
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal-free visible-light carbonylation of alkyl iodides to amides via consecutive photoinduced electron transfer
Fatima
Akhssas
a,
Guillaume
Chu
a,
Meriem
El Malamy
a,
Naomi
Illi
a,
Jasmine
Hertzog
 b,
Bertrand
Vileno
c,
Nolwenn
Le Breton
c,
Michael
Badawi
d,
Miguel
Ponce-Vargas
de,
Philipp
Gotico
*f,
Alexandre
Vasseur
d,
Zakaria
Halime
*g,
Christophe
Werlé
*dh and
Ibrahim
Abdellah
*a
b,
Bertrand
Vileno
c,
Nolwenn
Le Breton
c,
Michael
Badawi
d,
Miguel
Ponce-Vargas
de,
Philipp
Gotico
*f,
Alexandre
Vasseur
d,
Zakaria
Halime
*g,
Christophe
Werlé
*dh and
Ibrahim
Abdellah
*a
aUniversité de Lorraine, CNRS, L2CM, F-57000 Metz, France. E-mail: ibrahim.abdellah@univ-lorraine.fr
bUniversité de Lorraine, LCP-A2MC, F-57000 Metz, France
cInstitut de Chimie de Strasbourg, CNRS UMR 7177, Université de Strasbourg, F-67000 Strasbourg, France
dUniversité de Lorraine, CNRS, L2CM, F-54000 Nancy, France. E-mail: christophe.werle@univ-lorraine.fr
eUniversité de Reims Champagne-Ardenne, Moulin de la Housse, 51687 Reims Cedex 02 BP39, France
fInstitute for Integrative Biology of the Cell, CEA, CNRS, Université Paris-Saclay, F-91191 Gif-sur-Yvette, France. E-mail: philipp.gotico@cea.fr
gUniversité Paris-Saclay, CNRS, Institut de Chimie Moléculaire et des Matériaux d’Orsay, F-91405, Orsay, France. E-mail: zakaria.halime@universite-paris-saclay.fr
hMax Planck Institute for Chemical Energy Conversion, Stiftstr. 34−36, 45470 Mülheim an der Ruhr, Germany. E-mail: christophe.werle@cec.mpg.de
First published on 8th January 2026
Abstract
A visible-light-driven, metal-free carbonylation of unactivated alkyl iodides is reported, enabling the direct synthesis of 35 structurally diverse amides in good to excellent yields. The reaction shows broad functional-group tolerance toward both amines and alkyl iodides, including bioisosteric motifs and complex natural product derivatives, underscoring its potential for late-stage functionalization. Mechanistic investigations combining flash photolysis, spectroelectrochemistry, irradiated thin-layer cyclic voltammetry, and EPR spectroscopy reveal a consecutive photoinduced electron transfer (ConPET) mechanism that diverges from conventional single-photon photoredox catalysis. DFT calculations elucidate the key radical carbonylation steps governing reactivity and selectivity. This sustainable and operationally simple method offers a transition-metal-free approach to carbonylation, expanding the toolbox for highly reducing transformations under mild conditions.
Introduction
The direct conversion of simple alkyl halides into complex molecules remains a defining challenge in synthetic chemistry. Their strong C–X bonds and highly negative reduction potentials render both oxidative addition and single-electron transfer intrinsically difficult, limiting the catalytic strategies capable of activating these feedstock reagents under mild conditions.1–5 Surmounting this kinetic and thermodynamic barrier would unlock a wide spectrum of bond-forming transformations—from cross-couplings to carbonylations and beyond—based on readily available building blocks.Amides exemplify the impact such activation strategies can have. They are among the most pervasive functional groups in chemistry, forming the structural backbone of peptides and proteins and occurring in countless pharmaceuticals, agrochemicals, and advanced materials.6–11 Despite their ubiquity, amide synthesis still largely depends on stoichiometric condensation of carboxylic acids and amines with coupling reagents such as carbodiimides or phosphonium salts, generating significant waste (Scheme 1a1).12 Catalytic alternatives have improved atom economy through organo- and transition-metal-catalyzed amidations or dehydrogenative couplings.12–14
 | ||
| Scheme 1 Overview of strategies for amide bond formation. (a) Traditional approaches: stoichiometric or catalytic condensation of carboxylic acids (or acyl halides) with amines, and transition-metal-catalyzed dehydrogenative coupling of alcohols. (b) Challenges in activating unactivated alkyl iodides by single-photon photoredox catalysis due to their highly negative reduction potentials. (c) Conceptual motivations: the consecutive photoinduced electron-transfer (ConPET) approach, in which a photocatalyst radical anion absorbs a second photon to reach an excited state of enhanced reducing power. (d) This work: metal-free visible-light aminocarbonylation of alkyl iodides using 4CzIPN and an in situ CO source under mild conditions; mechanistic studies combine spectroscopy, electrochemistry, and DFT. | ||
Aminocarbonylation—the three-component coupling of CO, an amine, and an organic halide—offers an elegant catalytic entry to amides in a single step (Scheme 1a2).15–21 While well established for aryl, benzyl, and allyl halides, the extension to unactivated alkyl halides remains difficult because oxidative addition to Pd is slow and often followed by β-hydride elimination.22 Copper catalysis can circumvent these issues through single-electron transfer (SET) to generate alkyl radicals, enabling carbonylation of otherwise inert alkyl iodides, though typically under strongly reducing or forcing conditions.23–25 Ni(0) complexes supported by strongly σ-donating ligands such as N-heterocyclic carbenes or bulky phosphines were also reported to react with primary and secondary alkyl iodides under relatively mild conditions. These cases, however, remain limited in scope because the resulting metal-alkyl intermediates are also prone to rapid β-hydride elimination, and because SN2-type oxidative addition is strongly disfavoured by steric and electronic factors. In addition, first-row transition metals generally favour single–electron pathways over the higher-energy two-electron oxidative addition.26–30 Co and Fe complexes predominantly operate through SET pathways, which can enable carbonylative aminocarbonylations. However, these systems generally require high CO pressures, typically in the range of 6 to 40 bar.31,32
Photoredox catalysis has brought new momentum to this field by merging light-driven radical generation with transition-metal catalysis (Scheme 1a3).33–39 Visible-light excitation has been shown to promote radical-assisted oxidative addition and reductive elimination at Pd under mild conditions, and related strategies have been extended to nickel and manganese catalysis.37–39 More recently, tandem two-photon cycles based on fac-Ir(ppy)3-type photocatalysts have enabled the cleavage of demanding C(sp3)–I bonds even under visible light irradiation (Scheme 1a4).40 Recent advances have demonstrated that metal-free photocatalytic strategies, employing organic photocatalysts or photochemical approaches, enable the carbonylative transformation of unactivated alkyl iodides using aryl formates as CO surrogates.41,42 This process generates phenolate while promoting the reduction of the iodoalkane to the corresponding alkyl radical.41
Nonetheless, the direct reduction of unactivated alkyl iodides (Ered < −2 V vs. SCE) remains beyond the capability of most single-photon photocatalysts (Scheme 1b), limiting their engagement in mild radical transformations.1,43
To transcend this fundamental energetic barrier, the concept of consecutive photoinduced electron transfer (ConPET) has emerged as a transformative strategy (Scheme 1c).44–50 In this two-photon process, a photocatalyst radical anion absorbs a second photon to access an excited state of dramatically enhanced reducing power, enabling reactions far beyond the thermodynamic reach of conventional photoredox catalysis. Building on these advances, we envisioned that combining ConPET activation with in situ CO generation could enable a metal-free aminocarbonylation of unactivated alkyl iodides under visible light.
Here we describe such a system employing the organic photocatalyst 4CzIPN (Scheme 1d). Carbon monoxide is produced safely in situ (1–2 bar) from methyldiphenylsilane carboxylic acid (“Silacogen”) in a sealed two-chamber reactor and consumed directly under visible light and ambient conditions. This operationally simple protocol affords a broad range of amides—including complex natural products and bioisosteres—in up to 85% yield within 2–8 h. Mechanistic investigations (in situ UV-vis spectroscopy, flash photolysis, spectroelectrochemistry, irradiated thin-layer cyclic voltammetry, and DFT) support a ConPET mechanism in which photoexcited 4CzIPN is reduced to 4CzIPN˙− and, upon re-excitation, activates demanding C–I bonds. Compared to single-photon photocatalysts such as 4DPAIPN, this system enables rapid, mild, and operationally safe aminocarbonylation under low CO pressure.42
Results and discussion
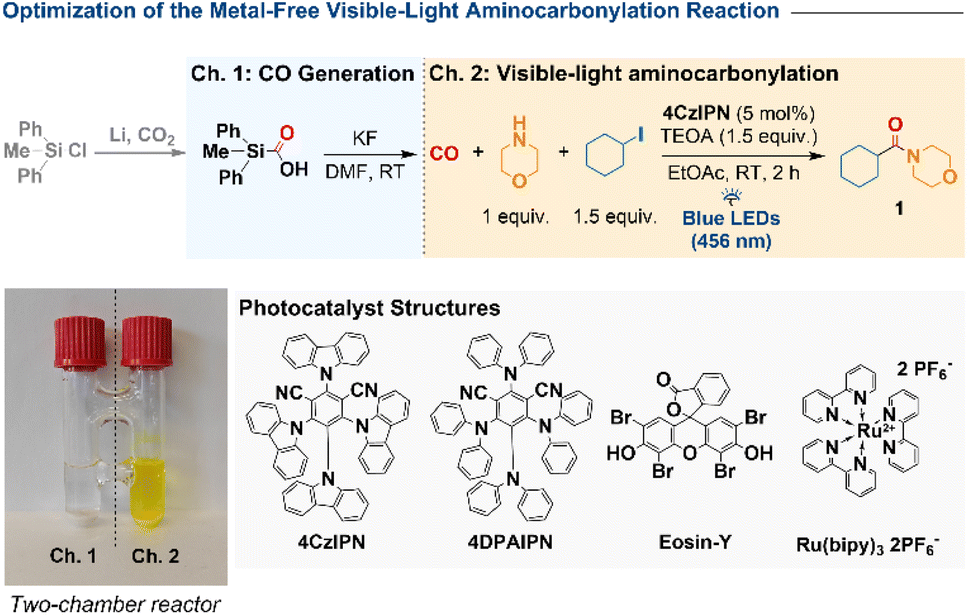
We commenced our investigation with the aminocarbonylation of iodocyclohexane and morpholine in a sealed two-chamber reactor (Table 1). Carbon monoxide was generated in chamber 1 from SilaCOgen—a bench-stable CO surrogate readily obtained from diphenylmethylchlorosilane and CO2—by treatment with KF in DMF.51,52 In chamber 2, iodocyclohexane, morpholine, 4CzIPN (5 mol%), and triethanolamine (TEOA) as a sacrificial electron donor were dissolved in ethyl acetate under argon and irradiated with a 50 W blue LED (456 nm, 50% intensity) at room temperature. After 2 h, amide 1 was obtained in 97% yield (GC-MS, hexadecane internal standard; entry 1). Control experiments highlighted the critical role of each component. Omission of TEOA reduced the yield to 37% (entry 2), and substitution with triethylamine or tributylamine led to diminished efficiency (79% and 67%, entries 3–4). The solvent proved decisive: EtOH, CH3CN, DMSO, and DMF afforded only 13–45% yield (entries 5–8), whereas acetone and 1,4-dioxane performed significantly better (71% and 84%, entries 9–10). Among the photocatalysts evaluated, 4CzIPN emerged as optimal. Eosin-Y delivered only 19% yield (entry 11), Ru(bpy)32+ was inactive (entry 12), and 4DPAIPN provided 60% (entry 13). No product was detected in the absence of photocatalyst or light (entries 14–15). Finally, when using substrates with very high negative reduction potentials, such as bromo- and chloro-cyclohexane, no products were obtained (entries 16–17).|
|
||
|---|---|---|
| Entry | Deviation from above reaction | Yield (%)b |
| a Chamber 1: SilaCOgen (0.60 mmol), KF (0.66 mmol), DMF (1.0 mL); chamber 2: morpholine (0.10 mmol), iodocyclohexane (0.15 mmol), 4CzIPN (5 mol%), TEOA (0.15 mmol), AcOEt (1.0 mL); 456 nm Kessil® blue LED, RT, 2 h. b GC-MS yield vs. hexadecane (see SI). | ||
| 1 | None | 97 |
| 2 | No TEOA | 37 |
| 3 | Et3N instead of TEOA | 79 |
| 4 | n-Bu3N instead of TEOA | 67 |
| 5 | EtOH as solvent | 13 |
| 6 | CH3CN as solvent | 28 |
| 7 | DMSO as solvent | 40 |
| 8 | DMF as solvent | 45 |
| 9 | Acetone as solvent | 71 |
| 10 | 1,4-Dioxane as solvent | 84 |
| 11 | Eosin-Y instead of 4CzIPN | 19 |
| 12 | Ru(bpy)3(PF6)2 instead of 4CzIPN | 0 |
| 13 | 4DPAIPN instead of 4CzIPN | 60 |
| 14 | No photocatalyst (LED light only) | 0 |
| 15 | No light | 0 |
| 16 | Bromocyclohexane instead of iodocyclohexane | 0 |
| 17 | Chlorocyclohexane instead of iodocyclohexane | 0 |
Substrate scope
With the optimized conditions in hand, we explored the scope of the visible-light aminocarbonylation (Scheme 2). Cyclic secondary amines such as morpholine, piperidine, tetrahydroisoquinoline, and pyrrolidine participated readily, furnishing the corresponding amides 1–4 in 61–85% yield. A Boc-protected piperazine delivered 5 in 59%, while a mono-substituted piperazine and diethylamine provided 6 and 7 in 51% and 57% yield, respectively. Primary amines were also suitable coupling partners, affording the corresponding amides 8–12 in up to 85% yield. Functionalized amines such as 2-aminoethanol and benzylamine were well tolerated, delivering amides 11 and 12, respectively, whereas propargylamine afforded product 10 in 67% NMR yield (40% isolated). | ||
| Scheme 2 Substrate scope of alkyl iodides and amines. (a) Chamber 1: SilaCOgen® (1.20 mmol), KF (1.32 mmol), DMF (2.0 mL); chamber 2: amine (0.20 mmol), alkyl iodide (0.30 mmol), 4CzIPN (5 mol%), TEOA (0.30 mmol), ethyl acetate (2.0 mL); 456 nm Kessil® blue LED, RT. Isolated yields unless otherwise noted. (b) NMR yields in parentheses (1H NMR with tetrachloroethane internal standard). (c) 2 h. (d) 4 h. (e) 8 h. (f) 4CzIPN (10 mol%). (g) Sila13COgen. (h) gram-scale (3 mmol of morpholine). | ||
Bioisosteric amines, including bicyclo[1.1.1]pentan-1-amine hydrochloride and its methyl-ester analogue, were compatible after in situ deprotonation with K2CO3, affording 16 and 17 in 50–78%. Strained heterocycles such as 2-oxa-6-azaspiro[3.3]heptane and 3-aminooxetane also underwent clean conversion, producing 18 and 19 in 74% yield.
Aromatic amines displayed lower reactivity: aniline remained unreactive, whereas N-alkylanilines required extended irradiation to furnish 13–15 in 42–75% isolated yield (up to 86% NMR yield).
The transformation was general with respect to the alkyl partner. Secondary iodides furnished the corresponding amides in 45–78% yield, encompassing cyclic, spirocyclic, and benzylic substrates (20–25). Tertiary iodides—including 1-iodoadamantane and tert-butyl iodide—underwent smooth coupling to give 26 and 27 (52–58%). Primary iodides exhibited substitution-dependent reactivity: heteroatom-substituted 2-(iodomethyl)tetrahydrofuran gave 28 in 72% NMR yield (37% isolated, the isolated yield being limited by purification), whereas (3-iodopropoxy)benzene, isobutyl iodide, and 1-iodooctane afforded 29–31 in 45–68%. Notably, substrates bearing unprotected hydroxyl or benzylic groups were well tolerated; 3-iodo-1-propanol and (2-iodoethyl)benzene yielded 32 and 33 in 80% and 75%, respectively.
The method was further extended to complex molecular scaffolds. Cholesterol- and stigmasterol-derived iodides underwent efficient aminocarbonylation under visible light (10 mol% 4CzIPN, 8 h irradiation), providing 34 and 35 in 57% and 67% yield, respectively. Carbon-13 labelling was achieved by replacing Silacogen with Sila13COgen, affording the labelled stigmasterol amide in 64% yield; 13C incorporation was confirmed by FT-ICR-MS (see the SI). A gram-scale experiment using the stigmasterol substrate further highlighted the practicality and scalability of the protocol, furnishing 35 in 68% isolated yield.
This method demonstrates the efficiency of this metal-free reaction, which proceeds without the need for a glove box, within a short reaction time (ranging from 2 to 8 hours) at room temperature. It is compatible with a broad range of alkyl iodides (primary, secondary, and tertiary), including complex molecules such as cholesterol and stigmasterol derivatives, as well as various amines “cyclic, acyclic, aromatic, and bioisosteric” thereby overcoming the substrate limitations observed in previous studies. Notably, the incorporation of isotopic 13CO using pure Sila13COgen achieves exceptional isotopic enrichment in natural products, reaching up to 99.7% (see SI). This represents a significant improvement over prior methods, which achieved enrichments of only 89% to 95%. Additionally, the CO source is derived exclusively from SilaCOgen, in contrast to previous approaches that relied on bubbling CO gas.42
Mechanistic studies
To gain mechanistic insights into the visible-light ConPET aminocarbonylation, a combination of photophysical, electrochemical, and spectroscopic techniques was employed. Stern–Volmer analysis revealed that both TEOA and morpholine can reductively quench the excited state of 4CzIPN to generate the radical anion 4CzIPN˙− (Fig. 1a). This observation accounts for the residual reactivity observed in the absence of TEOA (Table 1, entry 2), when morpholine is present at high concentration. The dynamic quenching rate constant, however, is an order of magnitude higher for TEOA (kq,TEOA = 3.11 × 108 M−1 s−1) than for morpholine (kq,morpholine = 5.07 × 107 M−1 s−1). At concentrations of 150 mM TEOA and 100 mM morpholine, the apparent quenching rates are 4.66 × 107 s−1 and 5.06 × 106 s−1, respectively, rationalizing the superior performance of TEOA, which efficiently generates 4CzIPN˙− while preserving morpholine as a nucleophile. | ||
| Fig. 1 (a) Stern–Volmer plot of [(τ0/τ) – 1] vs. concentration to determine dynamic reductive quenching by TEOA and morpholine. Data: quenching of TADF emission at 514 nm of 30 µM 4CzIPN in Ar-saturated ethyl acetate, laser flash photolysis (λexc = 430 nm, E = 11 mJ). (b) In situ spectroscopy of Ar-saturated MeCN, 45 µM 4CzIPN with 45 mM TEOA under blue irradiation (463 nm; black → red: 0 → 80 s). (c) Spectroelectrochemical generation of 4CzIPN˙− in Ar-saturated MeCN at −1.40 V vs. SCE. (d) Experimental EPR spectra of TEOA and 4CzIPN in the dark (black spectrum), under illumination (λ = 475 nm, blue spectrum), and corresponding simulation (red spectrum), taking into account six 14N nuclei with hyperfine coupling constants of 0.19, 0.17, 0.13, 0.08, 0.02, and 0.017 mT, respectively. Worthy of note, the two weakest values only contribute to the linewidth of the simulated spectra and are consistent with the optimized structures and SOMO distributions obtained by DFT (Fig. S3). | ||
Under catalytically relevant conditions, in situ absorption spectroscopy of 4CzIPN/TEOA in Ar-saturated MeCN revealed the growth of a new species upon 463 nm irradiation, characterized by diagnostic bands at 324, 336, and 470 nm (Fig. 1b; similar results in ethyl acetate, Fig. S4). UV-vis spectroelectrochemistry at −1.40 V vs. SCE, slightly beyond the first reduction wave, reproduced the same spectral features (Fig. 1c), assigning this species to the radical anion 4CzIPN˙−.
Continuous wave electron paramagnetic resonance (EPR) spectroscopy performed at room temperature under irradiation at 475 nm further corroborated the formation of 4CzIPN˙− (Fig. 1d). While no EPR signal was observed in the dark, a characteristic EPR fingerprint was detected as soon as the light was switched on (Fig. 1d, blue spectrum). The observed spectra are in agreement with those previously reported for 4CzIPN˙− and derivatives,44 obtained with hyperfine coupling to six nitrogen 14N nuclei (Fig. 1d, red spectrum). The observed signal also supports the role of morpholine acting as a sacrificial agent by transferring an electron to the photocatalyst (Fig. S2), explaining its partial consumption and the associated reduction in yield.
Single-photon photoredox cycles, as established for related catalysts such as 4DPAIPN, typically proceed through a radical anion (Ered ≈ −1.5 V vs. SCE) that is not sufficiently reducing to activate unactivated alkyl iodides (Ered < −2 V vs. SCE).40,42 In contrast, our results support a consecutive photoinduced electron transfer (ConPET) pathway,46,47,53 in which 4CzIPN˙− absorbs a second photon to form a highly reducing excited radical anion (4CzIPN˙−*), capable of cleaving C(sp3)–I bonds and initiating the aminocarbonylation.
To exclude the possibility that ground-state 4CzIPN˙− acts as the active reductant, we performed irradiated thin-layer cyclic voltammetry (CV)—to our knowledge, its first application in a ConPET aminocarbonylation (Fig. 2). CV of 4CzIPN (Fig. 2, black CV) exhibited a reversible first reduction with E1/2 = −1.23 V vs. SCE (4CzIPN/4CzIPN˙−). Addition of iodocyclohexane in the dark (Fig. 2, red CV) produced no enhancement of current at this wave, indicating that the electrochemically generated 4CzIPN˙− alone is insufficiently reducing. Current growth due to halide reduction appeared only beyond −1.70 V vs. SCE. Under 463 nm irradiation (Fig. 2, blue CV), however, a pronounced current increase at the first wave was observed, consistent with in situ photoexcitation of 4CzIPN˙− to its excited state (4CzIPN˙−*) or formation of a functionally equivalent solvated electron. Although the excited radical anion is ultrashort-lived and nonemissive,46 the light-induced catalytic wave provides compelling electrochemical evidence for its enhanced reducing character.54 Photoinduced detachment to solvated electrons,46,48,55 previously proposed for similar systems, may also contribute; our CV experiments cannot distinguish between these parallel pathways.
 | ||
| Fig. 2 Cyclic voltammograms (CVs) of 0.5 mM 4CzIPN in Ar-saturated MeCN with 0.1 M NBu4PF6 (black), with addition of 50 mM iodocyclohexane in the dark (red), and under irradiation of blue LED (blue). Dotted gray line shows background reduction of 50 mM iodocylohexane on the glassy carbon electrode. Inset shows a magnified part of the changes in the first redox peak. | ||
Together, the photophysical and electrochemical results support the ConPET sequence depicted in Scheme 3: (1) single-photon excitation of 4CzIPN; (2) reductive quenching by TEOA (or morpholine) to yield 4CzIPN˙−; (3) subsequent photoexcitation of 4CzIPN˙− to its excited state (4CzIPN˙−*) (or, alternatively, generation of a solvated electron); and (4) reduction of the alkyl iodide to produce the corresponding alkyl radical.
 | ||
| Scheme 3 Proposed ConPET mechanism for alkyl radical generation by 4CzIPN (PC). Reductive quenching of photoexcited 4CzIPN by TEOA forms 4CzIPN˙−, which, upon second-photon excitation, reaches an excited state capable of reducing unactivated alkyl iodides to alkyl radicals. | ||
This result contrasts with the recently reported mechanism involving a single photon absorption by the 4DPAIPN photocatalyst for the carbonylation of alkyl halides.42
The use of Eosin-Y resulted in only a 19% yield, which can be attributed to its insufficient reducing potential (Ered ≈ −1.1 V).56 The calculated energy difference (ΔE) of −54.9 kcal mol−1 for Eosin-Y indicates a relatively low electron affinity, consistent with the inefficient formation of a stable radical anion. This suggests that Eosin-Y does not effectively participate in the ConPET cycle, thereby explaining the observed low yield. In contrast, 4CzIPN exhibits a more negative energy difference (ΔE = −73.9 kcal mol−1), reflecting a higher electron affinity and enabling the formation of a more stable radical anion. This stability facilitates its participation in the ConPET cycle, leading to efficient re-excitation and a higher quantum yield in the photocatalytic process. For 4DPAIPN, the calculated ΔE of −66.9 kcal mol−1 indicates some capacity to form a radical anion; however, this species is less stable than the one derived from 4CzIPN. This lower stability correlates with its limited efficiency in the ConPET cycle compared to 4CzIPN (see SI).
The radical pathway of the carbonylation step was established through diagnostic control experiments. Addition of the radical scavenger TEMPO completely suppressed product formation (Scheme 4a). Furthermore, the use of cyclopropylmethyl iodide afforded the ring-opened amide 36 (Scheme 4b), a typical radical-clock outcome. These findings confirm that, once formed, the alkyl radical engages in a free-radical carbonylation sequence rather than a closed-shell pathway.
 | ||
| Scheme 4 Probing the radical mechanism. (a) TEMPO-trapping experiment: addition of TEMPO suppresses product formation. (b) Cyclopropylmethyl iodide undergoes ring-opening aminocarbonylation to yield the unsaturated amide 36, consistent with a radical pathway. | ||
Density-functional theory (DFT) calculations provided additional insight into the reaction profile following alkyl-radical formation (Fig. 3). The initially generated cyclohexyl radical (1a) rapidly adds to carbon monoxide to give the corresponding acyl radical (1b). This step proceeds with an activation barrier of 11.9 kcal mol−1 (TS1) and is slightly exergonic (ΔG = −0.8 kcal mol−1). Subsequently, (1b) undergoes iodine-atom transfer from iodocyclohexane to regenerate the cyclohexyl radical (1a), passing through TS2 with a barrier of 16.4 kcal mol−1 to afford the acyl iodide (1c), located +3.2 kcal mol−1 above (1b). This sequence suggests a radical chain-propagation mechanism in which the alkyl radical is continuously regenerated. Next, a nucleophilic attack of morpholine on acyl iodide 1c, accompanied by iodide departure, proceeds through TS3 (ΔG‡ = 11.3 kcal mol−1) to give the cationic intermediate 1d, which subsequently undergoes hydrogen abstraction (Int1) to yield the final amide (1e). The computed free-energy profile indicates that (1e) is 7.7 kcal mol−1 lower in energy than the starting radical (1a), confirming both the thermodynamic driving force and the overall feasibility of the transformation.
 | ||
| Fig. 3 Gibbs free energy (ΔG) profile for the proposed reaction path. DFT calculations performed at the M06-2X/6-311++G** level of theory, considering acetonitrile as the solvent within the Polarizable Continuum Model (PCM). | ||
Unlike morpholine, aniline fails to yield any carbonylation product. While aniline exhibits lower nucleophilicity (Naniline: 12.6–13.0) compared to morpholine (Nmorpholine: 15.6),57 this difference alone does not fully account for its lack of reactivity relative to other amines. DFT calculations reveal that the computed activation free energies (ΔG‡) for nucleophilic addition to the carbonyl group are comparable across the four amines investigated: aniline (12.9 kcal mol−1), ethylaniline (13.2 kcal mol−1), methylaniline (14.1 kcal mol−1), and morpholine (11.3 kcal mol−1). However, the stark contrast in observed reactivity is primarily governed by the thermodynamic stability of the resulting tetrahedral intermediates.
Morpholine demonstrates both a moderately low activation barrier and a product/intermediate free energy slightly lower than that of the reactants (ΔG = −1.0 kcal mol−1), indicating a kinetically accessible and thermodynamically favorable reaction pathway. Conversely, aromatic amines exhibit positive free energies of reaction (ΔG = +8.9, +4.0, and +6.9 kcal mol−1 for aniline, ethylaniline, and methylaniline, respectively), signifying thermodynamically disfavored intermediate formation that inhibits the reaction under standard conditions. This effect is most pronounced for aniline: despite a comparable activation barrier, the tetrahedral intermediate is highly unstable (ΔG = +8.9 kcal mol−1), effectively precluding product formation. Collectively, these findings underscore that both nucleophilicity and the stability of the tetrahedral intermediate dictate the reactivity of amines in this transformation.
Conclusions
We have developed a metal-free, visible-light-driven aminocarbonylation of unactivated alkyl iodides that affords a broad range of amides under mild conditions. The transformation tolerates diverse primary and secondary amines, sterically hindered and functionalized alkyl iodides, and complex scaffolds derived from bioisosteres and natural products, rendering it particularly attractive for late-stage functionalization and medicinal chemistry applications. Key practical advantages include the use of an inexpensive organic photocatalyst (4CzIPN) and a bench-stable, low-pressure CO source, which enhance operational safety and scalability compared to pressurized CO systems. Mechanistic studies combining in situ UV-vis spectroscopy, flash photolysis, spectroelectrochemistry, and irradiated thin-layer cyclic voltammetry support a consecutive photoinduced electron transfer (ConPET) pathway. Following initial single-photon reduction of 4CzIPN, a second excitation of the radical anion generates an exceptionally reducing excited state capable of activating demanding C(sp3)–I bonds. These observations exclude a simple single-photon process and establish ConPET as a general strategy for achieving extreme photoreductive potentials under mild, metal-free conditions. Overall, this work demonstrates that organic photoredox catalysis can rival and even surpass transition-metal systems in challenging carbonylation chemistry, offering a safe, scalable, and operationally simple route to amides while broadening the conceptual scope of highly reducing radical transformations.Author contributions
F. A., G. C., M. E., and N. I. conducted the photocatalytic experiments and performed the associated data analysis. J. H. carried out the FT-ICR-MS and HRMS analyses. B. V. and N. L. carried out the EPR experiments and analyzed the results. M. B. and M. P. V. performed the DFT calculations. A. V. provided technical support for the execution of experiments and data analysis. P. G. conducted the flash photolysis and electrochemistry analyses, including mechanistic studies. Z. H., C. W., and I. A. conceived the project and oversaw the experimental work. All authors contributed to the writing of the manuscript and have approved its final version.Conflicts of interest
The authors declare no conflicts of interest.Data availability
The data supporting this article are included in the supplementary information (SI). Supplementary information: experimental and syntheses procedures, characterisations including NMR, EPR, electrochemistry, spectroelectrochemistry, in situ UV-vis spectroscopy, DFT calculations. See DOI: https://doi.org/10.1039/d5sc09778a.Acknowledgements
This work was supported by the French National Research Agency (ANR) through the “Chaire de professeur junior (CPJ)” grant awarded to I.A. (ANR-23-CPJ1-0051-01), as well as by the Eurométropole de Metz. The authors gratefully acknowledge the support of the Max Planck Society. This work was also partly supported by the French France 2030 program “Initiative d'Excellence Lorraine (LUE),” reference ANR-15-IDEX-04-LUE. Additional funding was provided by the French research infrastructure INFRANALYTICS (FR2054) for EPR spectroscopy facilities. The authors extend their gratitude to the Centre de Calcul Régional ROMEO (http://romeo.univ-reims.fr) for computational resources. This work was partially supported by the MassLor research infrastructure at the University of Lorraine, for mass spectrometry analyses, with special thanks to F. Dupire.References
- T. Constantin, B. Górski, M. J. Tilby, S. Chelli, F. Juliá, J. Llaveria, K. J. Gillen, H. Zipse, S. Lakhdar and D. Leonori, Science, 2022, 377, 1323–1328 CrossRef CAS PubMed.
- L. Caiger, H. Zhao, T. Constantin, J. J. Douglas and D. Leonori, ACS Catal., 2023, 13, 4985–4991 CrossRef CAS.
- T. Constantin, M. Zanini, A. Regni, N. S. Sheikh, F. Juliá and D. Leonori, Science, 2020, 367, 1021–1026 Search PubMed.
- J. Corpas, M. Alonso and D. Leonori, Chem. Sci., 2024, 15, 19113–19118 Search PubMed.
- Z. Zhang, B. Górski and D. Leonori, J. Am. Chem. Soc., 2022, 144, 1986–1992 CrossRef CAS PubMed.
- E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org Chem., 2020, 4641–4651 CrossRef CAS.
- V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed.
- M. Winnacker, Eur. J. Lipid Sci. Technol., 2023, 125, 1–9 CrossRef.
- P. Zhang, C. B. Duan, B. Jin, A. S. Ali, X. Han, H. Zhang, M. Z. Zhang, W. H. Zhang and Y. C. Gu, Adv. Agrochem, 2023, 2, 324–339 CrossRef CAS.
- V. Froidevaux, C. Negrell, S. Caillol, J. P. Pascault and B. Boutevin, Chem. Rev., 2016, 116, 14181–14224 CrossRef CAS PubMed.
- J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS.
- R. M. De Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 Search PubMed.
- C. Gunanathan, Y. Ben-David and D. Milstein, Science, 2007, 317, 790–792 CrossRef CAS PubMed.
- A. Kumar, N. A. Espinosa-Jalapa, G. Leitus, Y. Diskin-Posner, L. Avram and D. Milstein, Angew. Chem., Int. Ed., 2017, 56, 14992–14996 CrossRef CAS PubMed.
- K. Rao, A. Sharma, G. K. Rathod, A. S. Barahdia and R. Jain, Org. Biomol. Chem., 2025, 23, 980–991 RSC.
- J. B. Peng, H. Q. Geng and X. F. Wu, Chem, 2019, 5, 526–552 CAS.
- M. Markovič, P. Lopatka, P. Koóš and T. Gracza, Org. Lett., 2015, 17, 5618–5621 CrossRef PubMed.
- J. R. Martinelli, D. M. M. Freckmann and S. L. Buchwald, Org. Lett., 2006, 8, 4843–4846 CrossRef CAS PubMed.
- P. Gotico, A. Del Vecchio, D. Audisio, A. Quaranta, Z. Halime, W. Leibl and A. Aukauloo, ChemPhotoChem, 2018, 2, 715–719 CrossRef CAS.
- S. Monticelli, A. Talbot, P. Gotico, F. Caillé, O. Loreau, A. Del Vecchio, A. Malandain, A. Sallustrau, W. Leibl, A. Aukauloo, F. Taran, Z. Halime and D. Audisio, Nat. Commun., 2023, 14, 4451 CrossRef CAS PubMed.
- W. Fang, Q. Deng, M. Xu and T. Tu, Org. Lett., 2013, 15, 3678–3681 CrossRef CAS PubMed.
- L. Wu, X. Fang, Q. Liu, R. Jackstell, M. Beller and X. F. Wu, ACS Catal., 2014, 4, 2977–2989 Search PubMed.
- F. Zhao, H. Ai and X. Wu, Angew. Chem., Int. Ed., 2022, 61, e202200062 CrossRef CAS PubMed.
- X. Yan, L. Fan, X. Zhang and G. Liu, Org. Chem. Front., 2022, 9, 6749–6765 Search PubMed.
- J. Ling, A. Bruneau-Voisine, G. Journot and G. Evano, Chem. - Eur. J., 2022, 28, e202201356 CrossRef CAS PubMed.
- T. Hatakeyama, T. Hashimoto, K. K. A. D. S. Kathriarachchi, T. Zenmyo, H. Seike and M. Nakamura, Angew. Chem., Int. Ed., 2012, 51, 8834–8837 CrossRef CAS PubMed.
- C. Sci and X. Hu, Chem. Sci., 2011, 2, 1867–1886 Search PubMed.
- S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 Search PubMed.
- T. Hatakeyama, T. Hashimoto, Y. Kondo, Y. Fujiwara and H. Seike, J. Am. Chem. Soc., 2010, 132, 10674–10676 Search PubMed.
- C. C. Cobalt-catalyzed, S. Miyaura, X. Ligands, L. R. Mills, D. Gygi, J. R. Ludwig, E. M. Simmons, S. R. Wisniewski, J. Kim and P. J. Chirik, ACS Catal., 2022, 12, 1905–1918 Search PubMed.
- K. Sun, G. Lu, W. Han and M. Beller, Angew. Chem., Int. Ed., 2025, 64, e202512346 CrossRef CAS PubMed.
- G. Xu, Z. Bao and X. Wu, J. Catal., 2025, 452, 116456 Search PubMed.
- Z. Wang, G. Fei, C. Xue and P. Xie, Mol. Catal., 2025, 574, 114859 Search PubMed.
- S. Sumino, A. Fusano, T. Fukuyama and I. Ryu, Acc. Chem. Res., 2014, 47, 1563–1574 CrossRef CAS PubMed.
- A. Fusano, S. Sumino, S. Nishitani, T. Inouye, K. Morimoto, T. Fukuyama and I. Ryu, Chem. - Eur. J., 2012, 18, 9415–9422 CrossRef CAS.
- T. Fukuyama, S. Nishitani, T. Inouye, K. Morimoto and I. Ryu, Org. Lett., 2006, 8, 1383–1386 CrossRef CAS PubMed.
- G. M. Torres, Y. Liu and B. A. Arndtsen, Science, 2020, 368, 318–323 CrossRef CAS PubMed.
- K. El Chami, Y. Liu, M. A. Belahouane, Y. Ma, P. L. Lagueux-Tremblay and B. A. Arndtsen, Angew. Chem., Int. Ed., 2023, 62, e202213297 CrossRef CAS PubMed.
- Y. H. Zhao, X. W. Gu and X. F. Wu, Org. Chem. Front., 2024, 11, 442–447 Search PubMed.
- J. A. Forni, N. Micic, T. U. Connell, G. Weragoda and A. Polyzos, Angew. Chem., Int. Ed., 2020, 59, 18646–18654 Search PubMed.
- E. Le Saux, M. Mathis, G. Panizzolo and B. Morandi, J. Am. Chem. Soc., 2025, 147, 39031–39035 CrossRef CAS PubMed.
- X. Liu, B. S. Portela, A. Wiedenbeck, C. H. Chrisman, R. S. Paton and G. Miyake, Angew. Chem., Int. Ed., 2024, 63, e202410928 CrossRef CAS PubMed.
- T. U. Connell, C. L. Fraser, M. L. Czyz, Z. M. Smith, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, J. L. Adcock, A. D. Scully, D. E. Gómez, N. W. Barnett, A. Polyzos and P. S. Francis, J. Am. Chem. Soc., 2019, 141, 17646–17658 CrossRef CAS.
- J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed.
- I. A. MacKenzie, L. Wang, N. P. R. Onuska, O. F. Williams, K. Begam, A. M. Moran, B. D. Dunietz and D. A. Nicewicz, Nature, 2020, 580, 76–80 CrossRef CAS PubMed.
- M. Villa, A. Fermi, F. Calogero, X. Wu, A. Gualandi, P. G. Cozzi, A. Troisi, B. Ventura and P. Ceroni, Chem. Sci., 2024, 15, 14739–14745 RSC.
- M. Villa, A. Fermi, F. Calogero, A. Gualandi, P. Franchi, M. Lucarini, B. Ventura, P. G. Cozzi and P. Ceroni, Angew. Chem., Int. Ed., 2025, 64, e202420009 CrossRef CAS PubMed.
- S. J. Horsewill, K. M. M. Sharrock, P. P. Fehér, J. M. Woolley, I. Pápai and D. J. Scott, Angew. Chem., Int. Ed., 2025, 64, e202506701 CrossRef CAS PubMed.
- I. Ghosh, T. Ghosh, J. I. Bardagi and B. König, Science, 2014, 346, 725–728 Search PubMed.
- K. B. Vega, A. H. Sánchez, T. Mandal, W. Silva, R. M. Gschwind, A. C. Neto, M. A. Barbosa Ferreira, B. König and M. W. Paixão, ACS Catal., 2025, 15, 18087–18096 CrossRef CAS.
- S. D. Friis, A. T. Lindhardt and T. Skrydstrup, Acc. Chem. Res., 2016, 49, 594–605 CrossRef CAS PubMed.
- S. D. Friis, R. H. Taaning, A. T. Lindhardt and T. Skrydstrup, J. Am. Chem. Soc., 2011, 133, 18114–18117 CrossRef CAS PubMed.
- B. Pfund and O. S. Wenger, JACS Au, 2025, 5, 426–447 Search PubMed.
- T. Casadei, A. Piccoli, D. Zeppilli, L. Orian, A. A. Isse and M. Fantin, ACS Catal., 2025, 15, 16938–16952 CrossRef PubMed.
- K. Reynolds, B. Johnston, B. Campbell, A. Li and D. Nocera, J. Am. Chem. Soc., 2025, 147, 31025–31033 Search PubMed.
- V. Srivastava and P. P. Singh, RSC Adv., 2017, 7, 31377–31392 Search PubMed.
- F. Brotzel, Y. C. Chu and H. Mayr, J. Org. Chem., 2007, 72, 3679–3688 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |