
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent progress in single-phase molecular multiferroic materials with ferroelectricity and ferroelasticity
Meng-Meng
Lun
a,
Meng-Meng
Sun
a,
Yong-Qiang
Wang
a,
Gao-Shang
Gong
a,
Mao-Cai
Wei
 a,
Yu-Ling
Su
a,
Da-Wei
Fu
*bc and
Zun-Qi
Liu
*c
a,
Yu-Ling
Su
a,
Da-Wei
Fu
*bc and
Zun-Qi
Liu
*c
aSchool of Electronics and Information, Zhengzhou University of Light Industry, Zhengzhou 450000, China
bInstitute for Science and Applications of Molecular Ferroelectrics, Key Laboratory of the Ministry of Education for Advanced Catalysis Materials, Zhejiang Normal University, Jinhua 321019, China
cChemical Engineering College, Xinjiang Agricultural University, Urumqi 830052, China. E-mail: dawei@seu.edu.cn; zunqi85@163.com
First published on 17th April 2026
Abstract
Single-phase multiferroic materials, characterized by the coexistence of ferroelectricity and ferroelasticity, have garnered significant attention due to the inherent coupling between polarization and strain. This coupling offers considerable potential for applications in multistate memory, high-sensitivity sensors and energy converters. Recently, molecular multiferroic materials have emerged as a research hotspot owing to their unique advantages, including structural diversity, facile tunability, mechanical flexibility, and environmental friendliness. Notably, these molecular multiferroics typically exhibit good mechanical flexibility and a pronounced response to applied stress, which facilitates the investigation of coupling effects between polarization and strain. However, existing reviews predominantly concentrate on a single ferroic property or specific material systems, resulting in a notable scarcity of systematic overviews of molecular multiferroic materials. This review aims to provide a comprehensive overview of recent progress relating to multiferroic materials that simultaneously possess ferroelectric and ferroelastic orders. We summarize key experimental findings, elucidate the relationship between molecular design strategy and material performance, and explore potential applications in flexible electronics, biomimetic sensing, and bio-integrated devices. This review is expected to shed new light on the future of this emerging field and promote cross-disciplinary cooperation and innovation in materials science, electronic engineering, biomedicine, and other related fields.
 Meng-Meng Lun | Meng-Meng Lun was born in April 1993 and received her PhD from Southeast University in 2024. She is currently a teacher and master's supervisor at the School of Electronics and Information, Zhengzhou University of Light Industry. Her research interests are in molecular ferroics (including ferroelectrics, ferroelastics and ferromagnetics) and photoluminescent materials. |
 Da-Wei Fu | Da-Wei Fu was born in November 1980. He is a young chief expert of the National Key Basic Research and Development Program (973 Program), a recipient of the National Excellent Young Scientists Fund, a new century talent of the Ministry of Education, and a professor and doctoral supervisor at Zhejiang Normal University and Xinjiang Agricultural University. His current research interest is in high-performance molecular dielectric/ferroelectric materials. |
 Zun-Qi Liu | Zun-Qi Liu was born in 1985 and received his PhD from Hokkaido University in Japan. He is currently a professor and doctoral supervisor at the Chemical Engineering College, Xinjiang Agricultural University. His research interests include the synthesis and characterization of functional materials, the study of ferroelectric and dielectric properties, and the comprehensive utilization of solid and liquid waste. |
1. Introduction
Ferroic materials have played a significant role in the advancement of human civilization. Among these, single-phase multiferroic materials, which exhibit both full ferroelectricity and full ferroelasticity, possess significant scientific and commercial potential.1–13 The field traces its origins to the discovery of the first molecular multiferroic, Rochelle salt (NaKC4H4O6·4H2O), by Joseph Valasek in 1920. Rochelle salt exhibits coexisting ferroelectric and ferroelastic orders within the temperature range of 255–297 K, and it is the earliest-reported single-phase molecular multiferroic with full ferroelectric and full ferroelastic orders.14,15 A second example, KH2PO4 (KDP), was discovered in 1935, displaying both ferroelectricity and ferroelasticity below 123 K.16 Subsequent research has uncovered additional molecular-based multiferroics.17 A unique characteristic of these materials is the coupling between the spontaneous polarization vector and the ferroelastic strain, implying that a change in one order parameter necessarily induces a cooperative change in the other. This inherent coexistence and coupling between polarization and strain can contribute to significant piezoelectric and electrostrictive effects, thereby enabling molecular multiferroics with full ferroelectricity and full ferroelasticity to show promising applications in high-density multi-state memory, highly sensitive sensors, and intelligent actuators.18–21Molecular materials offer distinct advantages, including structural diversity, a richness of components, and facile chemical modification and tunability.9,22 Molecular multiferroic materials can be constructed from a wide variety of structural building blocks, including organic cations (e.g., protonated amines and quaternary ammonium salts), inorganic anions or metal centers (e.g., transition metals, halide ions, and cyanide groups), and flexible organic ligands. Compared to inorganic single-phase multiferroics, such as (NH4)2Cd2(SO4)3, Ni3B7O13I, and Gd2(MoO4)3, which often suffer from mechanical rigidity and intrinsic brittleness, molecular multiferroics typically feature good mechanical flexibility and enable a pronounced response to applied stress. This facilitates the investigation of coupling effects between polarization and strain. Furthermore, they can be synthesized via low-temperature solution methods and exhibit good biocompatibility, biodegradability, and environmental friendliness.23 These properties position them as a promising material platform for next-generation flexible electronics and implantable medical devices, aligning with the principles of green chemistry.24,25
The intrinsic electromechanical coupling in these materials arises because the reversal of ferroelectric polarization is typically accompanied by lattice deformation that alters strain, while ferroelastic strain can facilitate the reorientation of building blocks and thereby affect the polarization state. This enables the control of the mechanical response via electric fields or the modulation of polarization states via stress.26 The coexistence of ferroelectric and ferroelastic order parameters involves intricate physical mechanisms relating to polarization-strain coupling, which can lead to emergent phenomena and innovative functionalities, including the negative piezoelectric effect, the electro-elastic effect, and the barocaloric effect. These capabilities enable novel applications in multi-state data storage, energy conversion, shape memory, and sensing.27 However, several challenges hinder practical application: the number of known single-phase multiferroics remains limited, the coexistence temperature of ferroelectricity and ferroelasticity is low, synergistic control via electric field and stress is difficult, and there is an insufficient understanding of coupling mechanisms.
This review aims to systematically summarize recent advances in molecular single-phase multiferroics with coexisting ferroelectric and ferroelastic orders. The review begins by discussing the physical foundations and mechanisms for the coexistence and coupling of these two order parameters from the perspective of symmetry conditions. Next, key experimental techniques for characterizing ferroelectricity, ferroelasticity, and their coupling effects in molecular materials will be introduced. Subsequently, representative molecular multiferroic systems reported in recent years, including coordination compounds and organic–inorganic hybrids, will be categorized and discussed, with an analysis of their design strategies, structures, and properties. Finally, based on a summary of existing research, the review will conclude with perspectives on current challenges and future areas of research focus in this field.
2. Fundamentals of the coexistence of ferroelectric and ferroelastic orders
2.1 The theory of full ferroelectric and full ferroelastic phase transitions
Ferroelectric materials are a class of functional compounds possessing two or more orientation states in the absence of an electric field that can be shifted from one to another of these states by an electric field. A fundamental feature of ferroelectrics is the breaking of spatial inversion symmetry, which limits these crystals to specific polar point groups: 1 (C1), 2 (C2), m (CS), mm2 (C2v), 4 (C4), 4mm (C4v), 3 (C3), 3m (C3v), 6 (C6), and 6mm (C6ν). Ferroelasticity serves as the mechanical analog of ferroelectricity, and it can be defined as a crystal having two or more orientation states in the absence of stress that can be switched by applying external stress, with the states differing in the spontaneous strain tensor at zero stress.28 A critical unifying principle for both ferroelectric and ferroelastic phase transitions is their inherent symmetry breaking: there is a transformation from a high-symmetry prototype phase to a lower-symmetry ferroic phase. This process invariably leads to the formation of distinct polarization and/or strain domains, each with specific orientations, as schematically depicted in Fig. 1.29 | ||
| Fig. 1 A schematic illustration of the coupling between ferroelectric and ferroelastic orders. | ||
In the late 1960s and early 1970s, the Japanese physicist Aizu Kêitsiro developed a systematic framework, based on phenomenological Landau theory and group theory, to classify all possible ferroic phase transitions. This theory introduced the concept of “prototype symmetry” and summarized the crystallographically allowed ferroic phase transitions, encompassing 88 species of ferroelectrics and 94 species of ferroelastics. The hallmark “Aizu notation” system describes a transition directly using the point group symbols of the high-symmetry and low-symmetry phases, with the standard notation being “[high-symmetry point group]F[low-symmetry point group]”.30–32 Among these species, 42 have been identified as fully ferroelectric and fully ferroelastic phase transitions, as shown in Fig. 2a, meaning all polarization and strain orientations of the ferroic phase can be fully controlled by an electric field or mechanical stress. In such systems, the prototype phase must be non-centrosymmetric, though not necessarily piezoelectric (e.g., the point group 432). These 42 species can be further categorized into four types based on the symmetry relations, as shown in Fig. 2a: (I) 4 species going from a chiral polar group to a chiral polar subgroup, (II) 13 species going from a chiral group to a chiral polar subgroup, (III) 9 species going from a polar group to a polar subgroup, and (IV) 16 species going from a non-centrosymmetric group to a polar subgroup. In such fully ferroelectric and fully ferroelastic crystals, the ferroelectricity and ferroelasticity are completely coupled: any change in the polarization vector is always accompanied by a change in the strain tensor, and vice versa. Therefore, this review specifically summarizes single-phase multiferroic materials with full ferroelectric and ferroelastic transitions.
 | ||
| Fig. 2 (a) The subdivision of the 42 fully ferroelectric and fully ferroelastic species. Schematic illustrations of the phase transition mechanisms in ferroics: (b) order–disorder type, (c) displacive type, and (d) mixed order–disorder and displacive type. | ||
2.2 Mechanisms driving full ferroelectric and full ferroelastic phase transitions
The mechanisms driving full ferroelectric and full ferroelastic phase transitions in reported single-phase molecular multiferroic materials are primarily categorized into order–disorder type, displacive type, and a synergistic combination of these. Taking the ferroelectric-paraelectric phase transitions of the inorganic oxides NaNO2 and BaTiO3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 33 and that of organic–inorganic hybrid perovskites (R-3AP)RbBr334 as examples, these three distinct transition mechanisms are shown in Fig. 2b–d.
33 and that of organic–inorganic hybrid perovskites (R-3AP)RbBr334 as examples, these three distinct transition mechanisms are shown in Fig. 2b–d.
Firstly, the order–disorder type transition is driven by the progressive alignment of molecular, ionic, or dipolar groups from a statistically disordered distribution among multiple equivalent positions into an ordered arrangement. In the high-temperature phase, dipolar or molecular units dynamically occupy multiple energetically equivalent orientations, resulting in a centrosymmetric structure. Upon cooling, these units become frozen into a specific orientation, giving rise to long-range order and symmetry breaking. As typified by NaNO2, such a compound transforms from a high-symmetry prototype phase to a low-symmetry ferroic phase upon cooling. This transition is predominantly driven by the “freezing” of disordered occupancies, such as the nitrogen atom within the NO2− anion, into an ordered state, as shown in Fig. 2b.
Secondly, the displacive type mechanism arises primarily from the relative displacement of ions within the lattice, generating an electric dipole moment and strain. For example, the relative displacement of O2− and Ti4+ ions induces spontaneous polarization in the inorganic ferroelectric BaTiO3 (Fig. 2c). For organic–inorganic hybrid systems, atomic displacement can lead to conformational changes in the organic moieties, thereby inducing a ferroic phase transition. Furthermore, Jahn–Teller distortion in the inorganic framework can create internal stress that induces spontaneous strain, while simultaneously causing separation between positive and negative charge centers, leading to spontaneous polarization.35
Thirdly, the phase transition mechanism in organic–inorganic hybrids generally arises from a synergistic effect combining the order–disorder transition of the organic components and displacive transition due to Jahn–Teller distortion in the inorganic anion framework.5 In hybrid perovskites, tilting or distortion of BX6 octahedra generally accompanies cation ordering, further contributing to symmetry lowering and ferroic phase transitions. As illustrated in Fig. 2d, the ferroic phase transition in 3D (R-3AP)RbBr3 originates from the dynamic order-to-disorder transition of organic R-3AP+ cations coupled with the deformation of the inorganic framework. In such fully coupled systems, any change in the polarization vector is always accompanied by a change in the strain tensor and vice versa.
3. Characterization of molecular multiferroics with ferroelectricity and ferroelasticity
Accurately characterizing the structure, ferroelectricity, and ferroelasticity of molecular-based single-phase multiferroic materials is crucial for understanding their structure–property relationships and coupling mechanisms, and for evaluating their application potential. This requires the integrated use of multiple complementary experimental techniques to probe the material's structural, electrical, and mechanical properties and their interrelation across different scales. The systematic characterization of a novel molecular material typically begins with dielectric constant measurements and differential scanning calorimetry (DSC) to establish the definitive phase transition temperature. This initial assessment is crucial for guiding subsequent structural investigations. Single-crystal X-ray diffraction (SC-XRD) is then utilized to determine the crystallographic structures of compounds at different temperature, specifically to verify if the transition mechanism is one of the 42 fully ferroelectric and fully ferroelastic species defined by Aizu.31 Once this conformity is established, detailed characterization of the ferroelectric and ferroelastic behaviors proceeds.3.1 Characterization of ferroelectricity
Characterizing ferroelectricity hinges on unequivocally demonstrating the existence of reversible spontaneous polarization. The primary and most direct technique for this purpose is the measurement of the polarization–electric field (P–E) hysteresis loop. By applying a cyclic electric field (E) to the material and measuring its resulting polarization (P), a distinct P–E loop is generated, allowing for the determination of critical ferroelectric parameters, including remanent polarization (Pr), saturation polarization (Ps), and the coercive field (Ec). For molecular crystals, the electric field is applied along the crystallographic polar axis to acquire the P–E loop. However, for small single crystals or thin films, this measurement often requires a ferroelectric tester equipped with a probe station.Piezoresponse force microscopy (PFM), a technique based on atomic force microscopy, enables the imaging and manipulation of ferroelectric domains at the nanoscale, as shown in Fig. 3a. Depending on the detection direction, PFM can be in vertical (VPFM) or lateral (LPFM) mode, corresponding to the out-of-plane and in-plane piezoresponse, respectively.36,37 It utilizes the inverse piezoelectric effect: when an AC voltage is applied to the sample surface via a conductive tip, ferroelectric domains undergo minute deformation (expansion/contraction or shearing) due to the piezoelectric effect. By detecting the amplitude and phase of this mechanical response, which is synchronized with the driving voltage using a lock-in amplifier, information about the piezoresponse strength and polarization direction is obtained. The PFM amplitude image displays the distribution of ferroelectric domains, while the PFM phase image distinguishes domains with opposite polarization directions (typically showing a 180° phase difference). More importantly, by applying a DC bias voltage through the tip, the domain switching process can be observed in situ, as shown in Fig. 3a(I–IX). This technique is a vital tool for studying domain structure dynamics and has been used to observe vortex domain structures in molecular ferroelectric materials.38–40 It is noteworthy that PFM demands high sample flatness, typically requiring smooth and uniform thin-film samples, which can be a limitation for materials with poor film-forming properties or whose thin-film phases differ from the bulk structure.
 | ||
| Fig. 3 (a) Configurations of a typical PFM system and the PFM setup. The evolution of ferroelectric domains for the molecular multiferroic material [FeII(C8-F-pbh)2] with temperature and electric field is illustrated in (I–IX). (b) A schematic illustration of ferroelastic domain characterization using PLM. The evolution of ferroelastic domains for FeII(C8-F-pbh)2 with temperature (X–XIII) and stress (XIV–XVII) is shown. Reproduced from ref. 36 and 41 with permission from Elsevier and Wiley, copyright 2015 and 2024. | ||
An indirect method to measure ferroelectricity is pyroelectric current measurement, which leverages the pyroelectric effect. When a ferroelectric material undergoes a temperature change (ΔT), charge (ΔQ) is released due to the variation of Ps with temperature. By cooling the sample from the paraelectric phase to the ferroelectric phase under two opposite poling fields and then heating it back to the paraelectric phase at a constant rate, the observation of a pair of opposite current peaks near the phase transition temperature proves that the spontaneous polarization can be reversed by an external field. Simultaneously, the temperature dependence of spontaneous polarization can be calculated from the pyroelectric data.
3.2 Characterization of ferroelasticity
Ferroelasticity originates from a symmetry-breaking structural transition that produces a spontaneous strain tensor. The different orientations of strain states lie in different ferroelastic twin domains. The core of characterizing ferroelasticity lies in demonstrating the existence of spontaneous strain that can be reversed by applied mechanical stress. The direct method is the measurement of the stress–strain (ε–σ) hysteresis loop, where applying a cyclic stress and measuring the strain response can give a typical ε–σ loop. However, this mechanical testing poses significant challenges for molecular ferroelastics, as bulk crystals often have limited stress tolerance.Polarized light microscopy (PLM) offers a non-contact, non-destructive method for observing ferroelastic domains. Because the spontaneous strain tensor is rigidly coupled with the optical indicatrix, the observation of birefringent domains normally indicates the presence of a ferroelastic phase.42–46 When a material is in its low-symmetry ferroelastic phase, different domain variants (also known as twin domains) possess distinct orientations of the optical indicatrix due to lattice distortion. Due to different crystallographic axis orientations, these domain regions exhibit different optical anisotropies, as shown in Fig. 3b. When linearly polarized light passes through these anisotropic regions, the difference in refractive indices causes a phase shift, altering the polarization state of the light. Therefore, under crossed polarizer light, alternating bright and dark regions correspond to ferroelastic domains with distinct strain tensors. In contrast, a material in its high-symmetry paraelastic phase shows uniform contrast under crossed polarizers. Researchers typically perform in situ observations of domain evolution with temperature under crossed polarizers, where the appearance and disappearance of domains across the phase transition serves as strong evidence for ferroelasticity (Fig. 3b(X–XIII)). Furthermore, applying stress to the sample while under observation allows for the in situ monitoring of ferroelastic domain wall motion, as shown in Fig. 3b(XIV–XVI). This method provides the most intuitive and convenient approach for observing ferroelastic domains, and it is applicable to both high-quality bulk single crystals and thin films, provided the sample surface is sufficiently flat.47 In nanoscale systems, atomic force microscopy (AFM) and piezoresponse force microscopy (PFM) can also be employed to visualize ferroelastic domain structures and domain wall motion.
3.3 Characterization of ferroelectric-ferroelastic coupling
The characterization of the coupling between ferroelectricity and ferroelasticity in single-phase molecular multiferroics has primarily been reported using PFM. This involves observing changes in ferroelectric domains while applying mechanical stress or detecting surface topographic changes via an atomic force microscope tip while applying an electric field. Significantly, for materials possessing both full ferroelectricity and full ferroelasticity, their ferroelectric domains are inherently identical to their ferroelastic domains. This direct correspondence ensures that any modification of the polarization vector P is always reciprocally linked to a change in the strain tensor ε. To advance the understanding of this coupled effect, our group has recently devised a novel technique: applying an electric field to the sample while conducting in situ observation of ferroelastic domain evolution using PLM.48 This method confirmed the coupled effect between ferroelectric and ferroelastic domains in the 2D multiferroic hybrid perovskite (cyclohexanemethylaminium)2PbCl4, enabling the demonstration of electric-field-induced strain in a molecular multiferroic system.4. Molecular multiferroics with ferroelectricity and ferroelasticity
The single-phase molecular multiferroic materials with ferroelectricity and ferroelasticity described in this review, including single molecules, perovskite structural hybrids and non-perovskite structural compounds, are summarized in Table 1. In addition, compounds that conform to one of the 42 species of full ferroelectric and full ferroelastic phase transitions defined by Aizu but that have not been characterized or investigated for ferroelasticity by the original authors are listed in Table S1.| Compound | n-D | Component | Phase transition | Polar/strain axis | P s (µC cm−2) | ε | Ref. |
|---|---|---|---|---|---|---|---|
| NaKC4H4O6·4H2O (Rochelle salt) | 0D |

|
222F2 (P212121–P21–P212121, Tc1 = 297 K, Tc2 = 255 K) | 2 | 0.25 | 14 | |
| KH2PO4 (KDP) | 0D |
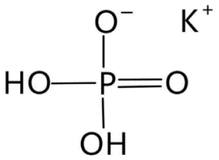
|
![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 2mFmm2 (I2d–Fdd2, Tc = 123 K) 2mFmm2 (I2d–Fdd2, Tc = 123 K) |
2 | 4.8 | 16 | |
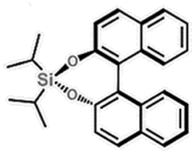
| (R)-BINOL-DIPASi (BINOL = 1,1′-bi-2-naphthol) | 0D |

|
2F1 (P21–P1, Tc = 362 K) | 2 | 1.6 | 0.027 | 49 |
| (S)-BINOL-DIPASi (BINOL = 1,1′-bi-2-naphthol) | 0D |
|
2F1 (P21–P1, Tc = 363 K) | 2 | 1.7 | 0.027 | 49 |
| FeII(C8-F-pbh)2 (C8-F-pbh = (1Z,N′E)-3-F-4-(octyloxy)-N′-(pyridin-2-ylmethylene)benzo-hydrazonate) | 0D |

|
222F2 (P212121–P21, Tc = 318 K) | 2 | 0.15 | 41 | |
| [(C6H5)3PCH3]FeBr4 | 0D |

|
222F2 (P212121–P21, Tc = 275 K, TN = 11 K) | 2 | 50 | ||
| PH4FeBr4 | 0D |
|
10.7 | >0.4 | 51 | ||
| [RFAO][ReO4] (RFAO = (4R,5R)-4-fluoro-1-azabicyclo[3.2.1]octane) | 0D |
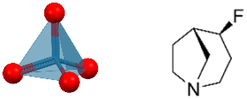
|
222F2, 432F2 (P21–C2221–P432, Tc1 = 350 K, Tc2 = 463 K) | 1 | 3.64 | 52 | |
| [SFAO][ReO4] (SFAO = (4S,5S)-4-fluoro-1-azabicyclo[3.2.1]octane) | 0D |

|
222F2, 432F2 (P21–C2221–P432, Tc1 = 350 K, Tc2 = 463 K) | 1 | 3.81 | 52 | |
| (C4H8NH2)3[SbCl5] (C4H8NH2+ = pyrrolidinium) | 1D |

|
mmmFmm2, mm2Fm, mm2F2 (Pmnb–Pmn21–Pn–P21Tc1 = 252 K, Tc2 = 247 K, Tc3 = 231 K) | 1, 2, 2 | 0.16 (217 K) | 53 | |
| [FPM][Fe3(µ3-O)(µ-O2CH)8] (FPM+ = 3-(3-formylamino-propyl)-3,4,5,6-tetrahydropyrimidin-1-ium) | 1D |

|
mm2Fm (Pca21–Cc–Cmc21, Tc1 = 426 K, Tc2 = 365 K) | 2 | c-Axis 4.64 (250 K) | 0.02369(365 K) | 54 |
| 4.55 (395 K) | 0.02385(426 K) | ||||||
| 4.46 (433 K) a-axis 4.01 (395 K) | |||||||
| [R-EQ]PbI3 (R-EQ+ = (R)-N-ethyl-3-quinuclidinol) | 1D |

|
622F2(s) (P6222–P21–P21, Tc1 = 380 K, T2 = 355 K) | 6 | 55 | ||
| C6H5N(CH3)3CdBr3xCl3(1−x) | 1D |

|
mm2Fm (Ama2–Cc, Tc = 453 K) | 2 | 3.6 | 0.215 | 20 |
| R-LIPF ([(R)-1-(4-chlorophenyl)ethylammonium]2PbI4) | 2D |

|
422F1 (P422–P1, Tc = 483 K) | 8 | 13.96 | 56 | |
| S-LIPF ([(S)-1-(4-chlorophenyl)ethylammonium]2PbI4) | 2D |

|
422F1 (P422–P1, Tc = 473.2 K) | 8 | 13.96 | 56 | |
| [(R)-PEDA]PbI4 (PEDA2+ = N-(1-phenylethyl)ethane-1,2-diaminium) | 2D |

|
222F2 (P212121–P21, Tc = 389 K) | 2 | 0.15 | 57 | |
| [(S)-PEDA]PbI4 | 2D |

|
222F2 (P212121–P21, Tc = 389 K) | 2 | 0.15 | 57 | |
| MFP ([cyclohexanemethylaminium]2PbCl4) | 2D |

|
4/mmmFmm2, 2mFmm2 (I4/mmm–γ-Pmc21–Cmc21, Tc1 = 413.2 K, Tc2 = 411.8 K, T3 = 316.2 K) |
2, 2 | 15.4 (300 K) | 48 | |
| 5.4 (316 K) | |||||||
| (S-3FQ)4RbEu(NO3)8 (S-3FQ+ = (S)-3-fluoroquinuclidinium) | 2D |

|
4F1 (P42–P1, Tc = 441 K) | 4 | 1.5 | 58 | |
| (R-3AP)RbBr3 (R-3AP2+ = (R)-3-ammoniopyrrolidinium) | 3D |

|
432F2(s) (P432–P21, Tc = 401 K) | 12 | 1.21 | 34 | |
| (R-M3HQ)2RbLa(NO3)6 (R-M3HQ+ = (R)-N-methyl-3-hydroxyl-quinuclidinium) | 3D |

|
23F3 (P213–R3, Tc = 278 K) | 4 | 0.32 | 19 | |
| (R-M3HQ)2NH4La(NO3)6 | 3D |

|
23F3 (P213–R3, Tc = 314 K) | 4 | 19 | ||
| (R-M3HQ)2RbEu(NO3)6 | 3D |

|
23F3 (P213–R3, Tc = 285 K) | 4 | 0.38 | 26 | |
| (S-M3HQ)2RbEu(NO3)6 | 3D |

|
23F3 (P213–R3, Tc = 284 K) | 4 | 26 | ||
| (R-M3HQ)2RbPr(NO3)6 | 3D |

|
23F3 (P213–R3, Tc = 280 K) | 4 | 0.32 | 59 | |
| MHyZn ([CH3NH2NH2]Zn(HCOO)3) | 3D |

|
![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) mF3, 3mF1(Rc–R3c–P1, Tc1 = 321 K, Tc2 = 178 K) mF3, 3mF1(Rc–R3c–P1, Tc1 = 321 K, Tc2 = 178 K) |
1, 6 | 3 | 60 | |
| (R-3HP)2RbBiBr6 (R-3HP+ = (R)-3-hydroxypyrrolidinium) | 3D |

|
2F1 (P21–P1, Tc = 394 K) | 2 | 1.94 (b-axis) | 61 | |
| 3.18 (c-axis) | |||||||
| R-3-FPC ([(R)-3-fluoropyrrolidinium]2[KFe(CN)6]) | 3D |

|
222F2 (C2221–P21, Tc = 430 K) | 2 | 9.4 | 62 | |
| S-3-FPC ([(S)-3-fluoropyrrolidinium]2[KFe(CN)6]) | 3D |

|
222F2 (C2221–P21, Tc = 428 K) | 2 | 62 |
4.1 Single-component molecular multiferroics
Guided by Aizu's31 group-to-subgroup principle, 42 species of materials are known to exhibit coexisting ferroelectric and ferroelastic phase transitions, with 17 of these involving transitions from a chiral group to a chiral subgroup. This highlights the efficacy of introducing crystallographic chirality via homochiral organic cations as a strategic approach for designing molecular multiferroics. Moreover, molecular homochirality imparts these materials with unique and attractive physical attributes, such as circularly polarized luminescence (CPL), chiroptical nonlinear optical effects, chiral-induced spin selectivity and CPL detection, making them highly relevant for advanced optoelectronic and spintronic applications. For example, the Xiong group49 introduced the organosilicon component of –Si(CH(CH3)2)2 as a ligand into axial-chiral 1,1′-bi-2-naphthol (BINOL) to construct a pair of enantiomeric molecular multiferroics, (R)-BINOL-DIPASi and (S)-BINOL-DIPASi, ingeniously combining the intrinsic polar and axial chirality of BINOL with a dynamic organosilicon component, as displayed in Fig. 4a. Structural characterization via SC-XRD confirmed that (R)-BINOL-DIPASi and (S)-BINOL-DIPASi exhibit a 2F1-type full ferroelectric and full ferroelastic phase transition at 362 K and 363 K, respectively. This transition is attributed to the order–disorder dynamics of the isopropyl groups within the organosilicon ligands. Electrical measurements, specifically P–E hysteresis loops, indicated spontaneous polarization (Ps) values of approximately 1.6 and 1.7 µC cm−2. Furthermore, PFM directly visualized the ferroic domain switching induced by a 110 V electric field, while PLM provided insights into the temperature-driven evolution of stripe-shaped ferroelastic domains. Nanoindentation measurements further reveal favorable mechanical softness (elastic modulus: ∼7 GPa). This work demonstrates the exceptional efficacy of axial chirality for constructing high-temperature single-component molecular multiferroics with good flexibility. | ||
| Fig. 4 (a) The design concept of (R)-BINOL–DIPASi and (S)-BINOL–DIPASi as single-component molecular multiferroic crystals. The stripe-shaped ferroelastic domains and rectangular ferroelectric domains of (R)-BINOL–DIPASi are imaged using PLM and PFM, respectively. (b) The crystallographic structure, symmetry and SOC of FeII(C8-F-pbh)2: the structure at 300 K (I), 330 K (II) and 80 K (IV). (III) The equatorial plane projection of the point group of C2 in the ferroelectric phase and D2 in the paraelectric phase, showing symmetry breaking. (V) Enlarged octahedrons of the FeIIN4O2 coordination sphere in the different phases, showing LS/HS states. (VI) The electronic structures of the FeII HS/LS states, showing d orbital breaking. Reproduced from ref. 49 and 41 with permission from American Chemical Society and Wiley, copyright 2022 and 2024. | ||
Integrating metal elements with organic components can also give rise to multifunctional coordination compounds. Recently, Ai et al.41 reported the molecular multiferroic crystal FeII(C8-F-pbh)2 (C8-F-pbh = (1Z,N′E)-3-F-4-(octyloxy)-N′-(pyridin-2-ylmethylene)benzo-hydrazonate), as shown in Fig. 4b, demonstrating the coexistence of ferroelectricity, ferroelasticity and spin-crossover (SCO) behavior within a single phase and representing a significant breakthrough integrating multiple order parameters. Due to H/F substitution, Tc of FeII(C8-F-pbh)2 (318 K) is much higher than that of the non-fluorinated parent compound (270 K), which enables room-temperature functionality. Structural analysis reveals that the phase transition, driven by the 180° flip-flop motion of the partial fluorobenzene moiety and the ordering of flexible alkyl chains, corresponds to symmetry breaking from a paraferroic phase (space group P212121) to a ferroic phase (space group P21), which is classified as a 222F2-type full ferroelectric and full ferroelastic phase transition. The reversible switching of ferroelectric and ferroelastic domains with temperature is directly visualized via PFM and PLM, respectively, as shown in Fig. 3. In addition, FeII(C8-F-pbh)2 displays a spin transition between high- and low-spin states, accompanied by d-orbital breaking within the t42ge2g and  configuration change of the octahedrally coordinated FeII center, as shown in Fig. 4b. This finding could further inspire the exploration of multiple-state molecular switches.
configuration change of the octahedrally coordinated FeII center, as shown in Fig. 4b. This finding could further inspire the exploration of multiple-state molecular switches.
4.2 Organic–inorganic hybrid multiferroics with ferroelectricity and ferroelasticity
4.2.1.1 Zero-dimensional organic–inorganic hybrid perovskites (0D OIHPs). Introducing homochirality into molecular ferroelectrics provides an effective route to realize chiral ferroelectric functionality. In 2025, Liao et al.52 reported a pair of molecular ferroelectric enantiomers, [RFAO][ReO4] and [SFAO][ReO4] (RFAO/SFAO = (4R,5R)/(4S,5S)-4-fluoro-1-azabicyclo[3.2.1]octane). This pair of ferroelectric enantiomers both crystallize in the chiral–polar point group 2 at room temperature and undergo two successive ferroelectric phase transitions with Aizu notations of 222F2 at 350 K and 432F2 at 463 K. The symmetry evolution from 2 to 222 and then to 432 is associated with the order–disorder behavior of the organic cations and anions within the crystal lattice. As a result, the enantiomeric crystals exhibit switchable spontaneous polarization together with a multistep second-harmonic generation circular dichroism (SHG-CD) response from a high to a low and finally to an inactive state. More importantly, spiral chiral ferroelectric domains were directly observed in the enantiomeric crystals, representing the first discovery of chiral domain configurations in enantiomeric ferroelectric materials and highlighting the intrinsic coupling between polarization and chirality.
4.2.1.2 One-dimensional organic–inorganic hybrid perovskites (1D OIHPs). Introducing homochirality into 1D chain-like OIHPs (ABX3, where A = organic cation, B = divalent metal ion, X = halogen) also serves as an effective strategy for constructing ferroelastic–ferroelectric multiferroics. In 2022, Fu et al.55 reported the first 1D homochiral hybrid perovskite, [(R)-N-ethyl-3-quinuclidinol]PbI3 ([R-EQ]PbI3), with the coexistence of full ferroelectricity and full ferroelasticity. At 380 K, [R-EQ]PbI3 undergoes a phase transition with the Aizu notation of 622F2(s), as shown in Fig. 5a, and it shows multiaxial ferroicity (six equivalent polarization and strain coupling orientations) at room temperature. Nevertheless, both the racemic analogue [Rac-EQ]PbI3 and the achiral parent compound [EQ]PbI3 (EQ = N-ethyl-quinuclidine) only experience the paraferroelastic-ferroelastic phase transition with the same Aizu notation of 6/mmmF2/m, and they possess six equivalent strain orientations in their ferroelastic phase.
 | ||
| Fig. 5 (a) The design of 1D multiferroic [R-EQ]PbI3 through introducing homochirality. Its stripe-shaped ferroic domains are imaged by PFM (I and II) and PLM (III and IV), respectively. (b) (V) Top: an illustration of the structural units of C6H5N(CH3)3CdBr3xCl3(1−x) in the two ferroelastic states and the structure confinement effect for ferroelectric/ferroelastic switching; the yellow and green arrows indicate the polarization directions. Bottom: photos of a bulk single crystal poled to the two ferroelastic states by an electric field, showing macroscopic shear strain in good correlation with the unit cell distortion. (VI) A view of the crystallographic structure along the 1D chain direction. (VII) The experimental setup for the shear strain and piezoelectricity measurements. (VIII) P–E loops of the solid solution series at a measurement frequency of 2 Hz. (IX) Unipolar S–E curves of the solid solution series at 2 Hz after pre-poling. Reproduced from ref. 55 and 20 with permission from American Chemical Society and Springer Nature, copyright 2022 and 2021. | ||
Researchers have long theorized that full ferroelectric and full ferroelastic coupling could induce the large deformation of a material, thereby generating giant piezoelectric responses. Significant experimental breakthroughs in this area remained elusive until Hu et al.20 achieved them through the halogen doping of an inorganic framework. Their investigation into a series of 1D solid-solution crystals, exemplified by C6H5N(CH3)3CdBr3Cl3(1−x) (Fig. 5b), revealed unprecedented electromechanical performance. Specifically, applying an electric field after electrode deposition on opposing faces of C6H5N(CH3)3CdCl3 single crystals resulted in substantial shear strain of up to 21.5%, a magnitude two orders higher than that found in conventional ferroelectric polymers and oxides. This exceptional performance is achieved through inorganic bond switching and enhanced by the structural confinement provided by large organic moieties, which effectively suppresses undesired 180° polarization switching. Furthermore, Br substitution in the hybrid ferroelectric C6H5N(CH3)3CdCl3 was employed to systematically tune phase-transition temperatures, mechanical properties, and piezoelectricity. Notably, at the Br-rich end of the solid solution, there is a sizable shear piezoelectric coefficient (d35) of up to 4830 pm V−1.
4.2.1.3 Two-dimensional organic–inorganic hybrid perovskites (2D OIHPs). Introducing homochirality into 2D OIHPs (AI2BX4/AIIBX4-type structure) in 2019, the Xiong group56 designed and synthesized the first pair of 2D lead-iodide perovskite enantiomers, [R- and S-1-(4-chlorophenyl)ethylammonium]2PbI4 (R- and S-LIPF), as shown in Fig. 6a, which show full ferroelectricity and full ferroelasticity at room temperature, whereas their racemic analogue [Rac-1-(4-chlorophenyl)ethylammonium]2PbI4 adopts the centrosymmetric space group P21/c. Notably, R-LIPF and S-LIPF undergo the ferroic phase transitions described by the Aizu notation of 422F1, and their transition temperatures are as high as 483 and 473.2 K, respectively. Using PLM, the reversible disappearance and reappearance of ferroelastic domains across the phase transition was clearly visualized. Furthermore, PFM measurements confirmed that the ferroelectric domains can be switched under an applied electric field, directly verifying the multiferroic nature. Additionally, both enantiomers possessed semiconductor characteristics, with a direct bandgap of 2.34 eV. Based on the homochiral strategy, Zeng et al.57 in 2021 achieved a further breakthrough by designing [(R)- and (S)-N-(1-phenylethyl)ethane-1,2-diaminium]PbI4 ([(R)- and (S)-PEDA]PbI4). They not only display a 222F2-type ferroelectric phase transition at ∼389 K but also demonstrate coupled ferroelastic domain switching and reversible thermochromism, and the crystals of [(R)- and (S)-PEDA]PbI4 manifest a color change from orange-yellow to orange-red driven by temperature. This study represents the first integration of ferroelectricity, ferroelasticity, and thermochromism in a single chiral perovskite, underscoring the versatility of molecular-level chiral engineering for creating multifunctional OIHPs. However, none of the aforementioned studies performed characterization of the coupling between ferroelectric polarization and ferroelastic strain in multiferroic OIHPs, such as observing electric-field-controlled ferroelastic domain switching via PLM or stress-modulated ferroelectric domain switching via PFM.
 | ||
| Fig. 6 (a) The approach of molecular design for 2D homochiral perovskite multiferroics. Packing views of the crystal structures of R-LIPF (lower left) and S-LIPF (lower right). (I) A topographic image and vertical PFM phase (II) and amplitude (III) images of R-LIPF thin film. Ferroelastic domain evolution of 1-R in a continuous heating and cooling process at 298 K (IV), 423 K (V), and 303 K (VI). (b) The design of the 2D multiferroic MFP through reducing molecular rigidity, and packing views of MFP in different phases; inset: stereosymmetric operation matching with the crystal lattice. (VII) Morphology, amplitude, and phase images of the 20 × 20 µm region, which are observed in the as-grown state and observed after applying a voltage of +120 V at the red rectangle wireframe and subsequently −120 V at the blue rectangle wireframe. (VIII) The observation of ferroelastic domain evolution controlled by electric and mechanical fields under a polarizing microscope. (IX) A schematic diagram of the evolution of ferroelastic domains under electric and stress fields. Reproduced from ref. 57 and 48 with permission from Wiley and American Chemical Society, copyright 2021 and 2024. | ||
Since the concept of multiferroicity was introduced in 1968, the coupled effect of polarization and strain in full ferroelectricity and full ferroelasticity had never been experimentally observed in OIHPs. Then, in 2024, our group reported a 2D multiferroic hybrid perovskite (cyclohexanemethylaminium)2PbCl4 (MFP) with full ferroelectricity and full ferroelasticity, as displayed in Fig. 6b.48 This study demonstrates that MFP undergoes the symmetry-breaking transition of 2mFmm2 type at 411.8 K, driven by dynamic reorientations of flexible organic cations. MFP exhibits large spontaneous polarization of 15.4 µC cm−2 at room temperature, whereas its rigid structural parent compound merely shows ferroelectricity. Importantly, the in situ switchable evolution of ferroelastic domains under either electric or mechanical fields is directly observed, representing the first experimental demonstration of coupled polarization-strain orientation-switching dynamics in hybrid perovskites.
4.2.1.4 Three-dimensional organic–inorganic hybrid perovskites (3D OIHPs). 3D OIHPs (ABX3-type structure) are constrained by Goldschmidt's tolerance factor, and multiferroicity in such structures has also rarely been reported so far. Zhang and colleagues34 introduced a homochiral organic cation, (R)-3-ammoniopyrrolidinium (R-3AP), as shown in Fig. 7a, into a rubidium-bromide framework, generating the multiferroic (R-3AP)RbBr3. (R-3AP)RbBr3 undergoes a reversible ferroelectric–ferroelastic phase transition at 401 K, identified by the Aizu notation 432F2(s). This transition is driven by the order–disorder transition of organic cations and Jahn–Teller distortion of the inorganic framework. (R-3AP)RbBr3 clearly exhibits multiferroicity, as evidenced by a saturation polarization of 1.21 µC cm−2, a reversible second harmonic generation switch, and observable, reversible ferroelastic domain evolution when viewed under a polarizing microscope.
 | ||
| Fig. 7 (a) The 3D multiferroic (R-3AP)RbBr3: packing views of (R-3AP)RbBr3 in the (I) ferroic and (II) paraferroic phase. (III) Its P–E hysteresis loop measured by the Sawyer–Tower method at 299 K. (IV) The domain structure of (R-3AP)RbBr3 at 308 K. (b) The 3D metal–organic framework MHyZn: packing views of MHyZn in the (V) ferroic and (VI) paraferroic phase. (VII) The spontaneous polarization of MHyZn determined from temperature-dependent pyroelectric current measurements. (VIII) The stripe-shaped ferroic domains of MHyZn at 110 K. Reproduced from ref. 34 and 60 with permission from Wiley and The Royal Society of Chemistry, copyright 2024 and 2018. | ||
c) to a polar phase (R3c) around 321 K, and a low-temperature ferroelectric and ferroelastic transition to a triclinic phase (P1) near 178 K, corresponding to the Aizu notation 3mF1. Structural analysis indicates that both transitions are driven by temperature-induced order–disorder transformations of the MHy+ organic cations. However, the coexistence of full ferroelectric and ferroelastic orders in MHyZn occurs well below room temperature, significantly limiting its practical applicability. Clearly, the careful selection of organic cations can yield new multiferroic formates, but the number of such compounds remains limited thus far.
 | ||
| Fig. 8 (a) The 3D nitrate-bridged rare-earth double perovskites (R- and S-M3HQ)2RbEu(NO3)6 (R1 and S1): (I) the design strategy and crystal structures of these perovskites. (II) (Q)2RbEu(NO3)6 at 293 K. (III) (MeQ)2RbEu(NO3)6 at 293 K. (IV) Rac at 293 K. (V) (S-M3HQ)2RbEu(NO3)6 at 253 K. (R-M3HQ)2RbEu(NO3)6 at 253 (VI) and 303 (VII) K. The NO3− ions are simplified as balls for clarity. (VIII) Ferroelastic domains of R1 in the LTP. (IX) CPL spectra of S1 (blue line) and R1 (red line) crystal samples excited by 397 nm. (X) Piezoelectric constant (d33)measurement of R1, using the quasi-static (Berlincourt) method. (XI) The characterization of ferroelectric domainal inversion. (b) Molecular engineering regulation achieving multipolar axes and out-of-plane polarization in a rare-earth hybrid double perovskite ferroelectric. (XII) PFM topography, amplitude, and phase images after the first electric poling with a tip bias of −220 V. (XIII) Single-crystal blocks and a red LED packaged with a 365 nm chip under daylight and ultraviolet light. (XIV) CPL spectra excited at 396 nm. Reproduced from ref. 58 and 26 with permission from Wiley and The Royal Society of Chemistry, copyright 2024 and 2022. | ||
Subsequently, by replacing the rare-earth ions with Eu3+ and Pr3+, the pair of enantiomeric compounds (R- and S-M3HQ)2RbEu(NO3)6 and (R-M3HQ)2RbPr(NO3)6 are synthesized.26 These derivatives maintain the same Aizu phase transition type and similar transition temperatures as the parent compound (R-M3HQ)2RbLa(NO3)6, while exhibiting comparable piezoelectric coefficients (d33 ≈ 103 and 102 pC N−1, respectively). Notably, (R- and S-M3HQ)2RbEu(NO3)6 simultaneously achieve CPL and a piezoelectric response, demonstrating the coupling between optical activity and ferroic order states, as shown in Fig. 8a. The most recent work introduced fluorine substitution at the organic cation to gain 2D (S-3FQ)4EuRb(NO3)8 (S-3FQ=(S)-3-fluoroquinuclidinium), shown in Fig. 8b, which undergoes a full ferroelectric and full ferroelastic phase transition with the Aizu notation of 4F1 at 411 K.59 (S-3FQ)4EuRb(NO3)8 successfully generates out-of-plane polarization and elevates the Curie temperature by 94 K compared to (S-3HQ)4EuRb(NO3)8 (S-3HQ = (S)-3-hydroxylquinuclidinium), and it retains CPL activity in a two-dimensional rare-earth double perovskite. Since there is much room for tailoring the structure by varying the organic cation, the alkaline metal ion, and the rare-earth ion, this series of works will lead to the discovery of novel rare-earth-based materials with excellent ferroelectricity, piezoelectricity, and other optoelectronic properties.
4.3 Non-perovskite structural multiferroics
Research progress relating to single-phase molecular multiferroics with non-perovskite structures is summarized here, including 0D packing structures, 1D corner-sharing octahedral structures, 3D organic hybrid bimetal halides with lonsdaleite topology and cyanido-bridged dimetallic complexes. | ||
| Fig. 9 (a) The design of the 0D multiferroic [(C6H5)3PCH3]FeBr4 through substituting the metal ion (I). (II) A zoomed-in view of [(C6H5)3PCH3]FeBr4 susceptibility. (III) A zoomed-in view of the magnetization of the compound with respect to the applied field at different temperatures. (b) The 1D trinuclear iron–oxo anionic chain complex [FPM][Fe3(µ3-O)(µ-O2CH)8]: the Newman projection and packing views of the FPM cation at LTP (IV and VII), ITP (V and VIII), and HTP (VI and IX). The “R” group stands for –CH2–C4H8N2. (X) The trinuclear iron–oxo anionic chain at 250 K. PFM amplitude (XI) and ferroelastic domain (XII) patterns of [FPM][Fe3(µ3-O)(µ-O2CH)8]. Reproduced from ref. 50 and 54 with permission from Wiley and American Chemical Society, copyright 2025 and 2024. | ||
Among 1D non-perovskite multiferroic compounds, the trinuclear iron–oxo anionic chain complex [FPM][Fe3(µ3-O)(µ-O2CH)8] (where FPM is 3-(3-formylaminopropyl)-3,4,5,6-tetrahydropyrimidin-1-ium) serves as a compelling example.54 As depicted in Fig. 9b, this complex features organic counterions formed from formamide groups acting as H-bond donors and acceptors linked to ammonium cations via alkyl chains, imparting significant conformational flexibility. This inherent conformational adaptability and the numerous H-bond binding sites within the FPM cation enable [FPM][Fe3(µ3-O)(µ-O2CH)8] to exhibit an unusual inverse temperature symmetry-breaking (ITSB) phase transition, alongside the coexistence of ferroelectricity and ferroelasticity, within the temperature range of 365–426 K. ITSB is driven by a two-step process: an initial conformational flip of the flexible FPM counterions at 365 K (LTP to ITP, Pca21 to Cc), followed by a conventional order–disorder transition at 426 K (ITP to HTP, Cc to Cmc21). These sequential transitions are precisely described by the mm2Fm species within the Aizu classification system. Crucially, the coexistence of ferroelasticity and ferroelectricity was conclusively evidenced by comprehensive characterization using PLM and PFM. Importantly, the complex not only exhibits multistep ferroelectric and ferroelastic switching but also presents remarkable nonlinear optical switching properties, broadening its potential functionalities.
 | ||
| Fig. 10 (a) The organic hybrid bimetal halide R-3HP with lonsdaleite topology: (I) the homochiral cation of (R)-3-hydroxypyrrolidinium. (II) The coordination polyhedral configuration of the inorganic framework viewed along the crystallographic bc-plane. (III) The basic 3D cavity structure at 293 K; H atoms are omitted for clarity. (IV) A topological perspective view in which the straight lines and circles represent the Br ion ligands and Rb/Bi atoms, respectively. A basic unit of lonsdaleite topology is highlighted in red, with boat-like and chair-like six-member rings in green and purple, respectively. (V) The P–E hysteresis loop and J–E curve along the b- and c-axes at room temperature. (VI) The ferroic domains at 300 K. (b) A pair of multiferroic chiral cyanido-bridged dimetallic complexes (R- and S-3-FPC): (VII) a pair of homochiral cations of (R and S)-3-fluoropyrrolidinium. (VIII) The coordination polyhedral configuration of the inorganic framework. (IX) The basic 3D cavity structure. (X) A topological perspective view. (XI) The PFM phase superimposed on a 3D topographic image for R-3-FPC thin films measured at 298 K. (XII) Ferroelastic domain patterns at 393 K. Reproduced from ref. 61 and 62 with permission from American Chemical Society and Wiley, copyright 2021 and 2022. | ||
5. Summary and outlook
Single-phase molecular multiferroics exhibit diverse applications due to the coupling between polarization and strain. For instance, in nonvolatile multistate memory, each storage unit can provide multi-bit information by combining polarization orientation and strain state, enabling electric-write/mechanical-read or mechanical-write/electric-read modes, thereby significantly enhancing storage density. In high-precision actuators and brakes, the piezoelectric effect coupled with ferroelastic strain allows for electric-field-controlled precise displacement. In highly sensitive sensors, minute stress changes can be converted into easily detectable electrical signals. In shape-memory devices, regulating the stable state of ferroelastic domains via an electric field enables electrically controlled shape-memory function, showing promise in fields such as micro-electromechanical systems (MEMS), robotics, and medical implants. How can such materials be efficiently designed and synthesized?Based on reported single-phase molecular multiferroic materials, a strategic approach focusing on composition and structural modulation is essential. For organic components, molecules with low symmetry and high motional or rotational freedom are preferred. Chemical modifications of the organic components include halogen substitution, the introduction of homochirality, the incorporation of organo-functional groups, the reduction of molecular rigidity, and the alteration of molecular conformation. For inorganic components, strategies including halogen substitution, metal-ion replacement, and doping with different elements to form solid solutions can be employed to modulate lattice distortion, electronic structure and phase transition behavior. These design principles collectively guide the synthesis of single-phase molecular multiferroics that meet the criteria for full ferroelectric and full ferroelastic coupling.
However, the development of this field still faces a series of challenges. First, single-phase molecular multiferroics that display strong polarization-strain coupling near room temperature remain scarce; most candidates operate at low temperatures or exhibit weak coupling. Second, the synthesis of molecular multiferroics still depends on trial-and-error approaches, falling far short of the function-by-design goal. There is an urgent need to establish an integrated database linking Aizu species, structures, and properties, and to combine first-principles calculations with machine-learning-driven high-throughput screening to discover superior candidates with high Curie temperatures and low coercive fields. Third, the theoretical foundations are incomplete. Extended theories capable of describing ferroelectric–ferroelastic and more complex order-parameter couplings (e.g., ferrotoroidicity) are required, alongside the incorporation of non-Hermitian physics, topological phase transitions, and the exploration of novel quantum-critical or nonequilibrium states. Moreover, precise dynamic control of coupled domains at the nanoscale remains technically limited; in situ microscopy techniques combined with phase-field simulations are needed to elucidate domain-wall dynamics and switching mechanisms. Finally, engineering-oriented research toward practical applications is crucial. Challenges related to material fabrication, device processing, fatigue, and reliability must be overcome to translate laboratory breakthroughs into viable devices.
Looking forward, interdisciplinary collaboration and rational design are expected to enable the creation and deployment of high-performance single-phase molecular multiferroics, achieving breakthroughs in both material performance and applications.
Author contributions
Meng-Meng Lun: conceptualization, investigation, data curation, visualization, writing – original draft. Meng-Meng Sun: investigation, data curation, writing – review & editing. Yong-Qiang Wang, Gao-Shang Gong, Mao-Cai Wei, Yu-Ling Su: investigation, writing – review & editing. Da-Wei Fu and Zun-Qi Liu: conceptualization, supervision, writing – review & editing, funding acquisition. All authors approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software, or code have been included, and no new data were generated or analysed as part of this review.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc10231f.
Acknowledgements
The authors gratefully acknowledge support from the Key Research Project of Higher Education Institutions (26B140017) and Science and Technology Research Project (252102210203, 232102230121) of Henan Province, and Natural Science Foundation of Henan Provincial (No. 262300422454).References
- H. Zheng and K. P. Loh, Adv. Mater., 2024, 36, e2308051 CrossRef PubMed.
- Q. Pan, Z. X. Gu, R. J. Zhou, Z. J. Feng, Y. A. Xiong, T. T. Sha, Y. M. You and R. G. Xiong, Chem. Soc. Rev., 2024, 53, 5781–5861 RSC.
- C. Y. Su, Y. F. Yao, Z. X. Zhang, Y. Wang, M. Chen, P. Z. Huang, Y. Zhang, W. C. Qiao and D. W. Fu, Chem. Sci., 2022, 13, 4794–4800 RSC.
- D. W. Fu, H. L. Cai, Y. Liu, Q. Ye, W. Zhang, Y. Zhang, X. Y. Chen, G. Giovannetti, M. Capone, J. Li and R. G. Xiong, Science, 2013, 339, 425–428 CrossRef CAS PubMed.
- Y.-Y. Tang, H.-Y. Ye, P.-F. Li, W.-Q. Liao, J.-X. Gao, X.-N. Hua, H. Cai, P.-P. Shi, Y.-M. You and R.-G. Xiong, Science, 2018, 361, 151–155 CrossRef.
- W. Q. Liao, D. Zhao, Y. Y. Tang, Y. Zhang, P. F. Li, P. P. Shi, X. G. Chen, Y. M. You and R. G. Xiong, Science, 2019, 363, 1206–1210 CrossRef CAS PubMed.
- Y. M. You, W. Q. Liao, D. Zhao, H. Y. Ye, Y. Zhang, Q. Zhou, X. Niu, J. Wang, P. F. Li, D. W. Fu, Z. Wang, S. Gao, K. Yang, J. M. Liu, J. Li, Y. Yan and R. G. Xiong, Science, 2017, 357, 306–309 CrossRef CAS PubMed.
- H. Y. Zhang and R. G. Xiong, Science, 2023, 381, 484–485 CrossRef CAS PubMed.
- P. Z. Huang, Z. Liu, L. K. Ye, H. F. Ni, J. Q. Luo, G. Teri, Q. Q. Jia, B. Zhuang, C. F. Wang, Z. X. Zhang, Y. Zhang and D. W. Fu, Nat. Commun., 2025, 16, 3071 CrossRef CAS PubMed.
- L. A. Gui, J. Chen, Y. F. Zhang, L. H. Li, J. R. Li, Z. B. Hu, S. Y. Zhang, J. Zhang, Z. Zhang, H. Y. Ye, Y. Peng, J. Ma and Y. Song, Angew. Chem., Int. Ed., 2025, 64, e202416380 CrossRef CAS PubMed.
- H. Zheng, A. Ghosh, M. J. Swamynadhan, Q. Zhang, W. P. D. Wong, Z. Wu, R. Zhang, J. Chen, F. Cimpoesu, S. Ghosh, B. J. Campbell, K. Wang, A. Stroppa, R. Mahendiran and K. P. Loh, Nat. Commun., 2024, 15, 5556 CrossRef CAS PubMed.
- Y. Liu, W. Guo, L. Hua, X. Zeng, T. Yang, Q. Fan, Y. Ma, C. Gao, Z. Sun and J. Luo, J. Am. Chem. Soc., 2023, 145, 16193–16199 CrossRef CAS PubMed.
- Q. Q. Jia, J. Q. Luo, Z. Y. Xue, J. S. Tang, W. Q. Qiu, C. F. Wang, Z. X. Zhang, H. F. Lu, Y. Zhang and D. W. Fu, Chin. Chem. Lett., 2025, 36, 110471 CrossRef CAS.
- J. Valasek, Phys. Rev., 1921, 17, 475–481 CrossRef CAS.
- J. Valasek, Phys. Rev., 1922, 20, 639–664 CrossRef CAS.
- G. Busch and P. Scherrer, Naturwissenschaften, 1935, 23, 737 CrossRef CAS.
- J. Jeong, H. S. Kim, G. Kwon, K. Jeong, H. Lee, J. H. Lee, M. Park, C. Lee, S. Yu, H. Kim, S. Im, K. Yoo, E. Lee and M. H. Cho, Adv. Mater., 2022, 34, e2108777 CrossRef PubMed.
- M. M. Lun, J. Q. Luo, Z. X. Zhang, J. Li, L. Y. Xie, H. F. Lu, Y. Zhang and D. W. Fu, Chem. Eng. J., 2023, 475, 145969 CrossRef CAS.
- C. Shi, J. J. Ma, J. Y. Jiang, M. M. Hua, Q. Xu, H. Yu, Y. Zhang and H. Y. Ye, J. Am. Chem. Soc., 2020, 142, 9634–9641 CAS.
- Y. Z. Hu, L. You, B. Xu, T. Li, S. A. Morris, Y. X. Li, Y. H. Zhang, X. Wang, P. S. Lee, H. J. Fan and J. L. Wang, Nat. Mater., 2021, 20, 612–617 CrossRef CAS PubMed.
- G. Feng, Q. Zhu, X. Liu, L. Chen, X. Zhao, J. Liu, S. Xiong, K. Shan, Z. Yang, Q. Bao, F. Yue, H. Peng, R. Huang, X. Tang, J. Jiang, W. Tang, X. Guo, J. Wang, A. Jiang, B. Dkhil, B. Tian, J. Chu and C. Duan, Nat. Commun., 2024, 15, 513 CrossRef CAS PubMed.
- Z.-T. Xia, H.-P. Chen, J.-C. Qi, H. Peng, X. Shen, Y.-J. Bai, Z.-Y. Wang, T.-E. Yang and W.-Q. Liao, Inorg. Chem. Front., 2025, 12, 2825–2832 RSC.
- T. Zhang, K. Xu, J. Li, L. He, D. W. Fu, Q. Ye and R. G. Xiong, Natl. Sci. Rev., 2023, 10, nwac240 CrossRef CAS PubMed.
- Y. F. Zhang, F. F. Di, P. F. Li and R. G. Xiong, Chem.–Eur. J, 2022, 28, e202102990 CrossRef CAS PubMed.
- L. Pan, H. F. Ni, P. Z. Huang, H. H. Li, M. J. Lin, G. Teri, J. Q. Luo, L. K. Ye, J. K. Xu, Q. Q. Jia, C. F. Wang, Z. Q. Liu, Z. X. Zhang, Y. Zhang and D. W. Fu, Angew. Chem., Int. Ed., 2026, e5334832 CAS.
- C. F. Wang, C. Shi, A. Zheng, Y. Wu, L. Ye, N. Wang, H. Y. Ye, M. G. Ju, P. Duan, J. Wang and Y. Zhang, Mater. Horiz., 2022, 9, 2450–2459 RSC.
- H. F. Ni, L. Pan, C. H. Du, L. C. Xu, J. C. Lou, P. G. Liu, H. N. Wang, C. F. Wang, Z. X. Zhang, D. W. Fu and Y. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202518937 CrossRef CAS PubMed.
- K. Aizu, J. Phys. Soc. Jpn., 1970, 28, 706–716 CrossRef CAS.
- J. Sapriel, Phys. Rev. B, 1975, 12, 5128–5140 CrossRef CAS.
- K. Aizu, J. Phys. Soc. Jpn., 1969, 27, 387–396 CrossRef CAS.
- K. Aizu, Phys. Rev. B, 1970, 2, 754–772 CrossRef.
- H. Schmid, J. Phys.: Condens. Matter, 2008, 20, 434201 CrossRef.
- R. E. Cohen, Nature, 1992, 358, 136–138 CrossRef CAS.
- T. Y. Ju, C. C. Fan, B. D. Liang, C. D. Liu, M. L. Jin, C. Y. Chai and W. Zhang, Adv. Funct. Mater., 2024, 34, 2316747 CrossRef CAS.
- I. B. Bersuker, The Jahn-Teller Effect, Cambridge Cambridge University Press, 2006 Search PubMed.
- J. Li, J.-F. Li, Q. Yu, Q. N. Chen and S. Xie, J. Materiomics, 2015, 1, 3–21 CrossRef.
- H. Y. Zhang, X. G. Chen, Y. Y. Tang, W. Q. Liao, F. F. Di, X. Mu, H. Peng and R. G. Xiong, Chem. Soc. Rev., 2021, 50, 8248–8278 RSC.
- H. Y. Zhang, X. J. Song, X. G. Chen, Z. X. Zhang, Y. M. You, Y. Y. Tang and R. G. Xiong, J. Am. Chem. Soc., 2020, 142, 4925–4931 CrossRef CAS PubMed.
- K. Ding, H. Ye, C. Su, Y. A. Xiong, G. Du, Y. M. You, Z. X. Zhang, S. Dong, Y. Zhang and D. W. Fu, Nat. Commun., 2023, 14, 2863 CrossRef CAS PubMed.
- J. Y. Li, T. Zhang, M. M. Lun, Y. Zhang, L. Z. Chen and D. W. Fu, Small, 2023, 19, e2301364 CrossRef PubMed.
- Y. Ai, Z. B. Hu, Y. R. Weng, H. Peng, J. C. Qi, X. G. Chen, H. P. Lv, X. J. Song, H. Y. Ye, R. G. Xiong and W. Q. Liao, Adv. Mater., 2024, 36, e2407822 CrossRef PubMed.
- H. P. Weber, B. C. Tofield and P. F. Liao, Phys. Rev. B, 1975, 11, 1152–1159 CrossRef CAS.
- M. M. Lun, C. Y. Su, J. Li, Q. Q. Jia, H. F. Lu, D. W. Fu, Y. Zhang and Z. X. Zhang, Small, 2023, 19, e2303127 CrossRef PubMed.
- Q. Q. Jia, G. Teri, Q. F. Zhou, J. Q. Luo, H. F. Ni, P. Z. Huang, P. G. Liu, L. Pan, C. F. Wang, Z. Liu, Z. X. Zhang, Y. Zhang and D. W. Fu, Angew. Chem., Int. Ed., 2025, 64, e202514669 CrossRef CAS PubMed.
- B. Zhuang, L. Pan, Z. L. Li, J. Y. Liu, Z. X. Zhang, K. Ding, Y. Zhang, Z. Q. Liu and D. W. Fu, ACS Mater. Lett., 2025, 7, 1540–1546 CrossRef CAS.
- J. Li, Y. Zhu, P. Z. Huang, D. W. Fu, Q. Q. Jia and H. F. Lu, Chem.–Eur. J., 2022, 28, e202201005 CrossRef CAS PubMed.
- C. Y. Su, H. G. Yi, H. F. Ni, G. W. Du, S. Q. Xia, Z. Q. Liu, Z. X. Zhang and D. W. Fu, Chem. Sci., 2025, 16, 23385–23393 RSC.
- Q. Q. Jia, G. Teri, J. Q. Luo, H. F. Ni, P. Z. Huang, M. M. Lun, Z. X. Zhang, Y. Zhang and D. W. Fu, J. Am. Chem. Soc., 2024, 146, 21120–21128 CrossRef CAS PubMed.
- Y. L. Zeng, Y. Ai, S. Y. Tang, X. J. Song, X. G. Chen, Y. Y. Tang, Z. X. Zhang, Y. M. You, R. G. Xiong and H. Y. Zhang, J. Am. Chem. Soc., 2022, 144, 19559–19566 CrossRef CAS PubMed.
- S. N. Li, R. J. Harrison, W. Li and M. A. Carpenter, Chin. J. Chem., 2025, 43, 3193–3198 CrossRef CAS.
- Y. Gao, M. Wu and P. Jena, Nat. Commun., 2021, 12, 1331 CrossRef CAS PubMed.
- X. J. Song, Y. Ai, X. G. Chen, Y. Qin, Y. Y. Tang, H. P. Lv, P. F. Li, H. Peng, Y. R. Weng, H. H. Chen, R. G. Xiong and W. Q. Liao, J. Am. Chem. Soc., 2025, 147, 16568–16577 CrossRef CAS PubMed.
- M. Ksiądzyna, A. Gągor, A. Piecha-Bisiorek, A. Ciżman, W. Medycki and R. Jakubas, J. Mater. Chem. C, 2019, 7, 10360–10370 RSC.
- L. Y. Zhan, Y. Zhou, N. Li, L. J. Zhang, X. J. Xi, Z. Q. Yao, J. P. Zhao and X. H. Bu, J. Am. Chem. Soc., 2024, 146, 5414–5422 CrossRef CAS PubMed.
- D. Fu, Z. Hou, Y. He, J.-C. Liu, H.-P. Lv and Y.-Y. Tang, Chem. Mater., 2022, 34, 3518–3524 CrossRef CAS.
- C. K. Yang, W. N. Chen, Y. T. Ding, J. Wang, Y. Rao, W. Q. Liao, Y. Y. Tang, P. F. Li, Z. X. Wang and R. G. Xiong, Adv. Mater., 2019, 31, 1808088 CrossRef PubMed.
- Y. L. Zeng, X. Q. Huang, C. R. Huang, H. Zhang, F. Wang and Z. X. Wang, Angew. Chem., Int. Ed., 2021, 60, 10730–10735 CrossRef CAS PubMed.
- N. Wang, Z. J. Xu, H. F. Ni, W. Luo, H. K. Li, M. L. Ren, C. Shi, H. Y. Ye, X. B. Fu, Y. Zhang and L. P. Miao, Angew. Chem., Int. Ed., 2024, 63, e202409796 CrossRef CAS PubMed.
- Q. Q. Jia, H. F. Lu, J. Q. Luo, Y. Y. Zhang, H. F. Ni, F. W. Zhang, J. Wang, D. W. Fu, C. F. Wang and Y. Zhang, Small, 2024, 20, e2306989 CrossRef PubMed.
- A. Sieradzki, M. Mączka, M. Simenas, J. K. Zaręba, A. Gągor, S. Balciunas, M. Kinka, A. Ciupa, M. Nyk, V. Samulionis, J. Banys, M. Paluch and S. Pawlus, J. Mater. Chem. C, 2018, 6, 9420–9429 RSC.
- L. He, P. P. Shi, L. Zhou, Z. B. Liu, W. Zhang and Q. Ye, Chem. Mater., 2021, 33, 6233–6239 CrossRef CAS.
- Y. Mao, X. G. Chen, Z. X. Gu, Z. X. Zhang, X. J. Song, N. Gu and R. G. Xiong, Angew. Chem., Int. Ed., 2022, 61, e202204135 CrossRef CAS PubMed.
- C. F. Wang, Y. Yang, Y. Hu, C. Ma, H. F. Ni, P. G. Liu, H. F. Lu, Z. X. Zhang, J. Wang, Y. Zhang, D. W. Fu, K. Zhao and Y. Zhang, Angew. Chem., Int. Ed., 2024, 63, e202413726 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |