
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModular assembly of chiral biaryl phosphoramidite (BPA) libraries by nickel catalysis
Xi Zhang†
,
Jingyi Bai†,
Yue Zhao ,
Minyan Wang and
Zhuangzhi Shi*
,
Minyan Wang and
Zhuangzhi Shi*
State Key Laboratory of Coordination Chemistry, Chemistry and Biomedicine Innovation Center (ChemBIC), School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210093, China. E-mail: shiz@nju.edu.cn
First published on 8th May 2026
Abstract
Chiral phosphoramidites have emerged as pivotal ligands in asymmetric catalysis, yet their synthesis has long been constrained by traditional de novo approaches. Here, we present a highly significant late-stage functionalization strategy, which facilitates the modular assembly of biaryl phosphoramidite (BPA) libraries. Leveraging P(III)-directed C–H activation by nickel catalysis, we have developed a versatile platform for facile modification of the chiral pocket within these BPAs, enabling rapid structural optimization and exploration of diverse chemical architectures. These formed ligand libraries have demonstrated exceptional performance across a spectrum of asymmetric palladium-catalysed reactions, underscoring their broad applicability and potential. Through a synergistic combination of experimental investigations and computational analyses, we have elucidated the underlying reaction mechanism with remarkable clarity. This research not only furnishes advanced synthetic tools for the preparation of phosphoramidite libraries but also sets a new benchmark for the design and synthesis of novel ligands using state-of-the-art synthetic methodologies.
Introduction
Transition-metal-catalysed asymmetric catalysis has profoundly reshaped modern synthetic chemistry, offering unparalleled precision and diversity in stereoselective transformations. Central to this progress is the strategic design of privileged chiral ligands, which provide exceptional control over regio-, diastereo-, and enantioselectivity.1 A pivotal milestone was achieved in 1994 with Feringa's development of MonoPhos, a groundbreaking chiral phosphoramidite ligand featuring a BINOL scaffold with a NMe2 motif (Fig. 1a).2 This ligand demonstrated unprecedented enantioselectivity in copper-catalysed conjugate additions of dialkyl zinc reagents to enones.3 This impressive achievement inspired extensive research into chiral phosphoramidite derivatives based on the BINOL framework, setting a new benchmark in asymmetric catalysis.4 However, the synthesis of phosphoramidites remains largely confined to traditional methods. These de novo approaches, which involve P–O or P–N bond formation through chlorophosphite synthesis, dichloroaminophosphine formation, or amine exchange, are often hindered by complex procedures, multi-step syntheses, and limited structural diversity.5 These challenges underscore the urgent need for more efficient and versatile synthetic methods to expand the accessibility of chiral phosphoramidite libraries and facilitate precise ligand optimization for targeted catalytic applications. | ||
| Fig. 1 Background and this discovery. | ||
Over the past decades, transition-metal-catalysed C–H activation has emerged as a transformative strategy in organic synthesis, offering unparalleled efficiency and selectivity in modifying carbon centers.6 Since the early 2000s, serendipitous discoveries have highlighted the potential of phosphoramidite ligands in this context (Fig. 1b). In 2003, Hartwig et al. reported a significant result where a metallacyclic iridium-phosphoramidite, formed via aliphatic C–H activation in situ, served as the active supporting ligand in iridium-catalysed kinetic asymmetric substitution of racemic allylic electrophiles.7 This discovery was further advanced by You and co-workers, who identified that the iridicycle is generated through aromatic C–H activation of the N-aryl group within the ligand, a key active species in many iridium-catalysed asymmetric allylic alkylation reactions.8 These findings were initially limited to stoichiometric transformations, where the metal and ligand are used in equimolar amounts. However, developing a catalytic process using small amounts of transition metals with stoichiometric phosphoramidites remains challenging due to the highly electron-rich and strongly coordinating nature of the P(III) center in these compounds.
Recent advancements have underscored the transformative potential of P(III)-directed C–H activation in catalytic processes.9 A pivotal development in this field has been the strategic incorporation of N–PR2 groups, which has demonstrated remarkable efficacy in enhancing regioselectivity and functional group tolerance (Fig. 1c). The attachment of the PtBu2 moiety to the nitrogen atom of indole facilitates C–H arylation at the C7 position with aryl bromides or carboxylic acid anhydrides, a regioselectivity that is often elusive with conventional directing groups.10 Moreover, the recent work by Evano and Lan has introduced a protocol for P(III)-directed ortho-selective C–H arylation of amino-phosphines using aryl iodides under rhodium catalysis.11 This method offers significant practical advantages, as the N-PR2 group can be readily removed under mild acidic conditions to yield NH-free products. Despite these advancements, current methodologies are predominantly constrained by their reliance on precious metal catalysts, and the removal of directing groups from the nitrogen atom does not fully leverage the inherent P(III) functionality.
Here, we present a highly efficient catalytic system for P(III)-directed C–H activation of phosphoramidites, which enables the modular assembly of chiral ligand libraries (Fig. 1d). This innovative approach utilizes a nickel catalyst to facilitate the efficient coupling of diverse aryl chlorides, constructing biaryl phosphoramidite (BPA) libraries with exceptional efficiency. The choice of nickel, a more abundant and cost-effective metal compared to iridium, significantly enhances the scalability and industrial applicability of this methodology.12 Additionally, the use of aryl chlorides, which are more economical and readily available than their bromo and iodo counterparts, highlights the practical and economic advantages of this approach. Notably, when Pd catalysis is employed with aryl iodides, a C–P coupling reaction of phosphoramidites occurs instead of C–H activation, resulting in the formation of P-chirogenic compounds.13 Through the implementation of late-stage functionalization strategies, we can modulate the chiral pocket of the formed BPA libraries, enabling rapid reaction optimization and the exploration of diverse chemical space.
Results and discussion
Reaction design
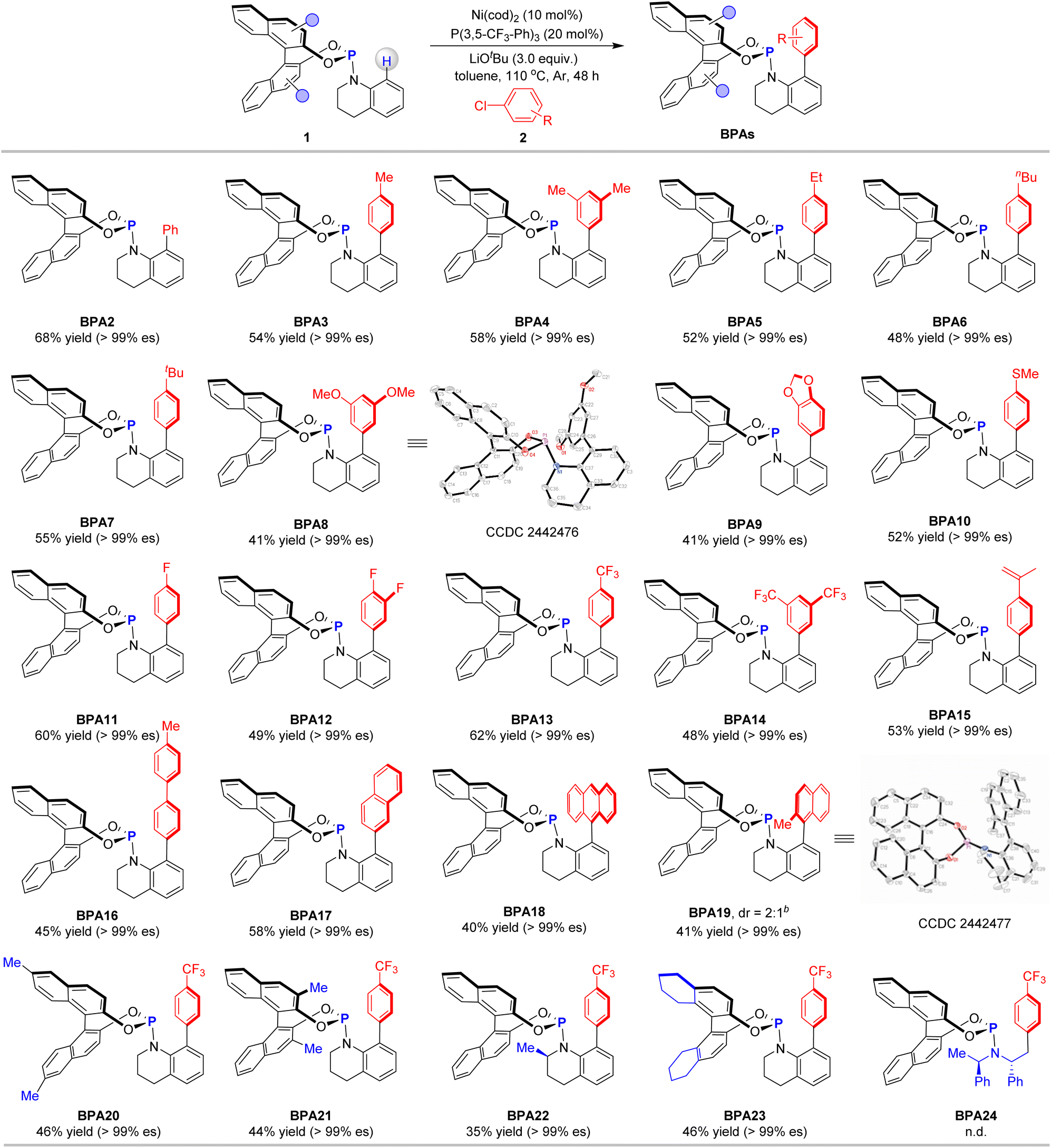
To optimize the reaction conditions, we initiated our study using enantiomerically enriched phosphoramidite 1a with 99% ee (enantiomeric excess) and aryl chloride 2a as model substrates, both of which are commercially available (Table 1). The reaction was first performed in the presence of [Ir(cod)Cl]2 (5.0 mol%) and LiOtBu (2.0 equivalents) in toluene at 110 °C for 48 hours in a N2 atmosphere. However, no desired product was observed, highlighting the significant difference between stoichiometric and catalytic processes (entry 1). Switching to [Rh(cod)Cl]2 instead of [Ir(cod)Cl]2 yielded a small amount of the desired product BPA1 (entry 2). Further screening of metal catalysts revealed that Ni(cod)2 significantly enhanced the reaction, increasing the yield of BPA1 to 42% (entry 3). Next, we explored the influence of ligands on the reaction. Using PPh3 as a ligand slightly improved the yield to 46% (entry 4). However, the electron-rich ligand P(2-OMe-Ph)3 was detrimental to the reaction (entry 5). In contrast, electron-deficient phosphine ligands such as P(2-F-Ph)3 favored product formation (entry 6). Further investigation showed that P(3-F-Ph)3 boosted the yield to 55% (entry 7), while P(4-F-Ph)3 reduced reactivity (entry 8). Interestingly, P(4-CF3-Ph)3 provided a higher yield of 52% (entry 9), and P(3,5-CF3-Ph)3 gave the best result, achieving a 64% yield of BPA1 (entry 10). We then examined the role of different bases. Replacing LiOtBu with KOtBu or NaOtBu drastically reduced the yield of BPA1, and Li2CO3 completely inhibited the reaction (entries 11–13). These results underscore the critical roles of both Li+ and OtBu− ions in the efficient conversion of the substrates. Solvent optimization studies demonstrated that replacing toluene with 1,4-dioxane led to a sharp decrease in reactivity (entry 14). We also screened reaction temperatures. Both increasing (130 °C) and decreasing (90 °C) the temperature resulted in reduced reactivity (entries 15–16). To further explore the impact of the catalyst, we tested other nickel catalysts, such as NiBr2, which exhibited much lower activity compared to Ni(cod)2 (entry 17). A control experiment confirmed that the reaction did not proceed in the absence of the catalyst, emphasizing its essential role (entry 18). Additionally, replacing aryl chloride 2a with aryl bromide 2a′ and iodide 2a″ resulted in significantly lower yields (entries 19–20), highlighting the unique reactivity of the C–Cl bond in this transformation. Notably, all the above reactions proceeded with excellent retention of chirality, affording the product BPA1 with 99% es.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | X | cat [M] (mol%) | L (mol%) | Base (equiv.) | Solvent | T (°C) | Yield of BPA1b (%) |
| a Reaction conditions: cat [M] (5.0–10.0 mol%), L (20.0 mol%), 1a (0.10 mmol), 2a (0.30 mmol), and base (0.30 mmol) in 1.0 mL of toluene, at 110 °C for 48 hours under nitrogen.b Isolated yield. | |||||||
| 1 | Cl (2a) | [Ir(cod)Cl]2 (5) | — | LiOtBu (3.0) | Toluene | 110 | 0 |
| 2 | Cl (2a) | [Rh(cod)Cl]2 (5) | — | LiOtBu (3.0) | Toluene | 110 | <5 |
| 3 | Cl (2a) | Ni(cod)2 (10) | — | LiOtBu (3.0) | Toluene | 110 | 42 |
| 4 | Cl (2a) | Ni(cod)2 (10) | PPh3 (20) | LiOtBu (3.0) | Toluene | 110 | 46 |
| 5 | Cl (2a) | Ni(cod)2 (10) | P(2-OMe-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 38 |
| 6 | Cl (2a) | Ni(cod)2 (10) | P(2-F-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 45 |
| 7 | Cl (2a) | Ni(cod)2 (10) | P(3-F-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 55 |
| 8 | Cl (2a) | Ni(cod)2 (10) | P(4-F-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 37 |
| 9 | Cl (2a) | Ni(cod)2 (10) | P(4-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 52 |
| 10 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 64 |
| 11 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | KOtBu (3.0) | Toluene | 110 | <5 |
| 12 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | NaOtBu (3.0) | Toluene | 110 | <10 |
| 13 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | Li2CO3 (3.0) | Toluene | 110 | 0 |
| 14 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | 1,4-Dioxane | 110 | <5 |
| 15 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 130 | 55 |
| 16 | Cl (2a) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 90 | 51 |
| 17 | Cl (2a) | NiBr2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | <10 |
| 18 | Cl (2a) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 0 | |
| 19 | Br (2a′) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 40 |
| 20 | I (2a″) | Ni(cod)2 (10) | P(3,5-CF3-Ph)3 (20) | LiOtBu (3.0) | Toluene | 110 | 18 |

Reaction scope
To comprehensively assess the versatility of the reaction, we initiated our investigation by examining the scope of aryl chlorides with phosphoramidite 1a (Table 2). The reaction involving chlorobenzene (2b) yielded BPA2 with a 68% yield and an es exceeding 99%. Aryl chlorides featuring alkyl substituents, including methyl (2c, 2d), ethyl (2e), n-butyl (2f), and tert-butyl (2g), proved to be compatible with the transformation, producing the desired products BPA2 to BPA7 in moderate yields while maintaining high enantioselectivities. Substrates with electron-donating groups, such as methoxy (2h), dioxole (2i), and methylthio (2j), underwent smooth reactions, yielding BPA8 to BPA10. The structure of product BPA8 was unequivocally confirmed through X-ray crystallographic analysis. Aryl chlorides bearing electron-withdrawing substituents, such as fluorine (2k, 2l) and trifluoromethyl (2m, 2n), also exhibited favorable outcomes, efficiently yielding BPA11 to BPA14. The reaction with aryl chloride 2o, which contains a sensitive olefinic group, was compatible, affording the corresponding product BPA15 in 53% yield with an es value surpassing 99%. Given the capability to rapidly modulate the steric and electronic properties of the ligand by incorporating diverse aryl motifs, substrates with biphenyl (2p) and naphthalen-2-yl (2q) reacted efficiently, yielding BPA16 and BPA17 with consistent enantiospecificity. The reaction involving the sterically hindered substrate 9-chloroanthracene (2r) with 1a afforded BPA18 in modest yield with excellent enantiospecificity. When 1-chloro-2-methylnaphthalene was employed as the substrate, the reaction yielded only 30% of the product. However, using the corresponding bromide 2s resulted in a 41% yield of BPA19, which comprised a pair of diastereomers in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. One of these diastereomers was successfully isolated, and its absolute configuration was unequivocally determined through X-ray crystallographic analysis. Using 1-chloro-4-(trifluoromethyl)benzene (2m) as the substrate, we further investigated the influence of substituents on the Binol moiety. When phosphoramidites 1b and 1c, bearing methyl groups at the 6, 6′ and 3, 3′ positions, respectively, were employed, the desired products BPA20 and BPA21 were successfully obtained. Additionally, phosphoramidite 1d, featuring a 2-methyl-1,2,3,4-tetrahydroquinoline motif, was compatible with the reaction, only yielding the aromatic C–H arylation product BPA22. Beyond the binol motif, the use of 8H-binol-derived phosphoramidite 1e in the reaction also resulted in the formation of BPA23 in 46% yield, with retained enantiospecificity. In contrast, ligands bearing amine-derived motifs—such as Feringa's ligand containing the bis(1-phenylethyl)amine moiety (BPA24)14 and Carreira's ligand—proved entirely ineffective under the current conditions. It is worth noting that only moderate yields were obtained for several of these products, primarily due to the susceptibility of the phosphoramidite ligands to oxidation, necessitating rapid chromatographic purification. In addition, the presence of trace water in the reaction system could lead to product hydrolysis, further impacting the yields.
:1 ratio. One of these diastereomers was successfully isolated, and its absolute configuration was unequivocally determined through X-ray crystallographic analysis. Using 1-chloro-4-(trifluoromethyl)benzene (2m) as the substrate, we further investigated the influence of substituents on the Binol moiety. When phosphoramidites 1b and 1c, bearing methyl groups at the 6, 6′ and 3, 3′ positions, respectively, were employed, the desired products BPA20 and BPA21 were successfully obtained. Additionally, phosphoramidite 1d, featuring a 2-methyl-1,2,3,4-tetrahydroquinoline motif, was compatible with the reaction, only yielding the aromatic C–H arylation product BPA22. Beyond the binol motif, the use of 8H-binol-derived phosphoramidite 1e in the reaction also resulted in the formation of BPA23 in 46% yield, with retained enantiospecificity. In contrast, ligands bearing amine-derived motifs—such as Feringa's ligand containing the bis(1-phenylethyl)amine moiety (BPA24)14 and Carreira's ligand—proved entirely ineffective under the current conditions. It is worth noting that only moderate yields were obtained for several of these products, primarily due to the susceptibility of the phosphoramidite ligands to oxidation, necessitating rapid chromatographic purification. In addition, the presence of trace water in the reaction system could lead to product hydrolysis, further impacting the yields.
Synthetic applications
The developed strategy has demonstrated remarkable efficiency in the synthesis of BPA libraries, representing a novel approach in the development of efficient chiral phosphoramidite ligands for catalysis and organometallic reactivity (Fig. 2). When the reaction of phosphoramidite 1a was scaled up to 2.5 mmol with PhCl (2b), the desired product BPA2 was obtained in 53% yield without significant loss in efficiency under standard conditions (Fig. 2a). Further investigations revealed that, even in the absence of the ligand, the corresponding product could still be obtained in 46% yield. This finding underscores that the primary role of the ligand is to enhance reaction efficiency rather than being a critical factor for the transformation itself. When 1.0 equivalent of BPA2 was allowed to react with a stoichiometric amount of [Rh(cod)Cl]2 or [Ir(cod)Cl]2, complexes BPA2-Rh and BPA2-Ir were obtained, respectively. X-ray crystallographic analysis revealed that the metal centers in both complexes are chelated solely by the phosphorus atom. For BPA2-Rh, the bond lengths and angles surrounding the Rh atom range from 2.118(4) to 2.3597(11) Å and 35.16(15) to 120.06(13)°, respectively. Similarly, the reaction with [Ir(cod)Cl]2 yielded a monometallic complex, BPA2-Ir, where the iridium center is chelated by P, C, and Cl atoms. The Ir-centered bond lengths and angles in BPA2-Ir range from 2.113(8) to 2.356(2) Å and 36.0(3) to 120.0(3)°, respectively. To better illustrate the steric repulsions at different regions of the formed ligands, the structural and spatial characteristics of the phosphoramidite complexes were systematically investigated using Multiwfn15 to generate detailed two-dimensional (2D) contour maps (Fig. 2b).16 The analysis revealed distinct differences in the spatial arrangements between the Rh and Ir complexes of 1a and their phenyl-substituted counterparts (BPA2-Rh and BPA2-Ir). The complexes 1a-Rh and 1a-Ir were found to create a relatively open and accessible environment for substrate binding. However, a significant structural modification was observed upon the introduction of a phenyl group into the ligand framework. The phenyl group in BPA2-Rh and BPA2-Ir was found to project into the southeast (SE) quadrant of the spatial environment, creating a pronounced steric effect around the metal center. As a direct consequence, the cavity around the metal that can accommodate the substrate becomes notably smaller. This reduction in the cavity size likely facilitates more pronounced ligand–substrate interactions, leading to the emergence of unique selectivity in the reaction system. | ||
| Fig. 2 Further investigations of the crystal model. | ||
Based on the above results, we then evaluated their potential in asymmetric catalysis (Fig. 3). We chose parent ligand 1a, with the representative products BPA1, BPA2, BPA13, BPA18, BPA21 and BPA23—each distinguished by unique steric and electronic properties, to generate a small ligand library and tested them for asymmetric catalysis. In our experimental investigation, we first evaluated the performance of our ligands in the asymmetric addition of phenylboric acid to aldehyde 25 (Fig. 3a).17 Initially, using the parent ligand 1a, the reaction yielded product 26 with a modest 46% yield and 43% ee. Subsequent screening of the developed ligands revealed that BPA1 emerged as the optimal ligand, significantly enhancing the reaction outcome to 52% yield and 90% ee. Building on these promising results, we extended our study to the enantioselective dearomative difunctionalization of indoles—a highly valuable strategy for constructing complex chiral N-heterocycles (Fig. 3b).18 The catalytic performance of these ligands was assessed in the palladium-catalysed enantioselective cycloaddition of indole 27 with phenylacetylene. Remarkably, ligand BPA2 demonstrated exceptional efficacy, delivering product 28 in 45% yield and 91% ee, thereby significantly outperforming the parent ligand 1a. Moreover, BPA13 exhibited a higher reactivity but low enantioselectivity, resulting in product 29 in 73% yield and 60% ee. To further explore the potential applications of BPAs, we investigated a Pd-catalysed dearomative arylvinylation of indoles 27, utilizing N-arylsulfonyl hydrazones as versatile coupling partners to access 3-vinylindolines derivative 29 (Fig. 3c).19 In contrast to the moderate enantioselectivity displayed by 1a, BPA1 proved to be highly effective, achieving an ee value of 90%. Furthermore, BPA23 also yielded a relatively good result, although it was not the optimal ligand. Finally, we applied the ligand library to a Pd-catalysed asymmetric sp3 C–H activation of compound 30 with4-OMe-iodobenzene to afford product 31 (Fig. 3d).20 Remarkably, ligand BPA2 significantly increased the enantioselectivity to 90% ee, representing a substantial improvement over the parent ligand 1a. These results unequivocally demonstrate the versatility and efficacy of the newly developed BPA libraries, as evidenced by their broad applicability and enhanced performance across a variety of challenging asymmetric transformations.
 | ||
| Fig. 3 Investigating some BPAs in asymmetric catalysis. | ||
Mechanistic studies
Subsequent mechanistic studies provided critical insights into the pathway of the Ni-catalysed C–H arylation (Fig. 4). Initial experiments employing phosphine 1a in conjunction with isotopically labeled substrate D-1a and PhCl (2b) revealed a significant primary kinetic isotope effect (KIE) of kH/kD = 2.15. This pronounced KIE value strongly implicates C–H bond cleavage as the rate-determining step in the catalytic cycle (Fig. 4a).21 Further mechanistic elucidation was achieved through a parallel competition experiment between aryl chloride 1b and 13b (5.0 equiv. each) in the presence of phosphoramidite 1a (1.0 equiv). The reaction yielded a mixture of BPA13 and BPA1 with more than 15/1 ratio (Fig. 4b). The pronounced selectivity toward the electron-deficient substrate 13b highlights the significantly accelerated reaction rates with electrophiles bearing electron-withdrawing groups, consistent with a mechanism driven by nucleophilic aromatic substitution.22 These experimental findings support the proposed mechanism, wherein oxidative addition of the aryl chloride to the Ni(0) complex is not the rate-determining step within the catalytic cycle. | ||
| Fig. 4 Mechanistic experiments. | ||
To elucidate the reaction mechanism in greater detail, we conducted comprehensive density functional theory (DFT) calculations using phosphoramidite 1a and aryl chloride 2a as model substrates (Fig. 5).23 The catalytic cycle initiates with a crucial step where the cod ligands of the Ni(cod)2 precatalyst are displaced by substrate 1a and the ligand, resulting in the formation of the highly reactive Ni(0) species INT1A. This species acts as the active catalyst in the subsequent transformations. Following this, INT1A coordinates with compound 2a, leading to the exothermic formation of complex INT2A, with an exothermic energy of 14.4 kcal mol−1. The oxidative addition of the C–Cl bond to the nickel center of INT2A proceeds through the transition state TS1A, which has an activation free energy of 13.8 kcal mol−1 (Fig. 5a). The base LiOtBu can potentially initiate two competing pathways for C–H metalation.24 Notably, the base-assisted outer-sphere concerted metalation-deprotonation pathway is energetically more favorable than the inner-sphere mechanism (14.6 kcal mol−1 for TS2A vs. 34.9 kcal mol−1 for TS2B, Fig. 5b). This significant energy difference strongly indicates that the base-assisted outer-sphere concerted metalation-deprotonation pathway is the predominant mechanism in this reaction. For the key transition state TS2A, the lengths of the C–Ni bond and the O–H bond, which are on the verge of formation, are measured to be 2.10 Å and 1.51 Å, respectively. Meanwhile, the length of the C–H bond about to be cleaved is determined to be 1.21 Å. The cleavage of the C–H bond represents the rate-determining step, which aligns with the experimentally observed kinetic isotope effect. The resulting nickelacycle intermediate INT5A undergoes reductive elimination via transition state TS3A, with a calculated barrier of 12.9 kcal mol−1, leading to the formation of intermediate INT6A. Finally, ligand exchange occurs, a process that efficiently regenerates the active Ni(0) species INT1A and simultaneously releases the desired product BPA1, thereby completing the catalytic cycle.
 | ||
| Fig. 5 Calculated energy profiles for the nickel-catalyzed P(III)-directed C–H arylation. All calculations were performed at the M06-D3/6-311+G(d,p)-SDD/SMD(toluene)//B3LYP-D3BJ/6-31G(d)-SDD level of theory using the Gaussian 09 software package. | ||
Conclusions
In summary, we developed a modular late-stage functionalization strategy for synthesizing BPA libraries, enabling efficient structural diversification of chiral phosphoramidite ligands. Leveraging nickel-catalysed P(III)-directed C–H activation, we achieved rapid optimization of the chiral pocket, resulting in ligand libraries with exceptional performance in palladium-catalysed asymmetric reactions. Combined experimental and computational insights clarified the reaction mechanisms, providing a foundation for future ligand design. This approach sets a new standard for ligand synthesis and opens avenues for developing next-generation chiral catalysts. Future work could expand this strategy to other ligand systems, explore new catalytic applications, and integrate computational tools for accelerated discovery.Author contributions
X. Z. performed the experiments, conducted the mechanistic studies, and analyzed the data. J. B. & M. W. performed the DFT calculations. Y. Z performed the crystallographic studies. Z. S. designed the study and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
CCDC 2442475 (BPA8), 2442477 (BPA19), 2442408 (BPA2-Rh), and 2442469 (BPA2-Ir) contain the supplementary crystallographic data for this paper.25a–dThe data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterizations, and analytical data of new compounds, and spectra of NMR and HPLC for new compounds (PDF). See DOI: https://doi.org/10.1039/d6sc00358c.
Acknowledgements
We would like to acknowledge financial support from the National Key R&D Program of China (2022YFA1503200), the National Natural Science Foundation of China (92361201, 22025104 and 22171134), the Natural Science Foundation of Jiangsu Province (BK20240059, BK20220033), and the Fundamental Research Funds for the Central Universities (020514380352).Notes and references
- (a) Q.-L. Zhou, Privileged Chiral Ligands and Catalysts, Wiley-VCH, Weinheim, 2011 CrossRef; (b) T. Imamoto, Chem. Rev., 2024, 124, 8657–8739 CrossRef CAS PubMed; (c) W. Fu and W. Tang, ACS Catal., 2016, 6, 4814–4858 CrossRef CAS; (d) T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS PubMed.
- R. Hulst, N. K. de Vries and B. L. Feringa, Tetrahedron: Asymmetry, 1994, 5, 699–708 CrossRef CAS.
- (a) B. L. Feringa, M. Pineschi, L. A. Arnold, R. Imbos and A. H. M. de Vries, Angew. Chem., Int. Ed., 1996, 35, 2374–2376 CrossRef; (b) B. L. Feringa, M. Pineschi, L. A. Arnold, R. Imbos and A. H. M. de Vries, Angew. Chem., Int. Ed., 1997, 36, 2620–2623 CrossRef CAS.
- (a) B. L. Feringa, Acc. Chem. Res., 2000, 33, 346–353 CrossRef CAS PubMed; (b) A. J. Minnaard, B. L. Feringa, L. Lefort and J. G. de Vries, Acc. Chem. Res., 2007, 40, 1267–1277 CrossRef CAS PubMed; (c) J. F. Hartwig and L. M. Stanley, Acc. Chem. Res., 2010, 43, 1461–1475 CrossRef CAS PubMed; (d) C.-X. Zhuo, C. Zheng and S.-L. You, Acc. Chem. Res., 2014, 47, 2558–2573 CrossRef CAS PubMed; (e) S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed; (f) B. M. Trost and G. Mata, Acc. Chem. Res., 2020, 53, 1293–1305 CrossRef CAS PubMed.
- J. F. Teichert and B. L. Feringa, Angew. Chem., Int. Ed., 2010, 49, 2486–2528 CrossRef CAS PubMed.
- (a) R. Giri, B.-F. Shi, K. M. Engle, N. Maugel and J.-Q. Yu, Chem. Soc. Rev., 2009, 38, 3242–3272 RSC; (b) C. G. Newton, S.-G. Wang, C. C. Oliveira and N. Cramer, Chem. Rev., 2017, 117, 8908–8976 CrossRef CAS PubMed; (c) C. Sambiagio, D. Schönbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnürch, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC; (d) Ł. Woźniak, J.-F. Tan, Q.-H. Nguyen, A. M. d. Vigné, V. Smal, Y.-X. Cao and N. Cramer, Chem. Rev., 2020, 120, 10516–10543 CrossRef PubMed; (e) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (f) C.-X. Liu, W.-W. Zhang, S.-Y. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2021, 143, 14025–14040 CrossRef CAS PubMed; (g) Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CrossRef CAS.
- (a) C. A. Kiener, C. Shu, C. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14272–14273 CrossRef CAS PubMed; (b) C. Shu, A. Leitner and J. F. Hartwig, Angew. Chem., Int. Ed., 2004, 43, 4797–4800 CrossRef CAS PubMed; (c) T. Graening and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 17192–17193 CrossRef CAS PubMed; (d) D. J. Weix and J. F. Hartwig, J. Am. Chem. Soc., 2007, 129, 7720–7721 CrossRef CAS PubMed; (e) H. Qin, J. T. Lowe and J. S. Panek, J. Am. Chem. Soc., 2007, 129, 38–39 CrossRef CAS PubMed; (f) S. T. Madrahimov, D. Markovic and J. F. Hartwig, J. Am. Chem. Soc., 2009, 131, 7228–7229 CrossRef CAS PubMed; (g) S. T. Madrahimov and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 8136–8147 CrossRef CAS PubMed; (h) T. W. Butcher, J. L. Yang, W. M. Amberg, N. B. Watkins, N. D. Wilkinson and J. F. Hartwig, Nature, 2020, 583, 548–553 CrossRef CAS PubMed.
- (a) W.-B. Liu, C. Zheng, C.-X. Zhuo, L.-X. Dai and S.-L. You, J. Am. Chem. Soc., 2012, 134, 4812–4821 CrossRef CAS PubMed; (b) X. Zhang, W. Liu, Q. Cheng and S.-L. You, Organometallics, 2016, 35, 2467–2472 CrossRef CAS; (c) S. Rieckhoff, J. Meisner, J. Kästner, W. Frey and R. Peters, Angew. Chem., Int. Ed., 2018, 57, 1404–1408 CrossRef CAS PubMed; (d) L. S. Hutchings-Goetz, C. Yang, J. W. B. Fyfe and T. N. Snaddon, Angew. Chem., Int. Ed., 2020, 59, 17556–17564 CrossRef CAS PubMed.
- (a) Z. Zhang, P. H. Dixneuf and J.-F. Soulé, Chem. Commun., 2018, 54, 7265–7280 RSC; (b) J. Zhang, L. Yao, J.-Y. Su, Y.-Z. Liu, Q. Wang and W. P. Deng, Green Synth. Catal., 2023, 4, 206–225 CAS; (c) Z. Li and Z. Shi, Acc. Chem. Res., 2024, 57, 1057–1072 CrossRef CAS PubMed.
- X. Qiu, P. Wang, D. Wang, M. Wang, Y. Yuan and Z. Shi, Angew. Chem., Int. Ed., 2019, 58, 1504–1508 CrossRef CAS PubMed.
- C. Jacob, J. Annibaletto, J. Peng, R. Bai, B. U. W. Maes, Y. Lan and G. Evano, Angew. Chem., Int. Ed., 2024, 63, e202403553 CrossRef CAS PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef CAS PubMed.
- A. Mondal, N. O. Thiel, R. Dorel and B. L. Feringa, Nat. Catal., 2022, 5, 10–19 CrossRef CAS.
- A. Mondal, V. Dašková, X. Chen, N. O. Thiel, G. Alachouzos and B. L. Feringa, Chem. Commun., 2025, 61, 6510–6513 RSC.
- T. Lu and F. Chen, J. Comput. Chem., 2012, 33, 580–592 CrossRef CAS PubMed.
- (a) P. Liu, J. Montgomery and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 6956–6959 CrossRef CAS PubMed; (b) G. Lu, C. Fang, T. Xu, G. Dong and P. Liu, J. Am. Chem. Soc., 2015, 137, 8274–8283 CrossRef CAS PubMed.
- K. Li, N. Hu, R. Luo, W. Yuan and W. Tang, J. Org. Chem., 2013, 78, 6350–6355 CrossRef CAS PubMed.
- R.-R. Liu, Y.-G. Wang, Y.-L. Li, B.-B. Huang, R.-X. Liang and Y.-X. Jia, Angew. Chem., Int. Ed., 2017, 56, 7475–7478 CrossRef CAS PubMed.
- R.-X. Liang, K. Wang, Q. Wu, W.-J. Sheng and Y.-X. Jia, Organometallics, 2019, 38, 3927–3930 CrossRef CAS.
- H.-R. Tong, S. Zheng, X. Li, Z. Deng, H. Wang, G. He, Q. Peng and G. Chen, ACS Catal., 2018, 8, 11502–11512 CrossRef CAS.
- (a) M. Gómez-Gallego and M. A. Sierra, Chem. Rev., 2011, 111, 4857–4963 CrossRef PubMed; (b) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066–3072 CrossRef CAS PubMed.
- (a) T. T. Tsou and J. K. Kochi, J. Am. Chem. Soc., 1979, 101, 6319–6332 CrossRef CAS; (b) C. N. Pierson and J. F. Hartwig, Nat. Chem., 2024, 16, 930–937 CrossRef CAS PubMed.
- (a) L. Ackermann, Acc. Chem. Res., 2020, 53, 84–104 CrossRef CAS PubMed; (b) N. Chatani, Acc. Chem. Res., 2023, 56, 3053–3064 CrossRef CAS PubMed; (c) Y. H. Liu, Y.-N. Xia and B.-F. Shi, Ni-catalysed chelation-assisted direct functionalization of inert C−H bonds, Chin. J. Chem., 2020, 38, 635–662 CrossRef CAS.
- (a) D. Balcells, E. Clot and O. Eisenstein, Chem. Rev., 2010, 110, 749–823 CrossRef CAS PubMed; (b) M. Garcia-Melchor, A. A. C. Braga, A. Lledós, G. Ujaque and F. Maseras, Acc. Chem. Res., 2013, 46, 2626–2634 CrossRef CAS PubMed; (c) D. L. Davies, S. A. Macgregor and C. L. McMullin, Chem. Rev., 2017, 117, 8649–8709 CrossRef CAS PubMed.
- (a) CCDC 2442475: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mzljr; (b) CCDC 2442477: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mzllt; (c) CCDC 2442408: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mzjcj; (d) CCDC 2442469: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mzlbk.
Footnote |
| † These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |