
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAnchoring and activation of catalytic sites on the clusters via intermolecular interactions
Feng
Qian†
a,
Zhihang
Zhao†
a,
Baoyu
Huang†
b,
Zhixiang
Xue
a,
Qinzhen
Li
 a,
Sha
Yang
a,
Jinsong
Chai
*a and
Manzhou
Zhu
*a
a,
Sha
Yang
a,
Jinsong
Chai
*a and
Manzhou
Zhu
*a
aSchool of Materials Science and Engineering and Centre for Atomic Engineering of Advanced Materials, Key Laboratory of Structure and Functional Regulation of Hybrid Materials of Ministry of Education, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, China. E-mail: chaijs@ahu.edu.cn; zmz@ahu.edu.cn
bDepartment Hunan Provincial Key Laboratory of Environmental Catalysis & Waste Recycling, College of Materials and Chemical Engineering, Hunan Institute of Engineering, 411104 Xiangtan, China
First published on 20th April 2026
Abstract
Constructing catalytically active sites on the clusters is a mainstream strategy to optimize their catalytic properties. However, it generally leads to unpredictable variations due to the notoriously sensitive relationship between the adduct group and the structure of metal nanoclusters. In this study, a strategy involving Au–Cd bimetallic clusters was proposed to achieve controllable modification of the cluster surface through Lewis acid-base specific binding between the recognition group and the cluster, thereby forming Au–Cd-pyridine units. Taking the ORR as a model reaction, the results show that the formed Au–Cd-pyridine catalytic sites can significantly enhance the catalytic activity. Furthermore, we explored the catalytic mechanism using ESI-MS, XPS, and SC-XRD, as well as DFT calculations, and revealed that the ternary catalytic centers regulate the adsorption behavior of OOH* intermediates (enhancement and change in adsorption mode from bridge-form to top-form). The pyridine-modified Au27Cd2 catalyst also demonstrates performance enhancement when extending its application to other electrocatalytic reactions, i.e. the NO3RR and HER. Finally, the Au16Cd3 cluster, which contained the Au–Cd-pyridine motifs, was prepared to further confirm the key role of Au–Cd-pyridine binding in enhancing catalytic activity.
Introduction
Clusters, as small-sized nanomaterials with well-defined atomic structures and structural modifiability, provide a platform for molecular design at the atomic level.1–5 Additionally, their precise atomic structure (including the electronic structure) enables the exploration of structure–property relationships, which provide pointers for the design of molecules.6–10 Taking catalytic applications as an example, various strategies, such as alloying, anchoring catalytic sites, desymmetrization, and ligand functionalization, can be employed to enhance their catalytic performance.11–17 The utilization of cluster-based catalysts has unravelled the “black box” issues in various catalytic reactions, pointing the way to the design of highly efficient catalysts.18–25 However, the structure of clusters is highly sensitive,26–28 and it is difficult to realize the controllable modification of the target structure, which limits the preparation of highly efficient cluster catalysts.Developing a strategy to achieve efficient and controllable modification of surface structures of clusters is expected to address the above issues. Specific recognition is an effective strategy to implement controllable modification of the cluster surface structure,29–31 and the challenge lies in how to insert and what kind of target recognition groups to insert. Recent reports on cluster catalysts have shown that most heteroatoms can be doped into gold clusters and exhibit Lewis acid activity, making an important contribution to the enhancement of cluster catalytic activity.32–36 Among the transition heterometallic atoms, Cd atoms show higher activity and can retain their Lewis acid character after doping into the clusters.37 In addition, Cd atoms are mostly reported to occupy the surface positions of the clusters38,39 (catalytic reactions are mostly carried out on the surface of the catalysts), and thus Cd atoms become a candidate for the insertion of target-recognizing groups into the clusters. Subsequently, Lewis bases can be introduced based on acid-base specific binding, thereby enabling effective and controllable modification of the cluster surface.40 In this regard, we hypothesized that this strategy could construct efficient cluster catalysts with targeted structural units.
Herein, the Cd atom was anchored to Au clusters as a target-recognition group, and pyridine activated it via acid-base-specific binding to construct effective catalytic sites (Scheme 1). The ORR catalytic activity of various atomically precise Au–Cd clusters was investigated. First, the catalytic activities before and after artificially adding pyridine into the catalytic system were compared, and the results showed that all the Au–Cd clusters displayed different degrees of improvement in the catalytic activities after the addition of pyridine. The comparison experiments of a series of gold clusters verified that the presence of the Cd atom plays a key role in the catalytic activity enhancement after pyridine addition. Using electrospray ionization-mass spectrometry (ESI-MS) and X-ray photoelectron spectroscopy (XPS), the specific binding between Cd and pyridine was characterized, instead of the original Cd–Cl bond. Furthermore, DFT calculations showed that Au–Cd-pyridine as a catalytically active site unit is critical for the ORR. Finally, we designed a Au16Cd3 catalyst with Au–Cd-pyridine units, and it shows the compared catalytic activity as expected with other Au–Cd clusters with the addition of pyridine.
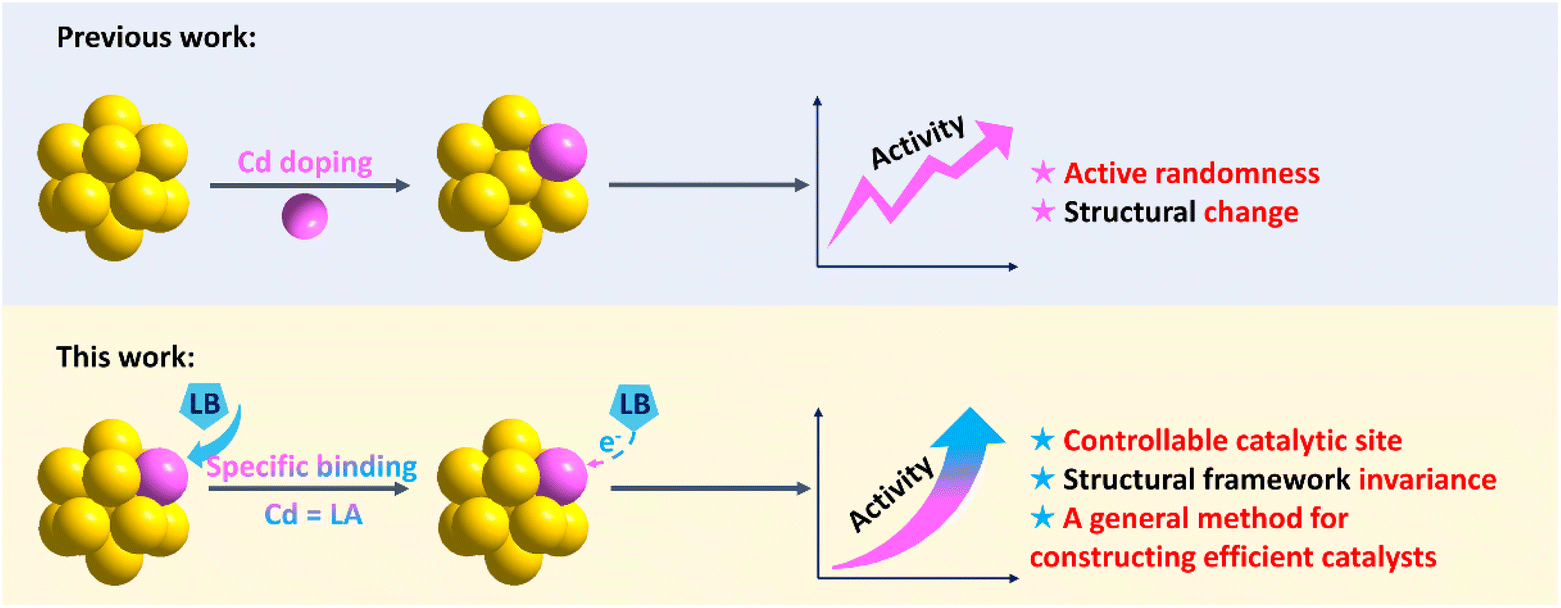 | ||
| Scheme 1 Comparison of the strategies for constructing effective catalytically active sites on the surface of nanoclusters (LA = Lewis Acid; LB = Lewis Base). | ||
Results and discussion
To enhance the catalytic activity of the clusters, the first strategy we employed was the introduction of pyridinophosphine as the functional ligand,41 and a new Au–Cd bimetallic nanocluster (NC) was obtained (the synthesis route is shown in Fig. S1). In its UV-vis spectrum, there are two broad peaks found at 600 and 850 nm, and a series of shoulder peaks at shorter wavelengths can also be observed (Fig. 1A). ESI-MS was used to characterize the composition of this cluster. As shown in Fig. 1B, a major peak at 4241.55 Da was found, which corresponded to [Au27Cd2(SAdm)16(PPh2Py)]2+ (HSAdm = 1-adamantanethiol, PPh2Py = diphenyl-2-pyridylphosphine), of which the isotopic pattern shows a good match with the calculated one (Fig. 1B, inset). Additionally, two weak peaks were also found at 4157.52 and 4259.03 Da, which can be assigned to [Au27Cd2(SAdm)15(PPh2Py)Cl]2+ (indicating the loss of one S-Adm ligand) and [Au27Cd2(SAdm)16(PPh2Py)Cl]2+, respectively. Importantly, the crystal structure of this NC was revealed by X-ray single-crystal diffraction (SC-XRD). It shows a formula of Au27Cd2(SAdm)16(PPh2Py)Cl (Au27Cd2 for short), which is consistent with the ESI-MS, XPS, nuclear magnetic resonance (NMR) and elemental analysis (EA) results (Fig. S2, S3, Tables S1 and S2). | ||
| Fig. 1 Characterization of Au27Cd2. (A) UV-vis spectrum and full crystal structure, (B) ESI-MS spectrum (M = Au27Cd2(S-Adm)16(PPh2Py)Cl), (C) framework (D) core and (E) peripheral protecting group of the Au27Cd2 cluster (color label: cyan/orange = Au, pink = Cd, yellow = S, green = P, bright green = Cl, light blue = N, and gray = C, and parts of C and all H atoms were omitted for clarity). | ||
Au27Cd2 was crystallized in the monoclinic P21/n space group (Table S1). As shown in Fig. 1C, Au27Cd2 is composed of a Au20Cd1 kernel, one Cd(SR)3Cl, four Au1(SR)2, one Au3(SR)4 motif, one bridging SR, and one pyridinophosphine ligand. The Au20Cd1 kernel is composed of one twisted icosahedron and three tetrahedral units via vertex-sharing mode, and two of these tetrahedral units are connected by sharing a gold atom (Fig. 1D). Remarkably, one of the Cd atoms was found to occupy one vertex of the icosahedron, which may be responsible for the distortion of the icosahedron. In addition, we further analyzed the cause of the distortion in the Au27Cd2 core. As shown in Fig. S4, the Au–Cd bond length in Au12Cd1 (M13) did not change significantly, indicating no substantial distortion in the region bonded to Cd. In contrast, the Au–Au bond lengths of the lower Au moiety opposite Cd exhibited considerable variations, ranging from the shortest 2.675 Å to the longest 3.662 Å, suggesting that the core distortion is mainly attributed to the distortion of the lower Au moiety. It is precisely this Au distortion that leads to the large variation in bond lengths of the three connected Au3 tetrahedra. Another Cd atom forms a Cd(SR)3Cl motif wrapping around the top of the NC, and the phosphine ligand was found to surround the bottom of the cluster. Three kinds of motifs, including four Au1(SR)2, one Au3(SR)4, and one bridging SR, were found to connect with both the icosahedron and tetrahedra (Fig. 1E).
Considering that Au27Cd2 is protected by functional pyridinophosphines and the doped heteroatoms, which have been reported to improve the catalytic activity of NCs,42,43 the ORR was employed to detect the catalytic activity of Au27Cd2. Before the electrochemical assessments, Au27Cd2 was loaded on activated carbon with a loading rate of 10% to form the Au27Cd2@C catalyst. The Au27Cd2@C catalyst was then tested by linear sweep voltammetry (LSV) under O2-saturated alkaline (0.1 M KOH) conditions. As shown in Fig. 2A, the current density at 1600 rpm is 3.77 mA cm−2 with a potential at −0.8 V. Additionally, the overall number of electrons transferred in the ORR process for the Au27Cd2@C catalyst was calculated to be 3.0, indicating a mix of two- and four-electron reduction processes (Fig. S5). For comparison, the ORR was carried out using a series of Au–Cd NCs with similar kernels (composed of an icosahedral and several tetrahedral units), and these NCs are diverse in the type of phosphine ligands but do not contain pyridinylphosphine (Fig. S6).44,45 Furthermore, to validate the methodology, the selected Au–Cd clusters were diverse, encompassing variations in charge, ligand type, and ligand functionalization. Additionally, the most critical considerations were ease of synthesis and stability during the catalytic process. As a result, the Au–Cd bimetallic catalysts exhibit comparable ORR catalytic activity regardless of their ligand type. As shown in Fig. S7–S11, the ORR current density of the Au–Cd bimetallic catalyst was in the range of 3.0–3.5 mA cm−2 (−0.8 V) at 1600 rpm. This result suggests that the ligand on the surface plays a small role in influencing the catalytic activity of the ORR. Importantly, in association with precise atomic structure analysis, we suggest that the main reason for the ineffectiveness of the pyridine for Au27Cd2 in promoting ORR performance may be caused by the long distance between the N atom of the pyridylphosphine ligand and the metal atom, which hinders their possible electron transfer.
 | ||
| Fig. 2 Electrochemical correlation test. (A) The linear sweep voltammetry (LSV) curves of the Au27Cd2 cluster at different rpm; (B) the Nyquist curve of the Au27Cd2@C catalyst before and after adding pyridine; (C) comparison of current density at 1600 rpm before and after the addition of pyridine on the Au27Cd2@C catalyst; (D) faradaic efficiencies of NH4+ for the Au27Cd2@C catalyst before and after adding pyridine; (E) LSV curves of the Au27Cd2@C catalyst before and after adding pyridine in 0.5 M K2SO4 solution containing 0.1 M NO3−; (F) Tafel plots of the Au27Cd2@C catalyst before and after adding pyridine; (G) HER polarization curves and (H) Tafel plots for the Au27Cd2@C catalyst before and after adding pyridine; (I) double-layer capacitance of the Au27Cd2@C catalyst before and after adding pyridine measured in 0.5 M H2SO4, where Δj/2 = (ja − jc)/2 (ja and jc denote the anode and cathode current density). | ||
To verify our speculation and also to make sure the pyridine works in catalysis, free pyridine was added to the catalyst before performing the ORR, as it can move arbitrarily and effectively come into contact with the NCs. The Nyquist curve of the Au27Cd2@C catalyst before and after adding pyridine is shown in Fig. 2B, and the charge transfer resistance is smaller for the latter. This suggests that the addition of pyridine enables a stronger electron transfer ability of the Au27Cd2@C catalyst in the ORR. The electrocatalytic ORR performance before and after the addition of free pyridine was evaluated with a series of Au–Cd bimetallic NC catalysts. As shown in Fig. 2C and S12, the current density of most Au–Cd bimetallic cluster catalysts was significantly increased after the introduction of free pyridine. In detail, the current densities at −0.8 V of Au27Cd2@C, Au19Cd3@C, Au24Cd1@C, and Au23Cd1@C increased from 3.77, 3.33, 3.28, and 2.94 mA cm−2 to 4.76, 4.02, 4.00, and 3.80 mA cm−2, respectively, showing an average increase of 0.82 mA cm−2 in the current density (Fig. S12) and a percentage increase > 20% (Table S3). On the other hand, the half-wave potentials of the above gold-cadmium clusters also shift positively upon the addition of pyridine (Table S4), once again confirming pyridine's influence on their catalytic activity. It is worth noting that Au30Cd1@C showed almost no change in current density after the addition of free pyridine. Considering that the Cd atom in the Au30Cd1 nanocluster is located in the kernel,45 this result is reasonable because it is difficult for the pyridine molecule to approach such an inside position. This suggests that the free pyridine can bind with the peripheral Cd atoms to form an effective catalyst for the ORR. For further verification, we introduced some Au NCs46–48 with a similar size to the above Au–Cd NCs. As shown in Fig. S13–S16, the relevant clusters were successfully prepared. Their catalytic results are displayed in Fig. S7, S17–S19 and Table S3. The advantage of Au–Cd bimetallic catalysts in ORR catalysis is noticeable. Moreover, pyridine can only lead to a slight change in the catalytic activity of Au NCs. Subsequently, a series of related complexes was prepared to compare with the catalytic activity of the NCs. As shown in Fig. S20–S24, the catalytic activity of these complexes and activated carbon is even lower than that of Au NCs. It is noteworthy that pyridine has almost no effect on their catalytic activity, and the pyridine on the surface may mask some of the sites, leading to a slight decrease in activity (Fig. S12 and Table S3). The above comparative results indicated that the overall structural frameworks of the NCs play an important role in the ORR and the binding site of pyridine is most likely located on the surface Cd atom of the NCs.
To validate our Lewis base-mediated surface cadmium activation strategy in the Au27Cd2 cluster, we evaluated diverse nitrogen-containing organic compounds (Fig. S25–S26) and also applied the Au27Cd2@C catalyst to multiple reaction systems (Fig. 2D–I and S27–S28), including the nitrate reduction (NO3RR) and hydrogen evolution reaction (HER). The nitrogen-containing organic compounds include piperidine, pyrrolidine, piperazine, diethylamine and triethylamine. They show different promotional effects, but none are stronger than pyridine. And, supplementing Au27Cd2@C with pyridine boosted NH4+ faradaic efficiency by 17% and current density by 47.80% at −1.2 V in the NO3RR, alongside a 92.12% current density increase at −0.6 V in the HER.
To demonstrate that the main influencing factor for pyridine to work is the existence of surface-doping Cd atoms, the structural stability of the Au27Cd2@C catalyst was characterized through XPS, transmission electron microscopy (TEM), and chronoamperometry (i–t curve), collectively confirming its robustness under catalytic conditions (Fig. S29–S31). Then, XPS tests were employed to investigate the valence states of the metal atoms in the NCs. As shown in Table 1, entry 1 and Fig. S32, the binding energies of Au 4f in the Au27Cd2@C catalyst before and after adding free pyridine was almost unchanged with a ΔE of 0.1 eV while the change in binding energy for Cd 3d was 0.25 eV, suggesting that it is the Cd atoms on the surface that bind to the free pyridine and undergo electron transfer. A similar behavior was observed in other Au–Cd NC catalysts (Table 1, entries 2, 3 and Fig. S33), except for Au30Cd1@C. As shown in Table 1, entry 4 and Fig. S33, the binding energies of both Au 4f and Cd 3d are similar in the Au30Cd1@C catalyst before and after the addition of pyridine. The unchanged binding energies of Au 4f were also found in monometallic Au NCs (Table 1, entries 5–7 and Fig. S34). From the XPS results, we suggest that the main reason for the enhancement in ORR performance for pyridine is most probably the electron transfer between the surface-occupying Cd atom in NCs and the N atom from pyridine (the effect of gold and pyridine is relatively weak). This is very meaningful for optimizing the catalytic activity of Au–Cd NCs, which can also lay a solid foundation for the catalytic application of the NCs. Furthermore, ESI-MS was employed to demonstrate the binding between pyridine and Au27Cd2. As shown in Fig. 3A and S35, a new peak attributed to [Au27Cd2 − Cl + py + 2Cs]2+ was found in the ESI-MS result of Au27Cd2 solution after the addition of pyridine (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio). This means the pyridine is present instead of the original Cl in Au27Cd2 to form the Au–Cd-Py unit.
:1 ratio). This means the pyridine is present instead of the original Cl in Au27Cd2 to form the Au–Cd-Py unit.
| Entry | Sample | Cd ΔE (eV) | Au ΔE (eV) |
|---|---|---|---|
| 1 | Au27Cd2@C | 0.25 | 0.1 |
| 2 | Au23Cd1@C | 0.3 | 0.05 |
| 3 | Au19Cd3@C | 0.25 | 0 |
| 4 | Au30Cd1@C | 0 | 0 |
| 5 | Au16@C | — | 0 |
| 6 | Au21@C | — | 0.1 |
| 7 | Au24@C | — | 0.05 |
 | ||
| Fig. 3 Characterization and DFT. (A) ESI-MS of Au27Cd2 NC solution after the addition of pyridine and (B) the DFT calculations of the free energies of the two catalysts of Au27Cd2 NCs and the pyridine group-modified Au27Cd2 ligand in the ORR process. (Color labels: yellow = Au, light purple = Cd, green = S, pink = P, gray = C, red = O, blue = N, and white = H). | ||
We introduced density functional theory (DFT) calculations to further investigate the reasons for the enhancement of the catalytic ORR reduction performance of Au27Cd2 NCs with the help of pyridine. The discrepancy between DFT calculated overpotentials and experimental measurements is often attributed to the inherent limitations of the Computational Hydrogen Electrode (CHE) model and other computational simplifications.49–51 Our calculations show that the overpotential for Au(111) is 1.24 V (Fig. S36), which is in excellent agreement with previous DFT studies by Deng et al.52 This benchmark demonstrates the reliability of our CHE model and confirms that the trends observed for our doped clusters are qualitatively valid, even if the absolute values for the clusters show some deviation due to their unique under-coordinated sites. Free energies of the initial Au27Cd2 and pyridine group-modified Au27Cd2 (Au27Cd2-py) in the ORR process were calculated. As shown in Fig. 3B, the calculations indicated that Au27Cd2-py has a lower overpotential than the Au27Cd2 catalyst, which is 0.88 and 1.05 eV, respectively. Detailed analysis reveals that the OOH* intermediate has a weaker interaction with Cd on Au27Cd2, which is achieved by comparing the intermediate to the base, leading to the maximum Gibbs free energy for the formation of the OOH* intermediate from O2. Therefore, this step is the potential-determining step of the reaction. In addition, simulations in the Au27Cd2 catalyzed system revealed that the O* intermediate shows a bridge adsorption configuration while OH* shows a top adsorption configuration, which leads to the fact that O* needs to break a chemical bond (O–Cd) when it gets H+ and e− to form the OH* intermediate, and results in another large Gibbs free energy barrier (1.03 eV). The comparison showed that the introduction of pyridine resulted in enhanced adsorption of Cd for OOH* intermediates on the Au27Cd2-py catalyst, which lowered the overpotential. Another important aspect is also reflected in the change in the adsorption mode from bridge-form adsorption to top-form adsorption during the formation of O* intermediates, which effectively lowers the Gibbs free energy barrier for the formation of their OH* intermediates. Meanwhile, we conducted Bader charge analysis of the pyridine-modified Au27Cd2 and calculated the charge differential density map (Fig. S37). We found that the introduction of pyridine molecules leads to significant charge redistribution. Specifically, pyridine acts as an electron donor, transferring a considerable amount of electron density (0.14 e) to the nanocluster. This electron injection directly alters the electronic structure of the active site, resulting in higher electron density. Meanwhile, we integrated the absolute ΔG values for *OOH, *O, and *OH adsorption as well as the zero-point energies and entropy corrections into Table S5 to ensure easy access and full reproducibility. This likely optimizes the adsorption strength of ORR intermediates (such as OOH*) on the active sites. Therefore, we believe that the essence of the pyridine-enhanced ORR reduction performance of Au27Cd2 nanocluster catalysts is to regulate the adsorption behavior of its intermediates in the ORR process through the pyridine ligand, which in turn facilitates the overall ORR.
To further confirm that Cd–pyridine binding may be the key to the formation of efficient catalysts, we constructed another new Au–Cd NC in which the peripheral cadmium atoms are coordinated with the nitrogen of the pyridine phosphine. As shown in Fig. S38A, this Au–Cd nanocluster exhibits a shoulder peak at 380–420 nm and a main peak at 512 nm. ESI-MS revealed that it exhibited a major peak at 2909.99 Da, which corresponds to [Au16Cd3(SAdm)9(PPh2Py)3Cl]2+. Additionally, a weak peak was found at 5855.01 Da, which can be assigned to [Au16Cd3(SAdm)8(PPh2Py)3Cl2]+ (Fig. S38B). And combining EA and NMR, we determined the molecular formula of this cluster to be Au16Cd3(SAdm)9(PPh2Py)3Cl3 (Fig S39 and Table S6). Furthermore, the crystal structure of Au16Cd3 was characterized by X-ray single-crystal analysis, which showed that the Au16Cd3 cluster crystallized in the triclinic P-1 space group (Table S7). Detailed structural analysis of Au16Cd3 is shown in Fig. 4A. Au16Cd3 has a Au13 kernel with an anti-cuboctahedral structure (Fig. 4B). Importantly, the core of this Au13 kernel is wrapped in two novel motifs, those are one “S–Au–Cd(N, Cl)–S” and two “S–Au–S–Cd(Cl)N” motifs (Fig. 4C). These two motifs are not found in the previously reported Au–Cd NCs. In addition to their compositional structure, the novelty is also present in the close distance of the heteroatom N from the metal atoms (Au and Cd), from which the formation of a Cd–N coordination bond can be observed.
 | ||
| Fig. 4 Structure and ORR performance of Au16Cd3. (A) Framework, (B) core and (C) peripheral protecting group of the Au16Cd3 cluster (color label: cyan/orange = Au, pink = Cd, green = P, bright green = Cl, light blue = N, and gray = C, and some C and all H atoms were omitted for clarity). (D) The LSV curves of the Au16Cd3 cluster at different rpm. (E) Koutecky–Levich plots of Au16Cd3 catalysis at different potentials. (F) The LSV curves of Au16Cd3 at 1600 rpm before and after the addition of pyridine. | ||
Since the free pyridine can transfer electrons with the Cd atom to enhance the catalytic activity of the NCs, Au16Cd3 exhibiting the Cd–pyridine interaction in its molecular structure, is predicted to have good catalytic activity. Therefore, the electrocatalytic ORR tests were conducted with Au16Cd3 as the catalyst. Similar to the other NCs in this study, Au16Cd3 was loaded on activated carbon with a loading rate of 10%. Then, the Au16Cd3@C catalyst was first tested by LSV under O2-saturated alkaline (0.1 M KOH) conditions. The current density of the Au16Cd3@C catalyst at different rotational speeds is shown in Fig. 4D, and the current density is 4.58 mA cm−2 at 1600 rpm with a potential of −0.8 V (vs. Ag/AgCl). It is noteworthy that the highest current density among the other Au–Cd NCs was measured to be 4.75 mA cm−2 assisted by free pyridine (Fig. 2C). In addition, the overall number of electrons transferred in the ORR process of the Au16Cd3@C catalyst was calculated to be 3.3, which is similar to other Au–Cd NCs (Fig. 4E). Furthermore, the free pyridine was added to the Au16Cd3@C catalyst; however, the change in current density before and after the addition was not significant (Fig. 4F) because the Cd atoms in Au16Cd3 have already coordinated with the pyridine group from the ligands. Moreover, its half-wave potential remains unchanged both before and after the addition of pyridine (Table S4). Finally, stability tests (Fig. S40 and S41) confirmed that the Au16Cd3@C catalyst exhibits good stability.
Conclusions
In summary, this study presents an approach to realize controllable modification of the Au–Cd cluster surface, that is, based on the Lewis acid-base specific binding between pyridine groups and metallic cadmium. Because the pyridine units were modified on the surface, the ORR catalytic activities of all modified clusters were enhanced. In detail, a small number of cadmium atoms were anchored on the surface of the gold cluster to construct the specific recognition groups for the subsequent surface modification. Free pyridine was then added to the system artificially to construct the Au–Cd–pyridine catalytic site based on the acid-base specific binding between pyridine and the metallic cadmium. This Cd–pyridine interaction was confirmed by ESI-MS, XPS and a series of comparative experiments. Furthermore, DFT calculations confirm Au–Cd–pyridine as the catalytic site in the ORR, and the modified pyridine optimizes its catalytic reaction pathway (enhanced adsorption of the OOH* intermediate and improved adsorption of the O* intermediate). Finally, we designed and synthesized another Au–Cd cluster in which the pyridine group from its ligand binds with the Cd atom directly via Cd–pyridine bonds. This Au–Cd cluster shows comparable catalytic activity to that of other Au–Cd NCs with the addition of free pyridine, which reaffirms the above conclusion. This study not only produces effective catalysts for the ORR but also provides a generalized approach to realize controllable modification of the cluster.Author contributions
All authors have approved the final version of the manuscript. J. C. and M. Z. conceived and designed the experiments. B. H. designed and performed the computational calculations. F. Q., Z. Z., and Z. X. synthesized and characterized the materials. Q. L. collected and solved the SCXRD data. F. Q. and S. Y. wrote and revised the manuscript. J. C. and M. Z. supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2381499 and 2381500 contain the supplementary crystallographic data for this paper.53a,bThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information: Fig. S1–S41 and Tables S1–S7 and crystallographic information for Au27Cd2 and Au16Cd3 (CIF). See DOI: https://doi.org/10.1039/d6sc00514d.
Acknowledgements
We acknowledge financial support from the National Natural Science Foundation of China (22371003, 22301001, 22575001, and U24A20480), the Anhui Provincial Natural Science Foundation (2508085MB032), and the Hunan Provincial Natural Science Foundation (2025JJ60119).References
- Z.-Y. Liu, Q.-B. Nie, B.-L. Han, R. K. Gupta, G.-L. Dong, G.-G. Luo, Z.-L. Yang and D. Sun, Chem. Soc. Rev., 2025, 54, 9092–9115 RSC.
- Z. Yang, Y. Wang, R. Zhang, T. Chen and J. Xie, Adv. Mater., 2025, 37, e08578 CrossRef CAS PubMed.
- M.-M. Zhang, T.-T. Liang, C. Zhang, H. Zhang, Y.-F. Li, Y. Li and S.-Q. Zang, Aggregate, 2025, 6, e70209 CrossRef CAS.
- M. Sera, S. Hossain, S. Yoshikawa, K. Takemae, A. Ikeda, T. Tanaka, T. Kosaka, Y. Niihori, T. Kawawaki and Y. Negishi, J. Am. Chem. Soc., 2024, 146, 29684–29693 CrossRef CAS PubMed.
- W.-L. Mu, L. Li, X.-Z. Cong, X. Chen, P. Xia, Q. Liu, L. Wang, J. Yan and C. Liu, J. Am. Chem. Soc., 2024, 146, 28131–28140 CAS.
- M. Bodiuzzaman, K. Murugesan, P. Yuan, B. Maity, A. Sagadevan, N. Malenahalli H, S. Wang, P. Maity, M. F. Alotaibi, D.-e. Jiang, M. Abulikemu, O. F. Mohammed, L. Cavallo, M. Rueping and O. M. Bakr, J. Am. Chem. Soc., 2024, 146, 26994–27005 CrossRef CAS PubMed.
- T.-H. Chiu, J.-H. Liao, Y.-Y. Wu, J.-Y. Chen, Y. J. Chen, X. Wang, S. Kahlal, J.-Y. Saillard and C. W. Liu, J. Am. Chem. Soc., 2023, 145, 16739–16747 CrossRef CAS PubMed.
- Q. Li, B. Huang, S. Yang, H. Zhang, J. Chai, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2021, 143, 15224–15232 CrossRef CAS PubMed.
- M. Zhang, S. Ji, Z. Zhang, M. Zhu, Q. Yao and J. Xie, CCS Chem., 2026, 8, 37–59 CrossRef.
- X.-H. Yue, W.-X. Chen, T. Yang, A. Muñoz-Castro, G. Frenking and Z.-M. Sun, Nat. Synth., 2023, 2, 423–429 CrossRef CAS.
- V. H. Do and J. M. Lee, Adv. Mater., 2025, 37, 2417516 CrossRef CAS PubMed.
- V.-H. Do and J.-M. Lee, Chem. Soc. Rev., 2024, 53, 2693–2737 RSC.
- Q. Li, T. Jiang, S. Yang, J. Chai, H. Yu and M. Zhu, Chem. Sci., 2025, 16, 19669–19676 RSC.
- S. Wang, C. Han, X. Chen, Y. Xiang, Q. Li, J. Chai, S. Yang, Y. Du, Q. Luo and M. Zhu, ACS Nano, 2025, 19, 25334–25341 CrossRef CAS PubMed.
- X. Zhu, W.-D. Si, K. Qu, L. Song, J. Wang, Y. Gong, D. Sun, X. Liu and A.-L. Wang, J. Am. Chem. Soc., 2025, 147, 44437–44446 CrossRef CAS PubMed.
- H.-Y. Zhu, X.-W. Wang, X.-Y. Chen, L.-Y. Li, Y.-X. Li, W. D. Yu, J. Yan and C. Liu, Chem. Sci., 2025, 16, 20389–20396 RSC.
- G. Li, G. Li, X. Liang, Y. Tian, J. Lu, X. Xu, X. Liu, J. Ma, S. Li and Y. Zhu, Chem. Sci., 2025, 16, 18642–18651 RSC.
- H. Zhao, C. Zhang, B. Han, Z. Wang, Y. Liu, Q. Xue, C.-H. Tung and D. Sun, Nat. Synth., 2024, 3, 517–526 CrossRef CAS.
- G. Johnson, M. Y. Yang, C. Liu, H. Zhou, X. Zuo, D. A. Dickie, S. Wang, W. Gao, B. Anaclet, F. A. Perras, F. Ma, C. Zeng, D. Wang, S. Bals, S. Dai, Z. Xu, G. Liu, W. A. Goddard and S. Zhang, Nat. Synth., 2023, 2, 828–837 CrossRef CAS.
- F. Ma, K. A. Abboud and C. Zeng, Nat. Synth., 2023, 2, 949–959 CrossRef CAS.
- Q. Yao, L. Liu, S. Malola, M. Ge, H. Xu, Z. Wu, T. Chen, Y. Cao, M. F. Matus, A. Pihlajamäki, Y. Han, H. Häkkinen and J. Xie, Nat. Chem., 2023, 15, 230–239 CrossRef CAS PubMed.
- K. Yonesato, D. Yanai, S. Yamazoe, D. Yokogawa, T. Kikuchi, K. Yamaguchi and K. Suzuki, Nat. Chem., 2023, 15, 940–947 CrossRef CAS PubMed.
- H.-H. Deng, K.-Y. Huang, Y. Zhong, Y. Li, H.-X. Huang, X.-Y. Fang, W.-M. Sun, Q. Yao, W. Chen and J. Xie, Chem. Sci., 2024, 15, 8922–8933 RSC.
- S. M. Han, M. Park, J. Kim and D. Lee, Angew. Chem., Int. Ed., 2024, 63, e202404387 CrossRef CAS PubMed.
- J. Chai, S. Yang, Y. Lv, H. Chong, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2019, 131, 15818–15821 CrossRef.
- X.-K. Wan, X.-S. Han, Z.-J. Guan, W.-Q. Shi, J.-J. Li and Q.-M. Wang, Nat. Commun., 2024, 15, 7214 CrossRef CAS PubMed.
- Q. Li, Y. Tan, B. Huang, S. Yang, J. Chai, X. Wang, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2023, 145, 15859–15868 CrossRef CAS PubMed.
- G. Deng, S. Malola, T. Ki, X. Liu, S. Yoo, K. Lee, M. S. Bootharaju, H. Häkkinen and T. Hyeon, Structural Isomerism in Bimetallic Ag20Cu12 Nanoclusters, J. Am. Chem. Soc., 2024, 146, 26751–26758 CrossRef CAS PubMed.
- X.-W. Qi, M. Tan, F.-Z. Zhang, L.-G. Liao, J. Zeng, Y.-T. Zhang, Z.-E. Hu, J. Li, S. Zhang and B.-J. Li, Adv. Funct. Mater., 2024, 34, 2407241 CrossRef CAS.
- J. Wang, M. Chen, Z. Zhang, L. Ma and T. Chen, Coord. Chem. Rev., 2023, 493, 215278 CrossRef CAS.
- L. Wang, X. Su, J.-H. Xie and L.-J. Ming, Coord. Chem. Rev., 2021, 454, 214326 CrossRef.
- W.-Z. Qiao, Y.-J. Wang, S. Li, R. Wang, J. Wu and S.-Q. Zang, CCS Chem., 2024, 6, 2131–2141 CrossRef CAS.
- S. Li, Q. Wu, X. You, X. Ren, P. Du, F. Li, N. Zheng and H. Shen, J. Am. Chem. Soc., 2024, 146, 27852–27860 CrossRef CAS PubMed.
- E. A. Romero, T. Zhao, R. Nakano, X. Hu, Y. Wu, R. Jazzar and G. Bertrand, Nat. Catal., 2018, 1, 743–747 CrossRef CAS.
- H. Guo, X. Sun, X. Yang, D. Zuo, J. Sun, Z.-A. Nan, X. Gong, S. Li, J. Wei, N. Zheng and H. Shen, ACS Nano, 2026, 20, 2353–2365 CrossRef CAS PubMed.
- Y. Tan, G. Sun, T. Jiang, D. Liu, Q. Li, S. Yang, J. Chai, S. Gao, H. Yu and M. Zhu, Angew. Chem., Int. Ed., 2024, 63, e202317471 CrossRef CAS PubMed.
- J.-Q. Fan, Y. Li, W. W. Xu and M.-B. Li, Angew. Chem., Int. Ed., 2024, 64, e202413861 CrossRef PubMed.
- J. Yang, F. Muckel, W. Baek, R. Fainblat, H. Chang, G. Bacher and T. Hyeon, J. Am. Chem. Soc., 2017, 139, 6761–6770 CrossRef CAS PubMed.
- L. Tang, A. Ma, C. Zhang, X. Liu, R. Jin and S. Wang, Angew. Chem., Int. Ed., 2021, 60, 17969–17973 CrossRef CAS PubMed.
- H. Zhang, Y. Li, L. Chen, Y. Yang, H. Lin, S. Xiang, B. Chen and Z. Zhang, Chem, 2022, 9, 242–252 Search PubMed.
- P. Li, Y. Jiao, Y. Ruan, H. Fei, Y. Men, C. Guo, Y. Wu and S. Chen, Nat. Commun., 2023, 14, 6936 CrossRef CAS PubMed.
- N. A. Arnosti, G. P. Wasser, Y.-C. Shen and M. F. Delley, J. Am. Chem. Soc., 2025, 147, 43487–43500 CrossRef CAS PubMed.
- K. Q. Zhong, F. Y. Yu, D. Zhang, Z. H. Li, D. H. Xie, T. T. Li, Y. Zhang, L. Yuan, H. Li, Z. Y. Wu and G. P. Sheng, Angew. Chem., Int. Ed., 2025, 64, e202500004 CrossRef CAS PubMed.
- C. Yao, C. Q. Xu, I. H. Park, M. Zhao, Z. Zhu, J. Li, X. Hai, H. Fang, Y. Zhang, G. Macam, J. Teng, L. Li, Q. H. Xu, F. C. Chuang, J. Lu, C. Su, J. Li and J. Lu, Angew. Chem., Int. Ed., 2020, 59, 8270–8276 CrossRef CAS PubMed.
- Y. Tan, Y. Lv, L. Xu, Q. Li, J. Chai, S. Yang, H. Yu and M. Zhu, J. Am. Chem. Soc., 2023, 145, 4238–4245 CrossRef CAS PubMed.
- S. Yang, S. Chen, L. Xiong, C. Liu, H. Yu, S. Wang, N. L. Rosi, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2018, 140, 10988–10994 CrossRef CAS PubMed.
- S. Chen, L. Xiong, S. Wang, Z. Ma, S. Jin, H. Sheng, Y. Pei and M. Zhu, J. Am. Chem. Soc., 2016, 138, 10754–10757 CrossRef CAS PubMed.
- D. Crasto, G. Barcaro, M. Stener, L. Sementa, A. Fortunelli and A. Dass, J. Am. Chem. Soc., 2014, 136, 14933–14940 CrossRef CAS PubMed.
- H. Zhang, H. Zhang, R. Wang, J. Lv, W. Huang, C. Guo and F. Yang, Catalysts, 2024, 14, 284 CrossRef CAS.
- J. Resasco, F. Abild-Pedersen, C. Hahn, Z. Bao, M. T. M. Koper and T. F. Jaramillo, Nat. Catal., 2022, 5, 374–381 CrossRef.
- E. Sargeant, F. Illas, P. Rodríguez and F. Calle-Vallejo, Electrochim. Acta, 2022, 426, 140799 CrossRef CAS.
- C. Deng, F. Li and Q. Tang, J. Phys. Chem. C, 2019, 123, 27116–27123 CrossRef CAS.
- (a) CCDC 2381499: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2ky4k7; (b) CCDC 2381500: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2ky4l8.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |