DOI:
10.1039/D5SC10104B
(Review Article)
Chem. Sci., 2026,
17, 9888-9911
Recent progress in iodate based nonlinear optical crystals
Received
24th December 2025
, Accepted 15th April 2026
First published on 17th April 2026
Abstract
Nonlinear optical (NLO) crystals are essential components in modern laser technology. For middle and far-infrared applications, iodates are regarded as highly promising materials due to their intrinsic non-centrosymmetric (NCS) structures, broad optical transparency, pronounced second-order NLO responses, and relatively high laser-induced damage thresholds (LIDTs). However, the tendency of spontaneous crystallization in favor of thermodynamically stable centrosymmetric (CS) phases poses a significant challenge to the rational design of iodates with a macroscopic NCS framework. This review systematically summarizes recent advances over the past decade in the rational designs and controlled syntheses of NCS iodates with significant SHG responses. Emphasis is focused on five key structural-modulation strategies: polymerization of different iodate anions, aliovalent substitution, fluorine substitution, and the synergistic integration of iodate anions with either planar π-conjugated units or tetrahedral building blocks. These approaches collectively enhance local polarization, tune the electronic structure, and optimize crystal-growth kinetics, thereby increasing the probability of forming NCS structures and promoting the development of high-performance iodate NLO crystals. Finally, future research directions and emerging trends in this field are outlined to provide a reference for the continued advancement of related research areas.
1 Introduction
Nonlinear optical (NLO) crystals are key materials for laser frequency conversion and spectral expansion, whose performance is crucial for the applicability of laser technology in various fields, including communication, medicine, precision measurement, and frontier scientific research.1–7 Currently, commercial NLO crystals, such as KTiOPO4 (KTP),8 KH2PO4 (KDP),9 and β-BaB2O4 (β-BBO),10 mainly operate in the ultraviolet (UV) to near-infrared range, but are inadequate for applications in the middle and far-infrared region (2–20 µm). Although commercial infrared NLO crystals (e.g., AgGaS2, ZnGeP2, and AgGaSe2) partially cover this spectral window, they suffer from inherent performance limitations: narrow bandgaps accompanied by low laser-induced damage thresholds (LIDTs) (AgGaS2) or inability to achieve phase-matching (AgGaSe2), or pronounced two-photon absorption near 1 µm (ZnGeP2).11–14
In response to these limitations, extensive research has focused on exploring new infrared (IR) NLO material families. Within chalcogenides, substantial research has yielded promising candidates that overcome traditional performance limitations. For instance, MgGa2Se4, the first defect-diamond-like (DCL) alkaline-earth metal selenide, exhibits a band gap of 2.96 eV, a high LIDT of 3.0 × AgGaS2, and a strong second-harmonic-generation (SHG) response of 0.9 × AgGaS2 with type-I phase-matching (PM).15 More notably, Li4MgGe2S7, the first alkali and alkaline-earth metal diamond-like (DL) IR NLO material, features a unique honeycomb-like framework that yields an exceptional band gap of 4.12 eV, which is the largest among quaternary metal chalcogenides, leading to a high LIDT of 7 × AgGaS2, while retaining a moderate SHG response of 0.7 × AgGaS2 with type-I PM.16
Despite these advances, chalcogenides still face inherent challenges. The trade-off between the band gap and nonlinearity persists: MgGa2Se4 offers stronger SHG intensity but a narrower band gap, while Li4MgGe2S7 achieves a wider band gap at the expense of SHG intensity. Additionally, issues such as two-photon absorption and difficulties in crystal growth further limit their practical application, motivating the search for alternative systems with more balanced performance.
Against this backdrop, iodates have emerged as an important class of materials for middle and far IR NLO crystals, owing to their distinct structural features and excellent comprehensive performance. Unlike chalcogenides, which achieve large SHG intensity but have lower LIDT, or halides, which prioritize LIDT at the expense of SHG, iodates achieve a favorable balance across multiple performance metrics. The stereochemically active lone pair (SCALP) electrons on the I5+ ion induce highly distorted trigonal pyramidal [IO3]− and see-saw shaped [IO4]3− units, which generate pronounced local polarization and hyperpolarizability. This characteristic not only favors the formation of non-centrosymmetric (NCS) crystal structures but also imparts large second-order NLO coefficients when these asymmetric units are aligned in their structures and significant birefringence. Furthermore, the low phonon energy of the I–O bonds enables broad optical transmission across key atmospheric windows, particularly in the 3–5 µm and 8–12 µm regions.17 This combination of strong SHG response and wide infrared transparency renders iodates promising candidates for mid-infrared laser technology.
Despite the notable advantages of iodates, their designs and syntheses still encounter two fundamental challenges. First, the SCALPs tend to adopt antiparallel dipole orientations, favoring the formation of thermodynamically more stable centrosymmetric (CS) structures, which makes the directed assembly of NCS phases particularly difficult. Second, even when the NCS structure is successfully realized, it remains essential to simultaneously optimize multiple performance parameters, including the NLO coefficient, band gap, birefringence (Δn), and LIDTs, to meet the requirements of practical materials and devices.18–24 Notably, the Δn is highly wavelength-dependent: for visible and near-IR applications, a moderate Δn (≥0.03) is sufficient, whereas for UV regions, the increased material dispersion demands a larger birefringence (≥0.07) to achieve PM. To address these challenges, researchers have recently developed a variety of effective structural-modulation strategies to systematically increase the probability of NCS crystallization while enhancing overall optical performance.
In fact, research in this field has progressed through a series of pivotal developments. In 2015, our group systematically reviewed the progress in metal iodate NLO crystals, focusing on systems with the NCS structures and notable SHG performance.25 In 2017, Guo et al. noted the potential of iodates for applications spanning the visible to mid-infrared spectrum in their review of middle and far-infrared NLO materials.26 In 2021, our group further summarized novel strategies for high-performance SHG materials, including a few iodates as representative examples to illustrate design approaches such as iodate polymerization and aliovalent substitution.27 In the same year, Wu et al. carried out a systematic theoretical and experimental analysis of structure–property relationships in metal iodates,28 while Guo et al. and Kong et al. reviewed the influence of SCALPs on SHG activity.29,30 Subsequent work has continued to advance the field. In 2023, the OK group demonstrated the application of aliovalent substitution strategies in d0-transition-metal/gallium iodates,31 and Yu et al. conducted a comprehensive survey of fluorine-containing iodate NLO crystals.17 Most recently, in 2025, Wang et al. also discussed the structure–property correlations of iodates in their review of advances in d0-transition-metal oxyfluorides.32 In conclusion, these contributions have significantly propelled the rational designs and performance optimizations of metal iodates.
Despite recent progress in exploring specific design strategies for iodate-based NLO materials, a systematic overview that consolidates these advances and elucidates their underlying structure–property relationships is still needed. This review highlights key advances over the past decade, centering on five core strategies: polymerization of [IO3]−/[IO4]3− units, aliovalent substitution, fluorine substitution, and combination of different promising units (π-conjugated and tetrahedral) (Fig. 1). By systematically reviewing the synthetic strategies and analyzing the structure–property relationships of representative compounds, we elucidate how these approaches help overcome symmetry constraints and balance multiple performance parameters. This work aims to provide a clear theoretical foundation and future directions for the rational design and controlled synthesis of high-performance iodate NLO crystals.
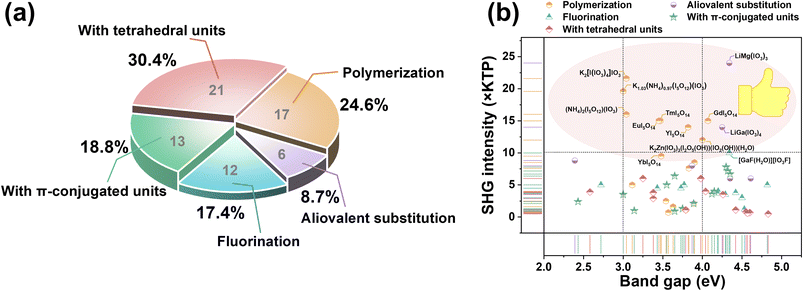 |
| | Fig. 1 (a) Statistical distribution of iodates obtained via various synthetic routes. (b) SHG intensity-band gap map for all screened crystals. | |
2 Polymerizations of different iodate anions
The I atom exhibits a wide range of oxidation states (e.g., +3 and +5) and diverse coordination geometries. For I5+, its SCALPs show pronounced stereochemical activity, triggering a second-order Jahn–Teller (SOJT) distortion that leads to significant polyhedral distortion. Specifically, the SCALPs occupy one side of the I atom, pushing the O atoms to the opposite side and forming highly distorted [IO3]− trigonal pyramids or “see-saw” shaped [IO4]3− units. These building blocks can further polymerize via O bridges to form a variety of polyiodate units, such as the [I2O5] dimer,33–35 the [I3O8]− trimer,36–38 pinwheel-shaped [I3O10]5−,39 the [I4O11]2− tetramer,40,41 the [I5O14]3− pentamer,42,43 and the 1D 1∞[I7O18(OH)]2− chain.44 Polyiodates not only increase the spatial density of functional units but also raise the local concentration of lone-pair electrons within the unit cell, thereby significantly enhancing the probability of NCS structure formation and favoring materials with strong optical anisotropy. In our previous work, a series of noble-metal iodates with excellent SHG responses were discovered, including α-NaAu(IO3)4 (1.17 × KTP at 2.05 µm),45 K2Au(IO3)5 (1.0 × KTP at 2.05 µm),46 and BaPd(IO3)4 (0.4 × KTP at 2.05 µm).47 These compounds contain [M(IO3)4] (M = Au3+, Pd2+) units, in which a square-planar [MO4] group shares O vertices with four [IO3]− units located on the same side of the plane. The square-planar [MO4] configuration has been shown to serve as an effective structure directing agent, promoting the ordered alignment of high-density SCALPs. Inspired by this, the I3+ cation possesses two lone pairs.48,49 To minimize electron–electron repulsion, its four ligands tend to adopt a coplanar arrangement, forming a square-planar [IIIIO4]5− unit that exhibits significant optical anisotropy. This section reviews the structural features and optical properties of iodates containing diverse polyanionic units. Representative polyiodates exhibiting notable SHG responses are summarized in Table 1.
Table 1 Optical properties of polyiodatesa
| Compounds |
Space group |
Anionic group |
SHG intensity |
Bandgap (eV) |
Birefringence |
PM/NPM |
Ref. |
|
Powder SHG data (Kurtz–Perry method) provide an initial indicator of NLO potential.52 N/A = not available or not reported.
|
| K2[I(IO3)4]IO3 |
Cmc21 |
Bowl-shaped [IIII(IVO3)4]− |
21.6 × KDP at 1064 nm |
3.04 |
0.358 at 543 nm |
PM |
48
|
| K1.03(NH4)0.97(I5O12)(IO3) |
Cmc21 |
Bowl-shaped [IIII(IVO3)4]− |
19.5 × KDP at 1064 nm |
3 |
0.405 at 546 nm |
PM |
49
|
| (NH4)2(I5O12)(IO3) |
Cmc21 |
Bowl-shaped [IIII(IVO3)4]− |
16 × KDP at 1064 nm |
3.04 |
0.431 at 546 nm |
PM |
49
|
| K2Zn(IO3)3(I2O5(OH))(IO2(OH))(H2O) |
P1 |
[I2O5(HO)] dimer and [IO3]− pyramid |
12 × KDP at 1064 nm |
4 |
0.055 at 532 nm |
PM |
35
|
| HBa2.5(IO3)6(I2O5) |
Fdd2 |
[I2O5] dimer and [IO3]− pyramid |
1.6 × KDP at 1064 nm |
3.63 |
N/A |
PM |
33
|
| K2Na(IO3)2(I3O8) |
Pc
|
[I3O8]− trimer and [IO3]− pyramid |
7.6 × KDP at 1064 nm |
3.83 |
0.055 at 1064 nm |
PM |
37
|
| γ-Cs2I4O11 |
P21 |
2D 2∞[I3O8]− layer and seesaw-shaped [IO4]3− |
5 × KDP at 1064 nm |
3.74 |
0.055 at 1064 nm |
PM |
40
|
| Ba4Ag5(IO3)6(I3O8)3(I4O11)2 |
Pnc2 |
[I3O8]− trimer and [I4O11]2− tetramer |
2.5 × KDP at 1064 nm |
3.54 |
0.082 at 1064 nm |
PM |
50
|
| C(NH2)3(I3O8)(HI3O8)(H2I2O6)(HIO3)4·3H2O |
P1 |
[I3O8]− trimer |
2.1 × KDP at 1064 nm |
3.89 |
0.06 at 550 nm |
PM |
51
|
| Bi4O(I3O10)(IO3)3(SeO4) |
R3c |
Pinwheel-shaped [I3O10]5− |
1.1 × KDP at 1064 nm |
3.79 |
N/A |
PM |
39
|
| ErI5O14 |
Pmn21 |
Semicircle-shaped [I5O14]3− |
0.7 × KDP at 1064 nm |
3.57 |
0.081 at 1064 nm |
PM |
42
|
| TmI5O14 |
Pmn21 |
Semicircle-shaped [I5O14]3− |
15 × KDP at 1064 nm |
3.47 |
0.092 at 1064 nm |
PM |
42
|
| EuI5O14 |
Cm
|
Semicircle-shaped [I5O14]3− |
15 × KDP at 1064 nm |
3.45 |
0.168 at 1064 nm |
PM |
42
|
| YbI5O14 |
Pmn21 |
Semicircle-shaped [I5O14]3− |
9.5 × KDP at 1064 nm |
3.48 |
0.106 at 1064 nm |
PM |
42
|
| YI5O14 |
Cm
|
Semicircle-shaped [I5O14]3− |
14 × KDP at 1064 nm |
3.82 |
N/A |
PM |
43
|
| GdI5O14 |
Cm
|
Semicircle-shaped [I5O14]3− |
15 × KDP at 1064 nm |
4.07 |
N/A |
PM |
43
|
| [o-C5H4NHOH]2[I7O18(OH)]·3H2O |
Ia
|
1D 1∞[I7O8(OH)]2− chain |
8.5 × KDP at 1064 nm |
3.9 |
N/A |
PM |
44
|
2.1
A
2[IIII(IVO3)4]IVO3 (A = K, NH4)
These iodate single crystals were synthesized via conventional hydrothermal methods at 160 °C for 3 days. In the preparation of K2[IIII(IVO3)4]IVO3, SeO2 is added to act as a mild reducing agent, converting a portion of I5+ to I3+, thereby enabling the co-incorporation of I in different oxidation states within the same lattice.48,49 K2[IIII(IVO3)4]IVO3, (NH4)2[IIII(IVO3)4]IVO3, and K1.03(NH4)0.97(I5O12)(IO3) all crystallize in the NCS polar space group Cmc21. In these structures, the I3+ ion is coordinated by four O atoms from distinct [IVO3]− groups, forming a square-planar [IIIIO4]5− unit. All four [IVO3]− groups are situated on the same side of this square plane, collectively assembling into a novel bowl-shaped polymeric unit [IIII(IVO3)4]− (Fig. 2a). Owing to the structure directing influence of the [IIIIO4]5− unit, the lone pairs of electrons in each [IVO3]− group adopt a highly consistent cis-orientation. This alignment imparts significant macroscopic polarity and remarkable optical anisotropy to the polymeric anion. To achieve charge balance, cations (K+ and NH4+) along with isolated [IVO3]− units occupy the interstitial spaces between these bowl-shaped assemblies (Fig. 2b). This ordered and highly polarized structural arrangement results in SHG responses, with measured intensity approximately 21.6, 16, and 19.5 times that of KDP, respectively. The planar square [IIIIO4]5− group can be effectively replaced by its structural analogs, [PdO4]6− and [AuO4]5−, to produce compounds with equally large SHG responses. The substitution requires aligning all [IO3]− units on the same side of the plane. This unilateral orientation serves as a necessary structural unit for generating the significantly high SHG response.
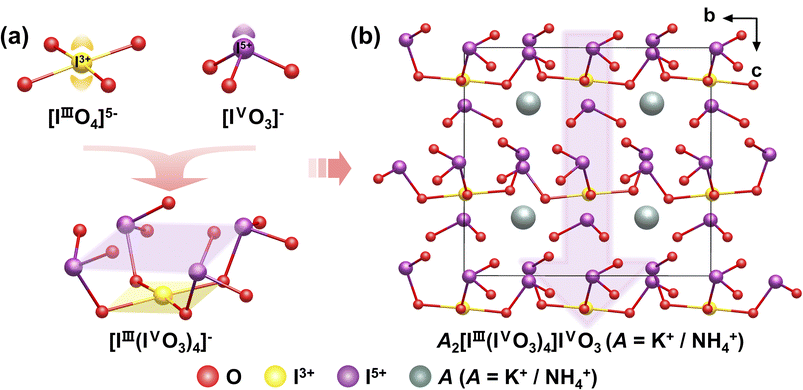 |
| | Fig. 2 (a) The [IIIIO4]5−, [IVO3]− and [IIII(IVO3)4]− units. (b) View of the structure of A2[IIII(IVO3)4]IVO3 (A = K+/NH4+) down the a-axis. Copyright 2025 and 2023 Wiley-VCH. | |
2.2 K2Zn(IO3)3(I2O5(OH))(IO2(OH))(H2O)
K2Zn(IO3)3(I2O5(OH))(IO2(OH))(H2O) was synthesized via mild hydrothermal conditions at 180 °C (3 days). The [ZnO6]10− octahedron acts as a central connector, being coordinated by one [IO3]− and one [I2O5(OH)]− unit to form a polar, “tumbler-like” [Zn(IO3)(I2O5(OH))] polyanion (Fig. 3a).35 Four additional [IO3]− groups attach through the four equatorial O atoms of the same octahedron, causing significant distortion and creating a top-heavy structure as the fundamental building block [Zn(IO3)5(I2O5(OH)]4− (Fig. 3b). Steric hindrance from [I2O5(OH)]− and hydrogen-bonding interactions ensure that the four equatorial [IO3]− groups are nearly identical in orientation and alignment. These units form a two-dimensional (2D) 2∞[Zn(IO3)3(I2O5(OH))]2− layer in the ab plane (Fig. 3c), with K+ ions, neutral [IO2(OH)] anions, and water molecules located in the interlayer space (Fig. 3d). This hierarchical structure reveals how the asymmetric and top-heavy configurations of I–O groups around the Zn2+-centered octahedral units, combined with their varied polymerization modes, create a structurally asymmetric environment that favors polar alignment. Although the [ZnO6]10− octahedral unit shows only a small distortion, the asymmetric and top-heavy I–O configurations around it, together, favor the polar alignment of the I–O groups, yielding a strong SHG response of 12 × KDP.
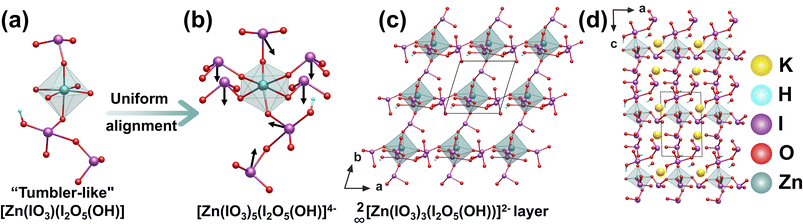 |
| | Fig. 3 (a) The “tumbler-like” [Zn(IO3)(I2O5(OH))] polyanions. (b) The fundamental building units [Zn(IO3)5(I2O5(OH)]4−. (c) The 2D 2∞[Zn(IO3)3(I2O5(OH))]2− layer. (d) The 3D structure of K2Zn(IO3)3(I2O5(OH))(IO2(OH))(H2O). Copyright 2023 American Chemical Society. | |
2.3 K2Na(IO3)2(I3O8)
K2Na(IO3)2(I3O8) was synthesized via a hydrothermal route at 220 °C for 4 days.37 The crystal structure can be described as a 3D network (Fig. 4c), where isolated [I3O8]− anions and two distinct [IO3]− groups are connected by 1D 1∞[K(1)O5]9− chains extending along the c-axis and 2D 2∞[K(2)NaO5]8− layers within the bc plane (Fig. 4a and b). The key feature is the well-aligned arrangement and additive superposition of dipole moments from all iodate units within the unit cell, resulting in a macroscopic polarization (Fig. 4d). This dipole superposition creates a macroscopic SHG response of 7.6 times that of KDP.
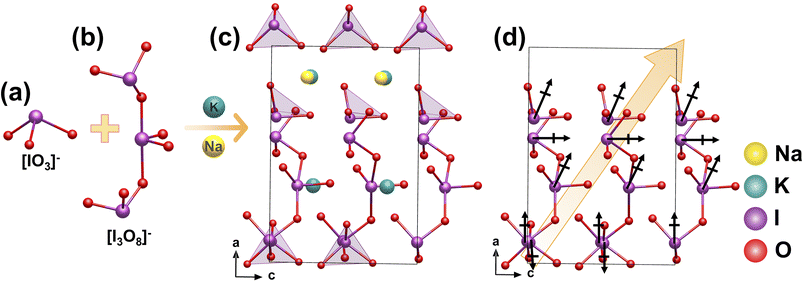 |
| | Fig. 4 (a) The [IO3]− pyramid. (b) The [I3O8]− trimer. (c) The structure of K2Na(IO3)2(I3O8). (d) The dipole moment directions of the I–O units (narrow black arrows) and the net polarization (wide orange arrow). Copyright 2020 American Chemical Society. | |
2.4 γ-Cs2I4O11
γ-Cs2I4O11 was prepared by using two hydrothermal methods: a one-step process with a H2O/TFA mixed solvent at 220 °C and a two-step process involving initial synthesis of β-Cs2I4O11 without TFA, followed by its conversion to the γ phase in TFA solution at 100 °C.40 The presence of TFA is critical for this conversion, as it acts as a structure-directing agent that promotes the protonation of O atoms in iodate groups, triggering the rearrangement of [IO4]3− polyhedra and driving the transformation of trans-[I3O10]5− trimers into their cis-[I3O10]5−. Both β- and γ-Cs2I4O11 feature a 2D 2∞[I3O8]− layer, yet they differ in the connectivity and spatial arrangement of their building units, which leads to distinct symmetries and NLO properties. In β-Cs2I4O11, two [IO3]− groups and one [IO4]3− unit are corner-shared to form a trans-[I3O8]− trimer (Fig. 5a). In contrast, the same units in γ-Cs2I4O11 adopt a cis-[I3O8]− trimeric configuration (Fig. 5b). These trimers extend along the ab plane, forming the 2D 2∞[I3O8]− layer, with Cs+ cations and remaining isolated [IO3]− anions located in the interlayer space (Fig. 5c). The transformation from the CS β phase to the NCS γ phase is primarily attributed to a geometric rearrangement of the iodate trimers, specifically, the conversion from a trans- to a cis-configuration. This structural change enables the dipole moments of the trimers in the γ phase to align along the b-axis and add constructively (Fig. 5d), yielding a macroscopic SHG response that is about 5 times that of KDP. This comparison highlights that controlling the spatial orientation of polar units is a critical structural-design strategy for breaking inversion symmetry and achieving strong SHG performance.
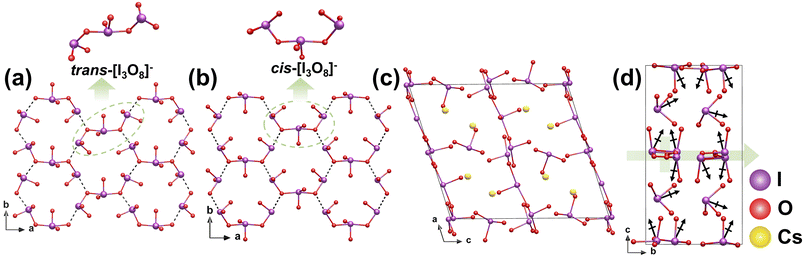 |
| | Fig. 5 The 2D 2∞[I3O8]− layers of β- and γ-Cs2I4O11 in the ab plane (a and b). Structure of γ-Cs2I4O11 in the ac plane (c). Local and net dipole moment directions of I–O polyhedra in a whole unit cell (d). Copyright 2023 American Chemical Society. | |
2.5 Bi4O(I3O10)(IO3)3(SeO4)
Colorless block single crystals of Bi4O(I3O10)(IO3)3(SeO4) were obtained via a hydrothermal reaction at 230 °C for 3 days in a highly acidic medium.39 Bi4O(I3O10)(IO3)3(SeO4) crystallizes in the polar space group R3c, featuring a unique 3D framework formed by the connection of various anionic units (Fig. 6b). In this structure, each [SeO4]2− tetrahedron coordinates to three Bi atoms through its O atoms, forming 18-membered rings (MRs) that extend in the ab plane and interconnect to form a continuous 2D 2∞[Bi4O(SeO4)]8+ layer. These ring-shaped channels are filled with pinwheel-shaped [I3O10]5− units and [IO3]− pyramids, which together with the rings form a 2D layered structure in the ab plane (Fig. 6a). Meanwhile, the [I3O10]5− units connect through Bi atoms, forming a second type of layer 2∞[Bi4O(I3O10)]5+ (Fig. 6c). These two different layered features are stacked along the c-axis and linked by shared O atoms, ultimately forming the overall 3D framework. The asymmetric polyiodate groups, with their SCALPs, disrupt CS and drive crystallization in the polar space group R3c, which exhibits enhanced polarization and a moderate SHG response of approximately 1.1 × KDP.
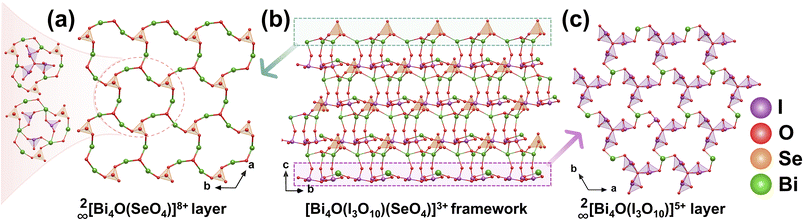 |
| | Fig. 6 (a) The 2∞[Bi4O(SeO4)]8+ layer; the left inset shows [I3O10]5− units and the [IO3]− pyramids filling the 18 MRs. (b) The [Bi4O(I3O10)(SeO4)]3+ framework. (c) The 2∞[Bi4O(I3O10)]5+ layer. Copyright 2020 Royal Society of Chemistry. | |
2.6 Ba4Ag5(IO3)6(I3O8)3(I4O11)2
Colorless plate crystals of Ba4Ag5(IO3)6(I3O8)3(I4O11)2 were synthesized via a hydrothermal reaction at 230 °C for 3 days.50 Ba4Ag5(IO3)6(I3O8)3(I4O11)2 crystallizes in the polar space group Pnc21. [AgOn] polyhedra exist in forms such as [AgO6]11− and [AgO4]7−. By connecting [I3O8]− trimers and isolated [IO3]− units, they assemble into a 2D layer of 2∞[Ag5(I3O8)3(IO3)4]2− anions parallel to the ac plane (Fig. 7b). [BaOn] polyhedra comprise [BaO8]14− and [BaO7]12− polyhedra. These interact with [I4O11]2− and [I3O8]− and [IO3]− anions. From a holistic structural perspective, the 2D 2∞[Ag5(I3O8)3(IO3)4]2− layers are embedded and stabilized within the 3D [Ba4(IO3)6(I3O8)3(I4O11)2]5− framework (Fig. 7c). Multiple iodate anions (isolated [IO3]−, [I3O8]− trimers, and [I4O11]2− tetramer) (Fig. 7a) and [AgOn] polyhedra synergistically enhance the macroscopic SHG response, exhibiting a moderate SHG performance of 2.5 times that of KDP.
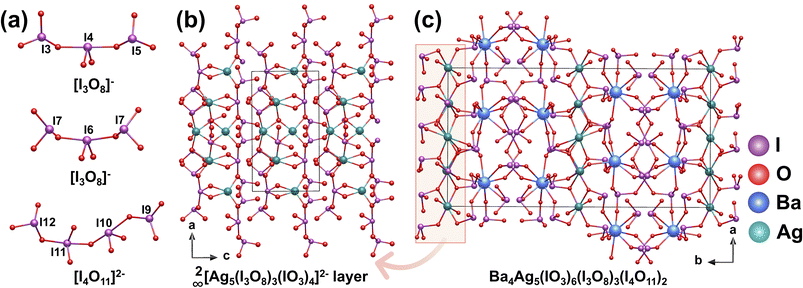 |
| | Fig. 7 (a) The [I3O8]− trimer and the [I4O11]2− tetramer. (b) The 2D 2∞[Ag5(I3O8)3(IO3)4]2− anionic layer. (c) The structure of Ba4Ag5(IO3)6(I3O8)3(I4O11)2 along the c direction. Copyright 2022 Royal Society of Chemistry. | |
2.7 REI5O14 (RE = Y, Eu, Gd, Tb, Dy, Ho, Er, Tm, and Yb)
A series of rare-earth polyiodates REI5O14 (RE = Eu, Er, Tm, Yb) were synthesized via H3PO4-mediated hydrothermal reactions, with EuI5O14 obtained at 230 °C and the other members (RE = Tm, Yb) formed at 180 °C. The family of REI5O14 compounds crystallizes in two distinct NCS polar space groups (Fig. 8a).42,43 For RE = Eu, Gd, and Y, the crystals crystallize in the polar space group Cm. In these structures, the I(1) atom exhibits half-occupancy disorder due to its special position on the m plane. One [I(1)O4]3− unit, two [I(2)O4]3− units, and two [I(3)O3]− units are bridged through O atoms, assembling into a novel semicircular [I5O14]3− polyanion (Fig. 8c). These polyanions are further interconnected through [REO8]13− polyhedra, forming 2D 2∞[REI5O14] layers. The overall crystal structure is constructed by the stacking of these layers along the b-axis (Fig. 8b). In contrast, for RE = Er, Tm, and Yb, the compounds crystallize in the polar NCS space group Pmn21. In these structures, all the I atom sites are fully occupied without positional disorder. One [I(1)O4]3− unit bridges two [I(2)O3]− groups to form a chain 1∞[I3O8]− polyanion. Two additional [I(3)O3]− groups are attached to this core via weak I–O interactions, resulting in a [(I3O8)(IO3)2]3− polyanion (Fig. 8d). These polyanions are interconnected through [REO8]13− polyhedra, forming 2D 2∞[REI5O14] layers that stack along the a-axis to generate the overall crystal framework (Fig. 8e). Among these compounds, the maximum SHG response reaches up to 15 × KDP, which primarily originates from the cooperative alignment of local dipole moments within the anionic units. In contrast, ErI5O14 exhibits a significantly weaker SHG efficiency of only 0.7 × KDP, which can be attributed to the competitive d–d and f–f electronic transitions of Er3+ ions that cause absorption of both the input and output light.
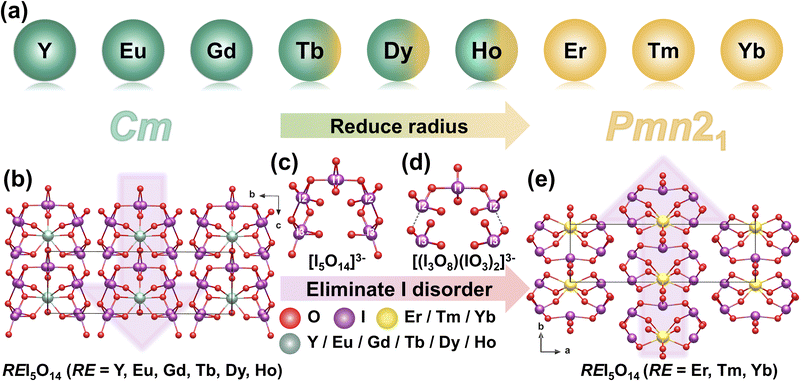 |
| | Fig. 8 (a) Decrease of the rare-earth ionic radius induces a space group transition in REI5O14 from Cm (Y, Eu, Gd, Tb, Dy, and Ho, with atomic disorder) to Pmn21 (Er, Tm, and Yb, disorder-free). (b) View of the REI5O14 (RE = Y, Eu, Gd, Tb, Dy, Ho) structure along the c-axis. (c) [I5O14] polyanion in REI5O14 (RE = Y, Eu, Gd, Tb, Dy, Ho). (d) [(I3O8)(IO3)2] polyanion in REI5O14 (RE = Er, Tm, Yb). (e) View of the REI5O14 (RE = Er, Tm, Yb) structure along the c-axis. Copyright 2024 Royal Society of Chemistry and 2019 Wiley-VCH. | |
2.8 [o-C5H4NHOH]2[I7O18(OH)]·3H2O
[o-C5H4NHOH]2[I7O18(OH)]·3H2O is isolated via slow evaporation of its aqueous precursor solution at room temperature. [o-C5H4NHOH]2[I7O18(OH)]·3H2O crystallizes in the polar space group Ia.44 Its structure features a novel 1D branched polyiodate chain 1∞[I7O18(OH)]2−, extending along the c-axis. This anion consists of seven distinct I atoms; the [I(1)O3(OH)]2−, [I(2)O4]3−, [I(3)O4]3−, and [I(4)O3]− units are interconnected through corner-sharing O atoms to form an [I4O11(OH)]3− tetramer (Fig. 9a). Simultaneously, the [I(5)O4]3−, [I(6)O4]3−, and [I(7)O3]− units are linked via corner-sharing O atoms, resulting in a trimer [I3O9]3− (Fig. 9b). Adjacent [I4O11(OH)]3− tetramers are bridged through I(1)–O(10)–I(4) linkage, forming a 1D [I4O10(OH)]− main chain. The [I3O9]3− trimers are subsequently grafted onto this main chain via I(4)–O(11)–I(5) bridges, collectively constructing the branched polyiodate chain 1∞[I7O18(OH)]2− (Fig. 9c). The 1D chains, aligned along the c-axis, are interconnected with lattice water molecules via hydrogen bonds, forming a 2D supramolecular layer. The protonated (o-C5H4NHOH)+ cations occupy the interlayer spaces, not only balancing the overall charge but also engaging in extensive hydrogen-bonding interactions with the 1∞[I7O18(OH)]2− chains. These hydrogen bonds serve as the primary interlayer interactions, further stabilizing the three-dimensional supramolecular framework (Fig. 9d). The compound exhibits an excellent SHG response (8.5 × KDP), which is attributed to the well-ordered alignment of the highly polarizable [IO3]− and [IO4]3− units within the 1∞[I7O18(OH)]2− polyiodate chain.
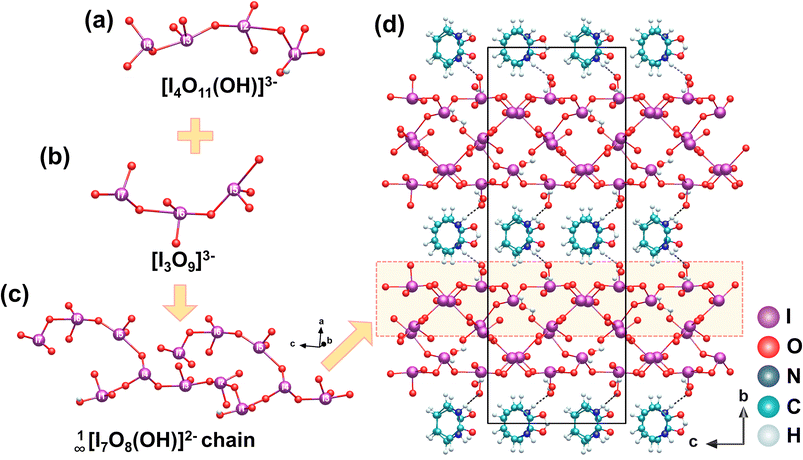 |
| | Fig. 9 (a) The [I4O11(OH)]3− tetramer. (b) The [I3O9]3− trimer. (c) The 1D 1∞[I7O18(OH)]2− branched chain along the c-axis. (d) The structure of [o-C5H4NHOH]2[I7O18(OH)]·3H2O. The hydrogen bonds are drawn as dotted lines. Copyright 2021 Wiley-VCH. | |
3. Aliovalent substitution
To further enhance the SHG performance and enrich the structural diversity of materials, our group has applied the well-established aliovalent substitution strategy to the iodate system, successfully developing a series of new NLO crystals with improved properties. This approach starts with a known parent compound that already exhibits a strong SHG response and then introduces co-substitution at two different anionic sites, or simultaneously at one cationic and one anionic site. Such substitutions effectively optimize the comprehensive performance. This section systematically discusses the key roles of vacancy-site, single-site, dual-site, and multi-site aliovalent substitution in the design of novel NLO crystals. Compounds with notable SHG responses obtained through this strategy are summarized in Table 2.
Table 2 Optical properties of iodates synthesized via aliovalent substitution
| Compounds |
Space group |
Anionic group |
SHG intensity |
Bandgap (eV) |
Birefringence |
PM/NPM |
Ref. |
| LiGa(IO3)4 |
P21 |
2D 2∞[Ga(IO3)6]3− layer |
14 × KDP at 1064 nm |
4.25 |
0.23 at 1064 nm |
PM |
53
|
| LiMg(IO3)3 |
P63 |
3D [Mg(IO3)6]4− framework |
24 × KDP at 1064 nm |
4.34 |
N/A |
PM |
54
|
| CsVO2F(IO3) |
Pna21 |
3D [VO2F(IO3)2]2− framework |
1.1 × KTP at 2.05 µm |
2.39 |
0.04 at 2.05 mm |
PM |
55
|
| α-Ba2[GaF4(IO3)2](IO3) |
Pna21 |
cis-[GaF4(IO3)2]3− and [IO3]− pyramid |
6 × KDP at 1064 nm |
4.61 |
N/A |
PM |
56
|
| β-Ba2[GaF4(IO3)2](IO3) |
P21 |
cis-[GaF4(IO3)2]3− and [IO3]− pyramid |
6 × KDP at 1064 nm |
4.35 |
N/A |
PM |
56
|
| PbFIO3 |
Iba2 |
[IO3]− pyramid |
8 × KDP at 1064 nm |
3.87 |
0.06 at 546.1 nm |
PM |
57
|
3.1 LiGa(IO3)4
Single crystals of LiGa(IO3)4 were obtained via a hydrothermal reaction at 220 °C for 3 days.53 Based on α-LiIO3 (Fig. 10a), a new material, LiGa(IO3)4, was successfully synthesized through partial substitution of Li+ ions by Ga3+ ions. In the crystal structure, Ga atoms coordinate with six [IO3]− groups, forming isolated 0D [Ga(IO3)6]3− units. [Ga(IO3)6]3− units further extend along the ab plane, connecting into a 2D layer 2∞[Ga(IO3)4]−while Li+ ions occupy the interlayer spaces, serving to balance charge (Fig. 10b). The partial substitution of Li+ (0.76 Å) by the smaller Ga3+ (0.62 Å) cation is accompanied by a contraction of the unit cell volume from α-LiIO3 to LiGa(IO3)4. With the number of [IO3]− groups per unit cell identical, the density of polar [IO3]− groups increases from 0.0148 Å−3 in α-LiIO3 to 0.0155 Å−3 in LiGa(IO3)4. As a result, the compound exhibits a strong SHG response of approximately 14 × KDP. Aliovalent substitution proves effective in tuning iodate structures and boosting NLO performance.
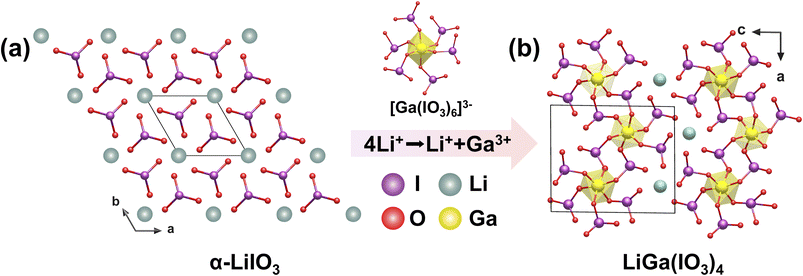 |
| | Fig. 10 (a) The structure of α-LiIO3 on the ab plane. (b) The 2D structure of LiGa(IO3)4 viewed down the b-axis. Copyright 2023 Wiley-VCH. | |
3.2 LiMg(IO3)3
Colorless prismatic crystals of LiMg(IO3)3 (P63) were synthesized via a hydrothermal reaction at 230 °C for 3 days in a dilute HCl medium.54 LiMg(IO3)3 can also be considered to be formed by substituting Ti4+ ions with Mg2+ ions in the parent compound Li2Ti(IO3)6. In the structure of Li2Ti(IO3)6 (P63), Li+ and Ti4+ occupy the 2b and 2a special sites, respectively. When Mg2+ replaces Ti4+, a Wyckoff site exchange occurs: Mg2+ occupies the original Li+ site and forms a [MgO6]10− octahedron. This alters the bonding pattern with the [IO3]− group, enhancing local asymmetric polarization. Concurrently, Li+ migrates to the higher-symmetry 2a site, acting as a counter-cation filling the 1D channels within the 3D framework (Fig. 11a and b). Mg2+ substitution for Ti4+ not only induces positional exchange but also increases the stacking density of [IO3]− groups, exhibiting strong SHG response (24 × KDP) and thereby enhancing effective SHG output.
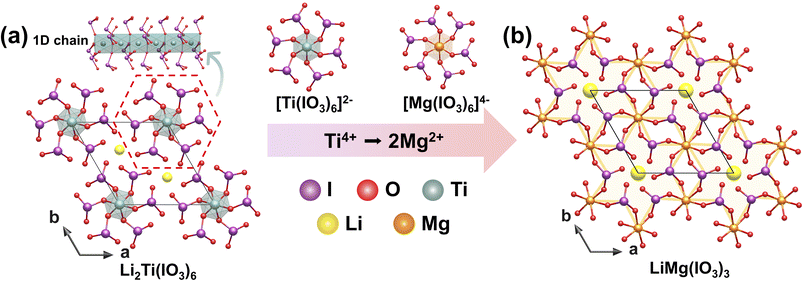 |
| | Fig. 11 Schematic atomic structure evolution from Li2Ti(IO3)6 (a) to LiMg(IO3)3 (b) via aliovalent substitution routes. Copyright 2019 Royal Society of Chemistry. | |
3.3 CsVO2F(IO3)
CsVO2F(IO3) crystals were hydrothermally synthesized at 160 °C for 72 hours.55 Through a dual-site aliovalent substitution strategy (Mo6+ + O2− → V5+ + F−), the [MoO6]6− octahedra in the parent compound CsMoO3(IO3) (Fig. 12a) were replaced by [VO5F]6− octahedra, preserving the original framework structure. In the resulting architecture, each [VO5F]6− octahedron coordinates with two [IO3]− groups, forming a cis-[VO3F(IO3)2]4− anionic unit. These units extend along the c-axis through shared O atoms, constructing a 1D chain structure with [IO3]− groups distributed on both sides. Adjacent chains are interconnected via [IO3]− groups, generating an extended structure with channel features, while Cs+ cations reside within the interchain voids, serving to balance charge and stabilize the overall structure (Fig. 12b). CsVO2F(IO3) exhibits remarkable NLO properties, demonstrating a strong SHG response of 1.1 times that of KTP (absorption edge: 525 nm; Eg = 2.36 eV), comparable to that of the parent compound CsMoO3(IO3) (∼1.2 × KTP). Theoretical analysis reveals that the ordered alignment and distinctive electronic structure of the cis-[VO3F(IO3)2]4− units play a critical role in achieving enhanced SHG efficiency.
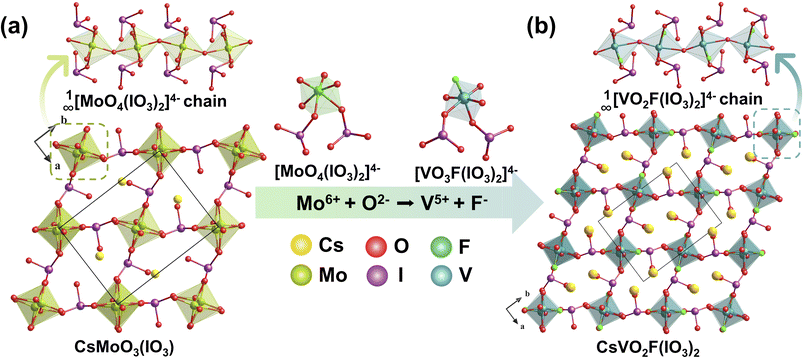 |
| | Fig. 12 Schematic atomic structure evolution from CsMoO3(IO3) (a) to CsVO2F(IO3)2 (b) via aliovalent substitution routes. Copyright 2020 Wiley-VCH. | |
3.4 α/β-Ba2[GaF4(IO3)2](IO3)
The formation of two distinct polymorphs (α/β-Ba2GaF4(IO3)2) under identical hydrothermal conditions is attributed to differences in the initial Ga3+ concentration, which influences the crystallization kinetics and the resulting arrangement of the anionic building units.56 Building upon the previously reported α- and β-Ba2[VO2F2(IO3)2] compounds by Poeppelmeier's group,58 the corresponding α- and β-Ba2[GaF4(IO3)2] phases were successfully synthesized via a three-site substitution strategy (V5+ + 2O2− → Ga3+ + 2F−) (Fig. 13a–d). All four compounds consist of 0D cis-[MOxF4−x(IO3)2]3− (x = 0, 2) anionic units, isolated [IO3]− groups, and Ba2+ counterions. The structural distinction between the α- and β-phases originates from different packing arrangements of the anionic units, resulting in crystallization in the polar space groups Pna21 and P21, respectively. Although the highly distorted [VO4F2]5− (Δd = 1.20 and 1.21) octahedra are replaced by less distorted [GaO2F4]5− (Δd = 0.32 and 0.44) octahedra, the polar topological characteristics of the framework are preserved. Consequently, the SHG responses of α- and β-Ba2[GaF4(IO3)2] are 6 times that of KDP, slightly lower than that of the parent compounds (9 × KDP). In contrast, the band gaps significantly increase to 4.61 eV and 4.35 eV, respectively, substantially wider than those of the parent phases (2.55 eV and 2.59 eV). Aliovalent substitution offers a rational synthetic strategy to effectively tune the structural dimensionality, polar unit density, and electronic configuration of iodates through precise site-specific replacement, thereby yielding crystalline materials with excellent NLO performance.
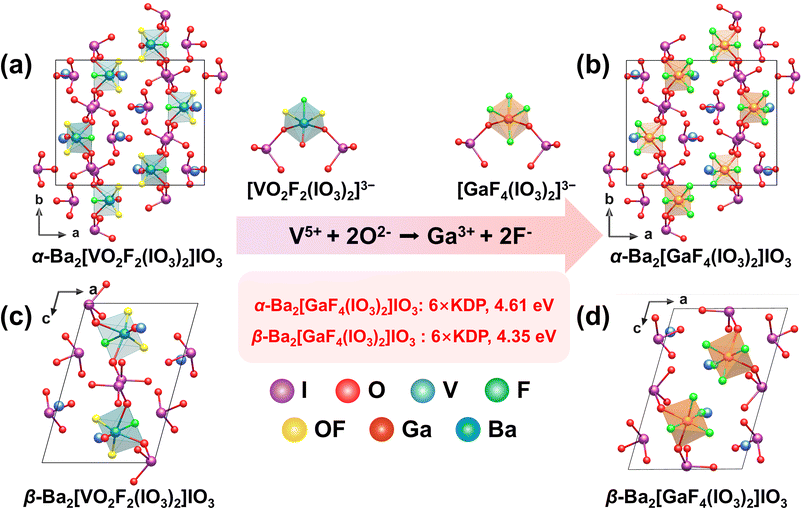 |
| | Fig. 13 Schematic atomic structure evolution from α/β-Ba2[VO2F2(IO3)2]IO3 (a) to α/β-Ba2[GaF4(IO3)2]IO3 (b) via aliovalent substitution routes. Copyright 2018 Wiley-VCH and 2018 American Chemical Society. | |
4. Fluorination substitution
Incorporating I–F bonds into iodates represents an effective strategy for tailoring their optical properties. The high electronegativity of the F atom helps to widen the optical band gap, while the resulting fluoro-iodate anions, such as the [IO3F]2− seesaw,59–62 [IO2F2]− seesaw,63,64 and [IOF4]− square pyramid,65 typically exhibit large polarizability anisotropy, which is conducive to enhancing the materials' SHG response. These anions can further polymerize via O bridges to form various aggregated structures ([I2O5F2]2− dimer, [I2O5F]− dimer, and [I3O7F3]2− timer).59,66 To date, the reported family of fluoroiodates remains relatively limited, with only compounds containing [IO3F]2−, [IO2F2]−, and [I3O7F3]2− confirmed to exhibit SHG activity. This section systematically discusses the structural characteristics and performance of NCS iodates containing these three types of fluoroiodate anionic units. Fluoroiodates with SHG activity are summarized in Table 3.
Table 3 Optical properties of fluoroiodates
| Compounds |
Space group |
Anionic group |
SHG intensity |
Bandgap (eV) |
Birefringence |
PM/NPM |
Ref. |
| [GaF(H2O)][IO3F] |
Pca21 |
Seesaw-shaped [IO3F]2− |
10 × KDP at 1064 nm |
4.34 |
N/A |
PM |
67
|
| CaCe(IO3)3(IO3F)F |
Pna21 |
Seesaw-shaped [IO3F]2− and [IO3]− pyramid |
5 × KDP at 1064 nm |
2.72 |
0.071 at 1064 nm |
PM |
68
|
| Rb2MoO2F3(IO2F2) |
Cmc21 |
[Mo(O2F3)(IO2F2)]2− |
5 × KDP at 1064 nm |
3.77 |
0.217 at 1064 nm |
PM |
69
|
| Cs2MoO2F3(IO2F2) |
Cmc21 |
[Mo(O2F3)(IO2F2)]2− |
4.5 × KDP at 1064 nm |
3.43 |
0.204 at 1064 nm |
PM |
69
|
| Rb2WO2F3(IO2F2) |
Cmc21 |
[W(O2F3)(IO2F2)]2− |
3.8 × KDP at 1064 nm |
4.42 |
0.166 at 1064 nm |
PM |
70
|
| Cs2WO2F3(IO2F2) |
Cmc21 |
[W(O2F3)(IO2F2)]2− |
3.5 × KDP at 1064 nm |
4.29 |
0.137 at 1064 nm |
PM |
70
|
| Cs2VOF4(IO2F2) |
Cmc21 |
[VOF4(IO2F2)]2− |
5 × KDP at 1064 nm |
4.82 |
0.088 at 1064 nm |
PM |
71
|
| RbIO2F2 |
Pca21 |
Seesaw-shaped [IO2F2]2− |
4 × KDP at 1064 nm |
4.2 |
N/A |
PM |
72
|
| CsIO2F2 |
Pca21 |
Seesaw-shaped [IO2F2]− |
3 × KDP at 1064 nm |
4.5 |
0.046 at 1064 nm |
PM |
73
|
| NH4IO2F2 |
Pca21 |
Seesaw-shaped [IO2F2]− |
1.2 × KDP at 1064 nm |
4.53 |
N/A |
NPM |
63
|
| Pb4O(IO3)3(I3O7F3)BF4 |
R3c |
Windmill-shaped [I3O7F3]2− |
4.5 × KDP at 1064 nm |
3.73 |
0.1 at 546.1 nm |
PM |
66
|
| Sr4O(IO3)3(I3O7F3)BF4 |
R3c |
Windmill-shaped [I3O7F3]2− |
3.7 × KDP at 1064 nm |
4.19 |
0.05 at 546.1 nm |
PM |
66
|
4.1 [GaF(H2O)][IO3F]
Wedge-shaped prismatic crystals of [GaF(H2O)][IO3F] were synthesized via a hydrothermal reaction at 230 °C for 67 hours in an HF-containing medium.67 [GaF(H2O)][IO3F] (Pca21) features a salt-inclusion like structure, built from 1D zigzag 1∞[GaF(H2O)]2+ chains and polar [IO3F]2− anions (Fig. 14a). These components assemble into a 2D layer 2∞[GaF(H2O)][IO3F] in the bc plane through Ga–O/F coordination bonds (Fig. 14b), with subsequent –ABAB– stacking along the a-axis, yielding a 3D framework (Fig. 14c). Crucially, the [IO3F]2− units are aligned unidirectionally along the c-axis, leading to constructive superposition of their local dipole moments. This configuration generates strong macroscopic polarity, which results in a remarkable SHG response of 10 × KDP. Moreover, the presence of fluorine atoms contributes to a wide optical bandgap of 4.34 eV, showcasing a valuable synergy between strong NLO activity and high transparency in the high-energy spectral region.
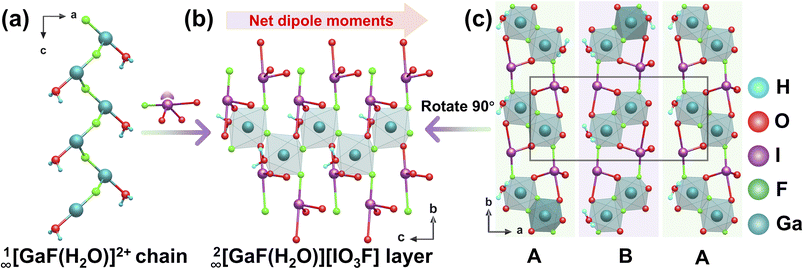 |
| | Fig. 14 (a) The 1∞[GaF(H2O)]2+ chain. (b) The 2∞[GaF(H2O)][IO3F] layer. (c) The structure of [GaF(H2O)][IO3F]. Copyright 2021 Royal Society of Chemistry. | |
4.2
A
2
MO2F3(IO2F2) (A = Cs, Rb; M = Mo, W)
A series of isostructural compounds A2MO2F3(IO2F2) (A = Cs, Rb; M = Mo, W) were synthesized under similar mild hydrothermal conditions (220–230 °C, 4 days), wherein hydrofluoric acid served as both a fluorinating agent and a crucial mineralizer to form the characteristic fluorinated anionic units simultaneously.69,70 These compounds all crystallize in the NCS polar space group Cmc21. Their structures include 0D [M(O2F3)(IO2F2)]2− polyanions (M = Mo, W), formed by combining fluorinated d0 transition metal [MO3F3]3− groups with fluoroiodate [IO2F2]− units through coordination bonds (Fig. 15a and b). In the crystal lattice, these [M(O2F3)(IO2F2)]2− polyanions align along the c-axis with a consistent polar orientation, while alkali metal cations (Cs+/Rb+) occupy interstitial sites to maintain charge neutrality (Fig. 15c). The strong SHG response of these materials results from the combined structure and the aligned arrangement of the [M(O2F3)(IO2F2)]2− anions. The [MO3F3]3− octahedra exhibit significant C4-type angular distortion, causing the M6+ cations to shift toward terminal O atoms and establishing a fixed local polarization direction. Meanwhile, the I5+ centers in the [IO2F2]− units, which possess SCALPs, create inherent asymmetry that further influences and amplifies the distortion direction of nearby [MO3F3]3− octahedra. Through this cooperative interaction, the dipole moments of all structural units add along the crystallographic c-axis, ultimately generating a strong NLO response at the macroscopic level (Fig. 15d).
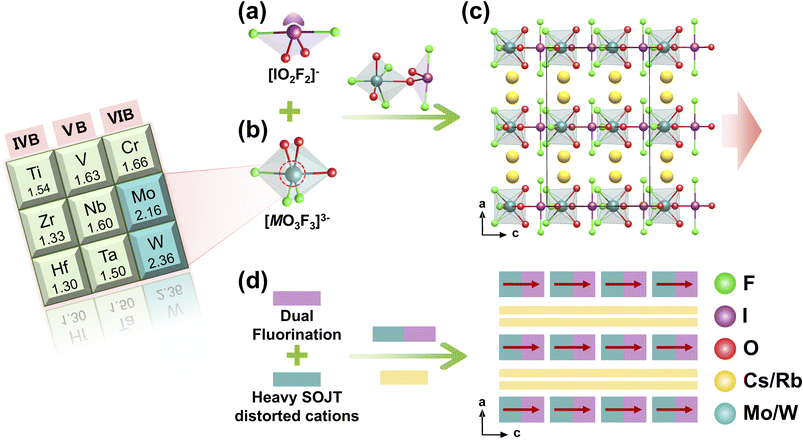 |
| | Fig. 15 (a) The [IO2F2]− polyhedron. (b) The [MO3F3]3− polyhedron. (c) The structure of A2MO2F3(IO2F2); the red arrow in the panel represents the net dipole moment of the structure. (d) Structural building unit description of the arrangement of 0D [MO2F3(IO2F2)]2 units and A+ cations (A = Rb, Cs; M = Mo, W); the red arrow in the panel represents the dipole moment of [M(O2F3)(IO2F2)]2− anions. Copyright 2022 Royal Society of Chemistry and 2021 American Chemical Society. | |
4.3
M
4O(IO3)3(I3O7F3)BF4 (M = Pb, Sr)
Single crystals of M4O(IO3)3(I3O7F3)BF4 (M = Pb, Sr) were synthesized via low-temperature solid-state reactions at 240 °C for 3 days.66M4O(IO3)3(I3O7F3)BF4 crystallizes in the NCS trigonal space group R3c, exhibiting a structurally complex 3D framework assembled from four distinct building units: conventional [IO3]− and [BF4]− anions, [M4O]6+ tetrahedra, and a previously unreported windmill-shaped [I3O7F3]2− fluoroiodate trimer (Fig. 16a–d). Structural analysis reveals that [BF4]− tetrahedra and [M4O]6+ tetrahedra interconnect via M–F and M–O bonds to form a 2D anionic 2∞[M4O(BF4)]5+ layer, characterized by 18 MRs. The [IO3]− and [I3O7F3]2− units occupy the interstitial spaces within these rings (Fig. 16e and f). These hybrid layers subsequently stack along the c-axis and are interconnected via Pb–O bonds, forming a 3D network (Fig. 16g). Detailed polarization analysis indicates that the local dipole moments associated with the [IO3]− and [I3O7F3]2− units exhibit superposition along the crystallographic c-axis, while undergoing substantial mutual cancellation within the ab plane due to their antiparallel arrangements. This anisotropic dipole alignment generates a net macroscopic polarization vector oriented predominantly parallel to the c-axis. Consequently, this NCS polar structure enables excellent comprehensive NLO performance, including strong SHG responses (4 and 3.5 × KDP, respectively) and wide optical bandgaps (3.73 and 4.19 eV), in contrast to Bi4O(I3O10)(IO3)3(SeO4) (in Section 2.5), which crystallizes in the same space group R3c but exhibits a significantly weaker SHG response of only 1.1 × KDP and narrower optical bandgap 3.79 eV. The superior comprehensive performance of M4O(IO3)3(I3O7F3)BF4 can be attributed to three key factors: (i) cation substitution (Pb2+ to Bi3+) modulates the electronic contribution to SHG, with Pb2+ showing a slight advantage over Bi3+ due to its more favorable lone-pair activity; (ii) tetrahedral anion substitution ([BF4]− to [SeO4]2−) preserves the overall framework topology; and (iii) most importantly, fluorine modification plays a dual critical role: it widens the band gap, while simultaneously contributing to the strong SHG response through the formation of asymmetric [I3O7F3]2− units. The SHG enhancement underscores the essential role of fluorine in achieving high-performance NLO materials.
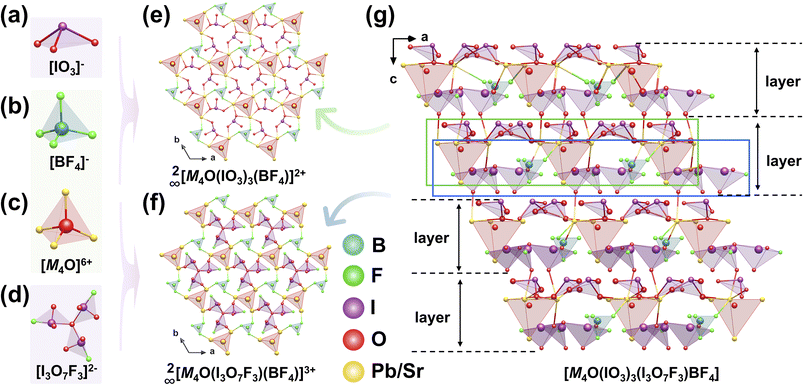 |
| | Fig. 16 (a) The [IO3]− polyhedron. (b) The [BF4]− polyhedron. (c) The [M4O]6+ polyhedron. (d) The [I3O7F3]2− polyhedron. (e) The 2∞[M4O(IO3)3(BF4)]2+ layer. (f) The 2∞[M4O(I3O7F3)(BF4)]3+ layer. (g) The 3D structure of M4O(IO3)3(I3O7F3)BF4 (M = Pb, Sr). Copyright 2021 American Chemical Society. | |
5. Combinations of the iodate groups with the π-conjugated units
π-Conjugated structural units are pivotal building blocks in the design of functional materials, and their essential characteristics can be summarized into two aspects: (i) their planar or nearly planar geometry; (ii) the presence of π-orbital electron clouds oriented perpendicular to the molecular plane. Due to electron delocalization, π-conjugated units exhibit higher hyperpolarizabilities and more pronounced polarizability anisotropy than non-conjugated structural moieties. Introducing π-conjugated groups into crystalline materials allows the synergistic exploitation of their electronic properties, enabling the design and synthesis of novel iodates with both strong SHG and outstanding birefringence. Representative [BO3]3−, [CO3]2−, and [NO3]− all exhibit favorable optical properties, owing to their wide band gaps, moderate birefringence, and good chemical stability.74–76 In recent years, with advances in research on organic-inorganic hybrid materials, the focus has expanded from traditional inorganic anions to organic π-conjugated cationic systems. Due to their larger conjugated systems and tunable molecular structures, these organic cations generally exhibit more pronounced polarizability anisotropy and hyperpolarizabilities compared to inorganic anions. In this section, we discuss five iodates containing representative π-conjugated units, while additional related compounds are summarized in Table 4.
Table 4 Optical properties of iodates containing π-conjugated groups
| Compounds |
Space group |
Anionic group |
SHG intensity |
Bandgap (eV) |
Birefringence |
PM/NPM |
Ref. |
| Na2[B4IO9](IO3) |
Cc
|
2D 2∞[B4IO9]− layer and [IO3]− pyramid |
7.8 × KDP at 1064 nm |
4.3 |
0.298 at 546 nm |
PM |
77
|
| Be2(BO3)(IO3) |
Pmc21 |
[IO3]− pyramid |
7.2 × KDP at 1064 nm |
4.32 |
0.172 at 1064 nm |
PM |
78
|
| Sc(IO3)2(NO3) |
R32 |
[IO3]− pyramid |
4 × KDP at 1064 nm |
4.15 |
0.348 at 546 nm |
PM |
79
|
| [C(NH2)3]2Mo2O5(IO3)4·2H2O |
Pna21 |
Boat-shaped [Mo2O5(IO3)4]2− |
5 × KDP at 1064 nm |
3.55 |
0.059 at 1064 nm |
PM |
80
|
| (C3H7N6)(IO3) |
Cc
|
[IO3]− pyramid |
6.7 × KDP at 1064 nm |
4.35 |
0.16 at 550 nm |
PM |
81
|
| α-(C4H5N2O)(IO3)·HIO3 |
Ia
|
[IO3]− pyramid |
6.4 × KDP at 1064 nm |
3.65 |
0.1 at 546 nm |
PM |
82
|
| (C7H4NO4)(IO3) |
Pna21 |
[IO3]− pyramid |
3.6 × KDP at 1064 nm |
4.12 |
0.35 at 546 nm |
PM |
83
|
| (C3N2I2H3)2Mo2O5(IO3)4·4H2O |
Pna21 |
Boat-shaped [Mo2O5(IO3)4]2− |
3.5 × KDP at 1064 nm |
3.00 |
0.254 at 550 nm |
PM |
84
|
| [C5H6O2N3]2[IO3]2 |
P21 |
[IO3]− pyramid |
2.4 × KDP at 1064 nm |
2.43 |
0.22 at 1064 nm |
PM |
85
|
| C(NH2)3(I3O8)(HI3O8)(H2I2O6)(HIO3)4·3H2O |
P1 |
[I3O8]− trimer |
2.1 × KDP at 1064 nm |
3.89 |
0.059 at 550 nm |
PM |
86
|
| (C3N2H5)2Mo2O5(IO3)4·3H2O |
Ama2 |
Boat-shaped [Mo2O5(IO3)4]2− |
1.3 × KDP at 1064 nm |
3.75 |
0.143 at 550 nm |
PM |
87
|
| Ce(IO3)2(NO3) |
P3121 |
[IO3]− pyramid |
1 × KDP at 1064 nm |
3.14 |
0.039 at 546 nm |
PM |
88
|
| β-(C4H5N2O)(IO3)·HIO3 |
Pca21 |
[IO3]− pyramid |
0.9 × KDP at 1064 nm |
3.65 |
0.19 at 546 nm |
PM |
82
|
5.1 Na2[B4IO9](IO3)
Na2[B4IO9](IO3), which crystallizes in the polar space group Cc with a unique 2D anionic layer framework, was synthesized via a boric acid melting reaction at 230 °C for 5 days.77 The fundamental building block consists of a [B4IO11]5− cluster (Fig. 17a), formed by three planar π-conjugated [BO3]3− groups, one [BO4]5− tetrahedron, and one [IO3]− trigonal pyramid. The borate groups form [B3O7]5− clusters, which are further bridged by additional [BO3]3− groups into a 2D layer, with [IO3]− groups hanging on the same side of the layer via I–O–B bridges, forming a novel 2∞[B4IO9]− layer with distinctive 18 MRs (Fig. 17b and c), creating a highly polarized structural configuration. The layers stack parallel along the c-axis, with the interlayer spaces occupied by Na+ cations and additional isolated [IO3]− groups (Fig. 17d). This well-designed framework enables synergistic alignment of two types of nonlinearly active units ([BO3]3− and [IO3]−), leading to a remarkable SHG response (7.8 × KDP) and an exceptionally large birefringence (0.295 at 546 nm).
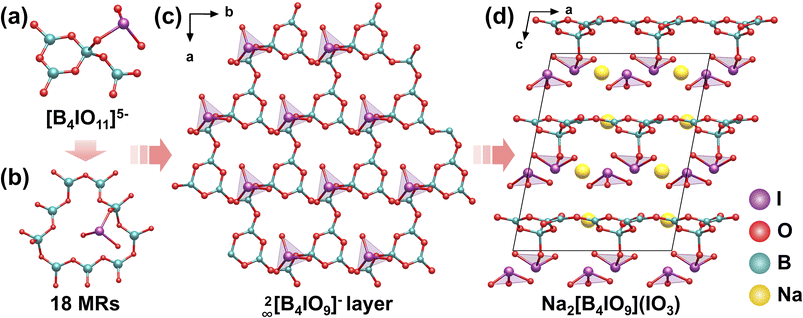 |
| | Fig. 17 (a) The [B4IO11]5− cluster. (b) The 18 MRs. (c) The 2∞[B4IO9]− layer. (d) View of the structure of Na2[B4IO9](IO3) down the b-axis. Copyright 2025 American Chemical Society. | |
5.2 Be2(BO3)(IO3)
Be2(BO3)(IO3) (BBIO) was obtained via a hydrothermal reaction at 200 °C for 5 days. It adopts a KBBF-type 2D layered structure (Fig. 18a), featuring a distinctive [Be2(BO3)(IO3)] composite layer composed of alternating 2∞[Be2BO5]3− layers and [IO3]− units.78 The 2∞[Be2BO5]3− layer maintains the fundamental connectivity of the 2∞[Be2BO3F2]− layer found in KBBF (R32) (Fig. 18d), and the ordered alignment of the [BO3]3− groups allows BBIO to inherit its inherent NLO capability. Unlike the 2∞[Be2BO3F2]− layer in KBBF, however, the 2∞[Be2BO5]3− layer in BBIO has a distinct corrugated morphology. This structural distortion not only creates new opportunities for crystal engineering but also generates undulating interlayer spaces that serve as crucial environments for embedding and arranging the [IO3]− units in an ordered manner. The [IO3]− groups connect to the corrugated 2∞[Be2BO5]3− layer through the terminal O atoms of the [BeO4]6− tetrahedra (Fig. 18b). The folded geometry of this polar borate layer further ensures a consistent polar orientation among all the [IO3]− units (Fig. 18c). At the same time, the corrugated structure permits the dipole moments of the [BeO4]6− tetrahedra to align coherently along the negative c-axis, a sharp contrast to the mutually canceled dipoles of the [BeO3F]5− units in KBBF. As a result, the overall dipole moment from both the [BeO4]6− tetrahedra and the [IO3]− groups adds constructively along the negative c direction, producing a strong SHG response approximately 7.2 times that of the KDP.
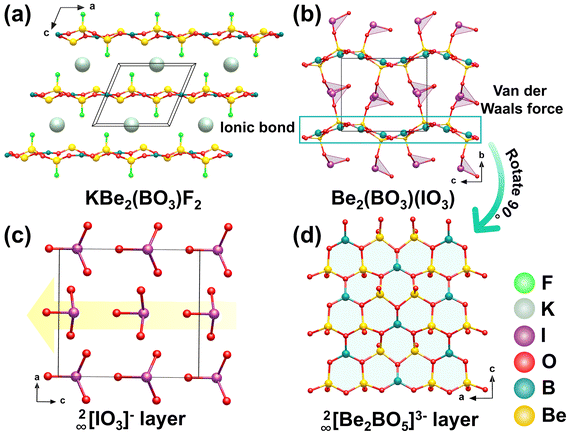 |
| | Fig. 18 Structural evolution from KBe2(BO3)F2 (KBBF) (a) to Be2(BO3)(IO3) (b). The 2∞[IO3]− layers (c) and the 2∞[Be2BO5]3− layers (d) in Be2(BO3)(IO3). The yellow arrow in the panel represents the dipole moment of the 2∞[IO3]− layers in Be2(BO3)(IO3). Copyright 2021 Wiley-VCH. | |
5.3 Sc(IO3)2(NO3)
Colorless block crystals of Sc(IO3)2(NO3) were obtained via a hydrothermal reaction at 160 °C for 3 days in a 50% HNO3 medium. [Sc(IO3)2(NO3)] features a distinctive layered framework, in which honeycomb-type 2∞[Sc(IO3)2] layers and 2D 2∞[NO3]− layers are stacked alternately along the c-axis in an -ABAB- sequence.79 Within the structure, each [ScO6]9− octahedron is connected to six surrounding [IO3]− units through shared O atoms, generating a 2D honeycomb-like network 2∞[Sc(IO3)2] in the ab plane (Fig. 19a–c). Notably, within the 2∞[Sc(IO3)2]+ layer, adjacent [IO3]− groups adopt a face-to-face arrangement across inter-layer gaps, while all [IO3]− units are aligned in a uniform orientation (Fig. 19d). These isolated [NO3]− units are well aligned within the quasi-2D layer (Fig. 19e). This configuration promotes the ordered superposition of anisotropic polarizability tensors and thus contributes to a potentially large birefringence. As a result, the compound exhibits one of the strongest SHG responses among nitrate-based materials (4 × KDP) and a remarkable birefringence of 0.348 at 546 nm, representing the largest value reported for inorganic oxide optical crystals in its class. The strategic fusion of functional groups, each contributing distinct optical advantages, establishes a powerful design paradigm for next generation high-performance nonlinear optical crystals.
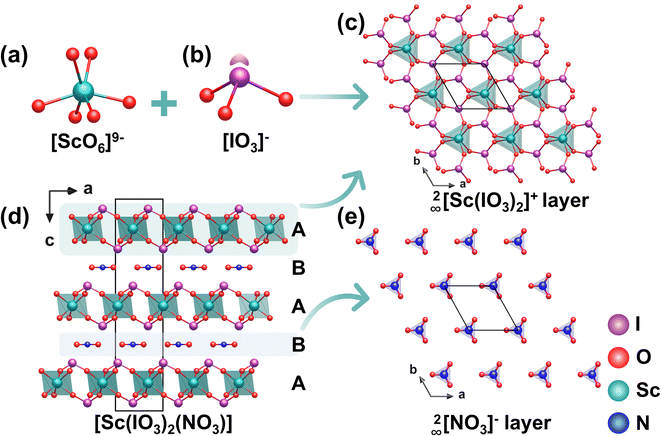 |
| | Fig. 19 (a) The [ScO6]9− octahedron. (b) The [IO3]− trigonal pyramid. (c) The 2∞[Sc(IO3)2]+ layer. (d) Crystal structure of Sc(IO3)2(NO3). (e) The 2D 2∞[NO3]− layer. Copyright 2020 Wiley-VCH. | |
5.4 [C(NH2)3]2Mo2O5(IO3)4·2H2O
[C(NH2)3]2Mo2O5(IO3)4·2H2O was synthesized under mild hydrothermal conditions at 85 °C for 4 days. It crystallizes in the polar orthorhombic space group Pna21.80 Its structure features a novel boat-shaped [Mo2O5(IO3)4]2− polyanionic cluster (Fig. 20b), constructed from two corner-shared [MoO6]6− octahedra and four [IO3]− units. Two [C(NH2)3]+ cations serve not only as charge-balanced counterions but also, through their extensive N–H⋯O hydrogen-bonding network, act as structure directing agents that organize multiple [Mo2O5(IO3)4]2− clusters around them (Fig. 20a and c). More importantly, the planar triangular geometry of the [C(NH2)3]+ cations, combined with steric hindrance and hydrogen-bonding guidance, causes these boat-shaped clusters to adopt a highly parallel alignment. This parallel arrangement allows the local dipole moments from the polar [IO3]− units and distorted [MoO6]6− octahedra within each cluster to add coherently along the c-axis, resulting in strong macroscopic polarization. Through this organic cation-templated, ordered assembly of functional inorganic clusters, the material shows excellent nonlinear optical response, with SHG about 5 times that of KDP. This work demonstrates how the shape and hydrogen-bonding ability of organic cations can be used to precisely control the spatial arrangement of inorganic clusters, improving overall optical properties.
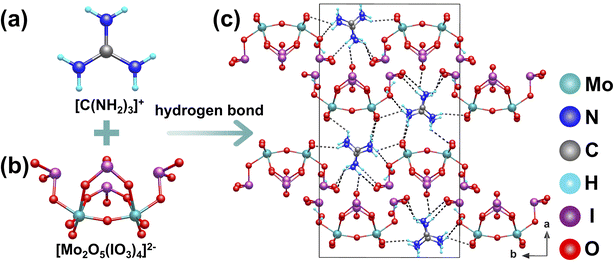 |
| | Fig. 20 (a) The [C(NH2)3]+ units. (b) The [Mo2O5(IO3)4]2− polyanion. (c) The 3D structure of [C(NH2)3]2Mo2O5(IO3)4·2H2O viewed down the c-axis. The black dotted lines represent hydrogen bonds. Copyright 2022 Royal Society of Chemistry. | |
5.5 (C3H7N6)(IO3)
Colorless rod-like crystals were obtained via a low-temperature (85 °C) solution reaction in a H3PO4–H2O mixed medium.81 The crystalline framework of (C3H7N6)(IO3) demonstrates a rational strategy for achieving symmetry breaking through the introduction of an expanded π-conjugated cation. Whereas the parent compound C(NH2)3(IO3) contains a C3-symmetric [C(NH2)3]+ cation forming a CS 3D network via hydrogen bonding (Fig. 21a–c), the protonated melamine cation [C3H7N6]+ exhibits a reduced symmetry from C3 to C2. This molecular-level symmetry reduction triggers a fundamental reconstruction of the hydrogen-bonding pattern: instead of forming a symmetrically compensated hydrogen-bond network, the [IO3]− anions establish directional N–H⋯O hydrogen bonds with specifically oriented N–H groups, achieving an aligned arrangement within the crystal lattice (Fig. 21d–f). The resulting hydrogen-bond reconstruction transforms local polar units into a macroscopically ordered architecture, ultimately accomplishing the transition from a CS to an NCS crystal (Cc) and generating a strong SHG response (6.7 × KDP) and a moderate birefringence (0.16 at 550 nm). This integrated strategy, combining asymmetric planar motifs (or chiral organic) units, constitutes a systematic and effective paradigm for the rational design of NCS iodate materials. The continued development of this molecular-level engineering approach is expected to unlock further structural and functional diversity. By precisely tailoring chiral environments, hydrogen-bonding networks, and spatial organization, future research can aim to effectively enhance the probability of NCS crystallization while optimizing the overall optical performance.
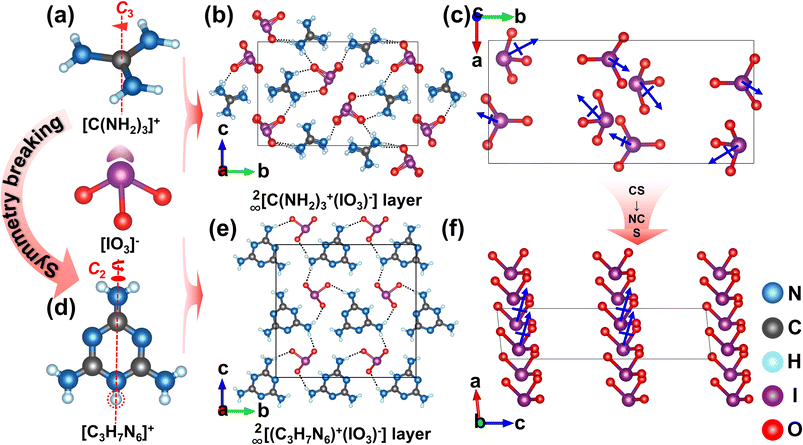 |
| | Fig. 21 (a) The [C(NH2)3]+ cation. (b) H-bond network of C(NH2)3IO3. (c) Arrangement of [IO3]− in C(NH2)3IO3. (d) The [C3H7N6]+ cation. (e) H-bond network of (C3H7N6)(IO3). (f) Arrangement of [IO3]− in (C3H7N6)(IO3). The black dotted lines represent hydrogen bonds; the arrow points in the direction of the dipole moment. Copyright 2025 Springer Link. | |
6. Synergy effects of the iodate groups with tetrahedral functional units
Tetrahedral anionic groups such as [PO4]3−, [SO4]2−, and [SeO4]2− represent an important class of SHG-active units. Despite their relatively low hyperpolarizability and small polarizability anisotropy, their incorporation into iodate systems enables effective synergistic modulation of structural and functional properties. These tetrahedral groups not only enrich the structural chemistry of iodates via their rigid topology and diverse coordination modes but also significantly widen the optical band gap of the resultant compounds, owing to their high bond energy and the presence of highly electronegative atoms. In contrast, iodate units themselves exhibit large hyperpolarizability and pronounced polarizability anisotropy, yet their band gaps are generally narrower. Therefore, the combination of tetrahedral anions with iodate groups creates a complementary system in which the large SHG coefficients and high polarizability anisotropy of iodates are synergistically coupled with the wide band gaps and structure directing capability of the tetrahedral units, leading to optimized overall NLO performance. This section presents representative examples of iodates containing tetrahedral [PO4]3−, [SO4]2−, and [SeO4]2− units, while other SHG-active iodates containing tetrahedral units are summarized in Table 5.
Table 5 Optical properties of iodates containing tetrahedral anionic groups
| Compounds |
Space group |
Anionic group |
SHG intensity |
Bandgap (eV) |
Birefringence |
PM/NPM |
Ref. |
| Nb2O3(IO3)2(SO4) |
P21 |
1D 1∞[Nb2O7(IO3)2]6− chain and [SO4]2− tetrahedron |
6 × KDP at 1064 nm |
3.25 |
0.22 at 1064 nm |
PM |
89
|
| Hg2(IO3)2(SO4)(H2O) |
C2 |
[IO3]− pyramid and [SO4]2− tetrahedron |
6 × KDP at 1064 nm |
3.98 |
0.138 at 546 nm |
PM |
90
|
| AgBi(SO4)(IO3)2 |
P1 |
2D 2∞[Bi(I2O4)O6]7− layer |
3.9 × KDP at 1064 nm |
2.58 |
N/A |
PM |
91
|
| 2D 2∞[Bi(IO3)O5]8− layer |
| [SO4]2− tetrahedron |
| Cd2(IO3)(PO4) |
Fdd2 |
[IO3]− pyramid and [PO4]3− tetrahedron |
4 × KDP at 1064 nm |
4.04 |
N/A |
PM |
92
|
| Cd1.62Mg0.38(IO3)(PO4) |
Fdd2 |
[IO3]− pyramid and [PO4]3− tetrahedron |
3.5 × KDP at 1064 nm |
4.26 |
N/A |
PM |
92
|
| [Ga(OH)2]3(IO3)(SeO4)·H2O |
P63mc |
[IO3]− pyramid and [SeO4]2− tetrahedron |
2.9 × KDP at 1064 nm |
3.38 |
N/A |
PM |
93
|
| [In(OH)2]3(IO3)(SeO4)·H2O |
P63mc |
[IO3]− pyramid and [SeO4]2− tetrahedron |
3.8 × KDP at 1064 nm |
3.38 |
N/A |
PM |
93
|
| Bi4O(I3O10)(IO3)3(SeO4) |
R3c |
[IO3]− pyramid and [SeO4]2− tetrahedron |
1.1 × KDP at 1064 nm |
3.79 |
N/A |
PM |
39
|
| Eu(IO3)(SO4)·3H2O |
P212121 |
[IO3]− pyramid and [SO4]2− tetrahedron |
1.1 × KDP at 1064 nm |
4.42 |
N/A |
NPM |
94
|
| Gd(IO3)(SO4)·3H2O |
P212121 |
[IO3]− pyramid and [SO4]2− tetrahedron |
0.8 × KDP at 1064 nm |
4.55 |
N/A |
NPM |
94
|
| Y(IO3)(SO4)·3H2O |
P212121 |
[IO3]− pyramid and [SO4]2− tetrahedron |
0.7 × KDP at 1064 nm |
4.6 |
0.118 at 532 nm |
PM |
94
|
| Na7(SeO4)3(IO3) |
P212121 |
[IO3]− pyramid and [SeO4]2− tetrahedron |
0.6 × KDP at 1064 nm |
4.57 |
0.087 at 546 nm |
PM |
95
|
| Na7(IO3)(SO4)3 |
P212121 |
[IO3]− pyramid and [SO4]2− tetrahedron |
0.5 × KDP at 1064 nm |
4.83 |
0.087 at 546 nm |
PM |
96
|
| Ln(IO3)(SeO4)(H2O)2·H2O |
P212121 |
[IO3]− pyramid and [SeO4]2− tetrahedron |
0.05–0.7 × KDP at 1064 nm |
N/A |
N/A |
N/A |
97
|
| (Ln = Gd, Dy, Ho, Er, Tm, Yb, Lu, Y) |
6.1 Nb2O3(IO3)2(SO4)
Nb2O3(IO3)2(SO4) was obtained through a two-step hydrothermal synthesis, using the intermediate Nb2O4(SO4) (prepared at 230 °C) as the precursor in a subsequent reaction at 210 °C.89 In the structure of Nb2O3(IO3)2(SO4), two Nb-centered octahedra share O atoms to form a [Nb2O9]8− unit (Fig. 22a). [IO3]− groups are attached to both sides of these chains, collectively constituting the [Nb2O7(IO3)2]6− structural units that extend along the chain direction (Fig. 22b). These chains are further interconnected via bridging O atoms, resulting in a 2D 2∞[Nb2O3(IO3)2]2+ layer (Fig. 22c). Isolated [SO4]2− tetrahedra occupy the interlayer spaces, functioning to balance charge and stabilize the framework (Fig. 22d). Given that the polarization effects of the [IO3]− units largely cancel each other out within the arrangement, the macroscopic second-order NLO response primarily originates from the superposition of dipole moments generated by the highly distorted [NbO6]7− octahedra. As a result, Nb2O3(IO3)2(SO4) exhibits a moderate SHG response, approximately 6 times that of KDP.
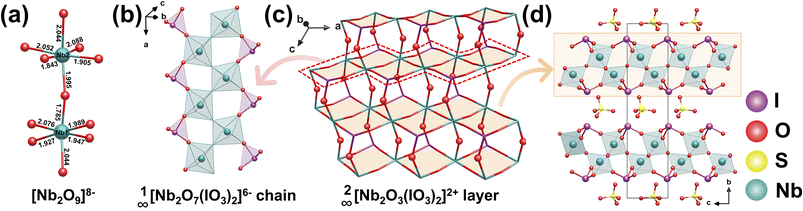 |
| | Fig. 22 (a) The [Nb2O9]8−. (b) The 1∞[Nb2O7(IO3)2]6− chain. (c) The 2∞[Nb2O3(IO3)2]2+ layer. (d) The structure of Nb2O3(IO3)2(SO4). Copyright 2019 Wiley-VCH. | |
6.2 Hg2(IO3)2(SO4)(H2O)
Hg2(IO3)2(SO4)(H2O) was synthesized via a hydrothermal reaction at 180 °C for 2 days.90 Its structure features two distinct types of layers: a 2∞[Hg(IO3)(H2O)]+ layer parallel to the ab plane (Fig. 23c), in which Hg2+ is coordinated by [IO3]− anions and water molecules, and a separate [SO4]2− anionic layer (Fig. 23a) intercalated between the Hg-iodate layers. These [SO4]2− layers are located in the interlayer region and interconnect the 2∞[Hg(IO3)(H2O)]+ layers via weak interactions, forming a 3D framework (Fig. 23b).The key structural units-polar [IO3]− groups, distorted [HgO8]6− polyhedra, and [SO4]2− tetrahedra with induced dipole components-all possess net dipole moments aligned along the b-axis. This coherent alignment leads to a strong SHG response of 6 × KDP, demonstrating that the ordered arrangement and synergistic interaction of multiple polar building blocks are crucial for achieving enhanced NLO performance.
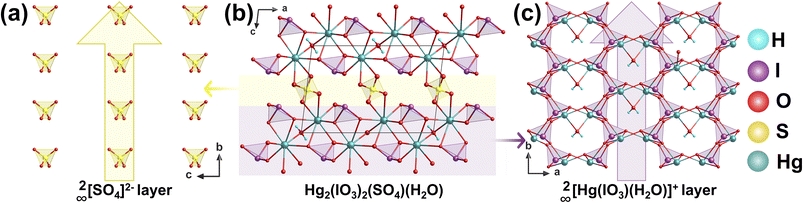 |
| | Fig. 23 (a) The 2∞[SO4]2− layer. (b) View of the structure of Hg2(IO3)2(SO4)(H2O) along the b-axis. (c) The 2∞[Hg(IO3)(H2O)]+ layer. Yellow and purple arrows indicate the polarity directions of [SO4]2−, [HgO8]14− and [IO3]−. Copyright 2022 Wiley-VCH. | |
6.3 AgBi(SO4)(IO3)2
AgBi(SO4)(IO3)2 was obtained via a hydrothermal reaction in a H2SO4 medium at 230 °C for 4 days.91 Its structure is constructed from Bi-centered layers through two distinct coordination modes. The [Bi(1)O8]13− polyhedra share O atoms with adjacent [IO3]− groups, generating a 2∞[Bi(1)(I2O4)O6]7− layer (Fig. 24a). Meanwhile, the [Bi(2)O8]13− polyhedra are corner-shared with [IO3]− units, forming a 2∞[(Bi(2)(IO3)O5]8− layer (Fig. 24c). These two types of layers are further interconnected via shared [IO3]− groups, assembling into 2∞[Bi2I4O15]4− bilayer units (Fig. 24b). These bilayer units are bridged along the c-axis by tetrahedral [SO4]2− anions, resulting in a 3D framework. Two Ag+ ions occupy the inter-bilayer spaces. The NLO properties of this compound originate predominantly from the polar [Bi2I4O15] bilayers, within which the dipole moments of the polar [IO3]− units are cooperatively aligned along the b-axis. This results in a macroscopically significant SHG effect, with an intensity 3.9 times that of KDP. The structure clearly illustrates how the assembly of layered polar units and 3D connections can achieve effective dipole summation and optimize NLO performance.
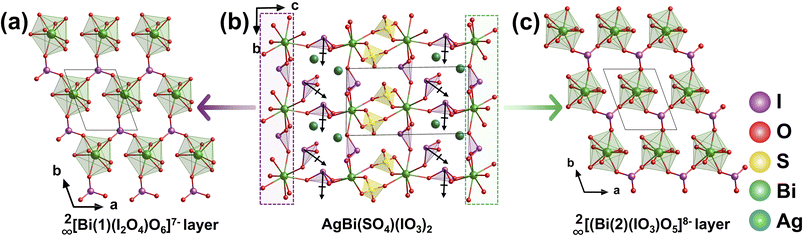 |
| | Fig. 24 (a) The 2∞[Bi(1)(I2O4)O6]7− layer. (b) The 3D framework of AgBi(SO4)(IO3)2. (c) The 2∞[Bi(2)(IO3)O5]8− layer. The black arrows indicate the dipole moment of [IO3]−. Copyright 2021 Royal Society of Chemistry. | |
6.4 CdxMg2−x(IO3)(PO4) (x = 2, 1.62)
The isostructural compounds Cd2(IO3)(PO4) and Cd1.62Mg0.38(IO3)(PO4), which crystallize in the NCS space group Fdd2, were obtained via hydrothermal reactions at 230 °C for 5 days.92 Using Cd2(IO3)(PO4) as an example, each Cd atom is coordinated by six O atoms from four [PO4]3− tetrahedra and two [IO3]− trigonal pyramids, forming a [CdO6] octahedron.
From a topological perspective, the 3D framework can be rationalized as an assembly of tetranuclear Cd4 cores, interconnected through [IO3]− and [PO4]3− bridging units. Each [IO3]− group links three adjacent Cd4 cores (Fig. 25a), whereas each [PO4]3− group connects four such cores (Fig. 25b). In turn, every Cd4 core is surrounded by eight [IO3]− and six [PO4]3− groups, which together generate Cd4(IO3)8/4(PO4)6/3 building units via corner- and edge-sharing (Fig. 25c). These units propagate in three dimensions to yield the complete polar framework (Fig. 25d). Notably, these materials exhibit strong SHG responses, measuring approximately 4 and 3.5 × KDP for Cd2(IO3)(PO4) and Cd1.62Mg0.83(IO3)(PO4), respectively.
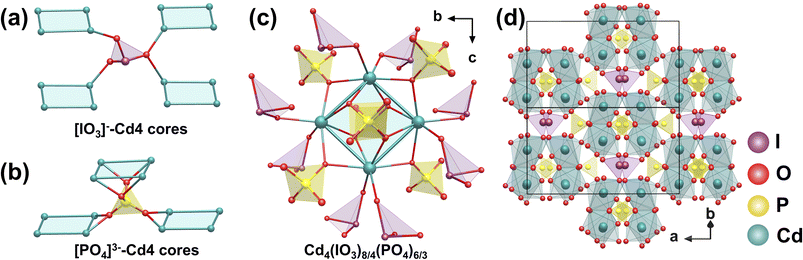 |
| | Fig. 25 (a) The group formed by [IO3]− and “Cd4 cores”. (b) The group formed by [PO4]3− and “Cd4 cores”. The Cd4(IO3)8/4(PO4)6/3 group (c) and the structure of Cd2(IO3)(PO4) along the diagonal direction (d). The blue lines do not represent the bonds, which could show the structure better. Copyright 2022 Royal Society of Chemistry. | |
6.5 [M(OH)2]3(IO3)(SeO4)·H2O (M = Ga, In)
[M(OH)2]3(IO3)(SeO4)·H2O (M = Ga, In) were synthesized via hydrothermal reactions at 230 °C for 2 days in a highly acidic H2SeO4 medium using Bi2O3 as a mineralizer. The compounds exhibit isostructural crystal structures. [MO2(OH)4]5− octahedra form trinuclear clusters [M3O6(OH)9]12− by sharing (OH)− ions (Fig. 26a).93 These clusters further interconnect via bridging O atoms, extending into a 2D layer 2∞[MO2(OH)2]3−. These layers exhibit a unique pore system featuring 3 MRs and 6 MRs. The [IO3]− and [SeO4]2− anions are uniformly distributed within the 3 MRs of the layer (Fig. 26b). The 2∞[MO2(OH)2]3− layers stack alternately along the c-axis in an -ABAB- sequence, with [IO3]− and [SeO4]2− groups positioned on opposite sides of each layer. The interlayer regions are occupied by lattice water molecules (Fig. 26c). Structural analysis reveals that the introduction of [SeO4]2− tetrahedra significantly regulates the arrangement of [IO3]− units, positioning them on the same side of the layer and orienting their intrinsic dipole moments into a common direction. This ordered arrangement enables effective synergistic superposition of the dipole contributions from each polar unit, resulting in these compounds reaching 2.9 and 3.8 times that of KDP.
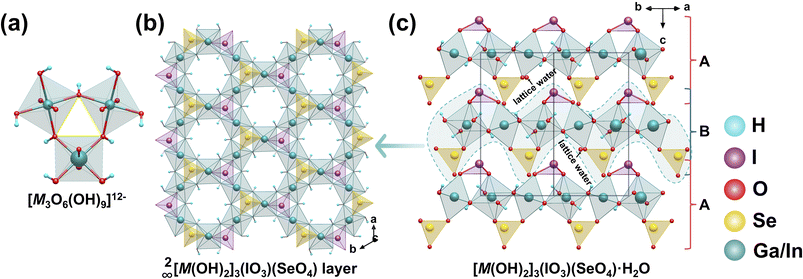 |
| | Fig. 26 (a) The [Ga3O6(OH)9]12−. (b) The 2∞[Ga(OH)2]3(IO3)(SeO4) layer. (c) The structure of [Ga(OH)2]3(IO3)(SeO4)·H2O. The H atoms of the lattice water molecules have been omitted for clarity. Copyright 2023 Royal Society of Chemistry. | |
7 Conclusions and perspectives
Based on a systematic review of recent synthetic approaches for novel iodates, this article highlights five representative strategies and their key advances in the field. From the existing research, the following general patterns and developing trends can be summarized:
(1) The polymerization of iodate groups can effectively increase the local density of lone-pair electrons within the unit cell, thereby significantly improving the likelihood of the NCS structure formation. This offers a vital chemical and structural foundation for designing and synthesizing polyiodates with exceptional NLO performance.
(2) Aliovalent substitution serves as an effective chemical modification strategy. Replacing specific structural units in the parent compound, including single-site, dual-site, multi-site, and vacancy positions, enables performance-oriented controllable synthesis and allows the fine-tuning of multiple key properties, such as NLO coefficients, band gap, and LIDTs.
(3) Introducing planar π-conjugated groups such as [BO3]3−, [NO3]−, and [C3H7N6]+ into iodate systems not only greatly enriches their structural chemistry diversity but also synergistically enhances the SHG efficiency and birefringence through improved electron delocalization and anisotropic polarizability.
(4) Incorporating tetrahedral units such as [PO4]3−, [SO4]2−, and [SeO4]2− into iodate primarily optimizes the optical-gap-related properties. The high bond energy of tetrahedral units and the high electronegativity of the atoms work cooperatively to promote a blue shift in the UV absorption edge and widen the optical band gap.
(5) The high electronegativity of the F atom contributes markedly to broadening the optical band gap of materials. Meanwhile, fluoroiodate anions such as [IO3F]2−, [IO2F2]−, [IOF4]−, and [IO2F4]3− generally exhibit excellent polarizability anisotropy, which favors the synergistic enhancement of the NLO response.
The analysis presented above highlights several general trends in the field and suggests promising avenues for future research:
(i) Expanding systems with π-conjugated units. To date, only two boroiodates have been reported, indicating significant limitations in current synthetic methodologies. Future efforts should expand the range of reaction media, including boric acid melting, solvothermal, and ionic-liquid techniques, to overcome the constraints of conventional synthesis. Given the diverse coordination geometries and marked polarizability anisotropy of B–O units, novel boroiodates are anticipated to feature structural innovation and breakthroughs in nonlinear optical performance. Research on organic–inorganic hybrid metal iodates remains in its early stages, with extensive room for exploration. The designability and diversity of organic components offer a critical opportunity for property modulation. By rationally selecting organic functional units with high polarizability anisotropy and large hyperpolarizability, hybrid materials can be directionally constructed that combine strong SHG effects with large birefringence, thereby advancing their potential applications in integrated photonics and frequency conversion.
(ii) Halogen-substituted iodates. Halogenated iodates represent a field with considerable room for exploration. Current research on fluoroiodates remains limited, with only eleven compounds reported to exhibit SHG activity, suggesting ample potential for structural diversification and performance tuning. The fact that all reported chloroiodates are SHG-inactive provides a clear direction: to expand research into this class and activate their NLO responses through structural design. For example, introducing chiral organic moieties effectively directs the crystallization of the material into the NCS space group. Bromoiodates remain unreported, which can be attributed to the thermodynamic instability associated with substituting the stronger I–O bonds with weaker I–Br bonds during synthesis. It is conceivable that iodate and linear units ([ICl2]−, [IBr2]−, and [I3]−) could be integrated within a single compound, creating a mixed-valence system. Such compositional and structural diversity may offer new avenues for tuning physical properties. This possibility remains largely unexplored and thus warrants systematic studies on how halogen substitution affects structure, polarization, and NLO performance. This research direction is expected to enrich iodate NLO materials.
(iii) Extended range of aliovalent substitutions. Current studies on anionic site substitution focus primarily on the replacement of O2− by F−. Future research should focus on substituting with halide ions of higher polarizability, such as Cl− and Br−. Such substitutions are expected to enhance the microscopic polarizability and macroscopic NLO response of iodates, while retaining structural stability, providing promising strategies for developing novel iodate materials with enhanced SHG performance.
(iv) Although hydrothermal methods remain the dominant synthetic route for constructing iodate systems, supplemented by boric acid melting and low-temperature solid-state reactions, the synthetic strategies are still limited. In the future, the systematic introduction of non-conventional approaches such as medium/low-temperature vacuum solid-state synthesis could break through the constraints of existing pathways, challenge traditional understanding of iodate structural formation, and thereby enable the discovery of iodate materials with novel topologies and functional properties, further enriching their structural chemistry.
(v) Enhanced integration of theoretical calculation and experimental synthesis. Strengthening the synergy between theory and experiment is crucial for advancing the field. Current experimental research still relies heavily on trial-and-error approaches, with limited controllability and goal-orientation in synthetic pathways. Therefore, it is necessary to establish a combined research paradigm that integrates dedicated iodate material databases, theoretical prediction, and high-throughput screening. Such databases would systematically catalogue known and hypothetical iodate structures, enabling the efficient evaluation of key properties-such as thermodynamic stability, NLO coefficients, and band gaps-and thereby guiding the targeted design and synthesis of materials. Deepening interdisciplinary collaboration between theoretical and synthetic chemists will facilitate a paradigm shift from “trial-and-error” exploration to “database-assisted, prediction-guided” research, accelerating the discovery and development of high-performance iodate NLO crystals.
(iv) Growth of large single crystals. The growth of large single crystals is a critical step toward the practical application of iodate NLO materials. Current studies are predominantly limited to microcrystalline or small-sized crystals, yet device-level applications demand bulk crystal dimensions. Many key optical properties, including SHG coefficients, refractive indices, and LIDTs, are measured on small crystals, and the accuracy and reliability of such measurements are often constrained by sample dimensions. To obtain practically meaningful performance data, it is necessary to grow high-quality single crystals on the millimeter to centimeter scale and systematically evaluate their optical homogeneity and NLO efficiency. Only after acquiring these key parameters can the practical potential of a material for applications such as laser frequency conversion and electro-optic modulation be accurately assessed, thereby bridging the gap between fundamental research and functional implementation in iodate systems. For the growth of large single crystals, iodates decompose before melting and are unsuitable for high-temperature melt techniques. As a result, they must be grown exclusively in low-to-medium temperature solution environments, including slow evaporation and low-temperature hydrothermal techniques, which have been widely employed for growing high-quality NLO crystals such as KDP and KTP.98,99 In both methods, the use of seed crystals is essential to guide nucleation and promote the growth of large, high quality single crystals. Crystal growth requires sufficient solubility of the compound in water or iodic acid solutions. Therefore, alkali metal iodates such as α-LiIO3, KIO3, and NaI3O8 are the most suitable for large crystal growth because they combine high solubility with well-established growth protocols. In contrast, transition metal iodates generally exhibit lower solubility, making crystal growth more challenging. Nevertheless, under carefully optimized hydrothermal conditions, compounds with sufficient solubility can still yield crystals of appreciable size. For example, NaVO2(IO3)2(H2O) has been successfully grown as single crystals via hydrothermal methods,100 demonstrating that crystal growth of transition metal iodates is achievable, albeit with greater complexity than their alkali metal counterparts.
Author contributions
YW Kang: investigation, visualization, writing – original draft & review & editing. CC Zhang: investigation. CL Hu: conceptualization, writing – review & editing. JG Mao: funding acquisition, supervision, conceptualization, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (22375201 and 22031009) and the Self-deployment Project Research Program of Haixi Institutes, Chinese Academy of Sciences (CXZX-2022-GH06).
Notes and references
- D. Cyranoski, Nature, 2009, 457, 953–956 CrossRef CAS PubMed.
- L. H. Nicholls, F. J. Rodríguez-Fortuño, M. E. Nasir, R. M. Córdova-Castro, N. Olivier, G. A. Wurtz and A. V. Zayats, Nat. Photonics, 2017, 11, 628–633 CrossRef CAS.
- L. Wu, S. Patankar, T. Morimoto, N. L. Nair, E. Thewalt, A. Little, J. G. Analytis, J. E. Moore and J. Orenstein, Nat. Phys., 2017, 13, 350–355 Search PubMed.
- S. Y. Niu, G. Joe, H. Zhao, Y. C. Zhou, T. Orvis, H. X. Huyan, J. Salman, K. Mahalingam, B. Urwin, J. B. Wu, Y. Liu, T. E. Tiwald, S. B. Cronin, B. M. Howe, M. Mecklenburg, R. Haiges, D. J. Singh, H. Wang, M. A. Kats and J. Ravichandran, Nat. Photonics, 2018, 12, 392–396 CrossRef CAS.
- J. Huang, A. Abudurusuli, Z. Yang and S. Pan, Small, 2025, 21, 2409997 CrossRef CAS.
- X. Dong, L. Huang and G. Zou, Eur. J. Inorg. Chem., 2025, 28, e2500302 Search PubMed.
- T. T. Tran, J. Young, J. M. Rondinelli and P. S. Halasyamani, J. Am. Chem. Soc., 2017, 139, 1285–1295 CrossRef CAS PubMed.
- J. D. Bierlein and H. Vanherzeele, J. Opt. Soc. Am. B, 1989, 6, 622–633 CrossRef CAS.
- Y. S. Liu, W. B. Jones and J. P. Chernoch, Appl. Phys. Lett., 1976, 29, 32–34 CrossRef CAS.
- C. T. Chen, B. C. Wu, A. D. Jiang and G. M. You, Sci. Sin B, 1985, 15, 235–243 Search PubMed.
- A. O. Okorogu, S. B. Mirov, W. Lee, D. I. Crouthamel, N. Jenkins, A. Y. Dergachev, K. L. Vodopyanov and V. V. Badikov, Opt. Commun., 1998, 155, 307–312 CrossRef CAS.
- G. D. Boyd, E. Buehler and F. G. Storz, Appl. Phys. Lett., 1971, 18, 301–304 CrossRef CAS.
- G. Boyd, H. Kasper, J. McFee and F. Storz, IEEE J. Quantum Electron., 1972, 8, 900–908 CrossRef CAS.
- K. L. Vodopyanov, F. Ganikhanov and J. P. Maffetone, Opt. Lett., 2000, 11, 841–843 CrossRef PubMed.
- P. Wang, Y. Chu, A. Tudi, C. Xie, Z. Yang, S. Pan and J. Li, Adv. Sci., 2022, 9, 2106120 CrossRef CAS PubMed.
- A. Abudurusuli, J. Huang, P. Wang, Z. Yang, S. Pan and J. Li, Angew. Chem., Int. Ed., 2021, 60, 24131–24136 CrossRef CAS PubMed.
- D. Gao, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Chin. J. Struct. Chem., 2023, 42, 100014 Search PubMed.
- Y. Kang, C. Yang, J. Gou, Y. Zhu, Q. Zhu, W. Xu and Q. Wu, Angew. Chem., Int. Ed., 2024, 63, e202402086 CrossRef CAS PubMed.
- C. Jin, Y. Li, J. S. Kim, J. Lim, H. Huang, C. Chen, J. Lee, Y. Heo, B. Zhang, J. I. Jang and K. M. Ok, Angew. Chem., Int. Ed., 2025, 64, e202512618 CrossRef CAS PubMed.
- Y. Kang and Q. Wu, Coord. Chem. Rev., 2024, 498, 215458 CrossRef CAS.
- F. Liang, K. Wu, H. Yu, D. Lu, Y. Wang, X. Li and H. Zhang, Angew. Chem., Int. Ed., 2025, 64, e202503341 CrossRef PubMed.
- D.-D. Zhou, C.-L. Hu, X.-W. Zhang, J.-G. Mao and F. Kong, Chem. Sci., 2024, 15, 19920–19927 RSC.
- C. Yang, Y. Kang, X. Wang, J. Gou, Y. Xiong, Z. Zhu, L. Chen and Q. Wu, Chem. Sci., 2024, 15, 15725–15730 RSC.
- F. Chen, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Chem. Sci., 2025, 16, 2015–2023 RSC.
- C.-L. Hu and J.-G. Mao, Coord. Chem. Rev., 2015, 288, 1–17 CrossRef CAS.
- S.-P. Guo, Y. Chi and G.-C. Guo, Coord. Chem. Rev., 2017, 335, 44–57 CrossRef CAS.
- J. Chen, C.-L. Hu, F. Kong and J.-G. Mao, Acc. Chem. Res., 2021, 54, 2775–2783 CrossRef CAS PubMed.
- F. Hou, D. Mei, M. Xia and Y. Wu, Coord. Chem. Rev., 2021, 444, 214038 CrossRef CAS.
- M. Yan, H.-G. Xue and S.-P. Guo, Cryst. Growth Des., 2021, 21, 698–720 CrossRef CAS.
- P.-F. Li, J.-G. Mao and F. Kong, Mater. Today Phys., 2023, 37, 101197 CrossRef CAS.
- Z. Bai and K. M. Ok, Coord. Chem. Rev., 2023, 490, 215212 CrossRef CAS.
- T. Wu, J. Liu, M. Liu, X. Li, M. Zhang, W. Cao, J. Gu and D. Wang, Coord. Chem. Rev., 2026, 549, 217347 CrossRef CAS.
- F.-F. Mao, C.-L. Hu, J. Chen, B.-L. Wu and J.-G. Mao, Inorg. Chem., 2019, 58, 3982–3989 CrossRef CAS PubMed.
- X. Xu, B.-P. Yang, C. Huang and J.-G. Mao, Inorg. Chem., 2014, 53, 1756–1763 CrossRef CAS PubMed.
- J. Chen, P. Yang, H. Yu, Z. Hu, J. Wang, Y. Wu and H. Wu, ACS Mater. Lett., 2023, 5, 1665–1671 CrossRef CAS.
- M.-Q. Lin, M.-F. Duan, J.-G. Mao and B.-P. Yang, New J. Chem., 2025, 49, 6103–6108 RSC.
- T. Abudouwufu, M. Zhang, S. Cheng, H. Zeng, Z. Yang and S. Pan, Chem. Mater., 2020, 32, 3608–3614 CrossRef CAS.
- X. Xu, C.-L. Hu, B.-X. Li, B.-P. Yang and J.-G. Mao, Chem. Mater., 2014, 26, 3219–3230 CrossRef CAS.
- F.-F. Mao, J.-Y. Hu, B.-X. Li and H. Wu, Dalton Trans., 2020, 49, 15597–15601 RSC.
- M.-L. Liang, M. Lacroix, C. Tao, M. J. Waters, J. M. Rondinelli and P. S. Halasyamani, Inorg. Chem., 2023, 62, 2942–2950 CrossRef CAS PubMed.
- G. L. Murphy, Y. Wang, P. Kegler, Y. Wang, S. Wang and E. V. Alekseev, Chem. Commun., 2021, 57, 496–499 RSC.
- Z. Li, Y. Shui, Z. Liang, L. Liu, J. Wan, X. Jiang, Z. Lin and H. Liu, Chem. Mater., 2024, 36, 10351–10360 CrossRef CAS.
- J. Chen, C. Hu, F. Mao, B. Yang, X. Zhang and J. Mao, Angew. Chem., Int. Ed., 2019, 58, 11666–11669 CrossRef CAS PubMed.
- Q.-Q. Chen, C. -L Hu, J. Chen, Y.-L. Li, B.-X. Li and J.-G. Mao, Angew. Chem., Int. Ed., 2021, 60, 17426–17429 CrossRef CAS PubMed.
- C. Huang, C.-L. Hu, X. Xu, B.-P. Yang and J.-G. Mao, Inorg. Chem., 2013, 52, 11551–11562 CrossRef CAS PubMed.
- X. Xu, C. Hu, B. Li and J. Mao, Chem. Eur J., 2016, 22, 1750–1759 CrossRef CAS PubMed.
- C.-F. Sun, C.-L. Hu, X. Xu and J.-G. Mao, Inorg. Chem., 2010, 49, 9581–9589 CrossRef CAS PubMed.
- M.-Q. Lin, C.-L. Hu, M.-F. Duan, B.-X. Li, J.-G. Mao and B.-P. Yang, Angew. Chem., Int. Ed., 2025, 64, e202504673 CrossRef CAS PubMed.
- N. Ma, J. Chen, B. Li, C. Hu and J. Mao, Small, 2023, 19, 2304388 CrossRef CAS PubMed.
- J. Chen, Q.-Q. Chen, F.-F. Mao, Z. Liu, B.-X. Li, X.-H. Wu and K.-Z. Du, Inorg. Chem. Front., 2022, 9, 5917–5925 RSC.
- D. Yan, M.-M. Ren, F.-F. Mao, Y. Ma, R.-L. Tang, B. Zhang, Y. Ma, X.-D. Zhang and S.-F. Li, Inorg. Chem., 2023, 62, 1323–1327 CrossRef CAS PubMed.
- S. K. Kutz and T. T. Perry, J. Appl. Phys., 1968, 39, 3798–3813 CrossRef.
- S. Yang, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, Small, 2024, 20, 2306459 CrossRef CAS PubMed.
- J. Chen, C.-L. Hu, F.-F. Mao, X.-H. Zhang, B.-P. Yang and J.-G. Mao, Chem. Sci., 2019, 10, 10870–10875 RSC.
- J. Chen, C. Hu, X. Zhang, B. Li, B. Yang and J. Mao, Angew. Chem., Int. Ed., 2020, 59, 5381–5384 CrossRef CAS PubMed.
- J. Chen, C. Hu, F. Mao, J. Feng and J. Mao, Angew. Chem., Int. Ed., 2019, 58, 2098–2102 CrossRef CAS PubMed.
- Y. Xu, C. Lin, D. Zhao, B. Li, L. Cao, N. Ye and M. Luo, Scr. Mater., 2022, 208, 114347 CrossRef CAS.
- H. Yu, M. L. Nisbet and K. R. Poeppelmeier, J. Am. Chem. Soc., 2018, 140, 8868–8876 CrossRef CAS PubMed.
- L. Zhu, M. Gai, W. Jin, Y. Yang, Z. Yang and S. Pan, Inorg. Chem. Front., 2021, 8, 3127–3133 RSC.
- M. Gai, T. Tong, Y. Wang, Z. Yang and S. Pan, Chem. Mater., 2020, 32, 5723–5728 CrossRef CAS.
- Y.-H. Wang, F.-Y. Li, D.-X. Jiao, Q. Wei, L. Wei and G.-Y. Yang, Inorg. Chem., 2023, 62, 17691–17696 CrossRef CAS PubMed.
- D. Wang, P. Gong, X. Zhang, Z. Lin, Z. Hu and Y. Wu, Dalton Trans., 2021, 50, 11562–11567 RSC.
- J. Huang, F. Guo, Z. Guo, J. Chen, B. Dai and F. Yu, Inorg. Chem., 2022, 61, 11803–11810 CrossRef CAS PubMed.
- M. Gerken, P. Hazendonk, A. Iuga, J. P. Mack, H. P. A. Mercier and G. J. Schrobilgen, J. Fluorine Chem., 2006, 127, 1328–1338 CrossRef CAS.
- R. R. Ryan and L. B. Asprey, Acta Crystallogr., Sect. B, 1972, 28, 979–981 CrossRef CAS.
- Y. Xu, Y. Zhou, C. Lin, B. Li, X. Hao, N. Ye and M. Luo, Cryst. Growth Des., 2021, 21, 7098–7103 CrossRef CAS.
- Q.-M. Huang, C.-L. Hu, B.-P. Yang, Z. Fang, Y. Lin, J. Chen, B.-X. Li and J.-G. Mao, Chem. Sci., 2021, 12, 9333–9338 RSC.
- N. Ma, C.-L. Hu, J. Chen, Z. Fang, Y. Huang, B.-X. Li and J.-G. Mao, Inorg. Chem. Front., 2022, 9, 5478–5485 RSC.
- Y. Hu, X. Jiang, C. Wu, Z. Huang, Z. Lin, M. G. Humphrey and C. Zhang, Chem. Mater., 2021, 33, 5700–5708 CrossRef CAS.
- Y. Hu, X. Jiang, T. Wu, Y. Xue, C. Wu, Z. Huang, Z. Lin, J. Xu, M. G. Humphrey and C. Zhang, Chem. Sci., 2022, 13, 10260–10266 RSC.
- M. Ding, H. Wu, Z. Hu, J. Wang, Y. Wu and H. Yu, J. Mater. Chem. C, 2022, 10, 12197–12201 RSC.
- Q. Wu, H. Liu, F. Jiang, L. Kang, L. Yang, Z. Lin, Z. Hu, X. Chen, X. Meng and J. Qin, Chem. Mater., 2016, 28, 1413–1418 CrossRef CAS.
- M. Zhang, C. Hu, T. Abudouwufu, Z. Yang and S. Pan, Chem. Mater., 2018, 30, 1136–1145 CrossRef CAS.
- H. Wu, Z. Wei, Z. Hu, J. Wang, Y. Wu and H. Yu, Angew. Chem., Int. Ed., 2024, 63, e202406318 CrossRef CAS PubMed.
- J. Song, C. Hu, X. Xu, F. Kong and J. Mao, Angew. Chem., Int. Ed., 2015, 54, 3679–3682 CrossRef CAS PubMed.
- Y. Lin, C.-L. Hu, Z. Fang, J. Chen, W.-J. Xie, Y. Chen, J.-P. Wang and J.-G. Mao, Inorg. Chem. Front., 2022, 9, 5645–5652 RSC.
- Y.-L. Deng, C.-L. Hu, X. Xu, M.-Z. Zhang and J.-G. Mao, J. Am. Chem. Soc., 2025, 147, 12858–12865 CrossRef CAS PubMed.
- G. Peng, C. Lin, H. Fan, K. Chen, B. Li, G. Zhang and N. Ye, Angew. Chem., Int. Ed., 2021, 60, 17415–17418 CrossRef CAS PubMed.
- C. Wu, X. Jiang, Z. Wang, L. Lin, Z. Lin, Z. Huang, X. Long, M. G. Humphrey and C. Zhang, Angew. Chem., Int. Ed., 2021, 60, 3464–3468 CrossRef CAS PubMed.
- W. Zeng, X. Dong, Y. Tian, L. Huang, H. Zeng, Z. Lin and G. Zou, Chem. Commun., 2022, 58, 3350–3353 RSC.
- Y.-W. Kang, C. L. Hu and J.-G. Mao, Sci. China Chem., 2025, 69, 1801–1806 CrossRef.
- Q.-Q. Chen, C.-L. Hu, M.-Z. Zhang, B.-X. Li and J.-G. Mao, Inorg. Chem., 2023, 62, 12613–12619 CrossRef CAS PubMed.
- M.-B. Xu, Q.-Q. Chen, B.-X. Li, K.-Z. Du and J. Chen, Chin. Chem. Lett., 2025, 36, 110513 CrossRef CAS.
- J.-J. Zhao, S.-F. Li, M.-H. Lv, J.-X. Wang, R.-L. Tang, H. Huang, B. Zhang and D. Yan, Inorg. Chem., 2025, 64, 807–812 CrossRef CAS PubMed.
- L. Zhang, X. Zhang, F. Liang, Z. Hu and Y. Wu, Inorg. Chem., 2023, 62, 14518–14522 CrossRef CAS PubMed.
- D. Yan, M.-M. Ren, F.-F. Mao, Y. Ma, R.-L. Tang, B. Zhang, Y. Ma, X.-D. Zhang and S.-F. Li, Inorg. Chem., 2023, 62, 1323–1327 CrossRef CAS PubMed.
- J.-J. Zhao, S.-F. Li, M.-H. Lv, J.-X. Wang, R.-L. Tang, H. Huang, B. Zhang and D. Yan, Inorg. Chem., 2025, 64, 807–812 CrossRef CAS PubMed.
- N. Ma, Y. Huang, C.-L. Hu, M.-Z. Zhang, B.-X. Li and J.-G. Mao, Inorg. Chem., 2023, 62, 15329–15333 CrossRef CAS PubMed.
- H. Tang, Y. Zhang, C. Zhuo, R. Fu, H. Lin, Z. Ma and X. Wu, Angew. Chem., Int. Ed., 2019, 58, 3824–3828 CrossRef CAS PubMed.
- Y. Li, M. Ji, C. Hu, J. Chen, B. Li, Y. Lin and J. Mao, Chem. Eur J., 2022, 28, e202200001 CrossRef CAS PubMed.
- H. Liu, Q. Wu, L. Liu, Z. Lin, P. S. Halasyamani, X. Chen and J. Qin, Chem. Commun., 2021, 57, 3712–3715 RSC.
- Q.-Q. Chen, C.-L. Hu, L.-J. Yao, J. Chen, M.-Y. Cao, B.-X. Li and J.-G. Mao, Chem. Commun., 2022, 58, 7694–7697 RSC.
- Q.-Q. Chen, C.-L. Hu, B.-X. Li and J.-G. Mao, Inorg. Chem. Front., 2023, 10, 3121–3130 RSC.
- S. Ma, L. Geng, B. Zhu and C. Meng, Dalton Trans., 2025, 54, 8183–8189 RSC.
- W. Song, J. Ren, J. Tan, L. Cao, X. Dong, L. Huang, D. Gao and G. Zou, J. Mater. Chem. C, 2023, 11, 9465–9471 RSC.
- M. Ding, H. Yu, Z. Hu, J. Wang and Y. Wu, Chem. Commun., 2021, 57, 9598–9601 RSC.
- M. Qie, J. Lin, F. Kong, M. A. Silver, Z. Yue, X. Wang, L. Zhang, H. Bao, T. E. Albrecht-Schmitt and J.-Q. Wang, Inorg. Chem., 2018, 57, 1676–1683 CrossRef CAS PubMed.
- C. Zhang, L. Huang, W. Zhou, G. Zhang, H. Hou, Q. Ruan, W. Lei, S. Qin, F. Lu, Y. Zuo, H. Shen and G. Wang, J. Cryst. Growth, 2006, 292, 364–367 CrossRef CAS.
- X. Zhuang, L. Ye, G. Zheng, G. Su, Y. He, X. Lin and Z. Xu, J. Cryst. Growth, 2011, 318, 700–702 CrossRef CAS.
- X. Xu, B.-X. Li, L.-X. Huang, B.-P. Yang, G. Zhang and J.-G. Mao, Inorg. Chem., 2023, 62, 1744–1751 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ac
*ac

























