
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective synthesis of aza-saccharins via anionic [1,4] Fries-type rearrangement of aryl sulfonimidoyl fluorides
Mario
Leypold
 *a,
Lorenzo
Poli
a,
Max
Earl
a,
Okky D.
Putra
b,
Karolina
Kwapien
a,
Richard J.
Lewis
a,
John J.
Murphy
a,
Marta
Passamonti
a,
Lena M.
von Sydow
a,
Victor
Spelling
c,
Ioannis
Asproudis
b,
Malvika
Sardana
c,
Claudia
Gatti
d,
Hikaru
Seki
a,
Thomas
Lemaitre
a,
Radvile
Juskaite
a,
Ranganath
Gopalakrishnan
a,
Stuart J.
Francis
a,
Cristina
Gardelli
a,
Per-Ola
Norrby
e and
Werngard
Czechtizky
a
*a,
Lorenzo
Poli
a,
Max
Earl
a,
Okky D.
Putra
b,
Karolina
Kwapien
a,
Richard J.
Lewis
a,
John J.
Murphy
a,
Marta
Passamonti
a,
Lena M.
von Sydow
a,
Victor
Spelling
c,
Ioannis
Asproudis
b,
Malvika
Sardana
c,
Claudia
Gatti
d,
Hikaru
Seki
a,
Thomas
Lemaitre
a,
Radvile
Juskaite
a,
Ranganath
Gopalakrishnan
a,
Stuart J.
Francis
a,
Cristina
Gardelli
a,
Per-Ola
Norrby
e and
Werngard
Czechtizky
a
aMedicinal Chemistry, Research and Early Development, Respiratory and Immunology, BioPharmaceuticals R&D AstraZeneca, Pepparedsleden 1, 43183 Mölndal, Sweden. E-mail: mario.leypold@astrazeneca.com
bEarly Product Development and Manufacturing, Pharmaceutical Sciences, R&D AstraZeneca, Pepparedsleden 1, 43183 Mölndal, Sweden
cEarly Chemical Development, Pharmaceutical Sciences, R&D AstraZeneca, Pepparedsleden 1, 43183 Mölndal, Sweden
dEarly Chemical Development, Pharmaceutical Sciences, R&D AstraZeneca, Macclesfield SK10 2NA, UK
ePredictive Science, Digital and Automation, Pharmaceutical Sciences, R&D AstraZeneca, Pepparedsleden 1, 43183 Mölndal, Sweden
First published on 28th April 2026
Abstract
A regioselective synthesis of aromatic and aliphatic aza-saccharins as cyclic sulfonimidamide derivatives is reported, starting from easily accessible aryl sulfonimidoyl fluorides and primary or secondary amines with broad functional group tolerance. The transformation is enabled by electron-withdrawing substituents and proceeds through an anionic [1,4] Fries-type rearrangement at cryogenic temperature, initiated by KHMDS-mediated ortho-deprotonation and rapid carbonyl migration. Mechanistic investigations indicate the formation of aryl sulfonimidoyl fluoridate anions, a distinct and previously unexplored motif in S(VI) and SuFEx chemistry. The involvement of these low-temperature-persistent intermediates is supported by cryogenic 19F/15N NMR studies of a 15N-labeled substrate, interception by acylation to give a stable adduct whose structure was confirmed by X-ray diffraction, and complementary DFT calculations indicating that the fluoridate anion is a viable low-energy intermediate. Regioselectivity follows predictable electronics-driven trends governed by meta-substituents, while selective aza-saccharin formation can be modulated by base choice and reagent addition order. Notably, the fluoridate anions are configurationally stable at sulfur, as demonstrated in a representative example, and subsequent amination via in situ SuFEx capture proceeds stereospecifically with inversion at sulfur. This synthetic method provides a mechanistically defined and stereocontrolled entry to chiral aza-saccharins with potential relevance to medicinal chemistry, agrochemical discovery and related disciplines.
Introduction
Within the pharmaceutical industry, medicinal chemists are confronted with the continuous task of improving the properties of bioactive compounds while simultaneously balancing efficacy with safety and tolerability. In their pursuit, a promising class of compounds known as sulfonimidamides has emerged as bioisosteric substitutes for widely used sulfonamides, thanks to their favorable blend of physicochemical and pharmacokinetic characteristics.1–3 These features have positioned sulfonimidamides as compelling motifs for addressing limitations associated with established sulfur(VI)-based scaffolds and for enabling therapeutic innovation. As a result, sulfonimidamides have attracted significant attention, leading to multiple patents since 2018 that highlight their utilization as inhibitors for NLRP-,4 STING-,5 and LXR-related disorders,6 and their potential for pesticidal applications.7In the realm of sulfonimidamides, an additional level of structural complexity can be achieved through rigidification in the form of cyclic analogs. Among these structures, aza-saccharins stand out as three-dimensional heterocycles with amine exit vectors positioned out of the plane, offering opportunities for diversification. Despite this promise, general synthetic access to aza-saccharins remains limited in the open literature. To date, our laboratories have disclosed the single general method for their synthesis relying on the ring closure of tert-butyldimethylsilyl-protected (TBS-protected) acyclic aryl sulfonimidamides onto a pre-installed ester moiety in ortho-position (Scheme 1A).8 According to our survey of the literature, this methodology was subsequently applied in 2020 patent disclosures, which describe the only additional examples of aza-saccharins, encompassing over 50 compounds evaluated as NLRP3 modulators (Scheme 1B).9,10 Given our interest in aza-saccharins for fragment-based hit finding and to broaden the synthetic access to this compound class, we developed an orthogonal strategy for the synthesis of aza-saccharins that employs an anionic [1,4] Fries-type rearrangement11,12 of aryl sulfonimidoyl fluorides, followed by sulfur(VI) fluoride exchange (SuFEx) chemistry.1u,13 The transformation proceeds via cryogenically stable aryl sulfonimidoyl fluoridate anions, which, to the best of our knowledge, have not been previously reported as intermediates or reactive motifs in SuFEx processes, and are intercepted in situ by amines to effect ring closure (Scheme 1C). The involvement of these fluoridate anions is supported by cryogenic 19F/15N NMR studies of a15N-labeled substrate, acylation trapping with X-ray characterization of a resulting adduct, and complementary computational simulations. In this study, we describe the regioselective synthesis of substituted aza-saccharins with its applicability to stereospecific protocols using easily accessible tert-butyloxycarbonyl-protected (Boc-protected) aryl sulfonimidoyl fluorides and amines with broad functional group tolerance.
 | ||
| Scheme 1 Methods for the synthesis of aza-saccharins and applications. | ||
Results and discussion
Reaction optimization
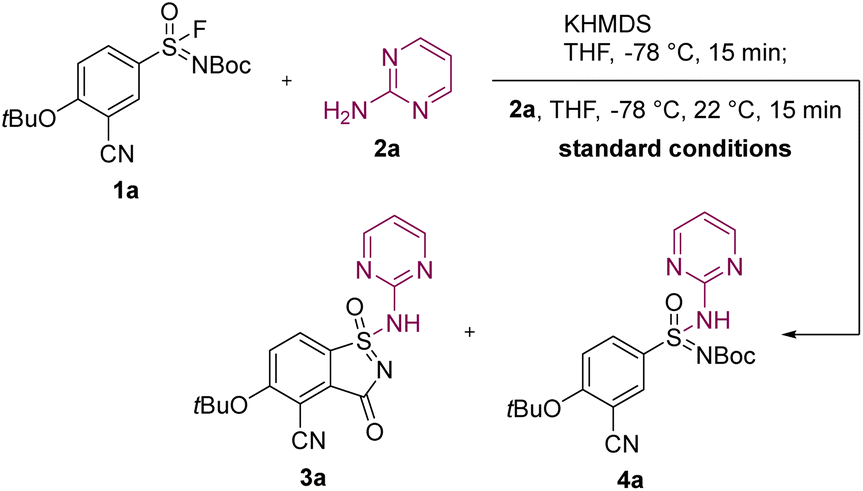
During our investigation into the formation of acyclic sulfonimidamides, we discovered a novel reactivity pattern upon the addition of potassium bis(trimethylsilyl)amide (KHMDS) to a solution containing sulfonimidoyl fluoride 1a and 2-aminopyrimidine (2a), resulting in the concomitant formation of aza-saccharin 3a and acyclic sulfonimidamide 4a. We hypothesized that favorable aza-saccharin formation could be facilitated by KHMDS pre-activation (KPA) of sulfonimidoyl fluoride 1a, which induces an anionic [1,4] Fries-type rearrangement to an ester analogue in ortho-position,14 akin to our previously established route (Scheme 1A), prior to the addition of amine. Under optimized reaction conditions, the treatment of sulfonimidoyl fluoride 1a (1 equiv.) with KHMDS (2.00 equiv.) at −78 °C, followed by the addition of 2-aminopyrimidine (2a, 1.20 equiv.), resulted in quantitative conversion and high isolated yield of aza-saccharin 3a (Table 1, Entry 1).15 Deviations from the optimized conditions revealed pronounced sensitivity to solvent, substrate structure, protecting group, reagent addition order, as well as base identity and equivalents. Among the ethers examined, tetrahydrofuran (THF) proved most effective, whereas methyl tert-butyl ether (MTBE) gave the lowest LCMS yield (Entry 2).16 Furthermore, the sulfonimidoyl chloride analogue of sulfonimidoyl fluoride 1a failed to furnish detectable aza-saccharin 3a and instead led to substrate degradation (Entry 3).17 In contrast, the methoxycarbonyl-protected (Moc-protected) aryl sulfonimidoyl fluoride still afforded the corresponding aza-saccharin 3a, albeit with diminished LCMS and isolated yields accompanied by increased side product formation (Entry 4).18 When KHMDS was added in the presence of amine 2a, preferential formation of acyclic sulfonimidamide 4a was observed (Entry 5). Interestingly, silazide bases with more Lewis-acidic counterions, such as lithium bis(trimethylsilyl)amide (LiHMDS) or sodium bis(trimethylsilyl)amide (NaHMDS),19,20 were found to be less reliable in the pre-activation of sulfonimidoyl fluoride 1a (Entries 6 and 7). While the origin of this counterion dependence is not fully established, it may reflect differences in ion pairing, aggregation and/or metal–substrate coordination of alkali-metal silazides under cryogenic conditions.21 Collectively, the strong dependence on reagent addition order and on the alkali-metal silazide employed provides a practical handle to control the chemoselectivity in the synthesis of aza-saccharins and acyclic sulfonimidamides. Finally, the optimized amount of KHMDS proved essential for achieving ideal results in aza-saccharin synthesis (compare Entries 1, 8 and 9).22|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | LCMS yield 3a, [4a] (%)b |
| a Reaction conditions: sulfonimidoyl fluoride 1a (75.0 µmol, 1 equiv.), KHMDS (150 µmol, 2.00 equiv.), THF, −78 °C, 15 min; 2-aminopyrimidine (2a, 90.0 µmol, 1.20 equiv.), THF, −78 °C, 22 °C, 15 min. b LCMS yield of Entry 1 verified via NMR experiment with 1,3,5-trimethoxybenzene as internal standard. c Isolated yield on a 400 µmol scale. d KHMDS (1 M in THF) used. n.d. = not detected. | ||
| 1 | None | >99 {89}c, [n.d.] |
| 2 | MTBEd | 81, [7] |
| 3 | Sulfonimidoyl chloride | Degradation |
| 4 | Moc | 68 {52}c, [n.d.] |
| 5 | KHMDS (2.00 equiv.) added last | 6, [92] |
| 6 | LiHMDS (2.00 equiv.) | 2, [95] |
| 7 | NaHMDS (2.00 equiv.) | 91, [n.d.] |
| 8 | KHMDS (1.50 equiv.) | 82, [n.d.] |
| 9 | KHMDS (2.50 equiv.) | 84, [n.d.] |
Substrate scope
The synthetic application of this chemistry to aza-saccharin formation was investigated using aryl sulfonimidoyl fluorides and their corresponding aza-saccharins (Scheme 2), while heteroaryl analogues were not examined. Various inductive electron-withdrawing substituents in the meta-position relative to the sulfonimidoyl fluoride moiety were compatible, and provided aza-saccharins 3 with complete (3b–3g) or high regioselectivity (3h![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3h′ = 92:8). Notably, cyclization in these substrates occurred at the sterically more demanding reaction site. Electron-withdrawing substituents in the para-position yielded aza-saccharins 3i–3k in fair yields, while ortho- and unsubstituted aza-saccharins 3l, 3m required higher temperatures due to less efficient KPA in the corresponding sulfonimidoyl fluorides under standard conditions.
:3h′ = 92:8). Notably, cyclization in these substrates occurred at the sterically more demanding reaction site. Electron-withdrawing substituents in the para-position yielded aza-saccharins 3i–3k in fair yields, while ortho- and unsubstituted aza-saccharins 3l, 3m required higher temperatures due to less efficient KPA in the corresponding sulfonimidoyl fluorides under standard conditions.
 | ||
| Scheme 2 aza-Saccharin formation: scope of aryl sulfonimidoyl fluorides. Reaction conditions: sulfonimidoyl fluoride 1 (400 µmol, 1 equiv.), KHMDS (800 µmol, 2.00 equiv.), THF, −78 °C, 15 min; 4-Me-6-Ph-pyrimidine-2-amine (2b, 480 µmol, 1.20 equiv.), THF, −78 °C, 22 °C, 15 min. aKPA: −78 °C, 4 h. bKPA: −50 °C, 15 min. cKPA: −30 °C, 30 min. Values correspond to isolated yields for the mixture of regioisomers. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms and minor parts are omitted for clarity. n.d. = not detected (below LCMS detection limit in reaction mixture). | ||
The use of bis-meta-substituted sulfonimidoyl fluorides enabled the synthesis of the corresponding aza-saccharins 3n–3r, and allowed mapping of relative cyclization trends based on observed regioselectivities influenced by substituents in the meta-position. The KPA of sulfonimidoyl fluorides appears to be governed by both inductive effects and polarization contributing to acidification, and potential stabilization of carbanions via negative hyperconjugation (vide infra).23 Leading to a productivity trend for substituents in the aza-saccharin synthesis of F > SOFNBoc > Cl ∼ Br ∼ CN > CF3 > H,20,24,25 these observations align better with the reactivity and regioselectivity patterns observed in the deprotonation of substituted arenes using strong potassium bases,26 yet complementary to the directed ortho-metalation with chelating lithium reagents.27
A limitation of the methodology was observed for aryl sulfonimidoyl fluorides bearing electron-donating substituents or extended π-systems. Under standard conditions, the 4-methoxy and 2-naphthyl derivatives failed to afford aza-saccharins 3s, 3t, respectively.20,28 These results indicate that reactivity under the current KPA conditions may require substrate-specific reoptimization, beyond the temperature adjustment used for less activated aryl sulfonimidoyl fluorides (aza-saccharins 3l, 3m).
The substrate scope exploration encompassing a wide range of amines and other nitrogen nucleophiles is presented in Scheme 3. 6-membered and 5-membered primary aromatic amines bearing heteroatom-substituted and sterically hindered motifs were tolerated to provide the corresponding aza-saccharins 3a, 3u–3ab in 54−90% yields. Of particular significance, the synthesis of aza-saccharin 3a could be readily accomplished on a 2.00 mmol scale without compromising the efficiency of the reaction. Importantly, this transformation displayed high functional group tolerance to enable direct access to functionalized aza-saccharins 3ac–3ah, even in the presence of acidic α-protons relative to the silazide base added, such as demonstrated in ketone- 3ai, lactam- 3aj, sulfone- 3ak, and phosphine oxide-containing aza-saccharin 3al. The successful formation of aza-saccharin 3am in 40% yield additionally expands the applicability of this methodology to secondary aromatic amines as reaction partners.
 | ||
| Scheme 3 aza-Saccharin formation: scope of amines and nitrogen nucleophiles. Reaction conditions: sulfonimidoyl fluoride 1a (400 µmol, 1 equiv.), KHMDS (800 µmol, 2.00 equiv.), THF, −78 °C, 15 min; amine 2 (480 µmol, 1.20 equiv.), THF, −78 °C, 22 °C, 15 min. aFor aliphatic amines: amine 2 (800 µmol, 2.00 equiv.). bNitrogen nucleophiles investigated on a 100 µmol scale with LCMS conversions reported. cAfter amine addition: 0 °C, 15 min. dKHMDS as NH2 surrogate: 40 °C, 2 h. Values correspond to isolated yields. Displacement ellipsoids are drawn at the 50% probability level. Hydrogen atoms and minor parts are omitted for clarity. n.d. = not detected (below LCMS detection limit in reaction mixture). | ||
Initially, aliphatic amines compromised the desired aza-saccharin formation. During the KPA of sulfonimidoyl fluorides, the silazide anion functioned exclusively as a base. However, at temperatures above −78 °C, it additionally displayed dual reactivity and acted as a nucleophile in the SuFEx reaction, leading to competition with aliphatic amines due to similar nucleophilicity in contrast to aromatic amines. To mitigate side product formation, a modified protocol was developed by increasing the amount of aliphatic amine added (2.00 equiv., Scheme 3). This modification proved effective, resulting in the synthesis of aza-saccharins 3an–3at in 20−53% yields.29 The free amino analogue aza-saccharin 3au was obtained in 56% yield using KHMDS directly as an NH2 surrogate. This orthogonal approach would allow access to additional regioisomers of the previously reported NLRP3 modulators when combined with the isocyanate-based modification described above (cf.Schemes 1B and 2). Furthermore, the successful isolation of aza-saccharin 3av, utilizing morpholine as a reaction partner, demonstrates the applicability of this synthetic protocol to secondary aliphatic amines.
In addition to aromatic and aliphatic amines, a set of other nitrogen nucleophiles was evaluated under the standard conditions. A representative hydrazine and sulfonamide did not furnish the corresponding aza-saccharins 3aw, 3ax, and instead led to notable side product formation. Selected mono-aza heterocycles delivered minor amounts of the pyrrole and indole aza-saccharins 3ay, 3az, which could not be isolated, suggesting that these scaffolds may become accessible with further optimization.30 The remaining N–H heteroaromatic nucleophiles tested were unreactive toward aza-saccharins 3ba–3bd, indicating that hydrolysis, observed during LCMS analysis rather than under experimental conditions, predominantly accounted for most reactions.20,31 Taken together, these observations underscore a limitation of the current protocol for these classes of nitrogen nucleophiles.
Mechanistic studies
A set of mechanistic experiments was conducted to support our mechanistic rationale in the KPA of aryl sulfonimidoyl fluorides prior to the addition of amines. Based on the observed regioselectivities and substituent-dependent reactivity trends, we postulated that inductive electron-withdrawing groups acidify the ortho-hydrogen atom adjacent to the sulfonimidoyl fluoride moiety, leading to the formation of the carbanionic intermediate 5a upon deprotonation with KHMDS (Scheme 4A).32,33 Rapid [1,4] Fries-type rearrangement via carbonyl migration within carbanion 5a would yield cryogenically stable aryl sulfonimidoyl fluoridate anion 6a, whose presence was supported by distinct 19F NMR (δ 112 ppm) and 15N NMR (δ 165 ppm) shifts in cryogenic NMR experiments utilizing 15N-labeled sulfonimidoyl fluoride 1a-15N. Treatment of intermediate 6a-15N with 2-aminopyridine (2c) provided aza-saccharin 3u-15Nvia SuFEx reaction and cyclization with full incorporation of the 15N-atom at the endocyclic position, whereas trapping of unlabeled intermediate 6a with acetyl chloride resulted in a clean N-acylation affording sulfonimidoyl fluoride 7a (compare with X-ray crystal structure of crystalline sulfonimidoyl fluoride 7b).20 | ||
| Scheme 4 Summary of mechanistic experiments for the formation of aza-saccharins. Reaction conditions: sulfonimidoyl fluoride (1 equiv.), KHMDS (2.00 equiv.), THF, −78 °C; 2-aminopyridine (2c, 1.20 equiv.), 4-Me-6-Ph-pyrimidine-2-amine (2b, 1.20 equiv.) or acyl chloride (6.00 equiv.), THF, −78 °C, 22 °C, 15 min. aReaction profile for the KPA of unlabeled sulfonimidoyl fluoride 1a was computed at the M06-2X/def2-TZVP level of theory with implicit solvent model (COSMO) for the treatment of THF. The relative Gibbs free energies ΔΔG195 are calculated at 195 K. Values correspond to isolated yields. Displacement ellipsoids are drawn at the 30% and 50% probability level. Hydrogen atoms and minor parts are omitted for clarity. | ||
Moderately acidic hydrogen atoms at benzylic positions ortho to the sulfonimidoyl fluoride were suspected to compete with deprotonation at the desired ortho-position, suppressing aza-saccharin formation (e.g., ortho-tolyl, Scheme 4B). Among the substituents investigated, only a meta-fluoro substituent of sulfonimidoyl fluoride 8a was acidifying enough to allow successful and selective aza-saccharin 9a formation while potentially stabilizing the deprotonated sulfonimidoyl fluoride 8avia negative hyperconjugation.23d,e Conversely, reactions involving the cyano and the unsubstituted derivative favored deprotonation at the benzylic position,12d forming sulfinamide/sulfonimidamide dimers 10b, 10c, respectively, as a mixture of stereoisomers.34
Kinetic isotope effect (KIE) experiments via intramolecular competition revealed a normal KIE in para-fluoro sulfonimidoyl fluoride 11a–d toward aza-saccharin 12a–d (Scheme 4Ca, kH/kD = 2.2). Its magnitude is consistent with a kinetically controlled, effectively irreversible deprotonation (carbon-hydrogen/deuterium bond cleavage) occurring in an asynchronous, rate-determining transition state followed by a rapid carbonyl migration.35 In contrast, bis-meta-dichloro sulfonimidoyl fluoride 11b–d exhibited an inverse KIE (Scheme 4Cb, kH/kD = 0.62) attributable to a pre-equilibrium isotope effect (EIE), in which a fast, reversible deprotonation precedes slower carbonyl migration, and therefore reflecting thermodynamic isotope partitioning in this pre-equilibrium rather than intrinsic bond cleavage kinetics. Consistent with these observations, density functional theory (DFT) calculations supported the substitution-dependent shift in the selectivity-determining step between meta- and para-substituted sulfonimidoyl fluorides.20 Additional evidence includes hydrogen/deuterium scrambling at the para-position of bis-meta-dichloro sulfonimidoyl fluoride 11c-d, indicating dynamic KPA behavior and sensitivity to substituent acidification (Scheme 4Cb). The coexistence of an inverse KIE together with deuterium exchange at nominally nonreactive carbon-hydrogen sites for meta-substituted sulfonimidoyl fluorides implies an increased barrier to carbonyl migration relative to ortho-deprotonation, rendering the deprotonation step reversible (compare the reaction profile for sulfonimidoyl fluoride 1a, Scheme 4A).36
During the exploration of chiral information preservation at the sulfur atom, stereoisomerically pure sulfonimidoyl fluoride 13SR (d.e. > 99.0%, e.e. > 99.0%, (S)-configuration at sulfur atom) afforded the corresponding aza-saccharin 14RR (d.e. > 90.6%, e.e. > 99.0%) without any erosion of optical activity of the benzylic stereogenic center (Scheme 4D).20 More importantly, upon KPA of sulfonimidoyl fluoride 13SR, SuFEx capture by 2-aminopyrimidine (2a) occurred with inversion of the stereogenic center at the sulfur atom,37 and therefore highlighted the suitability of this transformation for stereospecific protocols. Minor losses in stereoinformation can be attributed to the degenerative nucleophilic attack of solvated fluoride ions originating from the released potassium fluoride.13c All results obtained from the cryogenic NMR spectroscopy, trapping experiment (Scheme 4A) and the stereospecific route (Scheme 4D) collectively validate the fluoro substituent being attached and thus chiral information at the sulfur atom being conserved throughout the entire KPA of sulfonimidoyl fluorides, further expanding the utilization of this synthetic methodology.
Conclusions
In conclusion, we have developed a regioselective method for the synthesis of aza-saccharins from aryl sulfonimidoyl fluorides and amines. This transformation displays broad applicability, accommodating electronically diverse substrates bearing inductive electron-withdrawing substituents as well as a range of amine partners, while maintaining high functional group tolerance. Mechanistic studies are consistent with an anionic [1,4] Fries-type rearrangement under strongly basic conditions, in which regioselective ortho-deprotonation is followed by rapid carbonyl migration to low-temperature-persistent aryl sulfonimidoyl fluoridate anions, revealing a distinct and previously unexplored mode of reactivity in S(VI) and SuFEx chemistry. The involvement of these fluoridate anions is supported by cryogenic 19F/15N NMR studies of a 15N-labeled substrate, interception by acylation with X-ray characterization of the resulting adduct, and complementary computational analysis. Notably, chiral information at the sulfur center is preserved during the rearrangement, and subsequent SuFEx-based amination proceeds stereospecifically with inversion at sulfur, enabling a controlled and predictable route to stereodefined aza-saccharins.Overall, this work expands the synthetic and mechanistic understanding of sulfonimidoyl fluorides and establishes a reliable entry to functionalized aza-saccharins. Future studies will assess whether the present methodology can be extended to heteroaryl sulfonimidoyl fluorides and their corresponding aza-saccharins, as well as to carbon- and oxygen-based nucleophiles. Concurrently, ongoing work in our laboratories is focused on investigating the physicochemical and pharmacokinetic properties of aza-saccharins and evaluating their potential as bioisosteric replacements for sulfonamides in medicinal chemistry, agrochemistry, and related disciplines.
Author contributions
M. L., L. P., M. E., J. J. M., M. S., C. Gatti, H. S., T. L. and R. J. performed the experiments, O. D. P., R. J. L., M. P., L. M. v. S., V. S. and I. A. carried out analytical characterizations, K. K. performed computational simulations, M. L., J. J. M. and P.-O. N. elaborated the reaction mechanism, and M. L., H. S., R. G., S. J. F., C. Gardelli and W. C. contributed to project planning and supervision.Conflicts of interest
The authors declare the following competing financial interest(s): M. L., M. E., O. D. P., K. K., R. J. L., J. J. M., M. P., L. M. v. S., V. S., I. A., M. S., C. Gatti, H. S., R. J., R. G., S. J. F., C. Gardelli, P.-O. N. and W. C. are employees of AstraZeneca and may own stock or stock options. The authors have no other relevant affiliations or financial conflicts with the subject matter discussed in the manuscript apart from those disclosed. L. P. and T. L. and declare no conflict of interest.Data availability
CCDC 2517062–2517074 contain the supplementary crystallographic data for this paper.38a–mAll experimental and computational data supporting the findings of this study are provided in the supplementary information (SI). Supplementary information: experimental procedures, NMR spectra, computational methods, and optimized geometries. See DOI: https://doi.org/10.1039/d6sc00432f.
Acknowledgements
The authors thank Dr. Yantao Chen (AstraZeneca, Sweden) and Dr. Igor Shamovsky (AstraZeneca, Sweden) for mechanistic discussions regarding the synthesis of sulfonimidamide derivatives, including aza-saccharins, and Prof. Dr. Magnus J. Johansson for proofreading the manuscript. L. P. and T. L. thank the Erasmus program, L. P. the Alsace region, and T. L. the Occitanie region for financial support during their internship stays at AstraZeneca, Sweden.Notes and references
- For different strategies regarding the synthesis of acyclic sulfonimidamides, see: (a) C. Liang, F. Robert-Peillard, C. Fruit, P. Müller, R. H. Dodd and P. Dauban, Angew. Chem., Int. Ed., 2006, 45, 4641–4644 CrossRef CAS PubMed; (b) C. Liang, F. Collet, F. Robert-Peillard, P. Müller, R. H. Dodd and P. Dauban, J. Am. Chem. Soc., 2008, 130, 343–350 CrossRef CAS PubMed; (c) C. Worch, I. Atodiresei, G. Raabe and C. Bolm, Chem. Eur J., 2010, 16, 677–683 CrossRef CAS PubMed; (d) Y. Chen and J. Gibson, RSC Adv., 2015, 5, 4171–4174 RSC; (e) J. Buendia, G. Grelier, B. Darses, A. G. Jarvis, F. Taran and P. Dauban, Angew. Chem., Int. Ed., 2016, 55, 7530–7533 CrossRef CAS PubMed; (f) F. Izzo, M. Schäfer, R. Stockmann and U. Lücking, Chem.–Eur. J., 2017, 23, 15189–15193 CrossRef CAS PubMed; (g) T. Q. Davies, A. Hall and M. C. Willis, Angew. Chem., Int. Ed., 2017, 56, 14937–14941 CrossRef CAS PubMed; (h) M. Wright, C. Martínez-Lamenca, J. E. Leenaerts, P. E. Brennan, A. A. Trabanco and D. Oehlrich, J. Org. Chem., 2018, 83, 9510–9516 CrossRef CAS PubMed; (i) H. Yu, Z. Li and C. Bolm, Angew. Chem., Int. Ed., 2018, 57, 15602–15605 CrossRef CAS PubMed; (j) E. L. Briggs, A. Tota, M. Colella, L. Degennaro, R. Luisi and J. A. Bull, Angew. Chem., Int. Ed., 2019, 58, 14303–14310 CrossRef CAS PubMed; (k) M. Bremerich, C. M. Conrads, T. Langletz and C. Bolm, Angew. Chem., Int. Ed., 2019, 58, 19014–19020 CrossRef CAS PubMed; (l) P. K. Chinthakindi, A. Benediktsdottir, P. I. Arvidsson, Y. Chen and A. Sandström, Eur. J. Org Chem., 2020, 3796–3807 CrossRef CAS; (m) T. Q. Davies, M. J. Tilby, J. Ren, N. A. Parker, D. Skolc, A. Hall, F. Duarte and M. C. Willis, J. Am. Chem. Soc., 2020, 142, 15445–15453 CrossRef CAS PubMed; (n) G. B. Craven, E. L. Briggs, C. M. Zammit, A. McDermott, S. Greed, D. P. Affron, C. Leinfellner, H. R. Cudmore, R. R. Tweedy, R. Luisi, J. A. Bull and A. Armstrong, J. Org. Chem., 2021, 86, 7403–7424 CrossRef CAS PubMed; (o) P. K. Tony and M. C. Willis, J. Am. Chem. Soc., 2021, 143, 15576–15581 CrossRef PubMed; (p) M. J. Tilby, D. F. Dewez, A. Hall, C. M. Lamenca and M. C. Willis, Angew. Chem., Int. Ed., 2021, 60, 25680–25687 CrossRef CAS PubMed; (q) G.-f. Yang, Y. Yuan, Y. Tian, S.-q. Zhang, X. Cui, B. Xia, G.-x. Li and Z. Tang, J. Am. Chem. Soc., 2023, 145, 5439–5446 CrossRef CAS PubMed; (r) D.-D. Liang, N. Lional, B. Scheepmaker, M. Subramaniam, G. Li, F. M. Miloserdov and H. Zuilhof, Org. Lett., 2023, 25, 5666–5670 CrossRef CAS PubMed; (s) J. A. Andrews, J. Kalepu, C. F. Palmer, D. L. Poole, K. E. Christensen and M. C. Willis, J. Am. Chem. Soc., 2023, 145, 21623–21629 CrossRef CAS PubMed; (t) S. Pan, F. F. Mulks, P. Wu, K. Rissanen and C. Bolm, Angew. Chem., Int. Ed., 2024, 63, e202316702 CrossRef CAS PubMed; (u) P. R. Athawale, Z. P. Shultz, A. Saputo, Y. D. Hall and J. M. Lopchuk, Nat. Commun., 2024, 15, 7001 CrossRef CAS PubMed; (v) S. Teng, Z. P. Shultz, C. Shan, L. Wojtas and J. M. Lopchuk, Nat. Chem., 2024, 16, 183–192 CrossRef CAS PubMed; (w) P. Wu, L. Ling, Y. Hu, S. Pan and C. Bolm, ACS Sustainable Chem. Eng., 2024, 12, 15875–15880 CrossRef CAS; (x) D. Deans, S. Iqbal, Z. Zhong, C. S. Begg, N. A. Anderson, S. Ferrer Cabrera and J. A. Bull, ChemistryEurope, 2025, 3, e202500163 CrossRef CAS.
- For the synthesis of cyclic sulfonimidamides, see: (a) B. E. Cathers and J. V. Schloss, Bioorg. Med. Chem. Lett., 1999, 9, 1527–1532 CrossRef CAS PubMed; (b) N. Pemberton, H. Graden, E. Evertsson, E. Bratt, M. Lepistö, P. Johannesson and P. H. Svensson, ACS Med. Chem. Lett., 2012, 3, 574–578 CrossRef CAS PubMed; (c) I. Sen, D. P. Kloer, R. G. Hall and S. Pal, Synthesis, 2013, 45, 3018–3028 CrossRef CAS; (d) P. K. Chinthakindi, A. Benediktsdottir, A. Ibrahim, A. Wared, C.-J. Aurell, A. Pettersen, E. Zamaratski, P. I. Arvidsson, Y. Chen and A. Sandström, Eur. J. Org Chem., 2019, 1045–1057 CrossRef CAS; (e) Y. Chen, J. Söderlund, G. Grönberg, A. Pettersen and C.-J. Aurell, Eur. J. Org Chem., 2019, 4731–4740 CrossRef; (f) A.-K. Bachon, A. Hermann and C. Bolm, Chem.–Eur. J., 2019, 25, 5889–5892 CrossRef CAS PubMed; (g) J.-H. Schöbel, M. T. Passia, N. A. Wolter, R. Puttreddy, K. Rissanen and C. Bolm, Org. Lett., 2020, 22, 2702–2706 CrossRef PubMed; (h) J.-H. Schöbel, W. Liang, D. Wöll and C. Bolm, J. Org. Chem., 2020, 85, 15760–15766 CrossRef; (i) J.-H. Schöbel, P. Elbers, K.-N. Truong, K. Rissanen and C. Bolm, Adv. Synth. Catal., 2021, 363, 1322–1329 CrossRef; (j) F. Krauskopf, K.-N. Truong, K. Rissanen and C. Bolm, Org. Lett., 2021, 23, 2699–2703 CrossRef CAS PubMed; (k) P. Wu, J. S. Ward, K. Rissanen and C. Bolm, Adv. Synth. Catal., 2023, 365, 522–526 CrossRef CAS; (l) K. Natarajan, S. Ravindra, V. R. P. Priya, R. Kataria and G. C. Nandi, Eur. J. Org Chem., 2024, 27, e202301217 CrossRef CAS; (m) T. Guo, L. Xu and J. Dong, Org. Lett., 2025, 27, 1356–1361 CrossRef CAS PubMed.
- For reviews on the synthesis and applications of sulfonimidamides, see: (a) P. K. Chinthakindi, T. Naicker, N. Thota, T. Govender, H. G. Kruger and P. I. Arvidsson, Angew. Chem., Int. Ed., 2017, 56, 4100–4109 CrossRef CAS PubMed; (b) G. C. Nandi and P. I. Arvidsson, Adv. Synth. Catal., 2018, 360, 2976–3001 CrossRef CAS; (c) M.-K. Wei and M. C. Willis, Synthesis, 2025, 57, 1429–1440 CrossRef CAS.
- For examples of sulfonimidamide inhibitors in NOD-like receptor, pyrin domain-containing protein (NLRP)-associated disorders, see: (a) J. Katz, W. Roush, H. M. Seidel, D.-M. Shen and S. Venkatraman, WO2020154499 A1, 2020 Search PubMed; (b) P. Gibbons, K. W. Lai, C. Nilewski, R. M. Pastor, S. T. Staben, C. Stivala, B.-Y. Zhu and H. Chen , WO2021150574 A1, 2021 Search PubMed; (c) K. W. Lai, C. Nilewski, R. M. Pastor and C. Stivala, WO2023004257 A1, 2023 Search PubMed; (d) D.-M. Shen, K. F. Byth, D. Bertheloot, S. Braams, S. Bradley and D. Dean, et al. , J. Med. Chem., 2025, 68, 5529–5550 CrossRef CAS PubMed.
- For a patent example of sulfonimidamide inhibitors in Stimulator of Interferon Genes (STING)-associated disorders, see: S. Venkatraman, W. R. Roush and H. M. Seidel, WO2020106736 A1, 2020.
- For a patent example of sulfonimidamide inhibitors in Liver X Receptor (LXR)-associated disorders, see: C. Gege, M. Birkel, E. Hambruch, U. Deuschle and C. Kremoser, WO2018188795 A1, 2018.
- For a patent example of sulfonimidamides in pesticidal applications, see: I. Sen, V. Sikervar, G. Rawal, A. Edmunds, S. Rendler, M. Muehlebach and D. Emery, WO2019076778 A1, 2019.
- This class of aza-saccharins was referred to as “cluster D”, see: Y. Chen, C.-J. Aurell, A. Pettersen, R. J. Lewis, M. A. Hayes, M. Lepistö, A. C. Jonson, H. Leek and L. Thunberg, ACS Med. Chem. Lett., 2017, 8, 672–677 CrossRef CAS PubMed.
- For patent examples of aza-saccharins as NLRP3 modulators, see: (a) J. Katz, W. Roush, H. M. Seidel, D.-M. Shen and S. Venkatraman, WO2020102100 A1, 2020 Search PubMed; (b) E. Gabellieri and J. Molette, WO2020254697 A1, 2020 Search PubMed.
- For a perspective regarding NLRP3 modulators mentioning aza-saccharins, see: N. Li, R. Zhang, M. Tang, M. Zhao, X. Jiang, X. Cai, N. Ye, K. Su, J. Peng, X. Zhang, W. Wu and H. Ye, J. Med. Chem., 2023, 66, 14447–14473 CrossRef CAS PubMed.
- For examples regarding an anionic [1,4] Fries-type rearrangement, see: (a) H. Kim, K. Inoue and J.-i. Yoshida, Angew. Chem., Int. Ed., 2017, 56, 7863–7866 CrossRef CAS PubMed; (b) M. Korb and H. Lang, Chem. Soc. Rev., 2019, 48, 2829–2882 RSC.
- For examples regarding regioselective Fries rearrangement, including directed Snieckus-Fries rearrangement, see: (a) K. J. Singh and D. B. Collum, J. Am. Chem. Soc., 2006, 128, 13753–13760 CrossRef CAS PubMed; (b) K. Groom, S. M. S. Hussain, J. Morin, C. Nilewski, T. Rantanen and V. Snieckus, Org. Lett., 2014, 16, 2378–2381 CrossRef CAS PubMed; (c) Y. Ma, R. A. Woltornist, R. F. Algera and D. B. Collum, J. Org. Chem., 2019, 84, 9051–9057 CrossRef CAS PubMed; (d) S. Ghinato, F. De Nardi, P. Bolzoni, A. Antenucci, M. Blangetti and C. Prandi, Chem. Eur J., 2022, 28, e202201154 CrossRef CAS PubMed.
- For the synthesis of sulfonimidoyl fluorides, see: (a) C. R. Johnson, K. G. Bis, J. H. Cantillo, N. A. Meanwell, M. F. D. Reinhard, J. R. Zeller and G. P. Vonk, J. Org. Chem., 1983, 48, 1–3 CrossRef CAS; (b) B. Gao, S. Li, P. Wu, J. E. Moses and K. B. Sharpless, Angew. Chem., Int. Ed., 2018, 57, 1939–1943 CrossRef CAS PubMed; (c) S. Greed, E. L. Briggs, F. I. M. Idiris, A. J. P. White, U. Lücking and J. A. Bull, Chem. Eur J., 2020, 26, 12533–12538 CrossRef CAS PubMed; (d) L. Wang and J. Cornella, Angew. Chem., Int. Ed., 2020, 59, 23510–23515 CrossRef CAS PubMed; (e) Y. Liu, Q. Pan, X. Hu, Y. Guo, Q.-Y. Chen and C. Liu, Org. Lett., 2021, 23, 3975–3980 CrossRef CAS PubMed; (f) A. Zogu, K. Ullah, S. Spanopoulos, E. Ismalaj, W. M. De Borggraeve and J. Demaerel, Angew. Chem., Int. Ed., 2024, 63, e202403797 CrossRef CAS PubMed; (g) M. Andresini, L. Marraffa, D. Serbetci, P. Natho, M. Colella, L. Degennaro and R. Luisi, Adv. Synth. Catal., 2025, 367, e202400908 CrossRef CAS; (h) H.-s. Huang, Y. Yuan, W. Wang, S.-q. Zhang, X.-k. Nie, W.-t. Yang, X. Cui, Z. Tang and G.-x. Li, Angew. Chem., Int. Ed., 2025, 59, e202415873 Search PubMed.
- For a successful example using KHMDS as a base in a Fries rearrangement, see: G. M. R. Boston, I. Frank and H. Butenschön, Helv. Chim. Acta, 2021, 104, e2100025 CrossRef CAS.
- An average recovery rate of 90% was determined for the preparative liquid chromatography purification. Isolated yields are reported uncorrected..
- For the complete solvent screen data, including different ethers and toluene, together with corresponding LCMS yields, see the SI..
- LCMS analysis of the reaction mixture for the sulfonimidoyl chloride analogue of sulfonimidoyl fluoride 1a was dominated by low-intensity and unassigned signals consistent with degradation, which precluded meaningful quantitation. Accordingly, Table 1 reports “degradation” and no LCMS yield was determined (cf. ref. 2d for a direct conversion of sulfonimidoyl chlorides with nitrogen nucleophiles)..
- The increased side product formation observed for the Moc analogue of sulfonimidoyl fluoride 1a is consistent with competitive nucleophilic reactions by the released methanolate anion under the reaction conditions. Although other carbamate protecting groups (e.g., benzyloxycarbonyl (Cbz), 9-fluorenylmethyoxycarbonyl (Fmoc), allyloxycarbonyl (Alloc)) were not examined, the Boc vs. Moc comparison, together with established nucleophilicity trends of carbamate-derived alkoxides in aprotic media, suggests that carbamates releasing alkoxides more nucleophilic than the Boc-derived tert-butanolate would likely promote analogous side reactions. Consequently, Boc was selected as the optimal protecting group. For nucleophilicity trends of alkoxides, especially between methanolate and tert-butanolate, see: (a) D. K. Bohme and L. B. Young, J. Am. Chem. Soc., 1970, 92, 7354–7358 CrossRef CAS; (b) J. M. Dust and E. Buncel, Can. J. Chem., 1991, 69, 978–986 Search PubMed.
- NaHMDS was found to activate only certain sulfonimidoyl fluorides, such as sulfonimidoyl fluoride 1a. NaHMDS was proven to be inefficient in the activation of para-bromo sulfonimidoyl fluoride 1i..
- See the SI for details..
- For studies regarding ion pairing, aggregation and/or metal–substrate coordination of alkali-metal silazide bases (LiHMDS, NaHMDS and KHMDS), see: (a) B. L. Lucht and D. B. Collum, J. Am. Chem. Soc., 1995, 117, 9863–9874 CrossRef CAS; (b) R. A. Woltornist and D. B. Collum, J. Org. Chem., 2021, 86, 2406–2422 CrossRef CAS PubMed; (c) J. A. Spivey and D. B. Collum, J. Am. Chem. Soc., 2024, 146, 17827–17837 CrossRef CAS PubMed.
- Side product formation in the KPA of sulfonimidoyl fluorides generally started at temperatures above −50 °C..
- For general trends regarding inductive effects, polarization, and stereoelectronic effects (hyperconjugative acceptor ability of σ-bonds) in substituted phenyl carbanions, see: (a) P. G. Wenthold and R. R. Squires, J. Mass Spectrom., 1995, 30, 17–24 CrossRef CAS; (b) P. B. M. Andrade and J. M. Riveros, J. Mass Spectrom., 1996, 31, 767–770 CrossRef CAS; (c) I. V. Alabugin and T. A. Zeidan, J. Am. Chem. Soc., 2002, 124, 3175–3185 CrossRef CAS PubMed; (d) Z. Tian and S. R. Kass, Chem. Rev., 2013, 113, 6986–7010 CrossRef CAS; (e) I. V. Alabugin, in, Stereoelectronic Effects: A Bridge Between Structure and Reactivity, John Wiley & Sons, 2016, pp. 214–235 CrossRef.
- The relative ranking of the sulfonimidoyl fluoride (SOFNBoc) group is based on experiments with the 3,5-difluoro and 3,5-dichloro sulfonimidoyl fluoride derivatives. While the aza-saccharin formation with the 3,5-difluoro sulfonimidoyl fluoride derivative was unproductive, the corresponding aza-saccharin was isolated in 61% yield under the use of the 3,5-dichloro sulfonimidoyl fluoride derivative .
- X-ray crystal structures of aza-saccharins 3n–3r derived from bis-meta-substituted sulfonimidoyl fluorides are provided in the Supporting Information and support the corresponding regioselectivity assignments..
- For the reactivity and regioselectivity in the deprotonation of substituted arenes with the Lochmann-Schlosser base, see: (a) G. Katsoulos, S. Takagishi and M. Schlosser, Synlett, 1991, 731–732 CrossRef CAS; (b) E. Marzi, F. Mongin, A. Spitaleri and M. Schlosser, Eur. J. Org Chem., 2001, 2911–2915 CrossRef CAS; (c) E. Marzi, A. Spitaleri, F. Mongin and M. Schlosser, Eur. J. Org Chem., 2002, 2508–2517 CrossRef CAS; (d) E. Marzi, J. Gorecka and M. Schlosser, Synthesis, 2004, 1609–1614 CAS; (e) M. Schlosser, Angew. Chem., Int. Ed., 2005, 44, 376–393 CrossRef CAS PubMed.
- For a review on the directed ortho-metalation with lithium bases, see: V. Snieckus, Chem. Rev., 1990, 90, 879–933 CrossRef CAS.
- The unsubstituted phenyl derivative gave trace conversion under standard conditions, however, aza-saccharin 3m was obtained in low yield upon temperature adjustment during KPA. For the 4-methoxy derivative, minor formation of aza-saccharin 3s was detected via LCMS analysis following KPA at −30 °C for 30 min, yet the product could not be isolated..
- A modified synthetic protocol was evaluated for aza-saccharin formation with aliphatic amines using catalytic amounts of 1-hydroxybenzotriazole (HOBt), however, no improvements in isolated yields were observed (cf. ref. 1x).
- Attempts to increase conversion toward aza-saccharins 3ay, 3az by longer reaction times and/or warming to 40 °C led to increased side product formation.
- A detailed summary of the additional nitrogen nucleophile screening experiments, including LCMS observations and side product profiles, is provided in the SI..
- For KHMDS acting both as a base and single-electron donor, see: N. Ogawa, Y. Yamaoka, K.-i. Yamada and K. Takusa, Org. Lett., 2017, 19, 3327–3330 CrossRef CAS PubMed.
- Electron paramagnetic resonance (EPR) spectroscopy did not reveal any radical intermediates in the KPA of sulfonimidoyl fluoride 1a.
- Corresponding 6-membered ring products could not be detected via LCMS analysis of the reaction mixture with sulfonimidoyl fluorides 8b, 8c, respectively. Their formation would theoretically occur via benzylic deprotonation, followed by an anionic [1,5] Fries-type rearrangement and analogous interception by amines to effect ring closure (cf. ref. 12d for a related anionic [1,4] Fries rearrangement).
- For kinetic isotope effect (KIE) experiments via intramolecular competition, see: (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed; (b) W. Zi, Y.-M. Wang and F. D. Toste, J. Am. Chem. Soc., 2014, 136, 12864–12867 CrossRef CAS PubMed; (c) Z. Mao and C. T. Campbell, ACS Catal., 2020, 10, 4181–4192 CrossRef CAS.
- For examples of kinetic isotope effects used to elucidate reaction mechanisms, including inverse kinetic isotope effects and pre-equilibrium isotope effects, see: (a) D. G. Churchill, K. E. Janak, J. S. Wittenberg and G. Parkin, J. Am. Chem. Soc., 2003, 125, 1403–1420 CrossRef CAS PubMed; (b) M. Gomez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef CAS PubMed; (c) X. Gao, X.-Y. Yu and C.-R. Chang, Phys. Chem. Chem. Phys., 2022, 24, 15182–15194 RSC.
- For stereospecific substitution at chiral S(VI) centers (including sulfonimidoyl fluorides), which is commonly consistent with an SN2-type displacement leading to inversion at sulfur, see: (a) S. Greed, O. Symes and J. A. Bull, Chem. Commun., 2022, 58, 5387–5390 RSC; (b) O. L. Symes and J. A. Bull, Org. Chem. Front., 2025, 12, 6681–6697 RSC.
- (a) CCDC 2517062: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6k0; (b) CCDC 2517063: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6l1; (c) CCDC 2517064: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6m2; (d) CCDC 2517065: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6n3; (e) CCDC 2517066: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6p4; (f) CCDC 2517067: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6q5; (g) CCDC 2517068: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6r6; (h) CCDC 2517069: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6s7; (i) CCDC 2517070: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6t8; (j) CCDC 2517071: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6v9; (k) CCDC 2517072: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6wb; (l) CCDC 2517073: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6xc; (m) CCDC 2517074: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2qh6yd.
| This journal is © The Royal Society of Chemistry 2026 |