amide probes. | ||
| Fig. 3 Extending the strategy to a full-color palette for PG labeling in live bacteria. (a) Top: synthetic route for FluoDA probes; bottom: chemical structures of the fluorophores in each FluoDA probe. The red stars represent the fluorophores. (b) Live-cell imaging of S. aureus, B. subtilis, and C. glutamicum labeled with the FluoDA probes shown in panel (a). The average SBR corresponding to each imaging result is indicated in the respective figures. Scale bars: 2 µm. | ||
We assessed the photophysical properties of the FluoDA probes. All probes showed λabs and λem values similar to their parent dyes (Fig. S8 and Tables 1, S2). Within the short-wavelength range, FluoDA400 and FluoDA480, incorporating coumarin and NBD as fluorophores, are non-fluorogenic fluorophores in HEPES buffer (Fig. S8a and S8b). Notably, SDS treatment shifts the protonation equilibrium of FluoDA400 toward its protonated state, inducing a 46 nm hypsochromic absorption shift and pronounced fluorescence quenching (Fig. S9). FluoDA504 forms aggregates that quench its fluorescence due to the strong aggregation tendency of BODIPY.42 Upon SDS treatment, these aggregates are disassembled, resulting in a significant 11.3-fold enhancement of emission (Fig. S8d and S8i).
Rhodamines are well-regarded fluorophores for cellular labeling and super-resolution microscopy, and N-alkylation of rhodamines enables tunable photophysical properties.43,44 For our FluoDA probes, we directly coupled the tetrafluorobenzenesulfonamide moiety to the carboxyl group at the spiro position of distinct rhodamine (Rho) derivatives, including Rho 110, Rho 6 G, Rho B, Rho 101, and silica-rhodamine (SiR). This structural design enables the probes to cover a wide range of the visible spectrum (500–650 nm).
Notably, sulfonamide substitution at the spiro position of these rhodamine-based FluoDA probes did not promote the formation of the non-fluorescent spiro-lactam form, as verified by the absorption of zwitterion form in dioxane/water mixtures (Fig. S8j–n).43,45 Specifically, only modest fluorogenicity with a fold change of less than 5 was observed for these probes following SDS treatment (Fig. S8c, S8e–h, and Table 1). In HEPES buffer, most FluoDA probes existed in the fluorescent soluble form with a supernatant/pellet ratio greater than 2 (Fig. S8i).
To validate the labeling efficiency of all FluoDA probes, we evaluated their performance in living S. aureus, B. subtilis, and C. glutamicum cells ranging from 1 to 10 µM, without implementing a washing step (Method section). Confocal microscopy revealed clear cellular outlines for all FluoDA probes, with both septal and peripheral PG distinctly visible (SBR > 8, Fig. 3b, and Table 1). Specifically, FluoDA500 achieved the highest SBR of 46.3 in S. aureus, while FluoDA648 reached the highest SBR of 121 in B. subtilis. Notably, in C. glutamicum, all FluoDA probes exhibited robust labeling efficiency with SBR values exceeding 30. This robust performance is likely attributed to either the high number of PBPs (9 PBP homologs) in C. glutamicum or the high catalytic activity of these PBPs.46 To further confirm the contribution of fluorination, we synthesized two control probes (4 and 5), which are the non-fluorinated counterparts of FluoDA400 and FluoDA560, respectively (Fig. S10a). Both control probes showed significantly lower SBR than their fluorinated counterparts (Fig. S10b and S10c). These data clearly demonstrate that our strategy allows the conversion of commonly used fluorescent dyes into wash-free fluorescent probes for PG labeling in bacteria for live-cell imaging.
Fluorobenzenesulfonamide moiety enables fast incorporation of photosensitizer into PG
It has been demonstrated that conjugates of photosensitizers and D-amino acids can act as potential antibacterial agents against antibiotic-resistant bacteria.25,47 Building on our platform that enhances probe incorporation efficiency into bacterial cell walls, we proposed that this strategy could also deliver other cargos, such as photosensitizers, into PG to boost antibacterial efficiency. We therefore designed FluoEosinY, in which the photosensitizer, Eosin Y,48 and D-amino amide moiety was bridged by the tetrafluorobenzenesulfonamide moiety (Fig. 4a). We evaluated FluoEosinY in community-associated MRSA (USA300) cells to assess its labeling performance and antibacterial efficacy. | ||
| Fig. 4 PG-targeting FluoEosinY induces membrane and DNA damage in MRAS cells upon light irradiation. (a) Chemical structure of the PG-targeting photosensitizer FluoEosinY, and schematic illustration of the workflow for in vitro photoinactivation assay. (b) Fluorescence microscopy images of live MRSA cells labeled with FluoEosinY, Potomac Red, and Hoechst 34580 with or without light irradiation. Arrows: membrane damage site; asterisks: abnormal DNA aggregation. Scale bars: 2 µm. (c and d) Quantitative analysis of percentage of membrane-disrupted cells (c) and the ratio of DNA-occupied area to total cell area (AreaDNA/Areacell, d) under conditions with or without light irradiation. Box plots display the mean (squares), standard deviations (boxes), and 5–95% percentiles (whiskers). n = 132 cells. (e) Real-time fluorescence monitoring of FluoEosinY-labeled cells after continuous irradiation with a 528 nm laser. Scale bars: 2 µm. (f) Quantitative analysis of (left) percentage of membrane disrupted cells and (right) AreaDNA/Areacell at different laser irradiation durations. Data presented as mean ± s.e.m., n = 105 cells. (g and h) Representative TEM images showing FluoEosinY-treated MRSA cell without (g) and with (h) light irradiation. White arrows indicate the abnormal DNA condensation, and the asterisk indicates the collapse of the bacterial cell membrane after photodynamic treatment. Scale bars: 200 nm. (i) Volcano plot of all the differentially expressed genes in the FluoEosinY-labeled MRSA cells with light irradiation comparing to those in dark. |log2 fold change| ≥ 1 and p < 0.05 were used to analyze the difference. Red: upregulation, blue: downregulation. n = 3, biological replicates. (j) GO annotation analysis of the differentially expressed genes in the FluoEosinY-treated MRSA cells. | ||
After 30 minutes incubation with FluoEosinY and washing to remove unbound probe, cell cultures were irradiated with green light (519/26 nm, 4.5 mW·cm−2) to activate ROS production (Fig. 4a). Cells were then stained with Potomac Red (a membrane probe49) and Hoechst 34580 (a DNA probe). Multi-color imaging revealed clear cell wall-associated labeling of FluoEosinY (Fig. 4b). Following green light irradiation, bright membrane-associated puncta or aggregates emerged in 92.5 ± 7.2% cells (arrows in Fig. 4b), greatly excess the fraction of cells without irradiation (12.1 ± 6.3%, Fig. 4c). The irradiation also caused notably compaction of nucleoids (asterisks in Fig. 4b), where the DNA area occupied only 19.2 ± 4.6% of the entire cell area, comparing to 70.5 ± 9.5% in cells without irradiation (Fig. 4d).
To investigate how FluoEosinY induces photodamage, we performed time lapse imaging of FluoEosinY-labeled MRSA cells under continuous 528 nm laser irradiation on a confocal microscope. After 60 seconds of irradiation, FluoEosinY-labeled MRSA cells showed striking disruptions in membrane continuity and chromosomal condensation, whereas unlabeled cells exposed to the same irradiation exhibited no obvious changes (Fig. 4e, f and S11). These morphological changes consistent with acute photodynamic damage to both the cell envelope and chromosomal structure.50 Transmission electron microscopy (TEM) images clearly showed envelope disintegration and electron-dense, compacted nucleoids in FluoEosinY-labeled cells following green-light irradiation, confirming our observations in fluorescence microscopy imaging (Fig. 4g and h).
RNA-seq of FluoEosinY-labeled MRSA with or without irradiation revealed 195 photoactivation-responsive genes (p < 0.05), enriched for membrane organization, DNA binding, oxidoreductase activity, and transcriptional regulation (Fig. 4i and j). These signatures indicate ROS-mediated disruption of membranes and intracellular homeostasis, with no significant changes in PG-related genes.
Given the observed membrane disruption, we hypothesized that FluoEosinY-mediated photodamage increases membrane permeability, potentially facilitating the penetration of other antibacterial drugs through the cell envelope to reach the cytosol. Pre-treating MRSA with light-activated FluoEosinY significantly reduced the minimum inhibitory concentrations (MICs) of multiple intracellular-targeting antibiotics, including kanamycin, chloramphenicol, ciprofloxacin and erythromycin (Table 2). Indeed, intracellular quantification of ciprofloxacin revealed a >2-fold increase in accumulation in FluoEosinY-labeled, light-irradiated cells compared to control cells (Fig. S12). This finding supports the notion that FluoEosinY-mediated irradiation induces membrane damage, thereby enhancing membrane permeability to ciprofloxacin. These results suggest that our PG-specific photosensitizer could be applied in combination therapy with classic antibiotics.
| Antibiotic | Antibiotic class | Mechanism of action | MIC (µg·ml−1) | |
|---|---|---|---|---|
| Drug only | Drug + FluoEosinY | |||
| Kanamycin | Aminoglycosides | Bind the 30S subunit of 16S rRNA, causing mistranslation and bacterial death | 64 | 4 |
| Chloramphenicol | Phenicols | Block translation by targeting the A site of the 50 S subunit | 16 | 4 |
| Ciprofloxacin | Fluoroquinolones | Disrupt DNA replication via DNA gyrase and topoisomerase IV inhibition | 256 | 8 |
| Erythromycin | Macrolides | Target 23S rRNA in the 50S subunit, halting translation and peptide elongation | 256 | 32 |
PG-specific photosensitizer potentiates antibacterial efficacy against multidrug-resistant bacteria
We next evaluated the antibacterial efficacy of FluoEosinY under green light irradiation using colony-forming unit (CFU) assays. After green-light irradiation, FluoEosinY-labeled MRSA cells showed dose-dependent inactivation, achieving a 99.99% reduction in viable counts at concentrations of 10 µM or higher (Fig. 5a). In the absence of light, FluoEosinY had no measurable impact on cell viability or growth rate, underscoring the critical role of photoactivation (Fig. 5a). Control experiments using the L-enantiomer of FluoEosinY (FluoEosinY(L)) and the parent dye Eosin Y also showed no phototoxicity under identical conditions (Fig. 5b and c), confirming that PG-specific incorporation is essential for efficient photodynamic bacterial inactivation. This specificity, whereby FluoEosinY rapidly and selectively targets bacterial PG, enables effective inactivation at low PS concentrations and with low-dosage light exposure.51 To further validate the broader applicability of this strategy to other multidrug-resistant bacterial pathogens, we extended our tests to vancomycin-resistant Enterococcus faecium (VRE). As expected, these experiments also revealed high and specific bacterial photodynamic inactivation (PDI) against VRE (Fig. S13a–c). Moreover, the FluoEosinY-based therapeutic regimen did not induce drug resistance in either MRSA or VRE cells even after ten consecutive treatment cycles (Fig. 5d and S13d). These findings demonstrate that FluoEosinY exerts potent, selective, and resistance-refractory antibacterial activity against clinically relevant pathogens. | ||
| Fig. 5 High antibacterial efficiency and specificity of FluoEosinY in vitro. (a) Quantitative analysis of MRSA viability after treatment with different concentrations of FluoEosinY and light irradiation. (b) Representative plate images for colony counting of MRSA after treatment with DMSO, Eosin Y, FluoEosinY(L), and FluoEosinY with or without light irradiation. (c) Quantitative analysis of MRSA viability corresponding to the colony counting results in (b). (d) PDI efficiency against MRSA, quantified as log10(N/N0), across 10 rounds of photodynamic inactivation. Data are presented as mean ± s.d., n = 3 independent samples. (e) Flow cytometry analysis showed high PI staining in MRSA cells with FluoEosinY labeling and light irradiation. (f) Fluorescence microscopy images of live MRSA cells labeled with FluoEosinY, and live HeLa cells either labeled with or without FluoEosinY. All imaging was performed using a confocal microscope under identical imaging parameters. Consistent contrast settings (0–20000) were maintained across all images. Scale bars: 5 µm. (g) Cell viability (%) of HeLa cells after treatment with 10 µM of Eosin Y, and different concentrations of FluoEosinY with or without light irradiation. Data are presented as mean ± s.d., n = 3 independent samples. | ||
Membrane integrity was further assessed using propidium iodide (PI) staining as PI binds nucleic acids but cannot penetrate cells with intact membranes. Strong PI fluorescence in FluoEosinY-labeled, light-irradiated MRSA, but not in dark controls or cells treated with Eosin Y or FluoEosinY(L) (Fig. S14). Flow cytometry validated these findings: FluoEosinY-labeled, irradiated cells exhibited high PI intensities comparable to isopropanol-killed cells, while other treatment groups showed low intensity profiles similar to untreated bacteria cells (Fig. 5e and S15). Notably, under the same labeling conditions, HeLa cells exhibited no significant labeling with Eosin Y or FluoEosinY, whereas clear labeling was observed in MRSA cells. (Fig. 5f and S16). Moreover, cell viability assessments verified that FluoEosinY caused minimal toxicity to HeLa cells in culture (Fig. 5g). These data support its potential as a safe and targeted antimicrobial agent.
Bacterial biofilms serve as a key pathogenic strategy that enables S. aureus to evade host immune clearance and conventional antibiotic treatment, thereby causing persistent and recalcitrant infections.52 FluoEosinY demonstrates excellent labeling efficiency toward S. aureus biofilms (Fig. S17). Upon green light irradiation, the PI fluorescence signal was significantly enhanced compared with unlabeled or non-illuminated samples, indicating severe disruption of bacterial membrane integrity in the biofilm. These results support FluoEosinY as a promising approach for noninvasive eradication of biofilm-associated S. aureus infections.
PG-specific photosensitizer promotes healing of MRSA-infected skin tissue
To further evaluate the in vivo antimicrobial photodynamic therapy (aPDT) potential of FluoEosinY, a full-thickness MRSA-infected skin wound model was established (Fig. 6a, Method section). Quantification of MRSA loads in infected wounds at 3 days post-operation showed that the bacterial density in the untreated MRSA-infected group reached 7.69 × 107 CFU mL−1, while that in the mupirocin group was 2.13 × 107 CFU mL−1. Following FluoEosinY-mediated aPDT, the MRSA density at the wound site decreased one order of magnitude to 7.93 × 106 CFU mL−1, corresponding to an 89.7% MRSA killing efficiency. Even without light irradiation, the FluoEosinY (−light) group exhibited antibacterial activity, with a bacterial density of 1.33 × 107 CFU mL−1, comparable to that of the mupirocin group (Fig. 6b and c). Wound healing was also quantified over the 9 days treatment period. All groups showed gradual healing of infected skin wounds, as reflected by the continuous reduction in wound size (Fig. 6d and e). Among these groups, the FluoEosinY (+light) and mupirocin groups exhibited the fastest wound recovery, with ∼35% of the original wound area remaining by day 9. In contrast, FluoEosinY without light irradiation did not promote wound healing compared with the untreated MRSA-infected group. | ||
| Fig. 6 High antibacterial efficiency of FluoEosinY-based aPDT treatment in vivo. (a) Schematic illustration of the establishment of a mouse model infected with MRSA and the workflow for evaluating in vivo antibacterial efficiency. (b) Representative plate images for colony counting of MRSA colonies isolated from infected wound tissue at day 3 post-operation. (c) Quantitative analysis of MRSA viability corresponding to the colony counting results in (b). Data presented as mean ± s.d., n = 3 independent samples. (d) Representative images of wound size at 1, 3, 7, and 9 days post-operation, demonstrating wound healing progression in five experimental mouse groups subjected to different treatments. One small division on the ruler is 1 mm. (e) Quantitative measurement of skin wound size from 1 day to 9 days post-operation across different groups. Data presented as mean ± s.d., n = 6 independent samples. *p < 0.05, **p < 0.01, and ns, not significant (p > 0.05). Two-tailed unpaired t-test. (f) Hematoxylin and eosin (H&E) staining results of wound tissues at 9 days post-operation, showing histological changes in different experimental groups: control group: no MRSA challenge; MRSA group: MRSA challenge without any treatment; mupirocin-treated group: MRSA challenge followed by mupirocin treatment; FluoEosinY – Light group: MRSA challenge treated with FluoEosinY but without light irradiation; FluoEosinY + Light group: MRSA challenge treated with FluoEosinY combined with light irradiation. Scale bars: 50 µm. | ||
Additional hematoxylin and eosin (H&E) staining was performed on wound tissues from each treatment group at 9 days post-wounding, with representative images presented in Fig. 6f. As shown in these histological sections, the mupirocin group and FluoEosinY (+light) group exhibited a more rapid resolution of inflammatory infiltration compared to the untreated control group. Furthermore, prominent fibroblast proliferation was observed in these two treatment groups, indicating accelerated wound healing and tissue repair processes. Notably, relative to the PBS control and mupirocin groups, the FluoEosinY (+light) group displayed significantly reduced intracellular edema in the epidermal region, alongside obvious hair follicle formation. Collectively, these histological and phenotypic observations validate the potential of FluoEosinY-mediated antimicrobial photodynamic therapy for the treatment of skin infections caused by antibiotic-resistant bacteria.
A systematic assessment of in vivo biosafety is essential to support the further clinical translation and practical application of FluoEosinY. To this end, we performed a detailed in vivo safety evaluation in mice under topical administration without light irradiation. Briefly, circular full-thickness wounds were established on the dorsal skin of 6–8 weeks-old female BALB/c mice. Each wound was topically administrated with 100 µL of 30 µM FluoEosinY for 1 h, while PBS-treated wounds were set as the blank control. All mice were subsequently monitored for 9 consecutive days. At the endpoint, hematological analysis revealed that all routine blood indexes in the FluoEosinY-treated group remained within the physiological range and were comparable to those in the control group (Fig. S18a–h). In parallel, critical serum biochemical markers, such as alanine aminotransferase (ALT), aspartate aminotransferase (AST), blood creatinine (CRE) and blood urea nitrogen (BUN), showed no aberrant changes, indicating no obvious hepatic or renal toxicity (Fig. S18i–l). In addition, morphological observation and H&E staining of major vital organs demonstrated no visible tissue injury or histopathological damage (Fig. S19). Collectively, these results confirm the favorable biocompatibility and biosafety of topically administered FluoEosinY under the tested conditions.
Discussion
In this study, we have developed a fluorine-optimized PG labeling strategy that capitalizes on a fluorine-substituted benzenesulfonamide moiety to enable the rapid, high-efficiency incorporation of metabolic probes into bacterial PG. The established structure–activity relationship demonstrates that increased fluorine substitution correlates with higher PBP-binding affinity. Fluorine substitution is known to withdraw aromatic electron density and alter the electronic properties of the benzene ring, while also strengthening overall hydrophobic characteristics. These combined effects collectively promote stable probe–PBP recognition.32,34Traditional FDAA probes, widely used for PG visualization, typically require high concentrations (ranging from hundreds of µM to tens of mM) and cumbersome washing steps to reduce background fluorescence. Some even need prolonged incubation (hours) for effective labeling.14,25 In contrast, our strategy not only allows to generate wash-free probes that achieved high labeling performance in live bacteria at as low as 1 µM within minutes, but also enables the development of a full-color palette of fluorescent probes spanning the visible light spectrum, supporting multi-color imaging of PG dynamics across diverse bacterial species. Furthermore, the broad versatility in fluorophore selection facilitates the application of advanced microscopic techniques, such as fluorescence lifetime imaging and stimulated emission depletion (STED) microscopy, in future studies.
Beyond PG imaging, we successfully extended this strategy to therapeutic applications by bridging the photosensitizer Eosin Y and D-amino amide moiety via the tetrafluorobenzenesulfonamide moiety, creating FluoEosinY, a PG-targeted photosensitizer that addresses key limitations of conventional aPDT: low specificity, off-target toxicity, and insufficient accumulation at infection sites. FluoEosinY enables rapid, specific accumulation in the bacterial cell wall and, upon light activation, generates localized ROS that cause severe membrane disruption, DNA condensation, and ultimately cell death. The light-dependent and PG-specific nature of FluoEosinY's antibacterial activity highlights its targeting precision and minimizes off-target effects to host cells. Moreover, the lack of resistance development after multiple treatment cycles underscores the potential of this approach to combat antibiotic- and multidrug-resistant pathogens. Additionally, FluoEosinY reduced the MICs of intracellular antibiotics by enhancing membrane permeability, highlighting its potential for combination therapy to tackle multidrug-resistant infections.
The in vivo validation in a mouse MRSA skin wound model further solidified the strategy's clinical relevance. FluoEosinY-mediated aPDT significantly reduced wound bacterial loads, matching the efficacy of mupirocin while offering superior tissue repair benefits such as faster resolution of inflammatory infiltration, reduced epidermal edema, and promoted hair follicle formation. This suggests that FluoEosinY not only clears pathogens but also modulates the host wound-healing response, a dual benefit rarely reported for aPDT agents. Notably, FluoEosinY exhibited minimal toxicity toward mammalian cells, addressing a major concern with conventional aPDT (off-target ROS damage to host tissues).53 The strict light dependence of its activity further enhances safety, as the PS remains biologically inert until activated, reducing the risk of systemic side effects in clinical use.
This study primarily focused on Gram-positive bacteria, in which the PG layer is fully exposed and readily accessible. In contrast, the identical labeling approach was ineffective for Gram-negative bacteria (Fig. S20), largely owing to the outer membrane that restricts probe penetration and cellular uptake.39 Future efforts will further explore the applicability of fluorobenzenesulfonamide-based probes in these Gram-negative species.
The skin wound model reflects superficial infections, but deep-seated or biofilm-associated infections present greater challenges. Given the modular nature of our molecular design, the EosinY moiety can be readily replaced with near-infrared (NIR) photosensitizers. This adaptability provides a clear and straightforward path for future iterations of the probe to overcome the tissue penetration limitations associated with visible light, thereby extending the applicability of this platform to the treatment of deep-seated infections.
In conclusion, this fluorine-optimized PG labeling strategy marks a significant advancement for the bacterial imaging and targeted photodynamic inactivation. By enabling rapid and specific PG labeling, this approach provides a promising solution to address the growing challenges for both basic research on bacterial physiology and clinical translation toward combating drug-resistant infections.
Experimental section
Synthesis of probes
The general synthetic procedures for the FluoDA probes were outlined in Fig. 3a. First, the tetrafluorobenzenesulfonamide moiety was coupled with common fluorophores, including coumarin, NBD, BODIPY, and rhodamines. Next, the amine group, released via deprotection of the tert-butyloxycarbonyl (Boc) group, was further conjugated with N-α-Fmoc-D-aspartic amide. Finally, removal of the 9-fluorenylmethyloxycarbonyl (Fmoc) group using piperidine yielded the final FluoDA probes. The detailed procedures for the synthesis of TMR-0F/2F/4F, FluoDA probes, and FluoEosinY are given in the SI Note.Electrostatic potential (ESP) maps
Density Functional Theory (DFT) with the B3LYP and 6-311 + G (d, p) functional/basis set combination was used to generate all images.54,55 Gaussian 16 was used for the numerical data derived from a quantum mechanical analysis, and GaussView was used to process the data and generate ESP maps.56,57 Each ESP map used consistent isovalues (0.0004) and color scaling from −0.02 (red) to 0.02 (blue) for all the compounds.Molecular docking
The crystal structure of saPBP4 (PDB ID: 5TW8) was prepared with the Protein Preparation Wizard in the Schrödinger suite,58 following the standard protocol. This included adding hydrogens, assigning bond orders, filling missing side chains and loops, optimizing the H-bond network, and minimizing the system with the OPLS-2005 force field until the root-mean-square deviation of heavy atoms converged to 0.30 Å.59 Water molecules that formed more than three hydrogen bonds with ligand or receptor atoms were retained. Owing to the high molecular flexibility (with more than 10 rotatable bonds) with the probe, which challenges traditional docking methods, we employed SurfDock, a deep learning-based approach that has demonstrated superior performance for flexible ligands.60 This method demonstrates superior performance for highly flexible ligands. The binding pocket and surface information were defined based on the co-crystallized ligand. We then sampled 40 ligand conformations using SurfDock and selected the top-ranked pose according to the model's scoring function as the final docked structure.In vitro fluorescence and absorbance spectra measurements
For fluorescence turn-on measurements, 2 µM probes were incubated in HEPES buffer (50 mM HEPES, 120 mM NaCl, pH = 7.3) with or without 0.2% SDS at 25 °C for 2 hours, in a final volume of 55 µL in non-binding 96-well plates (In vitro scientific). Fluorescence emission spectra were recorded on a microplate reader (Spark 20 M, Tecan). The detailed parameters, including excitation and emission wavelength, bandwidth, and step size, were summarized in Table S3.For UV-vis absorption measurements, each probe (2 µM) was individually incubated in HEPES buffer (50 mM HEPES, 120 mM NaCl, pH 7.3) with or without 0.2% SDS at 25 °C for 2 h. The absorbance spectra were recorded using a 700 µL quartz cuvette on a U3900H spectrophotometer (Hitachi).
Measurements of absorbance spectra in water–dioxane mixtures
Solutions of the rhodamine-based probes (2 µM) were prepared in a series of water–dioxane mixtures with dioxane from 10% to 80%, v/v. Absorbance spectra were recorded using a 96-well plate (in vitro scientific) on a microplate reader (Spark 20 M, Tecan). All measurements were performed at 25 °C. All measurements were carried out at 25 °C. Absorbance values were normalized to their respective maximum absorbance. Finally, the normalized absorbance was plotted against the dielectric constant of the corresponding water-dioxane mixture.Quantum yield measurements
The quantum yields for fluorescence were calculated by comparison of the integrated area of the corrected emission spectrum of the samples with that of a solution of quinine sulfate in 0.05 M H2SO4 (Φ = 0.52),61 fluorescein in 0.1 M NaOH (Φ = 0.91),62 rhodamine 6G in ethanol (Φ = 0.94),63 rhodamine B in HEPES buffer (Φ = 0.31),64 rhodamine 101 in methanol (Φ = 1.0),65 Nile blue in ethanol (Φ = 0.27).66 The concentration of the reference was adjusted to matching the absorbance of the test sample. Quantum yield of the probe (2 µM) was measured in HEPES buffer (50 mM HEPES, 120 mM NaCl, 0.2% SDS, pH = 7.3). The quantum yields were calculated with the expression in eqn (1). Ax and As are the absorbance of probes in HEPES buffer and the standard in corresponding solvent, respectively. Sx and Ss are integrated fluorescence emission corresponding to probes and the standard, respectively. nx and ns are the refractive indexes of the solvent for probes and the standard. Φs is the quantum yield of the standard. The detailed excitation and emission wavelength for each probe were detailed in Table S4.
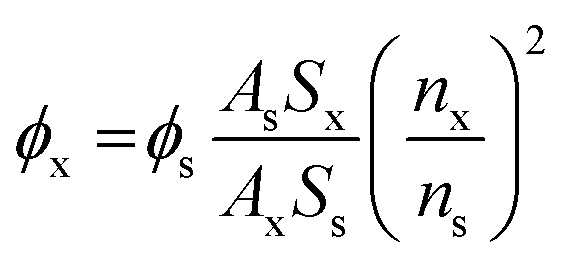 | (1) |
Dynamic light scattering (DLS) measurements
Two µM probes were incubated in HEPES buffer (50 mM HEPES, 120 mM NaCl, pH = 7.3) with and without 0.2% SDS at 25 °C for 2 h, respectively. The particle size of the buffer was then measured using a Zetasizer µV system.Quantification of the aggregation
Two µM probes were incubated in HEPES buffer (50 mM HEPES, 120 mM NaCl, pH 7.3) at 25 °C for 2 h. The pellet and supernatant fractions were then separated via centrifugation at 16000×g for 1 h at 25 °C. After incubation in HEPES buffer (50 mM HEPES, 120 mM NaCl, 0.2% SDS, pH 7.3) at 25 °C for 1 h, the fluorescence intensity of both fractions was measured using a microplate reader (Spark 20 M Tecan).
Plasmid construction
DNA fragments encoding SaPBP1[R65–S713], SaPBP2 [M59–S716], SaPBP3 [A46–K691], and SaPBP4 [Y21–Q383], each lacking the predicted transmembrane domain, were amplified from the S. aureus NCTC8325 genome using the appropriate primers. The resulting PCR products were cloned into pET51b(+) vector to generate recombinant constructs harboring DNA sequencing encoding an N-terminal Strep-tag and a C-terminal His-tag (Table S5). Point mutations were introduced using the Mut Express II Fast Mutagenesis Kit V2 (Vazyme) with the corresponding primers (Table S6) and all mutations were verified by Sanger sequencing.Purification of PBPs and their variants
The SaPBPs, and their variants were expressed in E. coli Rosetta strain. The bacterial culture was incubated at 37 °C to reach an optical density at 600 nm (OD600) of 0.8. The protein expression was induced by adding 0.5 mM isopropyl β-D-thiogalactopyranoside (IPTG) and allowed to grow at 16 °C overnight while shaking at 220 rpm. After 20 h, the bacteria were harvested by centrifugation at 4000×g for 10 min and lysed by sonication in the presence of 1 mg mL−1 lysozyme and 1 mM phenylmethylsulfonyl fluoride (PMSF). The cell lysate was cleared by centrifugation (50000×g, 30 min, 4 °C) and the supernatant was collected. All proteins were first purified by Ni-NTA affinity chromatography (Cytiva), followed by Strep-Tactin purification (IBA Lifesciences), both performed according to the respective manufacturers' protocols. The eluent containing SaPBPs was concentrated using Amicon Ultra Centrifugal Filter (30 kDa MWCO) and the concentration was determined by measuring the absorbance at 280 nm. The purified protein was diluted to the desired concentration with PBS buffer (10 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH = 7.4), aliquoted, flash-frozen in liquid nitrogen, and stored at −80 °C. The activity of SaPBPs was confirmed via labeling with β-lactam-fluorophore conjugates,67 with subsequent verification by SDS-PAGE (Fig. S21).
Transpeptidase activity assay
In a low-protein binding 1.5 mL tube, 1 µL of the tripeptide–coumarin conjugate (PC, 77.4 mM in DMSO) was added to the Kahne buffer (12.5 mM HEPES, 2 mM MnCl2, 0.25 mM Tween-80, 120 mM NaCl, pH = 7.4). SaPBPs were then added to adjust the final concentration of SaPBPs to 0.4 mg mL−1 and the mixture was incubated at 25 °C for 30 min. Subsequently, 0.5 µL of TMR-0F/2F/4F stock solution (1.8 mM in DMSO) was introduced, bringing the total volume to 60 µL, and incubation continued at 25 °C for 1 h. The reaction was stopped by immediate precipitation of the SaPBP with 500 µL ice-cold methanol. After centrifugation at 16000×g for 2 min, the supernatant was transferred to a clean tube and analyzed by reversed-phase HPLC (mobile phase of 0.1% TFA in water/acetonitrile, a 5–80% acetonitrile gradient over 35 min), monitoring absorbance at 540 nm. The enzymic products were validated by HRMS (Fig. S22).
Fluorescence polarization assay (FP)
Fifty nM TMR-0F/2F/4F or TADA was incubated with varying amounts of SaPBPs for 1 hour at 25 °C in the Kahne buffer supplemented with 0.5 mg·mL−1 BSA. Assays were performed in non-binding black 96-well plates (in vitro scientific) with a final volume of 55 µL and were measured on a microplate reader (Spark 20 M, Tecan) with excitation at 535 nm (bandwidth, 10 nm) and emission 585 nm (bandwidth, 10 nm). Fluorescence polarization (FP) was calculated according to eqn (2).
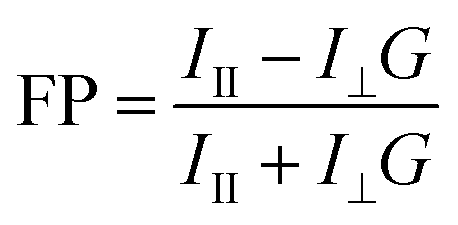 | (2) |
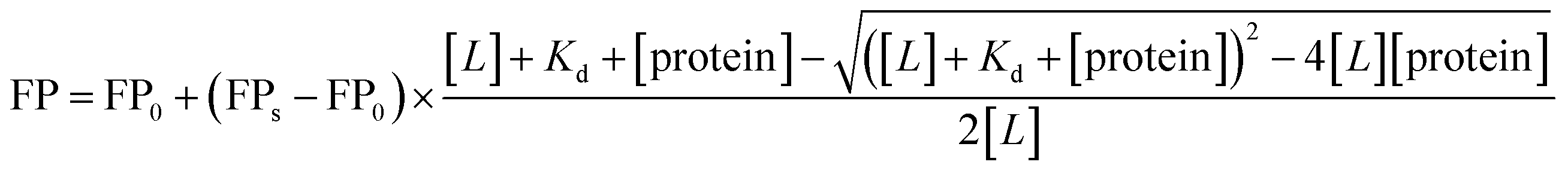 | (3) |
Isothermal titration calorimetry (ITC)
ITC experiments were performed using an ITC200 microcalorimeter instrument (MicroCal). The cell was loaded with 240 µL of 10 µM probe dissolved in PBS. SaPBP4 (150 µM, 40 µL) prepared in PBS buffer (pH = 8.12) was titrated into the cell through a syringe (2 µL per injection, except for the first injection of 1 µL). The titration curves were fitted to a one-site binding model using MicroCal PEAQ-ITC Analysis Software to obtain the dissociation constant (Kd), enthalpy change (ΔH), and entropy change (ΔS).Strains and growth conditions
Bacterial strains and their corresponding growth conditions are listed in Table S7. Frozen stocks (−80 °C in 30% glycerol-supplemented medium) were streaked onto fresh agar plates containing the corresponding growth medium and incubated overnight at specified temperature (30 °C or 37 °C). Single colonies were then inoculated into 3 mL of the appropriate liquid medium and grown with shaking (220 rpm) at the specified temperature (30 °C or 37 °C).Bacteria growth curves
The S. aureus (NCTC8325), S. aureus (USA300 LAC), and E. faecium (ATCC 51559) cells were grown to the stationary phase and then diluted in fresh TSB or BHI medium to an OD600 of ∼0.02, respectively. TMR-4F or FluoEosinY was supplemented to final concentrations of 0 µM (only DMSO), 1, 3, 10, 30, or 100 µM. 0.1 mL of each diluted cultures were transferred into a 96-well microplate to measure absorbance at 600 nm every 10 min for 12 h using a Tecan Spark 20 M plate reader at 37 °C, with shaking between measurements to ensure proper mixing.Probe labeling and imaging preparation
For live-cell imaging, 3 mg of low-melting-point agarose was dissolved in 100 µL of the appropriate growth medium by heating at 70 °C for 1 h, then maintained at 50 °C. Probe solutions were prewarmed to 50 °C and mixed with the molten agarose to obtain a 1.5% gel containing the desired final probe concentration (Table S7). Approximately 0.8 µL of live bacterial culture was applied to the gel pad, and cells were allowed to settle and grow at 30 °C or 37 °C for 1 h before confocal imaging.For fixed-cell imaging, exponentially growing cultures were centrifuged at 10000×g for 1 min at 25 °C and resuspended in 200 µL of fresh medium. Probe was added to reach the final concentration listed in Table S8, followed by incubation with rocking at the appropriate temperature for 1 h. To terminate labeling and fix the cells, 460 µL of prechilled 100% ethanol was added first, followed by the subsequent addition of 1 mL of cold 70% ethanol. The mixture was incubated on ice for 15 min, then washed three times with 1× PBS to remove residual ethanol and unlabeled probe. The fixed cells were resuspended in 20–50 µL of 1× PBS to a final OD600 of approximately 3.0. For imaging, 0.8 µL of the fixed cell suspension was applied to a gel pad prepared with 2% agarose in 1× PBS.
Labeled probe hydrolysis curve measurement
Probe labeling was performed as described for fixed-cell imaging. After labeling, S. aureus cells were washed twice with 1× PBS to remove unbound probes and resuspended in TSB medium for continued incubation at 37 °C with rocking to allow probe hydrolysis. Cells were fixed at different hydrolysis time points (0, 10, 30, and 60 min) using the same ethanol fixation procedure described above and imaged immediately. Cell segmentation was performed using Cellpose 4.0,68 and mean cellular fluorescence intensities were quantified for all cells at each time point to generate probe hydrolysis curves.Photosensitizer labeling and irradiation process
Exponentially growing bacterial cells were incubated with FluoEosinY for 30 min at 37 °C. Cells were then washed twice with 1× PBS to remove unlabeled probe and resuspended in 1× PBS to a final OD600 of approximately 1.0. The adjusted cultures were transferred into a 96-well microplate, with each well containing 0.1 mL of the culture. To evaluate the phototoxicity of the photosensitizer against bacterial cells, half of the wells were irradiated with green light (519/26 nm, 4.5 mW·cm−2) for 30 min; the remaining wells were maintained in the dark and served as controls.Image acquisition
Confocal imaging was conducted on a Leica STELLARIS confocal microscope, equipped with an HC PL APO 100× OIL CS2 objective (NA = 1.4), powered by white light lasers and HyD S Detectors. The specific microscopy parameters, including excitation power and wavelengths, and emission wavelengths, are thoroughly summarized in Table S8.Signal-to-background ratio (SBR) calculation
To calculate the fluorescence intensity at the septum and the background, a circular ROI with a 4-pixel radius in the center of each septum and a close-by region outside of the cells was selected. The integrated gray scale value of these two regions was calculated in Fiji69 as the signal intensity (S) and background (B), respectively. The SBR was calculated by dividing S by B.Fluorescence imaging of FluoEosinY-labeled cells
For endpoint imaging, methicillin-resistant S. aureus (MRSA, USA300 LAC) cells labeled with FluoEosinY were either irradiated with green light (519/26 nm, 4.5 mW·cm−2) for 30 min or kept in the dark as a control. Both groups were stained with Potomac Red (10 µM) and Hoechst 34580 (10 µg mL−1) at 37 °C for 30 min. After staining, cells were collected by centrifugation (10000×g, 1 min), washed, and resuspended in 1× PBS. An aliquot (0.8 µL) of the stained cell suspension was mounted on a PBS-based agarose gel pad and imaged using a Leica STELLARIS confocal microscope. Fluorescence signal from FluoEosinY, Potomac Red, and Hoechst 34580 was acquired using the settings detailed in Table S8.
For real-time imaging, MRSA cells labeled with FluoEosinY (no irradiation) were stained under the same conditions as above. After staining and resuspension in 1× PBS, 0.8 µL of the cell suspension was applied to a PBS-based agarose gel pad. Time-lapse imaging was performed using a Leica STELLARIS confocal microscope under continuous 528 nm laser excitation. Fluorescence signal from Potomac Red and Hoechst 34580 was collected at defined time intervals (0, 30, 60, 90, and 120 s) as described in Table S8.
Quantification of membrane compromise and DNA-to-cell area ratio
To assess membrane integrity, cells exhibiting compromised membranes were counted from fluorescence images acquired in the membrane dye channel and divided by the total cell number. The cell and DNA-occupied area were calculated by automated segmentation in the membrane and DNA stained channel using Cellpose 4.0.68 For each cell, the ratio of DNA-to-cell area was calculated.TEM imaging of FluoEosinY-labeled MRSA cells
MRSA cells labeled with FluoEosinY were either irradiated with green light (519/26 nm, 4.5 mW·cm−2, 30 min) or kept in the dark as a control. Both groups were fixed in 2.5% glutaraldehyde in 1× PBS at 4 °C overnight. After washing with 0.1 M phosphate buffer (pH = 8.0), cells were post-fixed with 2% osmium tetroxide at 4 °C overnight in the dark. Samples underwent dehydration through a graded ethanol series (30%, 50%, 70%, 80%, 95%, 100%), with each step carried out at 25 °C for 20 min. Afterward, they were incubated overnight in 70% ethanol at 4 °C. Dehydrated samples were washed three times with acetone, then infiltrated with epoxy resin-acetone mixtures of increasing concentrations (25%, 50%, 75%), with each concentration step maintained for 24 h. Subsequently, cells were incubated in 100% epoxy resin at 25 °C for 24 h, followed by overnight polymerization at 70 °C. Ultrathin sections were first stained with uranyl acetate and then with lead citrate, followed by imaging using a FEI TECNAI G2 F20 transmission electron microscope.Transcriptomic analysis of FluoEosinY-labeled MRSA cells
MRSA cells labeled with FluoEosinY were either irradiated with green light (519/26 nm, 4.5 mW·cm−2, 30 min) or kept in the dark as a control. The transcriptomes of the two groups were subjected to analysis at Bioyi Biotechnology Co., Ltd. Briefly, total RNA was extracted using TRIzol reagent, and RNA quality and integrity were assessed before rRNA depletion. Sequencing libraries were prepared using the ALFA-SEQ RNA Library Prep Kit II and sequenced on the Illumina NovaSeq X Plus platform (150 bp paired-end reads). After quality filtering with fastp, clean reads were aligned to the reference S. aureus genome. Differential gene expression analysis was performed using DESeq2, and gene ontology (GO) enrichment analysis was conducted using ClusterProfiler.Minimum inhibitory concentration (MIC) assay
The MIC assay was performed according to Clinical and Laboratory Standards Institute (CLSI) guidelines with minor modifications. Antibiotics (kanamycin, chloramphenicol, ciprofloxacin, erythromycin) were distributed in 96-well plates by sequential dilution. MRSA cells with or without prior PDI treatment were adjusted to ∼106 CFU mL−1 and added to each well. Plates were incubated at 37 °C for 24 h. The MIC was defined as the lowest antibiotic concentration that completely inhibited visible bacterial growth after incubation.Determination of intracellular antibiotic accumulation after PDI treatment
MRSA cells with or without prior PDI treatment were incubated with ciprofloxacin (200 µg mL−1) at 25 °C under gentle rotation for 2 h to allow antibiotic uptake. After two washes with 1× PBS to remove extracellular antibiotic, cells were resuspended in 1 mL of 1× PBS and adjusted to an OD600 value of 5.0. The cells were re-centrifuged, and then resuspended in 400 µL of water–methanol solution (4:6, v/v). Subsequently, the cells were lysed via bead-beating for 15 min, followed by ultracentrifugation to collect the supernatant. This entire extraction cycle was repeated twice, and the three batches of supernatants were pooled together. The pooled supernatant was subjected to HPLC analysis to determine the intracellular concentration of ciprofloxacin.
Bacterial viability staining assay
MRSA (USA300) and VRE (ATCC 51559) cells labeled with FluoEosinY were either irradiated with green light (519/26 nm, 4.5 mW·cm−2, 30 min) or kept in the dark as a control. Both groups were stained with PI at a final concentration of 30 µg mL−1 and incubated at 37 °C for 30 min. The cells were then harvested via centrifugation and resuspended in 1× PBS. For confocal imaging, 0.8 µL of the stained bacterial suspension was placed on a PBS-based agarose gel pad. Fluorescence signals from the FluoEosinY and PI were acquired according to the parameters detailed in Table S8. For flow cytometry analysis, the fluorescence intensity of PI was measured by collecting over 2000000 events per sample. Data were presented as overlaid histograms of fluorescence intensity (height), and PI fluorescence was detected using the PC5.5 channel (690/50 nm).
In vitro antimicrobial assay
MRSA and VRE cells labeled with FluoEosinY were either irradiated with green light (519/26 nm, 4.5 mW·cm−2, 30 min) or kept in the dark as a control. After the above treatment, both groups were adjusted to an OD600 value of 1.0, then serially diluted 105-fold in 1× PBS. A 50 µL aliquot of the diluted bacterial suspension was spread onto solid LB agar plates, which were then incubated at 37 °C for 20 h. Colony-forming units (CFU) were quantified by counting visible colonies, and the CFU count was used to reflect bacterial viability after treatment.Photodynamic inactivation resistance assays
To assess the development of resistance to photodynamic inactivation (PDI) using FluoEosinY, ten consecutive rounds of PDI treatment were performed. After each cycle of green light irradiation, the cell cultures were plated with series of dilution for CFU calculation. A single survived colony was cultured in fresh medium at 37 °C, 220 rpm for 20 h. When the OD600 reaches 1.0, the culture was then subjected to the next round of PDI under identical FluoEosinY concentration and irradiation conditions. Three independent assays were conducted.Biofilm irradiation and imaging
MRSA cells were inoculated into 96-well plates containing TSB + 0.5% glucose and cultured for 60 h to allow the formation of mature biofilms. After washing with 1× PBS to remove non-adherent bacteria, biofilms were then incubated with or without 30 µL FluoEosin Y for 30 min. Following incubation, samples were irradiated with green light (519/26 nm, 4.5 mW cm−2) for 20 min. After irradiation, biofilms were stained with PI and imaged using confocal laser scanning microscopy.Cell culture
HeLa cell lines were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) with L-Glutamine (Solarbio), 1 mM pyruvate supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Lonsera). All cells were grown at 37 °C, 5% CO2.Cell viability assay
Cell viability of FluoEosinY was evaluated using the CCK-8 Cell Proliferation and Cytotoxicity Assay Kit (Beyotime Biotech Inc). HeLa cells were seeded into sterile 96-well plates at 100 µL per well and pre-incubated for 24 hours at 37 °C under 5% CO2. After removing the medium by gently washing with HBSS, 110 µL of fresh medium containing 10 µM Eosin Y or FluoEosinY at 2, 5, 10, 20, or 30 µM were added to each well and the plate was incubated for 30 min. Subsequently, 10 µL of CCK-8 reagent was added to each well, followed by another 1 h incubation at 37 °C. The absorbance at 450 nm was measured with a microplate reader.Evaluation of labeling specificity of FluoEosinY towards HeLa cells
HeLa cells were co-incubated with 1 µg mL−1 Hoechst 33342 and 10 µM Eosin Y, FluoEosinY, or acEosin Y (a cell-permeable Eosin Y derivative),70 for 30 min, respectively. MRSA cells were incubated with 10 µM FluoEosinY for 30 min. After washing with 1× PBS twice, the cells were imaged using Leica STELLARIS confocal microscope under identical imaging parameters.
Mice
Female BALB/c mice aged six weeks were used for all experiments in vivo. Mice were housed in a specific pathogen-free environment with a 12 h light/dark cycle at 22 ± 2 °C. All animals handling procedures were approved by the Institutional Animal Care Committee of the Institute of Health and Medicine, Hefei Comprehensive National Science Center (Permit No.: IHM-AP-2025-070).In vivo antimicrobial assay
Circular wounds (8 mm in diameter) were created on the dorsal surface of BALB/c mice, and 50 µL of an MRSA suspension (1 × 108 CFU mL−1) was applied to each wound. The inoculum was retained using surgical tape (Hynaut, China) for secure fixation. After 24 h, wounds were rinsed with 10 mM 1×PBS and treated with 100 µL of either FluoEosinY (30 µM) or mupirocin (48.75 µg mL−1, a clinically used antibiotic for cutaneous bacterial infections). For the aPDT intervention, the FluoEosinY (+light) group was irradiated with green LED light (519/26 nm, 4.5 mW·cm−2) for 20 minutes, paused for 10 min, and then subjected to a second 20 min irradiation to induce ROS generation for antibacterial activity; in contrast, the FluoEosinY (−light) group was maintained in the dark. Wound photographs were captured at each predetermined treatment time point (days 1, 3, 7, and 9 post-treatment). The CFU in wound tissues was quantified at 3 days post-treatment. Additionally, H&E staining was conducted on wound tissue samples collected from each group at 9 days post-treatment.Biosafety evaluation in vivo
Circular wounds (8 mm in diameter) were created on the dorsal skin of 6–8 weeks old female BALB/c mice. Each wound was treated with 100 µL of FluoEosinY (30 µM) for 1 hour, with PBS alone serving as the control. On day 9 post treatment, whole blood and plasma were collected for routine hematological analysis including blood parameters (WBC, RBC, HGB, PLT, HCT, LYMPH, MONO, NEUT) as well as ALT, AST, BUN, and CRE. Mice were then euthanized, and major organs (heart, liver, spleen, lungs, and kidneys) were excised, fixed overnight in 4% paraformaldehyde, paraffin embedded, and sectioned for hematoxylin and eosin (H&E) staining and histological examination.Histological analyses
Mouse skin tissues were collected, fixed in 4% paraformaldehyde for 24 h, embedded in paraffin, and sectioned. Paraffin sections were stained with hematoxylin and eosin (H&E) to detect skin morphological changes. Mice skin tissues were dehydrated with 30% sucrose solution, paraffin-embedded, sectioned (5 µm thick), and subsequently dewaxed. Sections were stained sequentially with haematoxylin for 3–8 minutes (Baso, China) and Eosin for 1–3 minutes (Baso, China). Following staining, sections underwent dehydration, mounting, and air-drying before microscopic examination and photography. All sections were observed using the TissueFAXS viewer (TissueGnostics, Austria).Statistics
Data for in vitro titrations were from three independent replicates and shown as the mean ± s.d. For all of the imaging experiments, the number of cells and repetitions per condition are detailed in Table S8. Statistical significance was assessed using Student's t-test with the corresponding p values displayed in the relevant figures.Large language model
During the preparation of this study, the authors used ChatGPT Version 4.0 to refine the language. Following the use of this tool, the authors reviewed and edited the content as necessary, and assume full responsibility for the content of this publication.Author contributions
Conceptualization: L. X., X. Y.; methodology: X. W., L. D., J. L., J. L., Y. Y., H, S., H, W., J. W., X. L., J. L., D. C., L. X., X. Y.; S. L.; investigation: X. W., L. D., L. X., X.Y., S. L. software: X. W, L. D., J. W., X. L., D. C.; formal analysis: X. W., L. D., L. X., X. Y.; writing – original draft: L. X., X. Y.; writing – review & editing: X. W., L. D., L. X., X. Y.; supervision: L. X., X. Y.; funding acquisition: H. W., L. X., X. Y.Conflicts of interest
The authors declare no competing interests.Data availability
The supporting data has been provided as part of the supplementary information (SI). Supplementary information: Fig. S1–S22, Tables S1–S8, NMR spectra and further experimental details. See DOI: https://doi.org/10.1039/d6sc00548a.Acknowledgements
The authors thank other members in Yang, and Xue's laboratory for their valuable advice and discussions. The authors thank Dr Ethan C. Garner, Dr Mariana G. Pinho, Dr Hongwu Qian, Dr Jiangliu Yu, and Dr Ting Xue for providing bacteria strains and plasmids. The authors thank Dr Jishen Zheng for the measurement of the mass spectra. The authors thank Dr Luke D. Lavis for Potomac dyes for the bacterial membrane labeling. The authors would like to acknowledge the Confocal Imaging Unit at the Core Facility Centre for Life Science, University of Science and Technology of China. The Laboratory research staff (Zhenbang Liu, M. S.) contributed valuable technical expertise and assistance to this project. This work was partially carried out at the Instruments Center for Physical Science, University of Science and Technology of China. This work was supported by the Center for Advanced Interdisciplinary Science and Biomedicine of IHM (QYPY20220016 to L. X. and BJ9100000056 to X. Y.), the National Natural Science Foundation of China (32270035 to X. Y. and 22377115 to L. X.), the Fundamental Research Funds for the Central Universities (WK9100000063 to X. Y., WK9100000077 to H. W.), Guangxi Bagui Excellent Young Scholars Fund to S. L. and China Postdoctoral Science Foundation (2024M763147, GZC20241650 to H. W.).References
- M. Naghavi, et al., Global burden of bacterial antimicrobial resistance 1990-2021: a systematic analysis with forecasts to 2050, Lancet, 2024, 404, 1199–1226 CrossRef PubMed.
- O. Kocaoglu and E. E. Carlson, Progress and prospects for small-molecule probes of bacterial imaging, Nat. Chem. Biol., 2016, 12, 472–478 CrossRef CAS PubMed.
- B. Zhang, et al., Synthesis of vancomycin fluorescent probes that retain antimicrobial activity, identify Gram-positive bacteria, and detect Gram-negative outer membrane damage, Commun. Biol., 2023, 6, 16 CrossRef PubMed.
- K. Y. Wang, et al., Recent advances in organic molecule fluorescent probes for microbial imaging, J. Mater. Chem. B, 2025, 13, 10700–10714 RSC.
- J. E. Hudak, D. Alvarez, A. Skelly, U. H. von Andrian and D. L. Kasper, Illuminating vital surface molecules of symbionts in health and disease, Nat. Microbiol., 2017, 2, 8 Search PubMed.
- L. Y. Lin, Y. H. Du, J. Song, W. Wang and C. Y. Yang, Imaging commensal microbiota and pathogenic bacteria in the gut, Acc. Chem. Res., 2021, 54, 2076–2087 CrossRef CAS PubMed.
- M. Klausen, M. Ucuncu and M. Bradley, Design of photosensitizing agents for targeted antimicrobial photodynamic therapy, Molecules, 2020, 25, 5239 CrossRef CAS PubMed.
- Y. F. Zheng, et al., Development and Applications of D-Amino Acid Derivatives-based Metabolic Labeling of Bacterial Peptidoglycan, Angew. Chem., Int. Ed., 2024, 63, 26 Search PubMed.
- E. Kuru, et al., In Situ Probing of Newly Synthesized Peptidoglycan in Live Bacteria with Fluorescent D-Amino Acids, Angew. Chem., Int. Ed., 2012, 51, 12519–12523 CrossRef CAS PubMed.
- A. J. F. Egan, J. Errington and W. Vollmer, Regulation of peptidoglycan synthesis and remodelling, Nat. Rev. Microbiol., 2020, 18, 446–460 CrossRef CAS PubMed.
- K. Bush and P. A. Bradford, beta-Lactams and beta-Lactamase Inhibitors: An Overview, Cold Spring Harb. Perspect. Med., 2016, 6, a025247 CrossRef PubMed.
- P. D. A. Rohs and T. G. Bernhardt, Growth and division of the peptidoglycan matrix, Annu. Rev. Microbiol., 2021, 75, 315–336 CrossRef CAS PubMed.
- X. Q. Fan, et al., Intravital imaging of translocated bacteria via fluorogenic labeling of gut microbiota in situ, Proc. Natl. Acad. Sci. U. S. A., 2025, 122, e2415845122 CrossRef CAS PubMed.
- E. Kuru, S. Tekkam, E. Hall, Y. V. Brun and M. S. Van Nieuwenhze, Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ, Nat. Protoc., 2015, 10, 33–52 CrossRef CAS PubMed.
- B. J. Ignacio, T. Bakkum, K. M. Bonger, N. Martin and S. van Kasteren, Metabolic labeling probes for interrogation of the host-pathogen interaction, Org. Biomol. Chem., 2021, 19, 2856–2870 RSC.
- Y. P. Hsu, et al., Fluorogenic D-amino acids enable real-time monitoring of peptidoglycan biosynthesis and high-throughput transpeptidation assays, Nat. Chem., 2019, 11, 335–341 CrossRef CAS PubMed.
- Y. P. Hsu, X. Meng and M. S. VanNieuwenhze, Methods for visualization of peptidoglycan biosynthesis, Methods Microbiol., 2016, 43, 3–48 CAS.
- L. H. Ding, et al., Fluorogenic Probes for Real-Time Tracking of Bacterial Cell Wall Dynamics with Nanoscopy, ACS Nano, 2025, 19, 14389–14403 CrossRef CAS PubMed.
- R. Wiench, J. Fiegler-Rudol, K. Grzech-Lesniak, D. Skaba and J. Arnabat-Dominguez, Photodithazine-mediated antimicrobial photodynamic therapy: A systematic review of efficacy and applications, Int. J. Mol. Sci., 2025, 26, 8049 CrossRef CAS PubMed.
- W. Y. Zhou, X. Q. Jiang and X. Zhen, Development of organic photosensitizers for antimicrobial photodynamic therapy, Biomater. Sci., 2023, 11, 5108–5128 RSC.
- E. Nestoros, A. Sharma, E. Kim, J. S. Kim and M. Vendrell, Smart molecular designs and applications of activatable organic photosensitizers, Nat. Rev. Chem., 2025, 9, 46–60 CrossRef CAS PubMed.
- Q. Yu, et al., Photochemical strategies toward precision targeting against multidrug-resistant bacterial infections, ACS Nano, 2024, 18, 14085–14122 CrossRef CAS PubMed.
- N. Kwon, H. Y. Weng, M. A. Rajora and G. Zheng, Activatable photosensitizers: From fundamental principles to advanced designs, Angew. Chem., Int. Ed., 2025, 64, e202423348 CrossRef CAS PubMed.
- Y. P. Hsu, G. Booher, A. Egan, W. Vollmer and M. S. VanNieuwenhze, D-amino acid derivatives as in situ probes for visualizing bacterial peptidoglycan biosynthesis, Acc. Chem. Res., 2019, 52, 2713–2722 CrossRef CAS PubMed.
- F. Hu, et al., Visualization and in situ ablation of intracellular bacterial pathogens through metabolic labeling, Angew Chem. Int. Ed. Engl., 2020, 59, 9288–9292 CrossRef CAS PubMed.
- E. Y. Yu, et al., One-step light-up metabolic probes for discrimination and killing of intracellular bacteria, Mater. Chem. Front., 2022, 6, 450–458 RSC.
- C. G. Bon, et al., Structural and kinetic analysis of the monofunctional Staphylococcus aureus PBP1, J. Struct. Biol., 2024, 216, 11 CrossRef PubMed.
- E. Sauvage, F. Kerff, M. Terrak, J. A. Ayala and P. Charlier, The penicillin-binding proteins: structure and role in peptidoglycan biosynthesis, FEMS Microbiol. Rev., 2008, 32, 234–258 CrossRef CAS PubMed.
- S. Han, et al., Structural basis for effectiveness of siderophore-conjugated monocarbams against clinically relevant strains of Pseudomonas aeruginosa, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 22002–22007 CrossRef CAS PubMed.
- C. Contreras-Martel, et al., Crystal structure of penicillin-binding protein 1a (PBP1a) reveals a mutational hotspot implicated in β-lactam resistance in Streptococcus pneumoniae, J. Mol. Biol., 2006, 355, 684–696 CrossRef CAS PubMed.
- H. Zakariassen, et al., Aromatic residues in the catalytic center of chitinase A from affect processivity, enzyme activity, and biomass converting efficiency, J. Biol. Chem., 2009, 284, 10610–10617 CrossRef PubMed.
- A. D. de Araujo, H. N. Hoang, J. Lim, J. Y. W. Mak and D. P. Fairlie, Tuning electrostatic and hydrophobic surfaces of aromatic rings to enhance membrane association and cell uptake of peptides, Angew Chem. Int. Ed. Engl., 2022, 61, e202203995 CrossRef CAS PubMed.
- R. P. Matthews, T. Welton and P. A. Hunt, Competitive pi interactions and hydrogen bonding within imidazolium ionic liquids, Phys. Chem. Chem. Phys., 2014, 16, 3238–3253 RSC.
- Z. Huang, et al., Host-Enhanced Phenyl-Perfluorophenyl Polar-pi Interactions, J. Am. Chem. Soc., 2020, 142, 7356–7361 CrossRef CAS PubMed.
- S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Fluorine in medicinal chemistry, Chem. Soc. Rev., 2008, 37, 320–330 RSC.
- P. L. Purder, et al., Deconstructing protein binding of sulfonamides and sulfonamide analogues, JACS Au, 2023, 3, 2478–2486 CrossRef CAS PubMed.
- V. Navratna, et al., Molecular basis for the role of Staphylococcus aureus penicillin binding protein 4 in antimicrobial resistance, J. Bacteriol., 2010, 192, 134–144 CrossRef CAS PubMed.
- S. Gautam, T. Kim and D. A. Spiegel, Chemical probes reveal an extraseptal mode of cross-linking in Staphylococcus aureus, J. Am. Chem. Soc., 2015, 137, 7441–7447 CrossRef CAS PubMed.
- Y. P. Hsu, et al., Full color palette of fluorescent D-amino acids for in situ labeling of bacterial cell walls, Chem. Sci., 2017, 8, 6313–6321 RSC.
- X. N. Fang, et al., Ether rhodamines with enhanced hydrophilicity, fluorogenicity, and brightness for super-resolution imaging, J. Am. Chem. Soc., 2025, 147, 22253–22267 CrossRef CAS PubMed.
- W. Vollmer, B. Joris, P. Charlier and S. Foster, Bacterial peptidoglycan (murein) hydrolases, FEMS Microbiol. Rev., 2008, 32, 259–286 CrossRef CAS PubMed.
- D. T. Zhai, W. Xu, L. Y. Zhang and Y. T. Chang, The role of “disaggregation” in optical probe development, Chem. Soc. Rev., 2014, 43, 2402–2411 RSC.
- N. Lardon, et al., Systematic tuning of rhodamine spirocyclization for super-resolution microscopy, J. Am. Chem. Soc., 2021, 143, 14592–14600 CrossRef CAS PubMed.
- L. D. Lavis and R. T. Raines, Bright ideas for chemical biology, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed.
- L. Wang, et al., A general strategy to develop cell permeable and fluorogenic probes for multicolour nanoscopy, Nat. Chem., 2020, 12, 165–172 CrossRef CAS PubMed.
- N. Valbuena, et al., Characterization of HMW-PBPs from the rod-shaped actinomycete Corynebacterium glutamicum: peptidoglycan synthesis in cells lacking actin-like cytoskeletal structures, Mol. Microbiol., 2007, 66, 643–657 CrossRef CAS PubMed.
- N. Li, et al., Metabolic labeling strategy boosted antibacterial efficiency for photothermal and photodynamic synergistic bacteria-infected wound therapy, ACS Appl. Mater. Interfaces, 2022, 14, 46362–46373 CrossRef CAS PubMed.
- B. R. Fan, W. Peng, Y. Zhang, P. S. Liu and J. Shen, ROS conversion promotes the bactericidal efficiency of Eosin Y based photodynamic therapy, Biomater. Sci., 2023, 11, 4930–4937 RSC.
- C. K. Spahn, et al., A toolbox for multiplexed super-resolution imaging of the E. coli nucleoid and membrane using novel PAINT labels, Sci. Rep., 2018, 8, 14768 Search PubMed.
- K. Huang, et al., Metallacage- based enhanced PDT strategy for bacterial elimination via inhibiting endogenous NO production, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, 7 Search PubMed.
- S. Benson, et al., Photoactivatable metabolic warheads enable precise and safe ablation of target cells in vivo, Nat. Commun., 2021, 12, 2369 CrossRef CAS PubMed.
- T. Lavoie, K. E. Daffinee, M. L. Vicent and K. L. LaPlante, Staphylococcus biofilm dynamics and antibiotic resistance: insights into biofilm stages, zeta potential dynamics, and antibiotic susceptibility, Microbiol. Spectr., 2025, 13, e0291524 CrossRef CAS PubMed.
- Z. J. Zhou, J. B. Song, L. M. Nie and X. Y. Chen, Reactive oxygen species generating systems meeting challenges of photodynamic cancer therapy, Chem. Soc. Rev., 2016, 45, 6597–6626 RSC.
- W. Kohn, A. D. Becke and R. G. Parr, Density functional theory of electronic structure, J. Phys. Chem., 1996, 100, 12974–12980 CrossRef CAS.
- J. R. Reimers, Z. L. Cai, A. Bilic and N. S. Hush, The appropriateness of density-functional theory for the calculation of molecular electronics properties, Ann. N. Y. Acad. Sci., 2003, 1006, 235–251 CrossRef CAS PubMed.
- M. J. Frisch, et al., Gaussian 16 Rev. A.03, Wallingford, CT, 2016 Search PubMed.
- R. Dennington, T. A. Keith and J. M. Millam, GaussView Version 6, 2016 Search PubMed.
- G. M. Sastry, M. Adzhigirey, T. Day, R. Annabhimoju and W. Sherman, Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments, J. Comput. Aided Mol. Des., 2013, 27, 221–234 CrossRef PubMed.
- E. Harder, et al., OPLS3: A force field providing broad coverage of drug-like small molecules and proteins, J. Chem. Theory Comput., 2016, 12, 281–296 CrossRef CAS PubMed.
- D. Cao, et al., SurfDock is a surface-informed diffusion generative model for reliable and accurate protein-ligand complex prediction, Nat. Methods, 2025, 22, 310–322 CrossRef CAS PubMed.
- R. A. Velapoldi and H. H. Tonnesen, Corrected emission spectra and quantum yields for a series of fluorescent compounds in the visible spectral region, J. Fluoresc., 2004, 14, 465–472 CrossRef CAS PubMed.
- J. Shen and R. D. Snook, Thermal lens measurement of absolute quantum yields Using quenched fluorescent samples as references, Chem. Phys. Lett., 1989, 155, 583–586 CrossRef CAS.
- M. Fischer and J. Georges, Fluorescence quantum yield of rhodamine 6G in ethanol as a function of concentration using thermal lens spectrometry, Chem. Phys. Lett., 1996, 260, 115–118 CrossRef CAS.
- A. K. Gaigalas and L. L. Wang, Measurement of the fluorescence quantum yield using a spectrometer with an integrating sphere detector, J. Res. Nat. Inst. Stand. Technol., 2008, 113, 17–28 CrossRef CAS PubMed.
- K. H. Drexhage, Fluorescence efficiency of laser-dyes, J. Res. Natl. Bureau Stand., Sect. A, Phys. Chem., 1976, 80, 421–428 CrossRef.
- R. Sens and K. H. Drexhage, Fluorescence quantum yield of oxazine and carbazine laser-dyes, J. Lumin., 1981, 24–5, 709–712 CrossRef.
- O. Kocaoglu and E. E. Carlson, Profiling of beta-lactam selectivity for penicillin-binding proteins in Escherichia coli strain DC2, Antimicrob. Agents Chemother., 2015, 59, 2785–2790 CrossRef CAS PubMed.
- M. Pachitariu, M. Rariden and C. Stringer, Cellpose-SAM: superhuman generalization for cellular segmentation, bioRxiv, 2025, 2025, 651001 Search PubMed.
- J. Schindelin, et al., Fiji: an open-source platform for biological-image analysis, Nat. Methods, 2012, 9, 676–682 CrossRef CAS PubMed.
- W. Chyan, H. R. Kilgore, B. Gold and R. T. Raines, Electronic and Steric Optimization of Fluorogenic Probes for Biomolecular Imaging, J. Org. Chem., 2017, 82, 4297–4304 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |