
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAdvances in electrochemical technologies for PFAS destruction
Yuqing
Dong
a,
Shuaiyu
Gao
a,
Yuelin
Zhao
a,
Genban
Sun
 *a and
Jihong
Yu
*ab
*a and
Jihong
Yu
*ab
aBeijing Key Laboratory of Energy Conversion and Storage Materials, College of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: gbsun@bnu.edu.cn
bState Key Laboratory of Inorganic Synthesis and Preparative Chemistry, College of Chemistry, International Center of Future Science, Jilin University, Changchun 130012, China. E-mail: jihong@jlu.edu.cn
First published on 16th March 2026
Abstract
Per- and polyfluoroalkyl substances (PFAS) are a class of man-made chemicals extensively employed in industrial processes, with their strong C–F bond energy conferring exceptional stability. However, this stability also leads to bioaccumulation and environmental persistence, posing threats to ecosystems and human health. Conventional physical separation technologies can only concentrate PFAS without completely destroying them. Against this backdrop, electrochemical technology has emerged as one of the most promising strategies for complete PFAS destruction, benefiting from its mild reaction conditions and controllable electron transfer. This review systematically summarizes the research progress on electrochemical PFAS degradation, comprehensively examining the degradation mechanisms and key influencing factors in electrochemical oxidation, reduction, and combined processes. It highlights the crucial roles of density functional theory (DFT) and molecular dynamics (MD) calculations in elucidating interfacial behaviors and atomic-scale C–F bond activation mechanisms. Addressing the bottlenecks of mass transfer limitations and incomplete defluorination encountered in practical applications, this paper prospectively points out the potential of microenvironmental regulation and the development of bifunctional materials for achieving in situ deep mineralization. Furthermore, it briefly explores the application prospects of artificial intelligence (AI)-enabled high-throughput screening technologies in accelerating the development of multi-objective electrode materials. Overall, this review constructs a comprehensive research framework spanning from fundamental bond cleavage mechanisms to macroscopic processes and data-driven optimization. It provides systematic strategies for the complete destruction of PFAS and offers theoretical references for the rational design of advanced environmental functional materials.
1 Introduction
Per- and polyfluoroalkyl substances (PFAS) exhibit exceptional thermochemical stability along with hydrophobic and oleophobic properties.1–3 These characteristics enable their extensive use in chemical manufacturing, electronics, medical devices, textiles, and consumer products.4,5 However, these same properties confer high environmental persistence and bioaccumulation in natural systems.6 PFAS residues have now been detected in aquatic environments, atmospheric compartments, and soils across multiple global regions.7 Consequently, PFAS are recognized as emerging persistent organic pollutants of particular concern in aquatic systems. Epidemiological studies confirm that PFAS exposure induces multi-organ toxicity, affecting the liver, kidneys, thyroid, and other systems.8 Given their environmental recalcitrance and significant ecotoxicity, PFAS pose systemic threats to ecosystem integrity and public health.9It is reported that approximately 450–2700 tons of perfluorooctane sulfonate (PFOS) enter wastewater systems globally each year.10 To mitigate environmental and health risks, multiple countries have legislated restrictions on PFAS production and usage.11 In 2023, China included PFOS in the List of Priority Controlled New Pollutants, stipulating that its import and export will be prohibited starting from 2024, and requiring that PFOS-containing waste be subject to full-process supervision as hazardous waste. Practically, cities like Shanghai have carried out projects to replace firefighting foam and advance drinking water treatment. Through nanofiltration and activated carbon adsorption, the concentration of PFOS in water has been successfully controlled below 0.5 µg L−1. Concurrently, the U.S Environmental Protection Agency has set the limit for perfluorooctanoic acid (PFOA) and PFOS in drinking water at 4 ng L−1, while Denmark enforces a stricter 2 ng L−1 threshold in relation to separate PFAS analytes.12 Significantly, the European Union amended its Persistent Organic Pollutants Regulation in April 2025, tightening unintentional trace contaminant limits for PFOS and its salts to 0.025 mg kg−1.13 Given the extreme environmental persistence and trace-level occurrence of PFAS in contaminated media, developing efficient removal technologies constitutes a critical scientific challenge in environmental remediation.14
In conventional PFAS treatment technologies, adsorption methods achieve PFAS enrichment through physicochemical interactions.15,16 Activated carbon exhibits >90% capture efficiency for long-chain PFAS thanks to its large specific surface area, while functionalized ion-exchange resins enhance adsorption capacity to 120 mg g−1.17 However, short-chain PFAS remain challenging to remove effectively due to their high aqueous solubility and low adsorption affinity, with competitive anions reducing ion-exchange efficiency by >50%.18 Additionally, spent adsorbents require energy-intensive thermal regeneration or hazardous waste disposal.16 Membrane separation technologies leverage molecular exclusion mechanisms, where reverse osmosis (RO) achieves >99% removal efficiency across the PFAS spectrum, and nanofiltration (NF) selectively retains >90% of long-chain PFAS. Their modular design facilitates large-scale water supply applications without chemical additives.19 Nevertheless, RO consumes 3–10 kW h per ton of water and generates concentrated PFAS waste streams, with membrane fouling causing 40–70% flux decline. NF demonstrates limited efficacy for short-chain PFAS, accompanied by substantial membrane replacement costs. Recently developed destructive technologies, though proven for PFAS degradation in laboratory settings, face significant practical limitations. Current methods face significant hurdles: sonolysis is energy-intensive, bioremediation yields less than 10% metabolic efficiency, and photocatalysis suffers from low quantum yields and organic matter interference. These techniques exhibit poor anti-interference capacity in complex aquatic matrices and lack validation at engineering scales, severely constraining their potential for full-scale implementation.20
Electrochemical techniques have evolved into a prominent research topic in PFAS degradation studies, thanks to their mild reaction conditions, precisely controllable redox capacity, and particularly its dual functions of pollutant separation and mineralization destruction.21 Electrocoagulation (EC) achieves indirect adsorption of PFAS through hydroxide flocs formed by the dissolution of metal anodes.22 Capacitive deionization (CDI), on the other hand, completes molecular-level concentration of PFAS driven by the electrostatic force of the electric double layer.23 However, EC and CDI can only realize the enrichment and concentration of PFAS, and cannot break their stable C–F bonds. By coupling electrochemical oxidation (EO) and electrochemical reduction (ER) processes, the cleavage of C–F bonds in PFAS can be achieved, ultimately mineralizing them into harmless products such as CO2 and F−. EO utilizes strong oxidizing species like hydroxyl radicals (·OH) and sulfate radicals (SO4˙−) generated by boron-doped diamond (BDD) or Ti4O7 anodes, or through a direct electron transfer (DET) mechanism, to efficiently degrade typical PFAS such as PFOA and PFOS. ER triggers H/F exchange reactions through DET or via hydrated electrons (eaq−) and hydrogen radicals (·H) generated at the cathode, gradually defluorinating to form low-fluorinated products.24 Current research frontiers focus on developing magnetic electrode materials to enable efficient recovery of adsorbents and the in situ coupled design of oxidation-reduction bifunctional electrodes. Meanwhile, benefiting from the development of advanced characterization techniques, the understanding of multi-scale mechanisms from atomic interfacial reaction pathways to macroscopic kinetics continues to deepen.25 Despite the significant advantages exhibited by electrochemical technology, there remains an urgent need for systematic reviews in this field to clarify three key challenges: the reaction pathways of PFAS degradation by EO and ER, the quantitative laws governing the structure–activity relationships of electrode materials, and the strategies for inhibiting interference from actual water matrices.26 This will provide theoretical guidance for the engineering application of such technologies.27
Meanwhile, this review comprehensively evaluates the efficacy and mechanisms of EO, ER, and their synergistic technologies. In terms of characterization and auxiliary research techniques, the review focuses on multi-scale analytical methods and emerging intelligent auxiliary tools, including microstructure analysis of electrode materials, tracking of interfacial reaction processes, free radical identification, DFT/MD theoretical calculations, and AI-enabled HTS. These combined methods not only help clarify atomic-level reaction pathways but also promote the efficient development of high-performance electrochemical PFAS degradation systems.
This review systematically summarizes the state-of-the-art progress in the electrochemical degradation of PFAS (Fig. 1). To bridge the gap between atomic level interactions and macroscopic performance, this review first highlights the critical roles of multi-scale analytical methods and theoretical calculations, specifically density functional theory (DFT) and molecular dynamics (MD), in unraveling complex interfacial behaviors and C–F bond activation mechanisms. Building upon this fundamental understanding, we comprehensively evaluate the efficacy and governing mechanisms of diverse electrochemical strategies. For EO, we elucidate the synergistic effects of DET and indirect radical oxidation, detailing specific degradation pathways on benchmark anodes alongside key operational parameters. Regarding ER, we systematically unpack the stepwise defluorination mechanisms mediated by DET, ·H, and eaq−, followed by rational design strategies for carbon-based and catalyst-supported cathodes.
 | ||
| Fig. 1 Schematic diagram of systematic study on electrochemical degradation of PFAS. | ||
Recognizing the inherent limitations of stand-alone processes, we further explore recent advancements in synergistic technologies that couple electrochemistry with adsorption, membrane separation, and photocatalysis. These combined systems demonstrate significant advantages in overcoming mass transfer bottlenecks, reducing specific energy consumption, and enhancing overall defluorination efficiency. Beyond traditional material design, this review incorporates the transformative potential of artificial intelligence (AI)-enabled high-throughput screening (HTS) in establishing a closed-loop framework for the rapid discovery and multi-objective optimization of next-generation electrodes. Finally, we prospectively outline the persistent challenges and emerging opportunities in the field. By advocating for the in-depth integration of electrochemical technologies with separation techniques and data-driven paradigms, this work aims to provide a robust scientific foundation and a forward-looking roadmap for developing highly efficient, practical technologies to ultimately eradicate these recalcitrant “forever chemicals” from the environment.
2 Methodologies of mechanism and pathway investigations
Electrochemical technologies have demonstrated the capability to achieve complete mineralization of PFAS with high economic and environmental compatibility, thereby emerging as a promising research frontier in the field of environmental remediation. This chapter presents a systematic review of the core characterization techniques employed in investigating electrochemical PFAS degradation.28 The discussion encompasses the theoretical interpretive frameworks integral to this research domain, including: (1) structural and interfacial characterization of electrode materials, (2) quantitative analysis of reaction intermediates, (3) mechanistic elucidation of free radical species, and (4) theoretical calculation method. Collectively, these methodologies enable a multi-scale understanding of electrochemical PFAS degradation by bridging experimental observations with theoretical frameworks, from atomic-level interfacial interactions to macroscopic reaction kinetics. Application of these characterization methodologies facilitates the revelation of degradation pathways, prediction of reaction energetics, and rational design of efficient, sustainable electrochemical technologies for PFAS abatement.292.1 Characterization of electrode materials
The type and properties of electrode materials are core determinants in the electrochemical degradation of PFAS. Analysis of their electrical conductivity, oxygen evolution potential (OEP), surface reactivity, structural characteristics, and stability provides critical guidance for electrode design. Scanning electron microscopy-energy dispersive spectroscopy (SEM-EDS) enables visual characterization of the microstructure and elemental distribution on electrode surfaces. Successful material synthesis, uniform elemental distribution, and the impact of structural features on catalytic activity can all be validated by SEM-EDS analysis.30 The in-depth analysis of the surface chemical state and crystal structure of electrode materials relies on X-ray photoelectron spectroscopy (XPS) and X-ray diffraction (XRD). XPS is a key tool for understanding the efficient defluorination mechanism, as it can analyze the electron transfer, adsorption behavior, and intermediate product evolution between electrodes and PFAS. XRD is used to determine the crystal structure of electrode materials and structural changes induced by doping. For example, when Ce3+ is doped into Ti4O7 electrodes, the shift of XRD diffraction peaks indicates that Ce3+ doping leads to lattice distortion and the generation of oxygen vacancies.31 The characteristic diffraction peaks (111) and (220) of BDD electrodes show an inverse relationship between the full width at half maximum and grain size, which directly affects the electrical conductivity of the electrodes.32,33 These structural features play a decisive role in the selective adsorption of PFAS.The characterization of electrochemical activity and interfacial properties of electrodes is primarily achieved through various voltammetric techniques and impedance spectroscopy. The Linear Sweep Voltammetry (LSV) curve, by measuring the electrode potential–current response, accurately defines the working potential window of the electrode and quantifies the OEP, thereby distinguishing the oxidation pathways for PFAS degradation. For instance, the Ti/BDD electrode exhibits an OEP of 2.8 V vs. SCE, and its high OEP enables preferential indirect oxidation via ·OH generated from water oxidation to mineralize long-chain perfluorocarboxylic acids (PFCAs).34 Cyclic Voltammetry (CV), through scan rate-dependent analysis, reveals the adsorption mechanism and electron transfer kinetics of PFAS on the electrode surface, which is crucial for evaluating the redox characteristics of electrodes. For example, the hexadecyltrimethylammonium bromide(CTAB)-functionalized CNT electrode shows a characteristic reduction peak of PFOS at −0.58 V vs. Ag/AgCl, and the peak current correlates linearly with the scan rate, confirming that cationic surfactants regulate the interfacial electron transfer of PFAS via electrostatic adsorption.35 Electrochemical Impedance Spectroscopy (EIS), by fitting equivalent circuit models, accurately characterizes the charge transfer resistance (Rct) and mass transfer processes at the electrode/solution interface. For instance, the F/Sb co-doped Ti/Sn–Sb/SnO2–F–Sb electrode reduces Rct to 85 Ω cm2 by forming Sn–O–F bond defects, enabling efficient electron transfer for PFOS degradation.36 The interfacial properties of electrodes are critical for PFAS electrochemical degradation. Future designs should balance high OEP, low impedance, and anti-dissolution performance of electrode materials to achieve efficient and stable PFAS mineralization.
2.2 Analysis of reaction intermediates
Precise quantification of defluorination efficiency and identification of intermediate products are key indicators for evaluating PFAS treatment technologies. The fluoride ion-selective electrode (F−–ISE) has emerged as the preferred method for continuous monitoring of F− release due to its advantages of simplicity and rapidity. Furthermore, ion chromatography (IC) offers high sensitivity and anti-interference capability, making it particularly suitable for the analysis of complex matrix samples.37 Liquid chromatography-tandem mass spectrometry (LC-MS/MS), with its superior separation and structural identification capabilities, identifies intermediates by resolving ion peaks. For example, the detection of short-chain PFOS anions in PFOS degradation provides direct evidence of a stepwise chain-shortening mechanism driven by ·OH radical attack on –CF2– moieties.38 The detection of intermediates confirms that degradation occurs via direct electron transfer-driven decarboxylation and C–F bond cleavage.39 Nuclear Magnetic Resonance (NMR) spectroscopy provides direct evidence of molecular structural changes during the defluorination process. For instance, 19F NMR analysis reveals a shift in the fluorine signal of the CF2 groups in PFOA, moving from −119 ppm to −116 ppm, attributed to altered electronic environments after decarboxylation. This signal shift indicates preferential cleavage of the C–F bonds adjacent to the carboxyl group, confirming the occurrence of decarboxylation.402.3 Free radical identification
Accurate identification of reactive free radicals generated during the electrochemical degradation of PFAS is crucial for elucidating the reaction mechanisms, as these radicals are key intermediates responsible for triggering the cleavage of C–F bonds. Electron Spin Resonance (ESR) spectroscopy, as the standard for direct detection of unpaired electrons, plays a core role in radical identification. The characteristic signals of Cl· and ·OH observed in ESR spectra demonstrate their critical roles in PFOA degradation, where Cl· triggers the initial reaction and ·OH participates in subsequent carbon chain cleavage.41 Radical quenching experiments indirectly validate the contribution of specific radicals through kinetic intervention. This approach involves introducing selective quenchers into the system and observing changes in degradation efficiency and pathways.42 The quenching experiments and ESR results corroborate each other, revealing the degradation pathways of PFAS and enabling efficient degradation.2.4 Theoretical calculation
Theoretical calculation methods such as DFT and MD can reveal the essence of reactions at the atomic/electronic scale.43 In the study of electrochemical degradation of PFAS, these methods can reveal the interaction between PFAS and electrodes as well as reaction energy barriers, and clarify the degradation mechanism. They can also predict the electronic structure and catalytic activity of electrode materials, thereby enabling the targeted design of high-efficiency catalysts.44 In recent years, theoretical calculations have become an important tool in the research on the electrochemical degradation of PFAS.45DFT calculations play an irreplaceable role in revealing the C–F bond cleavage mechanism, reaction energy barriers, and reaction pathway prediction of PFAS.46 By constructing electrode-pollutant composite models, DFT can accurately simulate the adsorption configurations and adsorption energies of PFAS on the electrode surface, map the reaction potential energy surface, and gradually resolve the reaction energy barriers while identifying the rate-determining step.42,47
For instance, by calculating the C–F bond dissociation energy (BDE), Souza et al. pointed out that in PFOA, the α-C–F bond adjacent to the carboxyl group is more prone to cleavage due to the inductive effect.48 In contrast, PFOS exhibits reduced reactivity of its distal C–F bonds owing to the electronic effect of the sulfonic group, which provides a theoretical basis for targeted degradation strategies.48 Bentel et al. revealed the difference of C–F bond stability of different PFAS from the perspective of bond energy through DFT calculations, and showed the configuration and interaction of PFAS combined with electrons from the perspective of spatial structure, which together provided theoretical calculation evidence for the chemical behavior of PFAS under the action of electrons (Fig. 2a and b).49 Zenobio et al. calculated the interaction between propylene and different PFAS using DFT, and confirmed that the interaction between propylene and PFOS and PFBS was more stable and the chemical activity of the formed system was lower, while the interaction activity with PFOA was relatively higher (Fig. 2c).44
 | ||
| Fig. 2 (a) Calculation of C–F BDEs. (b) Structure of the adducts. Reproduced with permission.49 Copyright 2019, American Chemical Society. (c) Frontier orbital distributions. Reproduced with permission.44 Copyright 2022, Elsevier. | ||
In terms of reaction kinetics, Biswas et al. employed a DFT-based elementary kinetic approach to simulate the eaq− mediated defluorination process. Their work revealed that the average free energy barriers of PFOA and PFOS are 2.70 kJ mol−1 and 8.16 kJ mol−1, respectively. This demonstrates that PFOS has lower reactivity due to a more compact solvation shell of solvated electrons, and for the first time, identifies that this process is governed by statistical kinetics rather than pure thermodynamics.50 Blotevogel et al., on the other hand, developed kinetic models for the reduction of PFAS by Fe0/Zn0via DFT, noting that the electron transfer step is the RDS of defluorination.51 In the context of material design, Mohamed et al. compared the adsorption energies of various PFAS on Fe0 surfaces using DFT, finding that functional groups exert a greater influence on adsorption than carbon chain length.52 The study by Wang et al. demonstrated that a higher Ti3+ content in Ti4O7 anodes can enhance the adsorption of PFOS and reduce electron transfer energy barriers. Meanwhile, only pores with a diameter larger than 1.03 µm exhibit electrochemical activity.53,54 Ma et al., by combining computational and experimental approaches, confirmed that MXene modification can strengthen electrostatic and hydrogen-bonding interactions between polyamide membranes and short-chain PFAS, achieving a synergistic improvement in selectivity and permeability.55 Furthermore, Zhang et al. uncovered via DFT that the PFOA degradation pathway proceeds sequentially through two steps: reduction by photogenerated electrons to form  radicals, followed by reaction with superoxide radicals to generate short-chain products. This work clarified the critical roles of electrons and superoxide radicals.56 Raghavan et al. also observed that PFOA radicals undergo spontaneous decarboxylation in a vacuum environment, while the aqueous solvation effect inhibits this process. Applying a potential of 3.08 V vs. the standard hydrogen electrode (SHE) reduces the decarboxylation activation energy by 0.34 eV and the reaction energy by 1.05 eV, with decarboxylation occurring before defluorination.57 DFT calculations have revealed the polarization effect of the interfacial electrostatic field on PFAS molecules: a strong electric field alters the dipole moment orientation of PFOA molecules, leading to polarization and stretching of the carbon chain, which reduces the C–F bond energy by up to 40%. Specifically, for the α-CF2 group, the energy barrier for nucleophilic attack by O2·− decreases from 1.78 eV to 0.92 eV, providing a theoretical explanation for the high defluorination efficiency observed experimentally.58 Collectively, these studies demonstrate the systematic application value of DFT, ranging from uncovering PFAS degradation mechanisms at the electronic scale to guiding the design of functional materials.59
radicals, followed by reaction with superoxide radicals to generate short-chain products. This work clarified the critical roles of electrons and superoxide radicals.56 Raghavan et al. also observed that PFOA radicals undergo spontaneous decarboxylation in a vacuum environment, while the aqueous solvation effect inhibits this process. Applying a potential of 3.08 V vs. the standard hydrogen electrode (SHE) reduces the decarboxylation activation energy by 0.34 eV and the reaction energy by 1.05 eV, with decarboxylation occurring before defluorination.57 DFT calculations have revealed the polarization effect of the interfacial electrostatic field on PFAS molecules: a strong electric field alters the dipole moment orientation of PFOA molecules, leading to polarization and stretching of the carbon chain, which reduces the C–F bond energy by up to 40%. Specifically, for the α-CF2 group, the energy barrier for nucleophilic attack by O2·− decreases from 1.78 eV to 0.92 eV, providing a theoretical explanation for the high defluorination efficiency observed experimentally.58 Collectively, these studies demonstrate the systematic application value of DFT, ranging from uncovering PFAS degradation mechanisms at the electronic scale to guiding the design of functional materials.59
MD simulations, by capturing the diffusion and mass transfer behaviors of PFAS at electrode interfaces, multi-component interactions, and material interfacial dynamics, reveal the adsorption mechanisms and time-dependent processes of PFAS at the mesoscopic scale.60 MD simulations further clarify how to facilitate PFAS degradation through environmental regulation or material design, thereby overcoming the limitations of methods like DFT in terms of system-scale and dynamic spatial effects.61
By combining MD and DFT calculations, Wang et al. discovered three binding modes between octadecyltrimethylammonium bromide (OTAB) and perfluoro-3-methylundecanoic acid (PFMeUPA). Among these, the electrostatic and hydrophobic synergistic effect accounted for the highest proportion and exhibited the lowest binding energy, which was identified as the dominant adsorption mechanism (Fig. 3a–c).53 Santiago et al. further validated this through MD simulations and electrosorption experiments, revealing that short-chain PFAS adsorb via both amino groups and fluorine-containing groups, with the strength of electrostatic interactions surpassing that of fluorophilic interactions.9 As shown in Fig. 3d and e, when there is no fluorine group in the material, the groups containing amine will agglomerate, making it difficult for PFAS to contact the binding site. However, when the proportion of fluorine groups increases to 31%, this agglomeration is prevented, the material structure is looser, and PFAS can find the binding site more easily.9
 | ||
| Fig. 3 (a–c) Three types of adsorption modes between OTAB and PFMeUPA.53 Copyright 2023, American Chemical Society. Snapshots from copolymer MD simulations: (d) PFBA/P(A) and (e) PFBA/P(A69F31). Reproduced with permission.9 Copyright 2023, American Chemical Society. | ||
From a thermodynamic perspective, the study by Willemsen et al. noted that PFAS adsorption is an entropy-driven process, and the entropy increase rises linearly with the number of CF2 segments. This indicates that the hydrophobic effect makes a significant contribution to the adsorption of long-chain PFAS.62
Additionally, atomic-scale MD simulations by Lamb et al. demonstrated that the pH of the solution exerts a strong influence on the aggregation and adsorption behaviors of PFAS. Under neutral conditions, the headgroups of PFAS undergo deprotonation, and electrostatic repulsion leads to the formation of micelle-analogous assemblies that impede adsorption. In contrast, at acidic pH, the headgroups become protonated, electrostatic repulsion is reduced, and PFAS are more susceptible to be fully adsorbed on the graphene surface in a stacked manner.63 Collectively, these studies establish a systematic understanding spanning molecular dynamic behaviors, interfacial interaction mechanisms, and external condition regulation-highlighting the crucial role of MD simulations in elucidating the environmental behaviors of PFAS.64
Theoretical calculations, leveraging the synergy and complementarity of DFT and MD, have emerged as pivotal tools for unraveling electrochemical PFAS degradation mechanisms and guiding rational technology design. DFT can resolve reaction pathways and activation barriers at the electronic scale, whereas MD captures dynamic interfacial behaviors and mass–transfer processes across broader spatiotemporal scales. Their integration bridges electron-transfer events with macroscopic kinetics, moving PFAS remediation from trial and error experimentation toward mechanism informed optimization.44 Importantly, these theory tools also provide a direct data foundation for AI-enabled HTS: automated DFT/MD workflows can generate standardized descriptors and labels, such as PFAS adsorption energies on candidate electrode surfaces, key C–F bond cleavage and electron transfer barriers, interfacial solvation/ion-pairing characteristics, and stability relevant energetics, which are difficult to measure comprehensively in experiments.65 When combined with high-throughput experimental outputs, such theory-derived features enable machine-learning models to learn structure activity relationships, prioritize promising materials and operating windows, and iteratively refine predictions through experimental validation.10
3 AI enabled high throughput screening for PFAS degradation
The exploration of electrode and catalyst remains a central bottleneck for electrochemical PFAS destruction because performance is jointly governed by PFAS adsorption at electrified interfaces, kinetic barriers of key steps, reactive-species generation, suppression of side reactions, and long-term stability in realistic matrices.66 As a result, conventional one by one material optimization is often inefficient and provides limited guidance for PFAS destruction.67 AI-enabled HTS offers an alternative by integrating large-scale experimental and theory derived datasets with machine learning to identify and validate high potential candidates in an iterative, closed loop manner.68In a PFAS oriented workflow, AI-enabled HTS starts from generating mechanism relevant data at scale and then uses models to guide the next experimental or computational steps.69 High throughput experiments can rapidly test material libraries and operating windows using standardized electrochemical protocols, while analytics such as LC-MS/MS and fluoride quantification provide performance labels beyond PFAS removal, including defluorination, current efficiency, and durability.70 In parallel, automated DFT/MD can supply descriptors that are difficult to obtain comprehensively from experiments, such as adsorption energies of representative PFAS on candidate surfaces, as well as energetics associated with electron transfer and C–F bond activation.71 Combining these experimental labels with theory derived descriptors enables supervised models to learn quantitative structure activity relationships and to prioritize candidates for validation.72
Fig. 4 illustrates the key idea of integrating intelligent proposal with rapid feedback. It contrasts the traditional search paradigm with a combined approach that uses a large language model and an evolutionary strategy to propose and refine candidate materials, followed by rapid preparation and high-throughput electrochemical testing.73–76 In a PFAS context, the same pipeline can be used to propose candidate anode/cathode compositions or surface modifications, then quickly test them under standardized PFAS degradation protocols, and iteratively improve the candidate list based on measured removal, defluorination, and stability.77
 | ||
| Fig. 4 (a) The traditional search method. (b) The Combined method using LLM and genetic algorithm. (c) Schematic diagram of high-throughput electrochemical testing. Reproduced with permission.76 Copyright 2025, Wiley-VCH GmbH. | ||
For example, when predicting the adsorption energy of PFOA on candidate electrode surfaces,78 a training set can be constructed based on DFT calculated adsorption energies across representative surfaces,79 defect states,80,81 and terminations,82–86 alongside concise material descriptors that capture compositional and surface electronic properties.87 The resulting model can screen large candidate pools and output a ranked shortlist of materials predicted to exhibit strong PFAS affinity.88 This shortlist can then be refined through targeted calculations and validated via electrochemical degradation experiments.89 Beyond single property predictions, the development of PFAS electrodes is intrinsically multi-objective.90–92 For EO anodes, a high OEP is required to suppress parasitic oxygen evolution and broaden the usable oxidation window, while adequate PFAS affinity promotes interfacial electron transfer and maintains a reactive near-surface zone.93,94 Therefore, model guided multi-objective screening aims to identify optimal tradeoffs rather than a single best solution.95–97
To enable multi-objective optimization in PFAS electrochemistry, AI-enabled HTS is typically built as a workflow that combines descriptor construction, predictive modeling, and candidate generation.98 Feature engineering remains essential because it translates intrinsic material properties into descriptors that are directly relevant to PFAS destruction, such as PFAS adsorption strength, OEP related characteristics for EO anodes, energetics associated with electron transfer and C–F activation, and stability proxies.99–102 Depending on the research objective, different Machine Learning (ML) models can be deployed for prediction, screening, and design.103 For example, supervised learning models trained on curated “composition/structure performance” datasets are frequently used to rapidly predict the performance of untested candidates.104
Fig. 5 highlights the complementary roles of different AI tools in this workflow. Domain adapted language models can support literature triage and structured extraction of PFAS relevant conditions and outcomes (Fig. 5a),69,105 while graph based learning can capture structure property relationships to predict adsorption-related descriptors and redox–relevant properties (Fig. 5b).70,106 In addition, generative models can propose new candidates under explicit performance constraints.107
 | ||
| Fig. 5 (a) Three methods to adapt LLMs for wastewater management. Reproduced with permission.69 Copyright 2025, Springer Nature. (b) The research workflow for HTS of high-performance electrolyte additive molecules. Reproduced with permission.70 Copyright 2025, Wiley-VCH GmbH. (c) LLMs-driven research framework for HER materials. Reproduced with permission.76 Copyright 2025, Wiley-VCH GmbH. | ||
Large Language Model (LLM) driven research frameworks can further assist knowledge base construction and experimental planning.93 Although some demonstrations have been developed for other electrocatalytic reactions, they illustrate a transferable literature to experiment pipeline that can be adapted to PFAS electrochemistry for accelerating information retrieval, suggesting plausible parameter ranges (Fig. 5c).76,108
The key advantage of AI-enabled HTS is its closed loop iteration.109 Through continuous feedback among experiments, computation, and modeling, the screening strategy and model accuracy can be progressively improved.110 In a typical cycle, an initial dataset is used to train a model that proposes high potential candidates. These candidates are then prioritized for high-throughput testing and higher fidelity calculations, and the resulting validation data are added back to update the model.111 Repeating this prediction validation update loop accelerates convergence toward materials that meet target performance criteria.112
Although challenges persist in terms of data, models, and practical applications, with the continuous advancement of AI technologies, the growing accumulation of data resources, and the deepening of interdisciplinary collaboration, AI-enabled HTS technology is expected to accelerate the development of PFAS remediation materials and provide more effective solutions for water resource protection and environmental pollution control.
4 Electrochemical oxidation method for PFAS degradation
Electrochemical oxidation (EO) is an advanced oxidation technology that directly or indirectly degrades pollutants through electrochemical driving.113,114 By applying an external electric field, it drives direct or indirect oxidation reactions of target pollutants at the anode interface, thereby achieving their degradation and mineralization. EO demonstrates distinct advantages for the removal of PFAS, which are characterized by exceptionally strong C–F bonds.115 The efficacy of this technology hinges critically on the selection of anode materials and the oxidation processes induced under high applied potentials. On one hand, specific anode materials can generate highly reactive, surface-adsorbed ·OH or other potent oxidizing species, depending on the electrolyte composition at their surfaces.116 On the other hand, anodes possessing high oxygen evolution overpotentials may facilitate the oxidation of PFAS molecules adsorbed onto their surface through a DET mechanism.117 These robust oxidation pathways effectively attack and cleave the exceptionally stable C–F bonds within PFAS molecules, resulting in their stepwise defluorination and ultimate mineralization into harmless or low-toxicity inorganic end products, such as CO2 and F− ions.118 The efficiency and reaction pathways of anodic oxidation are profoundly dependent on key factors, including the catalytic activity and stability of the electrode material, the composition of the electrolyte, and operational parameters.65 A deep understanding of the mechanisms underlying these influencing factors and their interactions is fundamental for optimizing EO processes, enhancing PFAS removal efficiency, and improving energy utilization. This section will systematically explore the core mechanisms, electrode materials, and key influencing factors in the latest research advancements regarding PFAS degradation via EO.4.1 EO degradation mechanism of PFAS
The core mechanisms for degrading PFAS via EO include direct oxidation and indirect oxidation.119 These two pathways can operate independently or coexist synergistically, depending on the characteristics of electrode materials, electrolyte composition, and operating conditions. Direct oxidation relies on the specific adsorption of PFAS on the anode surface, where DET between the electrode and the adsorbed pollutants cleaves the C–F bonds. Indirect oxidation depends on strong oxidizing species generated at the anode, such as ·OH and SO4˙−. These radicals diffuse into the bulk solution to attack PFAS molecules non-selectively, with efficiency determined by the radical yield and lifetime.120The hydrophobic perfluorinated chains of PFAS easily bind to the hydrophobic regions of electrodes through electrostatic interactions, van der Waals forces, hydrogen bonds, etc., enhancing adsorption stability and optimizing electron transfer pathways.116 Huang et al. revealed that in the amorphous Pd/Ti4O7 system, π conjugation further increased the adsorption energy from −22.3 kJ mol−1 to −38.6 kJ mol−1. EPR and free radical probe experiments confirmed that almost no free radicals, like ·OH, were generated during the degradation process, indicating that the degradation mainly occurred through direct anodic oxidation. Therefore, the increase in adsorption energy directly affects the efficiency of electron transfer.39
The adsorption energy of PFAS with electrodes is crucial, and the high OEP of electrode materials is also key. Only when the applied anode potential exceeds the redox potential of PFAS, DET will occur, which effectively overlaps between the highest occupied molecular orbital (HOMO) of PFAS and the conduction band level of the electrode with high oxygen evolution potential, thus forming a direct electron transfer channel. Electrons are transferred directly from the adsorbed PFAS molecule to the anode.41 Schaefer et al. verified that when the potential exceeds the oxidation potential of PFOS, adding the strong ·OH scavenger tert-butanol has little effect on the degradation rate of PFOS. This is completely consistent with the DFT-predicted DET pathway that “does not require ·OH mediation”.121
Wang et al. elaborated on the mechanism of electrochemical degradation of PFOA using a Ti3+/TiO2-NTA anode (Fig. 6). Due to the pKa value of PFOA being approximately 0, under the electrolysis experimental conditions, PFOA molecules are almost completely ionized into C7F15COO−.39 This anion loses an electron through DET on the anode surface, generating an unstable C7F15COO˙ radical, marking the initiation of the degradation process (eqn (1)). Notably, Ti3+ self-doping significantly enhances the electrical conductivity of TiO2 nanotubes, while Ti–F bonds formed on the anode surface promote the DET reaction through coulombic forces.122 The generated C7F15COO˙ radical immediately undergoes a Kolbe decarboxylation reaction, producing a 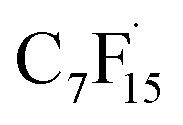 radical and CO2 (eqn (2)). This perfluoroalkyl radical
radical and CO2 (eqn (2)). This perfluoroalkyl radical  is attacked by ·OH generated near the anode, forming thermodynamically unstable C7F15OH (eqn (3)). Subsequently, C7F15OH undergoes intramolecular rearrangement (eqn (4)) and hydrolysis (eqn (5)), breaking a CF2 unit and releasing HF. Through repeated “unzipping” cycles, PFOA is ultimately mineralized into CO2 and HF. The experiment detected six short-chain PFCAs as intermediates, whose concentrations first increased and then decreased, confirming they serve as degradable intermediates. After 90 minutes of electrolysis, the system achieved remarkable mineralization: the total organic carbon (TOC) removal rate reached 93.3%, the defluorination rate was 74.8%, and an average of 12.8 F− ions were released per degraded PFOA molecule.122 These data strongly demonstrate that the electrochemical system efficiently converts PFOA into inorganic end products.
is attacked by ·OH generated near the anode, forming thermodynamically unstable C7F15OH (eqn (3)). Subsequently, C7F15OH undergoes intramolecular rearrangement (eqn (4)) and hydrolysis (eqn (5)), breaking a CF2 unit and releasing HF. Through repeated “unzipping” cycles, PFOA is ultimately mineralized into CO2 and HF. The experiment detected six short-chain PFCAs as intermediates, whose concentrations first increased and then decreased, confirming they serve as degradable intermediates. After 90 minutes of electrolysis, the system achieved remarkable mineralization: the total organic carbon (TOC) removal rate reached 93.3%, the defluorination rate was 74.8%, and an average of 12.8 F− ions were released per degraded PFOA molecule.122 These data strongly demonstrate that the electrochemical system efficiently converts PFOA into inorganic end products.
| C7F15COO− → C7F15COȮ + e− | (1) |
 | (2) |
 | (3) |
| C7F15OH → C6F13COF + H+ + F− | (4) |
| C6F13COF + H2O → C6F13COO− + 2H+ + F− | (5) |
 | ||
| Fig. 6 Proposed degradation pathways of PFOA by electrochemical oxidation. Reproduced with permission.122 Copyright 2021, Elsevier. | ||
Through experimental and theoretical calculations, Song et al. confirmed that under NaCl-containing conditions, Cl· is the key initial step triggering the degradation reaction of PFOA rather than DET.42 The core mechanism involves the synergistic action between Cl· and ·OH. Experimental results demonstrate that under these conditions, PFOA degradation efficiency reached 89.4–94.9% within 480 minutes, with a corresponding defluorination rate of 38.7–44.1%. The specific degradation pathway is illustrated in Fig. 7a. Gibbs free energy (ΔG) calculations revealed that the ΔG for the Cl·-initiated reaction was 65.57 kJ mol−1, significantly lower than that for the ·OH-initiated reaction (162.65 kJ mol−1). Consequently, the first step of PFOA degradation is predominantly triggered by Cl·, generating the intermediate C7F15COO· (eqn (6)). In contrast, the same reaction without radical involvement exhibited a prohibitively high ΔG of 693.34 kJ mol−1, unequivocally demonstrating that radicals are essential for effective PFOA degradation (Table 1). The resulting C7F15COO· subsequently undergoes decarboxylation. This step exhibits an extremely low activation energy barrier (ΔG‡) of only 1.72 kJ mol−1, facilitating rapid carboxyl group removal from the long-chain PFOA to yield the perfluoroalkyl radical ·C7F15, thereby initiating the carbon chain cleavage process. The ·C7F15 radical, synergistically with ·OH, progressively undergoes chain-shortening, defluorination, and hydrolysis reactions, ultimately forming short-chain PFCAs intermediates and further mineralizing. In summary, within this mechanism, Cl· primarily facilitates the low-energy-barrier initial activation, while ·OH governs the subsequent low-energy-barrier reactions involving chain cleavage, defluorination, and hydrolysis. The complementary functions of these two radicals collectively enable the highly efficient mineralization degradation of PFOA. This study provides a novel theoretical foundation and insights for utilizing electrochemical technologies to remove PFAS from the environment.42
| C7F15COO− + Cl˙ → C7F15COO˙ + Cl− | (6) |
 | (7) |
| S2O82− + ·OH → SO4˙− + HSO4− + 0.5O2 | (8) |
 | ||
| Fig. 7 (a) Scheme of the potential energy surface for the degradation of PFOA. Reproduced with permission.42 Copyright 2023, American Chemical Society. (b) Profile of the potential energy surfaces for the electro-oxidation of C7F15COO−. Reproduced with permission.123 Copyright 2022, American Chemical Society. | ||
| The possible reaction | ΔG (kJ mol−1) | ΔG‡ (kJ mol−1) |
|---|---|---|
| C7F15COO− → C7F15COO˙ | 693.34 | |
| C7F15COO− + Cl˙ → C7F15COO˙ + Cl− | 65.57 | 80.93 |
| C7F15COO− + ·OH → C7F15COO˙ + OH− | 162.65 | 330.16 |

|
−141.28 | 1.72 |

|
−412.92 | No barrier |
| C7F15OH + 2H2O → C6F13COF + HF + 2H2O | 20.81 | 55.92 |
| C6F13COF + 2H2O → C6F13COOH + HF + H2O | −39.08 | 114.29 |
Chen et al., through DFT simulations and experimental verification, proposed that the electrochemical degradation of PFOA is dominated by a direct oxidation pathway driven by holes. It achieves rapid mineralization through the “decarboxylation–hydroxylation–carbon chain cleavage” cycle and does not produce the short-chain PFCAs recognized traditionally.123Fig. 7b presents the reaction free energy change profile. The black parts represent the steps shared by the major and minor pathways, the red part represents the major pathway, and the blue part represents the minor pathway. In the major pathway, the ΔG‡ of each step is extremely low (6.24, 1.7, 5.11, 13.3 kJ mol−1), and the ΔG continuously decreases, with the reaction proceeding rapidly and spontaneously: during decarboxylation, ΔG drops sharply by – 145.9 kJ mol−1, highlighting its core triggering role; in the alkyl radical hydroxylation stage, ΔG drops directly from – 145.9 to – 565 kJ mol−1, with no obvious energy barrier and completion in an instant. In contrast, in the hydrolysis minor pathway, the transition states (such as TS RIV-b, TS RV-b) have high energy and large ΔG‡, the reaction is slow, and only a small amount of short-chain PFCAs are generated. Overall, it verifies the degradation mechanism that “hole oxidation is the core driving force, ·OH assists, and hydrolysis is secondary”, and clearly presents the thermodynamic and kinetic differences as well as the primary–secondary relationships of different pathways.
Zhao et al. innovatively proposed the electrochemical activation mechanism of persulfate (PS) in the BDD/PS system for the degradation of PFAS.124 The electrooxidation of PFAS initiates from DET on the surface of BDD. Subsequently, under the action of reactive oxygen species (ROS), structural units of PFAS are gradually removed, shortening the carbon chain, and ultimately mineralizing into CO2, H2O, and F− (Fig. 8a). On one hand, PS can be directly activated to generate SO4˙− (eqn (7)). On the other hand, ·OH produced by the electrolysis of water can also react with PS to form SO4˙− (eqn (8)). Compared with ·OH, SO4˙− has a longer half-life and can efficiently degrade PFAS within a wide pH range (2–8). Meanwhile, its strong electrophilicity makes it more suitable for attacking the hydrophobic perfluorocarbon chains of PFAS. In addition, the BDD anode can promote the conversion of PS into a highly reactive transition state PS* through discharge, enabling the degradation of PFAS via a non-radical pathway. After activation by the BDD anode, PS destroys the C–F bonds of PFAS with great force through a synergistic action mode of radical and non-radical pathways. Pierpaoli et al. also explained the electrochemical oxidation reaction pathways of PFOA and PFOS, and the results are shown in Fig. 8b. PFOA undergoes direct electron transfer to generate radicals, followed by decarboxylation, and then undergoes gradual chain scission and defluorination upon interaction with oxygen or hydroxyl radicals. PFOS undergoes defluorination first, and then the sulfonic acid group decomposes, and the carbon chain is gradually shortened through reactions with water and hydroxyl radicals. Eventually, both are converted into inorganic products such as CO2, HF, and SO42−.125
 | ||
| Fig. 8 (a) Proposed degradation pathway of PFOA and PFOS in the BDD/PS system. Reproduced with permission.124 Copyright 2024, Elsevier. (b) The possible reaction pathway for the PFOA and PFOS. Reproduced with permission.125 Copyright 2020, Elsevier. | ||
In direct oxidation, PFAS molecules adsorb onto the electrode surface and undergo DET. This process aids in breaking the C–F bonds, but it requires overcoming significant adsorption free energy barriers and high electrode potentials. In contrast, indirect oxidation achieves the deep mineralization of PFAS through non-selective radical attacks. However, it still faces challenges in effectively cleaving C–F bonds. Given the strength of C–F bonds, direct and indirect oxidation often act in concert. Taking the BDD electrode as an example, it efficiently degrades the adsorbed PFAS via the DET mechanism on its surface, while the ·OH radicals generated at the anode oxidize the pollutants in the solution phase. This synergistic mechanism of direct and indirect oxidation enhances the overall degradation efficiency by up to 40% compared to single–action pathways.114 Future research should focus on the kinetics of C–F bond cleavage, the interfacial electron transfer mechanism in synergistic systems, and the transformation pathways of highly stable intermediate products.
4.2 Investigation of parameters in EO for PFAS degradation
Core parameters of different anode materials, such as electron transfer capacity, distribution of catalytic active sites, ·OH generation efficiency, OEP, interfacial reaction kinetics, and chemical stability, directly regulate the mineralization efficiency of PFAS and the distribution of intermediate products. An ideal anode material must simultaneously satisfy the following requirements: a high OEP to suppress competitive oxygen evolution reactions, excellent chemical stability to adapt to long-term operation in complex electrolyte environments, and optimized surface properties to enhance PFAS adsorption affinity and reduce electron transfer impedance. For these reasons, materials such as BDD,127 Magnéli phase Ti4O7, PbO2, and SnO2 have become the focus of current research. A systematic analysis of their performance differences (the comparison of PFAS removal efficiencies shown in Table 2), action mechanisms, and application potential is of key significance for advancing the development of EO technologies.128
| PFAS | Initial concentration | Anode material | Cathode material | Electrolyte | pH | Current density (mA cm−2) | Applied potential (V) | PFAS removal (%) | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Perfluorooctanoic acid (PFOA); perfluorooctane sulfonate (PFOS); perfluorohexane sulfonate (PFHxS); perfluorobutanoic Acid (PFBA); hexafluoropropylene oxide dimer Acid (GenX); 6:2 fluorotelomer carboxylic acid (6:2 FTCA); perfluorobutane sulfonate (PFBS); perfluorohexane sulfonate (PFHxS); perfluorohexanoic acid (PFHxA); perfluoropentanoic acid (PFPeA); perfluoroheptanoic acid (PFHpA); perfluorononanoic acid (PFNA); perfluorodecanoic acid (PFDA); perfluoroundecanoic Acid (PFUnA); 6:2 fluorotelomersulfonic acid (6:2 FTS). | |||||||||
| PFOA | 1 mg L−1 | BDD | Ti | Na2SO4 + NaCl | 3–7 | 40 | 25 | 94 | 33 |
| PFOS | 89.1 | ||||||||
| PFHxS | 88.1 | ||||||||
| PFOA | 50 µM | BDD | Ti | PS | 3.8 | 40 | 99.9 | 124 | |
| PFOS | |||||||||
| PFOS | 100 mg L−1 | BDD/SnO2–F | Pt | NaCl | 3 | 30 | 95.6 | 129 | |
| PFOA | BND | Pt | Na2SO4 | 4.8 | 4 | 2.5 V vs. SCE | 99.3 | 130 | |
| PFOA | 20 mg L−1 | BDD | Stainless steel | Na2SO4 | 7 | 10 | 97.9 | 131 | |
| PFBA | 65.6 | ||||||||
| GenX | 84.9 | ||||||||
| 6:2 FTCA | 99.4 | ||||||||
| GenX | 0.25 mmol L−1 | BDD | Gr–Ni–foam | K2SO4 | 3 | 16 | 92.2 | 132 | |
| PFOA | 0.1 mg L−1 | BDD | Stainless steel | K2HPO4 + H3PO4 | 7 | 25 | 89 | 125 | |
| PFOS | 84 | ||||||||
| PFOA | 1350 ng L−1 | BDD | Stainless steel | 7.8 | 75 | 80 | 125 | ||
| PFOS | 3280 ng L−1 | 78 | |||||||
| PFOA | 0.1 mg L−1 | BDD | Stainless steel | K2HPO4 + H3PO4 | 7 | 75 | 99.9 | 125 | |
| PFOS | 84 | ||||||||
| PFOA | 100 mg L−1 | Ti/SnO2–Sb | Ti | NaClO4 | 5 | 10 | 9 | 90.3 | 133 |
| PFOA | 100 mg L−1 | Ti/SnO2–Sb/PbO2 | Ti | NaClO4 | 5 | 10 | 9 | 91.1 | 133 |
| PFOA | 100 mg L−1 | Ti/SnO2Sb/MnO2 | Ti | NaClO4 | 5 | 10 | 9 | 31.7 | 133 |
| PFOS | 20 µmol L−1 | (Ti1−xCex)4O7 | Stainless steel | Na2SO4 | 20 | 2.5 | 98.9 | 31 | |
| PFOS | 20 µmol L−1 | Ti4O7 | Stainless steel | Na2SO4 | 20 | 2.5 | 86.2 | 31 | |
| PFOS | 0.0929 mmol L−1 | Ti/TiO2-NTs/Ag2O/PbO2 | Ti | NaClO4 | 30 | 74.87 | 134 | ||
| PFBS | 5.39 | ||||||||
| PFHxS | 15.43 | ||||||||
| PFOS | 0.0929 mmol L−1 | Ti/PbO2 | Ti | NaClO4 | 30 | 41.59 | 134 | ||
| PFOS | 0.0929 mmol L−1 | Ti/TiO2–NTs/PbO2 | Ti | NaClO4 | 30 | 50.87 | 134 | ||
| PFOS | 0.2 µM | BRGO sponge | Spontex | Na2HPO4/NaH2PO4 | 7.2 | 230 | 7.6 V vs. SHE | 67 | 37 |
| PFOA | 48.9 | ||||||||
| PFBA | 16.6 | ||||||||
| PFBS | 38.6 | ||||||||
| PFHxS | 49.1 | ||||||||
| PFHxA | 32.8 | ||||||||
| PFOA | 0.5 mM | Ti4O7 | Stainless steel | NaClO4 | 5 | 3.9 | 99.9 | 135 | |
| PFOA | 0.5 mM | Ce–PbO2 | Stainless steel | NaClO4 | 5 | 3.5 | 99.5 | 135 | |
| PFOA | 0.5 mM | BDD | Stainless steel | NaClO4 | 5 | 3.8 | 98.8 | 135 | |
| PFOS | 0.1 mM | Ti4O7 | Stainless steel | NaClO4 | 5 | 3.9 | 93.1 | 135 | |
| PFBA | 100 mg L−1 | Ce–PbO2 | Ti | NaClO4 | 20 | 31.8 | 136 | ||
| PFPeA | 41.4 | ||||||||
| PFHxA | 78.2 | ||||||||
| PFHpA | 97.9 | ||||||||
| PFOA | 96.7 | ||||||||
| PFOA | 2.0 µM | Ti4O7 | Stainless steel | Na2SO4 | 7 | 5 | 3.322 V vs. SHE | >90 | 47 |
| PFOS | |||||||||
| PFOA | 4.14 mg L−1 | Ti4O7 REM | Stainless steel | K2HPO4 | 3.3 V vs. SHE | 99.9 | 137 | ||
| PFOS | 5 mg L−1 | Ti4O7 REM | Stainless steel | K2HPO4 | 7 | 3.6 V vs. SHE | 99.9 | 137 | |
| PFOA | 50 mg L−1 | Ti3+/TiO2-NTA | Ti | Na2SO4 | 3–11 | 2 | 98.1 | 122 | |
| PFOA | 50 mg L−1 | Ti/SnO2–Sb–Bi | Ti | NaClO4 | 4.7 | 22 | 3.37 V vs. SCE | 99 | 138 |
| PFNA | 0.25 mmol L−1 | Ti/SnO2–Sb–Ce | Ti | NaClO4 | 3 | 10 | 95.8 | 34 | |
| PFDA | 88.7 | ||||||||
| PFNA | 0.25 mmol L−1 | Ti/SnO2–Sb/Ce–PbO2 | Ti | NaClO4 | 3 | 10 | 97.1 | 34 | |
| PFDA | 92.2 | ||||||||
| PFNA | 0.25 mmol L−1 | Ti/BDD | Ti | NaClO4 | 3 | 10 | 98.7 | 34 | |
| PFDA | 96 | ||||||||
| GenX | 10 mg L−1 | N-GS/CeO2@Ti4O7 | Stainless steel | NaClO4 | 20 | 3.55 | 97 | 139 | |
| GenX | 10 mg L−1 | Ti4O7 | Stainless steel | NaClO4 | 20 | 3.55 | 86.8 | 139 | |
| GenX | 10 mg L−1 | Ti/RuO2–Ir7 | Stainless steel | NaClO4 | 20 | 3.55 | 50 | 139 | |
| PFOS | 2.0 µM | Ti4O7 REM | Stainless steel | Na2SO4 | 40 | 1.6–3.5 V vs. SHE | 90 | 54 | |
| PFOA | 20 ppm | Ti4O7 REM | Carbon felt | Na2SO4 | 28.6 | 14 | 92 | 140 | |
| PFOS | 50 mg L−1 | 3DG-PbO2 | Stainless steel | Na2SO4 | 7 | 30 | 96.2 | 38 | |
| PFOA | 0.12 mM | Ti4O7/Pd | Ti | Na2SO4 | 7.2 | 10 | 6 | 86.7 | 39 |
| PFUnA | 0.093 ng L−1 | Ti–ZnO | Stainless steel | None | 7.23 | 20 | 46.43 | 141 | |
| PFDA | 0.055 ng L−1 | 53.25 | |||||||
| PFNA | 0.046 ng L−1 | 45.45 | |||||||
| PFOA | 5.43 ng L−1 | 65.84 | |||||||
| PFHpA | 3.52 ng L−1 | 58.03 | |||||||
| PFHxA | 2.77 ng L−1 | 49.31 | |||||||
| PFPeA | 2.73 ng L−1 | 44.79 | |||||||
| PFBA | 2.95 ng L−1 | 40.74 | |||||||
| PFOS | 4.74 ng L−1 | 63.42 | |||||||
| PFHxS | 3.64 ng L−1 | 39.66 | |||||||
| PFBS | 0.86 ng L−1 | 38.95 | |||||||
| PFOA | 100 mg L−1 | Ti/SnO2–Sb2O5/PbO2-PVDF | Ti | NaClO4 | 3 | 40 | 5–15 | 92.1 | 142 |
| PFOS | 100 mg L−1 | Ti/Sn–Sb/SnO2 −F–Sb | Ti | NaClO4 | 3 | 20 | 99.6 | 36 | |
| PFOA | 100 µmol L−1 | Ti | CNTs-graphene | 3.6 | 0.6 | 96.9 | 143 | ||
| PFOS | 97 | ||||||||
| 6:2 FTS | 20 mg L−1 | Ti/SnO2–Sb2O5–Bi2O3 | NaClO4 | 6.94 | 6.8 | 5 | 26.8 | 144 | |
| PFOS | 5.5 | ||||||||
BDD electrodes demonstrate significant advantages in PFAS treatment due to their wide potential window, high OEP, and unique dual degradation mechanism combining DET with synergistic indirect ·OH oxidation.121 The BDD anode itself has high chemical stability and is not easily corroded. As shown in Fig. 9a, Zhao et al. compared the surface structure of the BDD anode in its original state with that after the EO process, clearly demonstrating the performance advantages of the BDD anode.124 Zhuo et al. prepared four kinds of electrodes, BDD, BDD/SnO2, BDD/PbO2 and BDD/SnO2–F, and their morphologies are shown in Fig. 9b respectively.129 Among them, BDD/SnO2-F has finer and dispersed surface particles due to the introduction of fluorine. This change in morphology and material is crucial to the degradation performance. Through finer dispersed particles, it increases the specific surface area and OEP, and can more efficiently adsorb and degrade 6:2 chlorinated polyfluoroether sulfonate (F-53B), so that its degradation rate of F–53B can reach 95.6% in 30 minutes, which is much better than other electrodes (Fig. 9c).129 Olvera-Vargas et al. confirmed through DFT calculations that GenX requires overcoming an energy barrier of 3.2 eV (E° = 3.65 V vs. SHE) for oxidation in acidic media. The upper limit of the BDD electrode's potential window (approximately 3.0 V vs. SHE) is significantly higher than that of Pt (1.9 V vs. SHE), thermodynamically satisfying the conditions for DET reactions.132 Simultaneously, at high potentials, BDD generates surface-adsorbed ·OH via the water discharge reaction, effectively enhancing indirect oxidation while suppressing electrode poisoning caused by intermediate product adsorption. This dual mechanism, “DET-initiated C–F bond cleavage followed by surface ·OH-mediated deep mineralization,” is key to its efficient PFAS degradation. Optimizing the electrode's microstructure is crucial for enhancing BDD degradation efficiency.145 Saleh et al. found that the degradation rate of PFBA-C4 was only 14% after 6 hours using a planar BDD electrode, whereas a mesh-plate composite electrode ((M + P)-BDD) achieved 97% degradation within 4 hours at 20 mA cm−2. This disparity stems from the three-dimensional mesh structure of (M + P)-BDD, which increases the specific surface area to promote greater ·OH generation through water ionization while reducing surface resistance, significantly optimizing reaction kinetics. For degrading PFHxA-C6, the removal rate achievable with P-BDD at 20–25 V was attained by (M + P)-BDD at only 6–8 V, effectively reducing voltage loss and energy consumption. This indicates that electrode structural design can fully unlock the application potential of BDD in PFAS degradation.146
 | ||
| Fig. 9 (a) SEM of BDD. Reproduced with permission.124 Copyright 2024, Elsevier. (b) SEM images of BDD, BDD/SnO2, BDD/PbO2, and BDD/SnO2–F, respectively. (c) Degradation ratios and the first-order kinetics analysis. Reproduced with permission.129 Copyright 2019, Elsevier. | ||
Compared with the costly BDD anode, titanium suboxide (TSO) has significant cost advantages (approximately $0.36/m2), a high OEP (>2.5 V vs. SHE), good electrical conductivity (∼1000 S cm−1), and excellent chemical stability. It is regarded as a powerful alternative to traditional anodes such as BDD in the field of EO degradation of PFAS. Moreover, the preparation process of TSO anodes usually avoids the use of toxic dopants like Sb and Pb, further reducing environmental risks.29,47 Under neutral pH conditions, the degradation selectivity of TSO for PFAS is generally higher than that of BDD electrodes, which helps reduce the oxygen evolution reaction (OER) side reaction. Theoretical calculations by Wang et al. showed that the energy barrier for C–S bond cleavage of PFOS on Ti4O7 is lower than that on Ti9O17. Experiments have confirmed that, benefiting from the higher Ti3+ content and optimized pore size, Ti4O7 anodes have more advantages in PFOS degradation efficiency and energy efficiency (Fig. 10a).54 A study by Gomri et al. using a Ti4O7-based reactive electrochemical membrane (REM) anode showed that increasing the current density and feed concentration, and reducing the REM flux can effectively improve the PFOA removal rate.140 Le et al. used an energy-saving Magnéli phase Ti4O7-REM anode to treat PFOA and PFOS, which not only achieved a high removal rate but also had an low energy consumption. The high specific surface area and flow-through operation of this technology significantly enhanced mass transfer and reaction rates.137
 | ||
| Fig. 10 (a) Energetics of PFOS with Ti4O7 and Ti9O17 clusters.54 Copyright 2021, Elsevier. (b) Ti4O7 ceramic materials. (c) Concentration changes during the electrooxidation process. Reproduced with permission.135 Copyright 2018, Elsevier. (d) SEM image of the fabricated (Ti1-xCex)4O7 (x = 3%) electrode and corresponding EDX elemental mappings, and the concentration change of PFOS oxidation. Reproduced with permission.31 Copyright 2021, American Chemical Society. | ||
The high overall porosity and interconnected porous network structure of Ti4O7 electrodes significantly promote mass transfer of PFAS and increase the reaction area. Combined with its large electroactive surface area and low local current density, high electrolysis efficiency can be achieved. The macroporous Magnéli phase Ti4O7 ceramic anode prepared by Lin et al. has an electroactive surface area 1302 times larger than its geometric area (Fig. 10b).135 This anode, which combines a high OEP (2.7 V vs. SCE), significant ·OH generation capacity, and DET capability, exhibits a higher oxidation rate for PFOA/PFOS than Ce-doped PbO2 and Ti/BDD electrodes(Fig. 10c).135 Despite these significant advantages, Ti4O7 electrodes still face challenges such as low interfacial charge transfer rate and insufficient ·OH yield.147 Ti4O7-based composite materials designed through heterostructure construction and defect engineering can further improve performance while maintaining stability and low-cost advantages. For example, Lin et al. introduced additional oxygen vacancies into Ti4O7 electrodes through Ce3+ doping, which optimized the electronic structure, reduced charge transfer resistance, promoted ·OH generation, and significantly enhanced electrocatalytic performance(Fig. 10d).31 The amorphous Pd-loaded Ti4O7 electrode developed by Huang et al. achieved efficient direct anodic oxidation of PFOA by enhancing electron transfer and pollutant binding capacity, and the degradation process was less susceptible to quenching by common anions.39
In the field of EO for PFAS degradation, element doping, material compositing, and porous structure design have emerged as core strategies to enhance anode performance. These strategies significantly strengthen the electrode's ability to degrade PFAS by precisely regulating the material's electronic structure, interfacial properties, and microtopography.
Element doping improves the degradation efficiency of PFAS by modifying the electronic structure and surface abilities of electrode materials. Rare earth element doping exhibits significant effects. For instance, Niu et al. found that doping rare earth elements into titanium-based lead dioxide electrodes can effectively increase the density of surface defects and the content of reactive oxygen species, promote the activation of water molecules and the generation of ·OH, while enhancing the corrosion resistance and stability of the electrode, thus greatly improving the adsorption and oxidation efficiency of PFAS. The prepared Ce-doped porous nanocrystalline PbO2 electrode showed superior performance in treatment capacity, degradation efficiency, and mineralization degree compared to the Ti/SnO2–Sb electrode when treating C4–C8 PFCAs.136 Non-metallic element doping is also effective. Liu et al. designed a B/N co-doped diamond (BND) electrode, which regulates the electronic structure through the synergistic effect of B and N.130 In addition, the PVDF-doped modified PbO2 electrode, by inhibiting grain growth and enhancing hydrophobicity, extended the electrode lifespan to 45.75 hours, and the PFOA degradation rate was improved to 92.1%.142
Composite electrodes achieve performance complementarity and synergistic enhancement by combining the characteristics of different materials. The combination of highly catalytically active metal oxides, such as MnO2 and RuO2, with conductive substrates like titanium mesh and graphite felt can synergistically promote the generation of ·OH while ensuring good conductivity and mechanical stability. The Ti3+ self-doped TiO2 nanotube array (Ti3+/TiO2-NTA) anode prepared by Wang et al., which combines self-doping with a nanotube structure, achieved a PFOA degradation rate of 98.1%, a TOC removal rate of 93.3%, a defluorination rate of 74.8%, and an energy consumption of only 7.6 Wh/L after 90 minutes of electrolysis at a current density of 2 mA cm−2.122 The Ti/SnO2–Sb–Bi anode and Ti/TiO2−NTs/Ag2O/PbO2 anode developed by Zhuo et al., by integrating various materials and constructing multi-layered interface architectures, electron transfer and the generation of reactive oxygen species are further facilitated, allowing for the efficient degradation of PFOA, PFOS, and their alternative compounds.134,138 The Ti/Sn–Sb/SnO2–F–Sb electrode prepared by Yang et al., through the co-doping of F and Sb combined with a Sn–Sb interlayer, extended the electrode lifespan from 80.1 h to 103.1 h, with a PFOS removal rate exceeding 99% within 120 minutes.36
Porous design enhances electrochemical reaction efficiency by increasing the specific surface area of electrodes and promoting mass transfer. The N-GS/CeO2@Ti4O7 heterojunction composite membrane electrode prepared by Liang et al. utilizes the porous structure and the “cementing” effect of CeO2 to achieve stable encapsulation of N-GS. Its high specific surface area (3.2 m2 g−1) and defect sites accelerate charge transfer, reducing the Rct to 5 Ω. In the EO of GenX, the degradation efficiency reaches 97% within 120 minutes, and the defluorination rate reaches 70–90% when treating actual wastewater.139 The three-dimensional (3D) graphene-lead dioxide (3DG-PbO2) composite anode developed by Duan et al. optimizes the crystal size of PbO2 through the 3D network structure of 3DG and enhances ·OH generation capacity. The degradation rate constant of PFOS by this anode is 2.33 times that of the pure PbO2 anode.38 In addition to such structural designs, porous electrodes composed of different material combinations also exhibit excellent performance.34,133
It is crucial to emphasize that high PFAS removal efficiency does not strictly equate to deep mineralization. Many modified anodes rapidly deplete parent PFAS molecules but yield low defluorination ratios, indicating the accumulation of recalcitrant short-chain intermediates. This discrepancy highlights the necessity of matching the electrode's microstructural design with the unique physicochemical properties of PFAS. Since PFAS are amphiphilic molecules with highly stable perfluorinated tails, optimizing the interfacial wettability of the electrode can forcibly drive these molecules into the Helmholtz layer, thereby enhancing DET. Furthermore, simply increasing the macroscopic surface area is insufficient. The rational design of hierarchical porous structures introduces a spatial confinement effect. These tailored microporous and mesoporous networks not only selectively enrich PFAS through hydrophobic interactions but also trap the generated ROS and intermediate short-chain fragments within the confined microenvironment. By significantly prolonging the residence time of intermediates near the active sites, this microstructural regulation effectively bridges the gap between superficial removal and ultimate defluorination.
The core role of chlorine-containing electrolytes (such as NaCl) is to enhance degradation by generating reactive chlorine species (Cl·, ClO·, HClO). Their oxidation potential (Cl·/Cl−: E° = 2.4 V vs. SHE) is higher than that of ·OH (E° = 2.0 V), enabling efficient attack on C–F bonds. Meanwhile, Cl− can react with ·OH to form ClOH˙−, which is then converted into Cl·, further enhancing the oxidizing capacity.129 However, their effectiveness is influenced by electrode materials. In the BDD electrode system, the presence of Cl− has less than a 20% impact on the degradation efficiency of PFOA/PFOS, because the generated perchlorates and other substances have limited competition with the DET pathway.121,133 Zhuo et al. found that on Ti/PbO2 electrodes, an appropriate amount of Cl− (<10 mM) increased the defluorination rate of F-53B from 72.9% to 77.4%, but high concentrations of Cl− could cause passivation of the surface of electrodes such as PbO2. However, a drawback of this system is the easy formation of toxic by-products like ClO4−, and when the Cl− concentration exceeds 10 mM, the amount of ClO4− produced is positively correlated with the degradation efficiency of PFAS.65
Inert electrolytes (such as NaNO3, NaClO4), due to the lack of participation of active anions, rely only on DET and indirect oxidation by ·OH, so the degradation rate of PFAS is usually lower than that in systems containing active electrolytes.130,131 Their advantage is that they can avoid the formation of toxic by-products like ClO4−, making them suitable for scenarios with high requirements for product purity.
Different from chlorine-containing electrolytes, the role of SO42− in sulfate systems (represented by Na2SO4) is dual. On one hand, SO42− may react with ·OH to form S2O82− with weaker oxidizing ability, resulting in a decrease in PFAS degradation efficiency.129 On the other hand, SO42− can be electrochemically activated to generate strongly oxidizing SO4˙− (E° = 2.5–3.1 V), and at this time, the degradation rate of PFOA is 2.3–3.4 times higher than that in systems with inert electrolytes (NaNO3, NaClO4).130
The core role of strong alkaline electrolytes (such as KOH, LiOH) is to enhance mass transfer by constructing a strong interfacial electric field. For example, high concentrations of OH− can accumulate in the nanopores of the electrode, forming a strong interfacial electric field, which increases the mass transfer coefficient of PFAS to the anode from 0.01 cm s−1 to 0.03 cm s−1.122 Extreme alkaline conditions (such as 8.0 M LiOH) can also compensate for the potential gap through the electric field, achieving complete defluorination of PFOS.148
In summary, different electrolyte systems affect the efficiency of EO degradation through differentiated mechanisms, and their selection needs to be comprehensively considered in combination with degradation efficiency, by-product risks, and practical application scenarios.
Unlike their long-chain counterparts, short-chain PFAS lack a substantial hydrophobic tail, rendering conventional adsorption mechanisms driven by hydrophobic interactions highly inefficient. Consequently, these highly soluble molecules easily escape the anodic oxidation zone before deep mineralization can occur. Nienhauser et al. confirmed that the longer the carbon chain, the easier the degradation of PFAS (Fig. 11a), and that the long-chain PFAS will reduce the electrode contact angle (Fig. 11c).149 However, in multi-component systems, the degradation of short-chain PFAS is significantly enhanced because they co-aggregate with long-chain homologues to form mixed micelles(Fig. 11b and d).149
 | ||
| Fig. 11 (a) BDD electrodes for single-solute solutions of each PFCA. (b) Degradation effect of multi-solute model wastewater matrix. (c) Sessile drop surface contact angle. (d) Schematic of hetero-micelle formation. Reproduced with permission.149 Copyright 2022, Elsevier. | ||
To address this challenge, electrode materials designed specifically for short-chain PFAS removal should balance hydrophobicity and hydrophilicity to facilitate both adsorption and diffusion. For example, increasing the surface area and incorporating functional groups such as hydroxyl, carboxyl, or sulfonic acid groups can enhance the interaction with short-chain PFAS, facilitating their concentration at the electrode surface. Additionally, the use of porous carbon materials or conductive polymers with controlled pore sizes can improve the mass transfer of short-chain PFAS, providing better access to reactive sites and improving degradation efficiency. The development of electrode materials tailored for short-chain PFAS degradation should focus on enhancing the adsorption efficiency while maintaining high electrochemical activity for the efficient generation of reactive species, such as hydroxyl radicals or sulfate radicals, that are effective in breaking down these more water-soluble contaminants.136
Functional groups directly affect the difficulty of C–F bond cleavage through electronic effects and spatial configurations. For example, Wang et al. found that on TSO electrodes, the degradation rate of PFOS containing sulfonic acid groups (-SO3H) is higher than that of PFOA with carboxylic acid groups (–CO2H). The core mechanism lies in the fact that the high electronegativity of –SO3H can activate adjacent carbon atoms, which is more conducive to the addition reaction of ·OH free radicals.47 Steric hindrance can inhibit degradation efficiency. GenX (C6) containing –CF3 branches has a significantly lower degradation rate (84.9%) than linear-structured PFOA (97.9%) because the branches hinder the contact between the –CO2H and the electrode surface. In contrast, 6:2 FTCA has a degradation rate of up to 99.4% due to the presence of C–H bonds in its molecule that are easily attacked by ·OH, showing the optimal performance.131
In multi-solute systems, the degradation rate of PFAS is generally higher than that in single-solute systems. This improvement is largely ascribed to the micellar structures formed by mixed PFAS on the electrode surface, which effectively reduces the electrode-solution contact angle. Consequently, the contact between short-chain PFAS and the electrode surface is significantly promoted, with their degradation rate increasing by up to 30% compared to that in single-solute systems.149 Even in complex mixed systems containing multiple types of PFAS, although the degradation of PFOA (C8) generates intermediates like PFBA(C4), the overall degradation rate of the system remains higher than that of the corresponding single-solute systems.149 In addition, the actual properties of polluted water sources have a significant impact on the electrochemical oxidation efficiency of PFAS. High electrolyte concentrations in actual industrial wastewater usually facilitate the generation of oxidants, thereby accelerating the degradation of PFAS, and their removal efficiency may even be higher than that in ultrapure water systems. However, when the water body contains excessively high concentrations of organic substances, these organics will compete with PFAS for active sites on the electrode surface or oxidants, thereby inhibiting the degradation efficiency of PFAS.149
In summary, the initial properties of PFAS regulate EO efficiency through multiple mechanisms. Long-chain PFAS enhance degradation kinetics by virtue of their high adsorption energy and the effect of improving interfacial wettability. The –SO3H group accelerates reactions by activating adjacent carbon atoms through its electronegativity. Mixed solute systems produce a synergistic effect through the formation of surface micelles. Considering the complex coexisting environment in actual water bodies, efficient removal of PFAS can be achieved by regulating extreme alkaline conditions or selecting specific electrolytes to strengthen the interfacial electric field or free radical pathways.
Compared with continuous electrolysis, pulsed electrolysis reconstructs the electrode microenvironment through periodic on–off cycles of current, which can promote the reorganization of the electrical double layer, renewal of the diffusion layer, and regeneration of ·OH, reduce electrode polarization, and thus show significant advantages. For example, the defluorination rate of PFOS can be increased to 1.5 times that of continuous electrolysis, achieving complete defluorination in only 60 pulse cycles, while energy consumption is significantly reduced by approximately 5.256 kWh m3. This is mainly attributed to the inhibition of ineffective side reactions and the optimization of charge transfer efficiency.148 Specific pulse parameters can further enhance degradation efficiency.
In summary, strategies such as precise regulation of *j*, adoption of pulsed electrolysis mode, and optimization of system resistance can synergistically improve PFAS degradation efficiency, promote deep removal of short-chain pollutants, and significantly reduce energy consumption. Future research should further explore the mechanisms by which different combinations of current properties affect the degradation of complex PFAS systems and promote the application and transformation of these optimization strategies in practical wastewater treatment engineering.
5 Electrochemical reduction method for PFAS degradation
As an emerging technology, electrochemical reduction (ER) can efficiently degrade C–F bonds in PFAS molecules through the dissociative electron transfer (DET) model or indirect reduction pathways, achieving deep defluorination and demonstrating low energy consumption potential. Compared with EO, ER has significant advantages in energy consumption control and defluorination selectivity, and is particularly suitable for the treatment of long-chain PFAS. Current research mainly focuses on overcoming key technical bottlenecks such as hydrogen evolution reaction (HER) competition, limitations of the electrochemical stability window of solvents, electrode surface passivation, secondary pollution, and low energy conversion efficiency. Optimization approaches include precise design of cathode materials, systematic analysis of reaction mechanisms, and scientific regulation of reaction conditions. The ER research on PFAS removal in recent years is shown in Table 3.| PFAS | Anode‖cathode | Electrolyte | pH | Reaction time (min) | Current density (mA cm−2) | Applied potential (V) | PFAS removal (%) | Ref. |
|---|---|---|---|---|---|---|---|---|
| a 2-(Trifluoromethyl)acrylic acid (TFMAA). | ||||||||
| PFOA (100 mg L−1) | BDD‖Fe3O4-rGA | Na2SO4 | 6 | 720 | 20 | 9.5 | 98.25 | 152 |
| PFOA (100 mg L−1) | BDD‖Cu-rGA | Na2SO4 | 6 | 720 | 20 | 9.5 | 98.91 | 152 |
| PFOA (100 mg L−1) | BDD‖rGA | Na2SO4 | 6 | 720 | 20 | 9.5 | 98.91 | 152 |
| PFOA (100 mg L−1) | BDD‖graphite | Na2SO4 | 6 | 720 | 20 | 9.5 | 81.68 | 152 |
| PFOA (100 mg L−1) | BDD‖Ti | Na2SO4 | 6 | 720 | 20 | 9.5 | 60.28 | 152 |
| PFOA (36 mM) | TiO2 mesh‖carbon paper molecular copper | MeCN + TBA ClO4 | 240 | 5 | 93 | 40 | ||
| PFOA (1 mM) | TiO2 mesh‖Au | KHCO3 | 9.5 | 840 | −1.80 V vs. Ag/AgCl | 41 | ||
| PFMeUPA (100 µM) | Ti sheet‖GAC | Na2SO4 | 420 | 10 | 100 | 61 | ||
| PFOS (100 µM) | 240 | 15 | 10 | |||||
| PFOA (10 mg L−1) | Pt‖Co–CN2–Fe2O3 modified gas diffusion electrode | Na2SO4 | 2 | 120 | −0.06 V vs. RHE | 96.1 | 153 | |
| PFOS (5 ppm) | Pt‖CNT-CTAB | NaClO4 | 11.5 | 120 | −0.58 V vs. Ag/AgCl | 92.2 | 35 | |
| PFOA (100 mg L−1) | Pt‖Rh/Ni | Fe(C5Me5)2 | 1440 | −1.25 V vs. Ag/AgCl | 50.38 | 154 | ||
| TFMAA (5 mg L−1) | Pt‖ZIF-67/C@CP | Na2HPO4·12H2O NaH2PO4·2H2O | 2880 | −2.29 × 10−3 | −1.2 V vs. Ag/AgCl | 99.66 | 155 | |
| TFMAA (5 mg L−1) | Pt‖CP | Na2HPO4·12H2O NaH2PO4·2H2O | 2880 | −2.29 × 10−3 | −1.2 V vs. Ag/AgCl | 18.98 | 155 | |
| TFMAA (5 mg L−1) | Pt‖C@CP | Na2HPO4·12H2O NaH2PO4·2H2O | 2880 | −2.29 × 10−3 | −1.2 V vs. Ag/AgCl | 92.31 | 155 | |
| PFMeUPA (5 mg L−1) | Pt‖MWCNT–COOH–OTAB | Na2HPO4·12H2O NaH2PO4·2H2O | 7 | 10.75 | −1.6 V vs. Ag/AgCl | 99.81 | 53 | |
| GenX (5 mg L−1) | BDD‖Cu | Na2SO4 | 120 | 13.5 | 66 | 156 | ||
| NaCl | ||||||||
| GenX (5 mg L−1) | BDD‖Ti | Na2SO4 | 120 | 13.5 | 52 | 156 | ||
| NaCl | ||||||||
| GenX (5 mg L−1) | BDD‖Au | Na2SO4 | 120 | 13.5 | 92 | 156 | ||
| NaCl | ||||||||
5.1 ER degradation mechanism of PFAS
In the ER of PFAS, the key difference between DET and indirect reduction lies in the electron transfer pathway and the dependence on catalysts.157 DFT calculations indicate that efficient ER of PFAS requires two key conditions: the applied potential is lower than −2 V (vs. SHE), and the reaction transition state energy barrier is less than 40 kJ mol−1.When investigating the ER mechanism of PFOA, Calvillo-Solís et al. first revealed through reaction kinetics analysis that this reaction follows a concerted reaction mechanism—electron transfer and C–F bond cleavage occur simultaneously (electron transfer coefficient α < 0.5), rather than a stepwise mechanism where electron transfer and bond cleavage proceed sequentially (α ≥ 0.5) (Fig. 12a). The realization of this concerted mechanism is closely related to the adsorption behavior of PFOA on the electrode surface: the potential energy curve in Fig. 12b shows that the adsorption of PFOA on the electrode surface can significantly reduce the reaction activation energy barrier, laying a thermodynamic foundation for the simultaneous occurrence of electron transfer and C–F bond cleavage. From the perspective of the specific reaction pathway on the gold electrode surface (Fig. 12c), PFOA first adsorbs on the inner Helmholtz plane of the electrode, then accepts electrons to undergo a reduction reaction, accompanied by C–F bond cleavage and defluorination; the generated intermediate species further interact with water or OH− to complete the subsequent transformation.41 Through theoretical analysis of frontier orbitals and electron density difference (Fig. 12d), the researchers revealed the orbital energy distribution of PFOA molecules, electron transfer sites, and the variation law of molecular electron density after electron injection from the perspective of electronic structure, which provides a key theoretical basis for clarifying the nature of electron transfer.41 Furthermore, the results of free energy calculation (Fig. 12e) show that at a potential of U = −2.84 V s. SHE, the free energy from PFOA reactants, transition state (TS) to reduction products, shows a gradual decreasing trend, which further verifies the feasibility of this concerted reduction pathway from a thermodynamic perspective.158,159
 | ||
| Fig. 12 (a) Two possible mechanisms. (b) Potential energy diagram. (c) Proposed degradation mechanism. (d) HOMO and LUMO orbitals and Electron density difference map. (e) Calculated Gibbs free energy potential energy surface. Reproduced with permission.41 Copyright 2024, American Chemical Society. | ||
In addition, a study by Wang et al. first confirmed that C–F bond cleavage can be achieved through DET alone, and clarified that the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C unsaturated structure in PFAS molecules is the key to promoting C–F bond cleavage because it is more likely to accept electrons, while saturated intermediates are difficult to further defluorinate, which provides a new basis for understanding the structure dependence of PFAS degradation.53
C unsaturated structure in PFAS molecules is the key to promoting C–F bond cleavage because it is more likely to accept electrons, while saturated intermediates are difficult to further defluorinate, which provides a new basis for understanding the structure dependence of PFAS degradation.53
Indirect reduction exhibits stronger universality in PFAS degradation. It relies on the electrolysis of water on the electrode surface to generate ˙H and eaq−, which achieve defluorination through hydrogen–fluorine exchange and hydrogenation reactions, requiring catalyst stabilization.49,160 Liu et al. via DFT calculations demonstrated that the P sites of crystalline cobalt phosphide (CoP) are more favorable for ˙H generation, providing key reactive species for dehalogenation (Fig. 13a). Fig. 13b shows that florfenicol (FLO) and its dehalogenated intermediates can stably adsorb on the CoP surface, and the higher the degree of dehalogenation, the stronger the adsorption, ensuring the continuous progress of the reaction. Fig. 13c presents that the free energy reaction pathway of FLO dechlorination on CoP is thermodynamically spontaneous throughout, and each step is easy to occur.160 Together, these three aspects reveal the intrinsic mechanism of CoP efficiently catalyzing FLO dehalogenation.155
 | ||
| Fig. 13 (a) DFT-calculated free energy pathways. (b) Adsorption energies of FLO and intermediates. (c) Free energy diagram. Reproduced with permission.160 Copyright 2019, American Chemical Society. (d) Proposed mechanisms for the HDF of PFOA on Rh/Ni cathode. Reproduced with permission.154 Copyright 2021, Elsevier. | ||
In the process of decomposing PFOA by ER using a Rh/Ni cathode, Zhu et al. verified that the core of the indirect reduction mechanism is hydrodefluorination (HDF). This mechanism does not involve the direct transfer of electrons to PFOA molecules; instead, it relies on active atomic hydrogen (H) generated in situ at the cathode as a reducing agent to indirectly mediate the cleavage and substitution of C–F bonds in PFOA (Fig. 13d). The H stored in the Rh lattice nucleophilically attacks the adsorbed PFOA molecules, gradually replacing the F atoms therein. Under the attack of H, the C–F bonds break, F− ions detach, and C–H bonds are formed. This stepwise defluorination process is driven by the electrons continuously supplied by the cathode to maintain the reaction momentum, ultimately promoting the gradual degradation of PFOA into short-chain fluorinated intermediates and F− ions.154
Tan et al. from Westlake University found that the mechanism dominated by eaq− is the core pathway for the current cathodic reduction of PFAS. As the strongest reducing agent, eaq− (E0 = −2.87 V vs. SHE) can efficiently break C–F bonds. Further clarification using Marcus' theory and DET calculations shows that the activation free energy of electron transfer (ET) is much higher than that of the defluorination step, confirming that ET is the rate-limiting step rather than the direct cleavage of C–F bonds. The activation free energy of ET depends on the molecular structure of PFAS. For example, the CF2COO− group and the long-chain region are more likely to accept electrons, while ether bonds or short chains will hinder electron transfer.161
Both DET and H pathways have also been confirmed to participate in the reaction synergistically. Grunfeld et al. found that the activated carbon cathode can directly transfer electrons to PFAS through surface functional groups at a potential of −1.4 V vs. SHE, while atomic hydrogen generated by hydrolysis assists in defluorination.61
The catalytic synergistic mechanism is enhanced through the surface modification of electrode materials, with the core being the regulation of reaction pathways by introducing catalytically active sites. For example, the molecular copper electrocatalyst designed by Sinha et al. can achieve a 93% degradation rate and 99% defluorination rate of PFOA through 4-hours cathodic controlled current electrolysis at room temperature, with a current-normalized pseudo-first-order rate constant as high as 3.32 L h−1 A−1, which is far superior to traditional high-temperature, high-pressure, or high-energy-consuming processes.40 The nickel catalyst designed by Doi et al. can drive the multi-step C–F bond cleavage of perfluoroalkyl aromatics under mild conditions of 60 °C without extreme reaction environments.162 The cobalt complex designed by Liu et al. can target the cleavage of low-bond-energy tertiary C–F bonds in branched PFAS, achieving 16/19 C–F bond cleavage.163 Different metal active centers exhibit varying effects on PFAS reduction. Therefore, it is necessary to further screen metals with high activity, such as exploring new transition metals or their combinations. By altering the oxidation state and ligand environment of metals, their adsorption and activation capabilities for C–F bonds in PFAS can be adjusted to enhance catalytic efficiency and selectivity. In addition, the catalytic performance can be optimized by controlling the morphology, particle size, and pore structure of the catalyst. Preparing nano-sized catalysts increases the specific surface area and the number of active sites; designing porous structures facilitates the diffusion and mass transfer of PFAS molecules, thereby improving reaction efficiency. Furthermore, constructing composite catalysts by combining materials with different functions, leveraging their respective advantages, enables efficient and selective reduction of PFAS.
5.2 Investigation of parameters in ER for PFAS degradation
Carbon-based materials have garnered significant attention owing to their low cost and scalability for practical applications. Activated carbon, one of the most commonly used cathode materials, possesses an enormous specific surface area and is capable of offering abundant active sites for PFAS adsorption and ET.164 However, the electrical conductivity of activated carbon is relatively limited. The granular activated carbon (GAC) cathode can achieve partial defluorination of 6:2 fluorotelomer without a catalyst at a potential of 10 V, but the defluorination rate is low.61 Carbon nanotubes (CNTs), on the other hand, enrich PFAS and electron donors through the confinement effect, promote the generation of hydrated electrons under UV synergy, and the defluorination rate can reach more than 80% and can be reused 5 times.165
Metal electrodes have also been extensively studied. Hughes et al. used a BDD anode and Cu, Ti, and Au cathodes, separating oxidation and reduction reactions through an H-type electrolytic cell containing an agar membrane. The results showed that electrochemical reduction contributed significantly to the total degradation of GenX, accounting for 52% with the Cu cathode, 66% with the Ti cathode, and 92% with the Au cathode.156
In recent years, modified electrodes have become a research hotspot. For example, cathodes modified with quaternary ammonium salt surfactants can surmount the electrostatic repulsion between anionic PFAS compounds and the cathode through hydrophobic interactions and electrostatic interactions, promote the adsorption of PFAS on the cathode surface, and significantly improve the reduction efficiency. Wang et al. studied the ER system equipped with a quaternary ammonium surfactant-functionalized cathode for the degradation of PFMeUPA. At −1.6 V (vs. Ag/AgCl), the cathode modified with OTAB achieved a removal rate of 99.81% and a defluorination rate of 78.67%. Among them, PFMeUPA is initially adsorbed on the modified cathode surface through hydrophobic and electrostatic interactions. DET induces the cleavage of C–F bonds, and the CC structure of PFAS contributes to the fragmentation of C–F bonds. The degradation pathway comprises sequential reductive defluorination and hydrogenation. This work uncovers the critical role of quaternary ammonium surfactants in electron transfer and electrocatalytic performance, providing an innovative strategy for remediating PFAS-contaminated groundwater.53
While surfactant modifications partially mitigate this issue by promoting hydrophobic interactions, achieving true defluorination requires a deeper microstructural intervention. Constructing 2D layered confinement structures, such as MXenes or hierarchically structured carbon networks, offers a microenvironment where local electrostatic fields and steric effects are drastically altered. These interlayer channels or molecular-level pores act as nanoreactors. When PFAS molecules intercalate into these confined spaces, the spatial restriction prevents the premature desorption of partially hydrodefluorinated intermediates. Consequently, the local concentration of both the target pollutants and reactive species is maximized, ensuring that stepwise C–F bond cleavage is driven to completion, ultimately advancing the process from preliminary parent compound removal to comprehensive structural destruction.
In addition to surfactant modification, researchers have further optimized electrode performance by means of loading nanocatalysts on the electrode surface and preparing composite material electrodes. Graphene, with its unique electronic structure and high specific surface area, can improve the electron transfer performance of electrodes and show good application prospects. For instance, loading transition metal nanoparticles on graphene electrodes can not only enhance the electrical conductivity of the electrodes but also provide more catalytically active sites, thereby improving the degradation capacity of PFAS. For example, Li et al. prepared reduced graphene oxide aerogels (rGA) using a microbubble template method, and loaded Fe3O4 and Cu nanoparticles to obtain Fe3O4-rGA and Cu-rGA as cathodes. These cathodes were combined with BDD electrodes to construct a heterogeneous Fenton-like system. Through the synergistic effect between rGA loaded with Fe3O4 or Cu NPs and BDD electro-oxidation, the abundant active sites promoted the generation of ·OH. In combination with the BDD anode, which has a high OEP, a wide electrochemical window, and an inert surface without secondary pollution, the synergistic effect of the two enabled the removal rate of PFOA to reach over 98% within 360 minutes and complete removal within 720 minutes, with the F− generation rate exceeding 60%. Moreover, the energy consumption and cost are relatively low.152
Zhang et al. explored that the ZIF-67 modified cathode could achieve a defluorination rate of 97.16% at a voltage of −1.2 V vs. Ag/AgCl. Its core advantage lies in the efficient catalytic generation of H by the Co–N structure, which promotes defluorination through the synergistic effect of direct reduction and H-mediated indirect reduction. The introduction of carbon black enhances the electrical conductivity and adsorption performance of the electrode. DFT calculations confirmed that the Co–N structure with low nitrogen content is more conducive to the generation and adsorption of H, providing a new method for the efficient removal of short-chain PFAS.155
Loading nanoparticles such as precious metals or transition metals on the electrode surface can utilize the high catalytic activity of metals to effectively reduce the activation energy of the reaction, accelerating the cleavage of C–F bonds and the mineralization of PFAS.166 In addition, after introducing non-precious metal catalysts into the electrode system, they can achieve efficient adsorption and catalytic oxidation of PFAS by virtue of their special electronic structure and surface properties.
The structural characteristics of PFAS molecules affect ER efficiency. Their specific functional groups significantly influence their electron-accepting capacity and reaction pathways. For example, PFAS containing CC structures can promote the cleavage of C–F bonds and accelerate the reduction reaction. In addition, intermediate free radicals generated in the ER can further react with PFAS molecules, promoting the defluorination process. The electron cloud distribution on the cathode surface and the spatial configuration of PFAS molecules directly affect ET efficiency and reaction selectivity. Theoretical calculations reveal the microscopic interaction mechanism between PFAS and the electrode surface by simulating molecular orbital interactions. At high concentrations, PFAS tend to form micelles/semi-micelles, which reduce mobility and hinder degradation. Therefore, it is necessary to control the concentration or optimize the electrode surface to enhance adsorption.
The type and concentration of electrolytes are crucial for solution conductivity, the generation of active species, and reaction kinetics. When Na2SO4 is used as the electrolyte, electrochemical activation can generate SO4˙− and ·OH, which participate in PFAS degradation. Inert electrolytes need to have a high ionic strength and a wide potential window. Studies have shown that Na2SO4 is more effective than NaClO4 and has less environmental impact. For example, Saleh et al. systematically studied the effects of 17 types of saltwater electrolytes on the electrocatalytic conversion of PFOA, confirming that the increase in electrolyte concentration and conductivity accelerates degradation. The addition of Cl− has no significant effect, while NO3− reduces efficiency. The degradation of PFOA is not affected by the pH of the bulk solution, and there is a good correlation between conductivity and degradation rate, providing an important basis for electrolyte optimization. Mixed electrolytes can balance cost and efficiency, but it is necessary to avoid the generation of chlorine-oxygen free radicals (·ClO) that may cause side reactions.167
The pH value affects the existing forms of PFAS and the charge properties of the electrode surface. Alkaline conditions inhibit HER, and OH− will participate in the reaction to generate superoxide free radicals, which affects the degradation products and degradation efficiency. Acidic conditions enhance hydrophobic interactions, and some PFAS are more likely to gain electrons and be reduced, but it is necessary to balance the dissociation state of PFAS.
A higher *j* can provide more electrons per unit time, which can accelerate the rate of the reduction reaction in the initial stage.33 However, an excessively high *j* will trigger side reactions such as HER, consuming electrons and reducing the energy efficiency of PFAS degradation.61 Studies by Ma et al. showed that when a direct current of 0.3 V was applied, the removal rate of PFOA reached 63.2%, and when the voltage was 0.8 V, the removal rate increased by 2.7%. The effective enhancement of PFAS removal was achieved by strengthening electron transfer.168
In addition, reaction temperature and other coexisting substances in the solution can also affect the degradation process. Increasing temperature can accelerate the molecular movement rate and promote mass transfer, but it is limited by electrode stability and energy consumption.61 However, excessively high temperatures may lead to intensified side reactions. Coexisting substances may compete with PFAS for active sites on the electrode surface and change the structure and properties of PFAS through chemical reactions, thereby affecting the degradation effect. Zeidabadi et al. explored the impact of organic and inorganic components such as natural organic matter (NOM), Cl−, NO3−, and HCO3− on the degradation process. The results showed that NOM and Cl− would inhibit PFAS degradation, while NO3− and HCO3− had little effect on degradation but significantly reduced defluorination efficiency.169
ER technology provides an efficient solution for PFAS degradation through the synergy of multiple pathways dominated by hydrated electrons, assisted by DET and atomic hydrogen. The design of electrode materials has expanded from precious metals to single-atom catalysts and carbon-based materials, taking both efficiency and cost into account. The optimization of electrolytes and the selection of potential windows can inhibit side reactions and improve defluorination selectivity. Future research should focus on developing anti-pollution and long-life electrodes, and further explore the impact mechanism of complex matrices in actual water bodies on the reduction pathway.
6 Combined technologies for PFAS degradation
In addressing PFAS pollution, single electrochemical technologies have various limitations, often facing issues such as high energy consumption, poor selectivity, numerous side reactions, and interference from coexisting substances.170 However, combined systems can effectively overcome these challenges by integrating different technologies with electro-oxidation or reduction, such as nanofiltration, adsorption,171 electrocoagulation,172 photocatalysis,173 Fenton's method,174 or through innovations in electrode/system design,175 These combinations not only reduce the overall cost but also lower the concentration of harmful by-products generated after PFAS degradation, achieving good treatment effects on groundwater with different conductivities and PFAS pollution of varying degrees,29Table 4 summarizes the research on the degradation of PFAS by the combination of electrochemical technology and other technologies in recent years.| Technologies | PFAS | Experiment conditions | Energy consumption | PFAS removal (%) | Defluorination (%) | Ref. |
|---|---|---|---|---|---|---|
| a Perfluoroalkyl sulfonic acids (PFSAs); perfluoroheptanesulfonic acid (PFHpS); perfluoropentanesulfonic acid (PFPeS); trifluoroacetic acid (TFA); perfluoropropanoic acid (PFPrA). | ||||||
| AO/Rotation | PFOA | BDD anode produced bubbles, 4.5 V/SHE, 9 h, 48 µM | 7 kWh m−3 log−1 | 99 | 93 | 176 |
| AO/Rotation | PFOA | BDD anode alone, 4.5 V/SHE, 9 h, 48 µM | 13 kWh m−3 log−1 | 99 | 82 | 176 |
| AO/Rotation | PFOA | BDD anode, 100 mL per min N2, 4.5 V/SHE, 5 h, 48 µM | 9.2 kWh m−3 log−1 | 99 | 98 | 176 |
| EC-EO | PFCAs | Anode EC/EO:Zn/Ti4O7, cathode: Stainless steel | >90 | 177 | ||
| PFSAs | ||||||
| NF-EO | PFHxA | BDD/BDD, 50 A m−2 20 °C 870 mg L−1 20 bar | 8.94 kWh m−3 log−1 | 98 | 178 | |
| GIC-adsorption-EO | PFOS | 20 mA cm−2 20 min | 13 kWh m−3 | 83 | 179 | |
| PFOA | Mixed system | 62 | ||||
| PFBA | 11 | |||||
| GIC-adsorption-EO | PFHpS | Anode: Graphite block | 7.8 kWh m−3 | 93.5 | 180 | |
| PFHxS | Cathode: perforated stainless steel | 85.7 | ||||
| PFPeS | Adsorption 20 min + regeneration 10 min | 71.2 | ||||
| PFBS | 19.7 V 1 A | 74.7 | ||||
| PFOA | 71.1 | |||||
| PFHxA | 36.4 | |||||
| PFBA | 2.6 | |||||
| Fenton-BDD | GenX | Cathode: Gr-Ni-foam, anode: BDD/Pt/FTO, 0.05 M K2SO4 | 92.2 | 89 | 132 | |
| 0.2 mM Fe2+, 8–16 mA cm−2 2–6 h | ||||||
| ZnO-EO | PFOA | Anode: Nano-ZnO-coated Ti | 65.84 | 141 | ||
| PFOS | Cathode: Stainless steel | 63.42 | ||||
| PFHpA | 20 mA cm−2 40 min | 58.03 | ||||
| PFHxA | 49.31 | |||||
| Fenton-EO | PFCAs | Anode: Ti/BDD | 4.43–39.43 kWh m−3 | 86.1–100 | 181 | |
| PFSAs | Cathode: Pt | |||||
| F–53B | NaCl 2 g L−1 5 mA cm−2 2 h | |||||
| Fenton-EO | PFCAs | Anode: Ti/IrO2 | 22.8–98.64 kWh m−3 | 54.5–98.1 | 181 | |
| PFSAs | Cathode: Pt | |||||
| F–53B | NaCl 2 g L−1 15 mA cm−2 3 h | |||||
| Redox-polymer ED-NF | TFA (C2) | Activated carbon cloth electrode | 89 | 100 | 20 | |
| PFPrA(C3) | Cellulose-based membrane filter | 86 | 76 | |||
| PFBA(C4) | 1.0 V 10 mL min−1 10 mM NaCl | 90 | ||||
| PFHxA(C6) | 89 | |||||
| PFOA(C8) | 90 | |||||
| Fenton-EO | PFOA | Anode: Ti4O7 | 92 | 182 | ||
| PFOS | Cathode: Carbon felt, 50 mM Na2SO4 5 h, 0.2 mM FeSO4 pH = 3, Drum in O2, 13 mA cm−2 | 100 | ||||
| EO-ES | PFOS | Anode: BRGO | 10.1 ± 0.7 kWh m−3 | 67 | 75.3 | 37 |
| PFOA | Cathode: Spontex | 48.6 | 74.1 | |||
| PFHxS | 10 mM Na2HPO4/NaH2PO4 | 49.1 | 81.1 | |||
| PFHxA | pH = 7.2 | 34.2 | 83.3 | |||
| UV-ER | PFNA | Anode: Pt–Ti, cathode: CNT-PS-35 | 35.4 | 183 | ||
| PFOA | 50 mM NaClO4 UV/-2 V | 33.5 | ||||
| PFOS | 2 h pH = 11.5 N2 | 20.4 | ||||
| PE-UV-EC | PFOS | Anode: [NiFe]-layered double hydroxide | 5.256 kWh m−3 | 100 | 148 | |
| Cathode: AvCarb MGL190, 8.0 M LiOH, pH = 14.9 | 83.3 | |||||
| PFOA | UN 254 nm 40 mW cm−2, +1.6 V vs. RHE | |||||
| One cycle = 1 min ON + 5 min OFF | ||||||
| PE-UV-EC | PFOS | Anode:[NiFe]–(OH)2, cathode: Nifethal 70 | 68.2 kWh m−3 | 100 | 100 | 184 |
| PFOA | 8.0 M LiOH, UN 254 nm, Eon = 1.6 V 30 s, Erev = −1.74 V 1 s | |||||
| PDC-BDD | PFOA | BDD/BDD, 7.518 g per L K2HPO4, 0.931 g per L KH2PO4 | 1.95 kWh m−3 | 56 | 145 | |
| On 300 ms + Off 300 ms 8V, 30 min 20 °C 300 rpm N2 | ||||||
| ER-EO | PFOA | Co–CN2–Fe2O3, -0.06 V pH = 2120 min, 0.05 M Na2SO4 | 96.1 | 95.9 | 153 | |
The treatment efficiency can be improved by adjusting the electrode layout or utilizing reaction by-products. For example, the AO/Rotation system proposed by Wan et al. rotates the electrode direction by 90°, leveraging hydrogen bubbles produced via the cathodic hydrogen evolution reaction to deliver PFOA to the anode, thereby enhancing its oxidative degradation. Experimental results demonstrated that at a N2 flow rate of 100 mL min−1, the defluorination efficiency increased from 31% to 100% within 3 hours. Notably, the energy consumption was merely half that of the direct anodic oxidation (AO) process, while secondary pollution induced by aerosol emission was effectively avoided. This system achieved performance breakthroughs and cost reduction merely by adjusting the spatial layout of electrodes and utilizing natural bubble behavior, without the need to develop new electrode materials (Fig. 14a–c).176
 | ||
| Fig. 14 (a) Schematic diagram of the AO system, AO/Rotation system. (b) Time profiles of PFOA removal. (c) TOC removal. Reproduced with permission.176 Copyright 2023, American Chemical Society. (d) Schematic illustration of the redox-polymer ED system. Reproduced with permission.20 Copyright 2024, Springer Nature. | ||
Other studies have combined multiple treatment processes to leverage the advantages of different technologies. Shi et al. adopted a method combining EC and EO to treat PFAS. First, the EC process removes pollutants such as organic matter and heavy metals in wastewater, and uses cations generated by the dissolution of the zinc anode to form hydroxyl complexes that adsorb PFAS.185 Under specific conditions, the generated foam can also promote the separation of PFASs, completing the initial concentration. In the subsequent EO stage, the Ti4O7 anode is used to degrade the concentrated PFAS through direct electron transfer and hydroxyl radicals. This process can efficiently separate, concentrate, and degrade PFASs in groundwater and wastewater.177 Because the traditional method needs to go through multiple separation processes such as adsorption, filtration, ion exchange, etc., and the long chain and ultra short/short chain PFAS should be treated separately, followed by defluorination, the process is very cumbersome. Kim et al. Comparatively studied the redox polymer electrodialysis system, which can treat PFAS while desalting through the synergistic effect of electrosorption and ion migration (Fig. 14d).20,37 Soriano et al. proposed a strategy of first separating via NF and then electrochemically degrading the NF concentrate. Using a DowFilm NF270 membrane under a pressure of 20 bar, the rejection rate of PFHxA reached 99.4%, and the volume was reduced by 5 times in the concentration mode. Subsequently, a commercial electrochemical cell with bipolar BDD electrodes was used to treat the NF retentate. With a current density of 50 A m−2, the degradation rate of PFHxA reached 98%, the total organic carbon removal rate exceeded 95%, and the energy consumption was 15.2 kWh m3. This demonstrates the considerable potential of the combination of membrane separation and electrochemical oxidation in addressing PFAS-contaminated water.178
The Trzcinski team conducted studies on the treatment of PFAS by combining Graphite Intercalation Compound (GIC) adsorption with electrochemical oxidation in 2023 and 2024, respectively. In 2023, targeting PFOS, they adopted the method of GIC adsorption combined with EO. The adsorption of PFOS by GIC conformed to the Langmuir model, with a capacity of 53.9 µg g−1, reaching equilibrium within 20 minutes. Adsorption was achieved through hydrophobic interactions and other mechanisms, resulting in a 99% removal rate of PFOS. However, the degradation produced short-chain by-products, making complete degradation difficult.180 In 2024, the research was extended to PFOA and PFBS. The adsorption capacities of GIC for the three substances varied significantly: 53.9 µg g−1 for PFOS, 22.3 µg g−1 for PFOA, and 0.985 µg g−1 for PFBS. PFOS could be completely degraded under a current of 28 mA cm−2, and the degradation rate of PFOA reached 98% at 25 mA cm−2. PFBS needed to be adsorbed first before degradation. The removal rate in actual water bodies decreased by 3–40%, but under a current of 40 mA cm−2, the removal rates of PFOS and PFOA could reach 95% and 68%, respectively. The advantage is that it clarifies that hydrated electrons on the GIC surface dominate the degradation and that it can handle various PFAS.179
Building upon the integration of adsorption and EO, such as the GIC-EO system, it is crucial to recognize that physically decoupling adsorption from degradation can still face mass transfer bottlenecks and the accumulation of recalcitrant short-chain intermediates. To truly bridge the gap between preliminary ‘removal’ and ultimate ‘mineralization’, future research must pivot towards the design of bifunctional materials that seamlessly integrate exceptional adsorption capacity with high electrocatalytic activity.
In this context, advanced porous frameworks, such as conductive molecular sieves and MXene-based carbon composites, represent a highly promising frontier. Unlike conventional adsorbents that merely concentrate pollutants, conductive molecular sieves can be engineered with precise pore architectures to selectively capture and confine PFAS molecules—including highly soluble short-chain variants—via size-exclusion and electrostatic interactions. Once anchored within these confined nanospaces, the structurally integrated conductive network facilitates rapid direct electron transfer and the localized generation of high-concentration ROS. This enrichment-degradation in situ coupling drastically prolongs the residence time of PFAS and their intermediate fragments near the catalytic active sites, effectively converting physical confinement into deep C–F bond cleavage. Similarly, 2D materials like MXenes offer tunable interlayer spacings and rich surface terminations, allowing them to act as dynamic nanoreactors that simultaneously intercalate PFAS and catalyze their destruction. Transitioning towards such bifunctional adsorbent-electrocatalysts will be a pivotal strategy for achieving complete PFAS mineralization in complex water matrices while minimizing overall energy consumption.
Other studies have focused on developing new synergistic reaction systems. Olvera-Vargas et al. investigated an electro-Fenton (EF) synergistic degradation system using graphene-coated nickel foam (Gr-Ni-foam) as the cathode and BDD as the anode for treating short-chain GenX. The degradation of GenX starts with DET on the BDD surface, followed by the continuous promotion of degradation by ·OH radicals generated by EF.132 Zhang et al. utilized a nano-ZnO-coated Ti electrode, combining the synergistic effect of EO and photocatalytic oxidation. The nano-ZnO generates additional ·OH, which synergistically enhances with the radicals produced by EO. Its rough nanostructure also increases the electrode surface area and catalytic active sites, improving the removal efficiency.141 Rao et al. developed a UV/CNT electrode system. First, PFAS is adsorbed on the electrode surface; the electrode provides instantaneous ET under negative potential to weaken the C–F bonds, and eaq− generated by UV photolysis of water attacks the weakened C–F bonds, significantly improving the defluorination efficiency of refractory sulfonates.183 Huynh et al. explored a Fenton-assisted EO process, where H2O2 generated at the cathode provides raw materials for the Fenton reaction, and the anode generates ·OH and strong oxidants. Fe3O4 is added as a catalyst to accelerate the Fenton reaction and reduce pH requirements. Meanwhile, Fe3+ near the anode can be regenerated into Fe2+ through electroreduction, maintaining the Fenton reaction cycle and increasing the radical yield181
In addition, some studies have explored new electrode materials. Duinslaeger et al. investigated a boron-doped graphene sponge anode, which exhibited a certain removal efficiency for short-chain PFAS under low-conductivity electrolytes and single-pass low-flux mode, with low energy consumption. Moreover, this electrode is electrochemically inert to Cl− and does not generate chlorate or perchlorate, providing possibilities for the electrochemical degradation of complex PFAS-containing wastewater and brine.37 Gomri et al. studied the EO combined with EF technology using Magnéli phase Ti4O7 as the anode, which achieved high degradation rates of PFOA and PFOS with significantly reduced energy consumption.182 In 2024, Meng et al. prepared [NiFe]-layered double hydroxide nanocatalysts via laser and immobilized them on a hydrophilic carbon fiber paper anode. In an 8.0 M LiOH aqueous solution, complete defluorination of PFOS was achieved through pulsed electrolysis combined with ultraviolet irradiation. The materials used are non-precious, improving the efficiency of electrical energy and capital expenditure.148,184 Han et al. studied a Fe2O3-assisted Co–CN2 configuration bifunctional single-atom catalyst, which can integrate reduction and oxidation processes, simultaneously inducing PFOA degradation and reducing O2 to H2O2. Under the condition of −0.06 V, 96.1% PFOA degradation rate and 95.9% defluorination rate were achieved within 120 minutes, confirming the key mechanism of the synergistic effect between reduction and oxidation processes.153
For practical engineering applications, Specific Energy Consumption (SEC) is a critical metric for evaluating the economic viability of PFAS degradation technologies. When treating real environmental water, where PFAS concentrations are usually at trace levels, the energy efficiency of various electrochemical processes differs significantly. While stand-alone EO can completely cleave PFAS chains, its direct application to raw water yields a prohibitively high SEC, often ranging from tens to hundreds of kWh m3. This is primarily because extreme trace concentrations cause severe mass transfer limitations, wasting most of the applied electrical energy on background electrolyte resistance and the competitive OER. Similarly, ER faces severe energy efficiency challenges. Although it avoids the OER, it requires high cathodic overpotentials to overcome C–F bond activation barriers, which inevitably triggers the energy-consuming HER.
In contrast, combined technologies offer a decisive energy-saving advantage through a “concentrate-and-destroy” strategy. By utilizing pre-treatment methods like membrane separation or physical adsorption, massive volumes of trace PFAS wastewater are concentrated into a significantly smaller volume of retentate or eluent. Consequently, the electrochemical module only treats this minimized volume, which exponentially enhances internal mass transfer efficiency and drastically reduces the energy wasted on parasitic side reactions.186
7 Conclusions
With advantages such as mild operating conditions and controllable electron transfer, electrochemical technologies have demonstrated significant potential for the degradation and remediation of PFAS. This review systematically summarizes the research progress of electrochemical technologies in PFAS destruction, exploring a research trajectory from atomic-scale bond-cleavage mechanisms and interfacial regulation in various electrochemical processes, to AI-enabled HTS, aiming to provide a reference for the further optimization of these technologies.Regarding specific technological systems, EO exhibits notable advantages in destroying long-chain PFAS through the synergistic effects of direct electron transfer and ROS. However, its efficacy is often limited by competitive OER and the high cost of anode materials. Conversely, ER provides an alternative pathway to overcome oxidative barriers and achieve hydrogen substituted defluorination, though practical applications must address the intense electrostatic repulsion at the cathode interface and the high activation energy of C–F bonds. To mitigate the limitations of single processes, combined technologies that integrate electrochemistry with separation techniques or in situ adsorption have demonstrated promising synergistic effects, offering more viable engineering solutions for the pre-concentration and deep degradation of PFAS in complex water matrices.
Although considerable progress has been made in enhancing PFAS degradation efficiency in recent years, several technical challenges remain in practical applications. Notably, it is crucial to recognize that a high parent compound removal rate does not strictly equate to complete mineralization. During the degradation process, the accumulation of short-chain intermediates frequently occurs. Because these short-chain species possess weaker hydrophobicity and encounter higher electrostatic repulsion barriers, effectively mitigating their escape to achieve deep defluorination remains a critical issue that current electrochemical technologies must address.
To address these challenges, future electrode material design might increasingly focus on the confinement effects of micro-interfaces and bifunctional synergy. Relying solely on macroscopic physical adsorption or solitary electrocatalysis often presents limitations in achieving deep mineralization. Developing bifunctional materials that seamlessly integrate specific adsorption-enrichment with high electrocatalytic activity represents a highly promising direction for in-depth exploration. Through size exclusion, hydrogen bonding, or electrostatic interactions within microscopic pores, such materials are expected to anchor short-chain PFAS in situ while simultaneously concentrating reactive species near the catalytic active sites. This modulation of the microenvironment helps alleviate mass transfer limitations and facilitates the transition of PFAS from physical surface enrichment to deep structural destruction.
Furthermore, navigating the vast and complex compositional space of materials via conventional trial-and-error methods is often excessively time-consuming. The introduction of AI-enabled HTS closed-loop systems offers a more efficient approach for the rapid development of multi-objective electrode materials. Future research could attempt to more tightly couple intrinsic mechanistic descriptors derived from theoretical calculations with machine learning models. This integration would allow for the simultaneous pursuit of high degradation activity while comprehensively balancing side-reaction suppression, target pollutant affinity, and long-term stability.
Overall, achieving the complete and harmless destruction of PFAS remains a long-term and complex endeavor. Narrowing the gap between ideal laboratory conditions and realistic, complex water matrices remains a focal point for future research. With a deepening understanding of microscopic confined mineralization mechanisms and the progressive application of data-driven design, electrochemical technologies are poised to play an increasingly active and robust role in the practical remediation of PFAS and other emerging contaminants.
Author contributions
Yuqing Dong: conceptualization, methodology, data curation, writing – original draft, writing – review & editing. Shuaiyu Gao: conceptualization, methodology. Yuelin Zhao: validation, data curation. Genban Sun: writing – review & editing, project administration, funding acquisition. Jihong Yu: conceptualization, methodology, writing – review & editing, project administration, funding acquisition.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analyzed as part of this review.Acknowledgements
We thank the Fundamental and Interdisciplinary Disciplines Breakthrough Plan of the Ministry of Education of China (JYB2025XDXM903), the National Natural Science Foundation of China (22288101, 22271018, 22571020), the ‘111 Center’ (B17020).Notes and references
- J. Wang, C. Cao, Y. Zhang, Y. Zhang and L. Zhu, Appl. Catal., B, 2021, 286, 119911 CrossRef CAS.
- S. Yin, J. F. López, C. Sandoval-Pauker, J. J. Calvillo Solís, S. Glass, A. Habib, W.-Y. Lee, M. S. Wong, P. J. J. Alvarez and D. Villagrán, Appl. Catal., Bc, 2024, 355, 124136 CrossRef CAS.
- M. Liao, S. Zhao, G. Zhan, J. Liang, Z. Li, F. Dong, Y. Pan, H. Li and L. Zhang, J. Am. Chem. Soc., 2025, 147, 3402–3411 Search PubMed.
- J. S. Ko, N. Q. Le, D. R. Schlesinger, D. Zhang, J. K. Johnson and Z. Xia, Sci. Rep., 2021, 11, 19014 CrossRef CAS PubMed.
- H. Song, L. Yan, J. Ma, J. Jiang, G. Cai, W. Zhang, Z. Zhang, J. Zhang and T. Yang, Water Res., 2017, 116, 182–193 Search PubMed.
- C. E. Schaefer, C. Andaya, A. Urtiaga, E. R. McKenzie and C. P. Higgins, J. Hazard. Mater., 2015, 295, 170–175 CrossRef CAS PubMed.
- C. E. Schaefer, S. Choyke, P. L. Ferguson, C. Andaya, A. Burant, A. Maizel, T. J. Strathmann and C. P. Higgins, Environ. Sci. Technol., 2018, 52, 10689–10697 CrossRef CAS PubMed.
- J. Hou, G. Li, M. Liu, L. Chen, Y. Yao, P. H. Fallgren and S. Jin, Chemosphere, 2022, 287, 132205 CrossRef CAS PubMed.
- A. Román Santiago, S. Yin, J. Elbert, J. Lee, D. Shukla and X. Su, J. Am. Chem. Soc., 2023, 145, 9508–9519 CrossRef PubMed.
- O. E. Beyioku, A. Gilboa and A. Ronen, Sep. Purif. Technol., 2025, 372, 133457 CrossRef CAS.
- R. Ahmad, X. Liu, H. N. Ilyas, P. Hanphaiboon, W. Wang, M. Noman, B. Pan and Y. Wang, Sep. Purif. Technol., 2025, 367, 132797 CrossRef CAS.
- J. Hu, S. Wang, J. Yu, W. Nie, J. Sun and S. Wang, Environ. Sci. Technol., 2021, 55, 1260–1269 Search PubMed.
- S. Verma, R. S. Varma and M. N. Nadagouda, Sci. Total Environ., 2021, 794, 148987 Search PubMed.
- M. S. Samuel, G. Kadarkarai, D. R. Ryan, S. T. McBeath, B. K. Mayer and P. J. McNamara, Sci. Total Environ., 2024, 942, 173736 Search PubMed.
- P. Gogoi, Y. Yao and Y. C. Li, ChemElectroChem, 2022, 10, e202201006 CrossRef.
- A. Menezes, L. C. Goveas, R. Vinayagam and R. Selvaraj, J. Water Process Eng., 2025, 69, 106621 CrossRef.
- N. Liu, Y. Li, M. Zhang, N. Che, X. Song, Y. Liu and C. Li, Sci. Total Environ., 2024, 946, 174223 CrossRef CAS PubMed.
- R. Singh, U. T. Uthappa, Y.-H. Ahn and S. S. Ray, Inorg. Chem. Commun., 2025, 178, 114614 CrossRef CAS.
- L. Jiang, R. Cao, Z. Huang, J. Jin, Z. Du and H. Li, Chem. Eng. J., 2024, 502, 158161 CrossRef CAS.
- N. Kim, J. Elbert, E. Shchukina and X. Su, Nat. Commun., 2024, 15, 8321 CrossRef CAS PubMed.
- Z. Lin, M. S. Ersan, S. Garcia-Segura, F. Perreault and P. Westerhoff, RSC Adv., 2024, 14, 15627–15636 RSC.
- J. Niu, H. Lin, C. Gong and X. Sun, Environ. Sci. Technol., 2013, 47, 14341–14349 CrossRef CAS PubMed.
- L. I. FitzGerald, J. F. Olorunyomi, R. Singh and C. M. Doherty, ChemSusChem, 2022, 15, e202201136 CrossRef CAS PubMed.
- A. L. Junker, J.-M. A. Juve, L. Bai, C. S. Qvist Christensen, L. Ahrens, I. T. Cousins, M. Ateia and Z. Wei, Environ. Sci. Technol., 2025, 59, 8939–8950 CrossRef CAS PubMed.
- J.-M. Arana Juve, B. Wang, M. S. Wong, M. Ateia and Z. Wei, Curr. Opin. Chem. Eng., 2023, 41, 100943 CrossRef.
- X. Chen, T. Yuan, X. Yang, S. Ding and M. Ma, Catalysts, 2023, 13, 1308 CrossRef CAS.
- M. K. Wilsey, T. Taseska, Z. Meng, W. Yu and A. M. Müller, Chem. Commun., 2023, 59, 11895–11922 RSC.
- A. Dube, S. J. Malode, H. Akhdar, A. N. Alodhayb and N. P. Shetti, Colloids Surf., B, 2025, 252, 114653 CrossRef CAS PubMed.
- S. Sharma, N. P. Shetti, S. Basu, M. N. Nadagouda and T. M. Aminabhavi, Chem. Eng. J., 2022, 430, 132895 CrossRef CAS.
- Z. Meng, M. K. Wilsey and A. M. Müller, ACS Catal., 2024, 14, 16577–16588 CrossRef CAS PubMed.
- H. Lin, R. Xiao, R. Xie, L. Yang, C. Tang, R. Wang, J. Chen, S. Lv and Q. Huang, Environ. Sci. Technol., 2021, 55, 2597–2607 CrossRef CAS PubMed.
- Q. Zhuo, S. Deng, B. Yang, J. Huang, B. Wang, T. Zhang and G. Yu, Electrochim. Acta, 2012, 77, 17–22 Search PubMed.
- S. Sukeesan, N. Boontanon and S. K. Boontanon, Environ. Technol. Innovation, 2021, 23, 101655 CrossRef CAS.
- H. Lin, J. Niu, J. Xu, H. Huang, D. Li, Z. Yue and C. Feng, Environ. Sci. Technol., 2013, 47, 13039–13046 Search PubMed.
- Y. Su, U. Rao, C. M. Khor, M. G. Jensen, L. M. Teesch, B. M. Wong, D. M. Cwiertny and D. Jassby, ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922 CrossRef CAS PubMed.
- B. Yang, J. Wang, C. Jiang, J. Li, G. Yu, S. Deng, S. Lu, P. Zhang, C. Zhu and Q. Zhuo, Chem. Eng. J., 2017, 316, 296–304 CrossRef CAS.
- N. Duinslaeger and J. Radjenovic, Water Res., 2022, 213, 118148 Search PubMed.
- X. Duan, W. Wang, Q. Wang, X. Sui, N. Li and L. Chang, Chemosphere, 2020, 260, 127587 CrossRef CAS PubMed.
- D. Huang, K. Wang, J. Niu, C. Chu, S. Weon, Q. Zhu, J. Lu, E. Stavitski and J.-H. Kim, Environ. Sci. Technol., 2020, 54, 10954–10963 CrossRef CAS PubMed.
- S. Sinha, A. Chaturvedi, R. K. Gautam and J. J. Jiang, J. Am. Chem. Soc., 2023, 145, 27390–27396 CrossRef CAS PubMed.
- J. J. Calvillo Solís, C. Sandoval-Pauker, D. Bai, S. Yin, T. P. Senftle and D. Villagrán, J. Am. Chem. Soc., 2024, 146, 10687–10698 CrossRef PubMed.
- D. Song, B. Qiao, X. Wang, L. Zhao, X. Li, P. Zhang, Y. Yao, H. Chen, L. Zhu and H. Sun, Environ. Sci. Technol., 2023, 57, 9416–9425 CrossRef CAS PubMed.
- J. Hu, X. Yang, X. Song, Y. Miao, Y. Yu, W. Xiang, M. Huang, W. Wu, K. Liang, S. Zhao and H. Liu, J. Hazard. Mater., 2024, 480, 136283 CrossRef CAS PubMed.
- J. E. Zenobio, O. A. Salawu, Z. Han and A. S. Adeleye, J. Hazard. Mater. Adv., 2022, 7, 100130 CAS.
- F. Jiang, M. Wu, Z. Zhu, C. Jiang, Z. Ai and J. Gong, Water Res., 2025, 284, 124049 Search PubMed.
- N. Loganathan, C. E. Schumm, M. K. O'Reilly and A. K. Wilson, Environ. Sci. Technol., 2025, 59, 14637–14648 CrossRef CAS PubMed.
- Y. Wang, R. D. Pierce, H. Shi, C. Li and Q. Huang, Environ. Sci.:Water Res. Technol., 2020, 6, 144–152 Search PubMed.
- B. B. de Souza and J. Meegoda, Sci. Total Environ., 2024, 926, 171738 CrossRef CAS PubMed.
- M. J. Bentel, Y. Yu, L. Xu, Z. Li, B. M. Wong, Y. Men and J. Liu, Environ. Sci. Technol., 2019, 53, 3718–3728 CrossRef CAS PubMed.
- S. Biswas and B. M. Wong, ACS ES&T Eng., 2023, 4, 96–104 Search PubMed.
- J. Blotevogel, R. J. Giraud and T. Borch, Chem. Eng. J., 2018, 335, 248–254 CrossRef CAS.
- M. S. Mohamed, B. P. Chaplin and A. A. Abokifa, ACS Omega, 2024, 9, 44532–44541 CrossRef CAS PubMed.
- Y. Wang, Y. Xiao, Y. Wang, Q. Lin, Y. Zhu, Z. Ni and R. Qiu, Environ. Sci. Technol., 2023, 57, 7578–7589 CrossRef CAS PubMed.
- Y. Wang, L. Li, Y. Wang, H. Shi, L. Wang and Q. Huang, Chem. Eng. J., 2022, 431, 13929 Search PubMed.
- J. Ma, Y. Wang, H. Xu, M. Ding and L. Gao, Process Saf. Environ. Prot., 2022, 168, 275–284 CrossRef CAS.
- X. Zhang, J. Nan, H. Zhang, P. Li and X. Lin, Water Res., 2025, 286, 124182 CrossRef CAS PubMed.
- S. Raghavan, B. P. Chaplin and S. Mehraeen, J. Phys. Chem. C, 2025, 129, 10027–10037 CrossRef CAS.
- Y. Wang, J. Zhang, W. Zhang, J. Yao, J. Liu, H. He, C. Gu, G. Gao and X. Jin, Angew. Chem., Int. Ed., 2024, 63, e202402440 CrossRef CAS PubMed.
- Y. Chen, Y. Yang, J. Cui, H. Zhang and Y. Zhao, J. Hazard. Mater., 2024, 465, 133260 CrossRef CAS PubMed.
- B. Yan, R. Alessandri, S. J. Marrink, L. S. Lee and J. Liu, Environ. Sci. Technol. Lett., 2025, 12, 626–631 CrossRef CAS.
- D. Ackerman Grunfeld, A. M. Jones, J. Sun, S. T. Le, R. Pickford, Q. Huang, M. Manefield, N. Kumar, M. J. Lee and D. M. O'Carroll, Environ. Sci.:Water Res. Technol., 2024, 10, 272–287 RSC.
- J. A. R. Willemsen and I. C. Bourg, J. Colloid Interface Sci., 2021, 585, 337–346 CrossRef CAS PubMed.
- B. G. Lamb and B. Ma, Nanoscale, 2025, 17, 10632–10643 RSC.
- S. Kancharla, A. Choudhary, R. T. Davis, D. Dong, D. Bedrov, M. Tsianou and P. Alexandridis, J. Hazard. Mater., 2022, 428, 128137 Search PubMed.
- Y. Yang, Front. Environ. Sci. Eng., 2020, 14, 85 Search PubMed.
- K. Wang, R. Wang, W. Shan, Z. Yang, Y. Chen, L. Wang and Y. Zhang, Environ. Sci. Technol., 2024, 58, 22766–22776 Search PubMed.
- F. Ecke, B. Ytrehus, M. Evander, B. Hörnfeldt, A. Leijon, J. Malmsten, A. Skrobonja and L. Ahrens, Sci. Rep., 2025, 15, 31003 Search PubMed.
- Y. Zhang, J. Liu, A. Moores and S. Ghoshal, Environ. Sci. Technol., 2020, 54, 4631–4640 Search PubMed.
- B. Xu, G. Wu, Z. Li, G. Xu, H. Zeng, R. Tong and H. Y. Ng, npj Clean Water, 2025, 8, 82 Search PubMed.
- G. Xu, Y. Li, J. Li, J. Li, X. Liu, C. Wang, W. Mai, G. Yang and L. Pan, Angew. Chem., Int. Ed., 2025, 64, e202511389 Search PubMed.
- C. S. Tshangana, S. T. Nhlengethwa, S. Glass, S. Denison, A. T. Kuvarega, T. T. I. Nkambule, B. B. Mamba, P. J. J. Alvarez and A. A. Muleja, npj Clean Water, 2025, 8, 41 Search PubMed.
- R. Sun, J. Cao, A. A. Dolatabad, L. Zhao, J. Mai, X. Zhang and F. Xiao, npj Emerg. Contam., 2025, 1, 10 Search PubMed.
- Y. Chen, Z. Guo, M. Angelaki, Y. Carreira Mendes Da Silva, J. Song, W. Wu, J. Lin, A. Gandolfo, J. Chen and C. George, J. Am. Chem. Soc., 2025, 147, 33754–33760 Search PubMed.
- D. Xia, H. Zhang, Y. Ju, H.-b. Xie, L. Su, F. Ma, J. Jiang, J. Chen and J. S. Francisco, J. Am. Chem. Soc., 2024, 146, 11266–11271 Search PubMed.
- Y. Zhang, Y. Zhi, J. Liu and S. Ghoshal, Environ. Sci. Technol., 2018, 52, 6300–6308 Search PubMed.
- Z. Fu, P. Huang, X. Wang, W. D. Liu, L. Kong, K. Chen, J. Li and Y. Chen, Adv. Energy Mater., 2025, 15, 2500744 Search PubMed.
- T. Araki, H. Ota, Y. Murata, Y. Sumii, J. Hamaura, H. Adachi, T. Kagawa, H. Hori, J. Escorihuela and N. Shibata, Nat. Commun., 2025, 16, 6526 Search PubMed.
- A. N. Garcia, Y. Zhang, S. Ghoshal, F. He and D. M. O'Carroll, Environ. Sci. Technol., 2021, 55, 8464–8483 Search PubMed.
- Y. Zhang, J. Tang, Z. Ni, Y. Zhao, F. Jia, Q. Luo, L. Mao, Z. Zhu and F. Wang, J. Phys. Chem. Lett., 2021, 12, 5279–5285 Search PubMed.
- W. Ding, T. Tang, X. Tan and Y. Huang, Environ. Sci. Technol., 2025, 59, 17899–17908 Search PubMed.
- Y. Wang, H. Xu and R. Li, Sci. Rep., 2025, 15, 21023 Search PubMed.
- S. Yaghoobian, M. A. Ramirez-Ubillus, L. Zhai and J.-H. Hwang, Chem. Sci., 2025, 16, 13564–13573 Search PubMed.
- Y. Zhang, A. Moores, J. Liu and S. Ghoshal, Environ. Sci. Technol., 2019, 53, 8672–8681 Search PubMed.
- W. Ding, X. Tan, G. Chen, J. Xu, K. Yu and Y. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 41584–41592 Search PubMed.
- M. Hattori, D. Saha, M. Z. Bacho and N. Shibata, Nat. Chem., 2025, 17, 1480–1487 Search PubMed.
- Y. Huang and A. A. Keller, ACS Sustainable Chem. Eng., 2013, 1, 731–736 Search PubMed.
- D. Wang, X. Liu, Z. Guo, W. Shan, Z. Yang, Y. Chen, F. Ju and Y. Zhang, Environ. Sci. Technol., 2024, 58, 20160–20171 Search PubMed.
- Y. Yang, J. Wu, Z. Luo, X. Jia, G. Jiang and F. Wang, J. Am. Chem. Soc., 2025, 147, 32537–32547 CrossRef CAS PubMed.
- Y. Shi, M. Sun, H. Shi, Z. Liang, B. Qiao, S. Zhao, X. Pu and D. Song, Sci. Bull., 2025, 70, 3058–3089 CrossRef CAS PubMed.
- Z. Wang, Y. Zhang, B. Liu, K. Wu, S. Thevuthasan, D. R. Baer, Z. Zhu, X.-Y. Yu and F. Wang, Anal. Chem., 2016, 89, 960–965 Search PubMed.
- M. Ahmadi, O. Adeleye, G. Vonalt and M. Nazemi, Environ. Sci. Technol., 2025, 59, 17952–17977 Search PubMed.
- Y. Wang, C. Tang, T. Xiao, F. Xiong, X. Zhou, K. Zhang and J. Gong, J. Energy Chem., 2025, 110, 728–739 Search PubMed.
- M. Rittiruam, J. Noppakhun, S. Setasuban, N. Aumnongpho, A. Sriwattana, S. Boonchuay, T. Saelee, C. Wangphon, A. Ektarawong, P. Chammingkwan, T. Taniike, S. Praserthdam and P. Praserthdam, Sci. Rep., 2022, 12, 16653 Search PubMed.
- E. R. Pezoulas, B. Tajdini, Y. Ko, A. A. Uliana, R. Giovine, H. Furukawa, H. Vatankhah, J. Börgel, K. C. Kim, C. Bellona and J. R. Long, J. Am. Chem. Soc., 2025, 147, 21832–21843 Search PubMed.
- J. Benavides-Hernández and F. Dumeignil, ACS Catal., 2024, 14, 11749–11779 Search PubMed.
- Y. He, J. Zhou, Y. Li, Y.-D. Yang, J. L. Sessler and X. Chi, J. Am. Chem. Soc., 2024, 146, 6225–6230 Search PubMed.
- R.-R. Liang, S. Xu, Z. Han, Y. Yang, K.-Y. Wang, Z. Huang, J. Rushlow, P. Cai, P. Samorì and H.-C. Zhou, J. Am. Chem. Soc., 2024, 146, 9811–9818 Search PubMed.
- D. J. Lea-Smith, F. Hassard, F. Coulon, N. Partridge, L. Horsfall, K. D. J. Parker, R. D. J. Smith, R. R. McCarthy, B. McKew, T. Gutierrez, V. Kumar, G. Dotro, Z. Yang, T. P. Curtis, P. Golyshin, S. Heaven, B. Jefferson, P. Jeffrey, D. L. Jones, K. Le Corre Pidou, Y. Liu, T. Lyu, C. Smith, A. Yakunin, Y. Zhang and N. Krasnogor, Nat. Commun., 2025, 16, 3538 Search PubMed.
- J. Wang and Y. Zhang, Next Mater., 2025, 9, 100993 Search PubMed.
- F. Dikmen, A. Demir, B. Özkaya, M. O. Raza, J. Rasheed, T. Asuroglu and S. Alsubai, Sci. Rep., 2025, 15, 25347 Search PubMed.
- I. Peivaste, S. Belouettar, F. Mercuri, N. Fantuzzi, H. Dehghani, R. Izadi, H. Ibrahim, J. Lengiewicz, M. Belouettar-Mathis, K. Bendine, A. Makradi, M. Horsch, P. Klein, M. E. Hachemi, H. A. Preisig, Y. Rezgui, N. Konchakova and A. Daouadji, Compos. Struct., 2025, 372, 119419 Search PubMed.
- R. Batra, L. Song and R. Ramprasad, Nat. Rev. Mater., 2020, 6, 655–678 CrossRef.
- A. O. C. Iroegbu, M. L. Teffo, E. R. Sadiku, R. Meijboom and S. P. Hlangothi, npj Clean Water, 2025, 8, 85 CrossRef CAS.
- N. Jeong, S. Park, S. Mahajan, J. Zhou, J. Blotevogel, Y. Li, T. Tong and Y. Chen, Nat. Commun., 2024, 15, 10918 Search PubMed.
- M. A. Ismail, A. G. Taki, S. Kumar, S. S. Sammen, A. Amari, A. Bongale, O. Kisi and A. Salem, Sci. Rep., 2025, 15, 9439 Search PubMed.
- K. Muta, K. Okamoto, H. Nakayama, S. Wada and A. Nagaki, Nat. Commun., 2025, 16, 416 Search PubMed.
- Z. Chen, Z. Wei, Z. Yang, Y. Chen and Y. Zhang, Environ. Sci. Technol., 2025, 59, 18990–19001 Search PubMed.
- Y. Liang, L. Yang, C. Tang, Y. Yang, S. Liang, A. Wang, J. Xu, Q. Huang and H. Lin, Nat. Commun., 2025, 16, 1972 Search PubMed.
- A. Nandy, A. Rana, N. Shibata and S. Banerjee, J. Am. Chem. Soc., 2025, 147, 22542–22549 Search PubMed.
- J. Yin, W. Li, H. Chen, J. Qiu, H. Feng, X. Xu, Q. Jin and X. Wang, ACS Catal., 2025, 15, 15754–15764 Search PubMed.
- Z. Chen, J. Chen, S. Tan, Z. Yang and Y. Zhang, Environ. Sci. Technol., 2024, 58, 2542–2553 Search PubMed.
- Y. Wu, K. Pei, J. Zhou, J. Xiong, Y. Liu, P. Li, F. Li, J. Wang, X. Liu, M. Deng and C. Wu, Sci. Rep., 2025, 15, 26141 Search PubMed.
- B. T.-W. Tan, N. H. H. Abu Bakar and H. L. Lee, J. Environ. Chem. Eng., 2025, 13, 114990 Search PubMed.
- M. Mirabediny, J. Sun, T. T. Yu, B. Åkermark, B. Das and N. Kumar, Chemosphere, 2023, 321, 138109 CrossRef CAS PubMed.
- J. Radjenovic, N. Duinslaeger, S. S. Avval and B. P. Chaplin, Environ. Sci. Technol., 2020, 54, 14815–14829 Search PubMed.
- S. T. McBeath, A. Serrano Mora, F. Asadi Zeidabadi, B. K. Mayer, P. McNamara, M. Mohseni, M. R. Hoffmann and N. J. D. Graham, Curr. Opin. Electrochem., 2021, 30, 100865 CrossRef CAS.
- S. Giannakis, K.-Y. A. Lin and F. Ghanbari, Chem. Eng. J., 2021, 406, 127083 Search PubMed.
- M. Gar Alalm and D. C. Boffito, Chem. Eng. J., 2022, 450, 138352 CrossRef CAS.
- J. López-Vázquez, C. S. Santos, R. Montes, R. Rodil, J. B. Quintana, J. Gäbler, L. Schäfer, F. C. Moreira and V. J. P. Vilar, Chem. Eng. J., 2024, 482, 148925 Search PubMed.
- M. Veciana, J. Bräunig, A. Farhat, M.-L. Pype, S. Freguia, G. Carvalho, J. Keller and P. Ledezma, J. Hazard. Mater., 2022, 434, 128886 Search PubMed.
- C. E. Schaefer, C. Andaya, A. Burant, C. W. Condee, A. Urtiaga, T. J. Strathmann and C. P. Higgins, Chem. Eng. J., 2017, 317, 424–432 CrossRef CAS.
- C. Wang, T. Zhang, L. Yin, C. Ni, J. Ni and L.-A. Hou, Chemosphere, 2022, 286, 131804 Search PubMed.
- Z. Chen, X. Wang, H. Feng, S. Chen, J. Niu, G. Di, D. Kujawski and J. C. Crittenden, Environ. Sci. Technol., 2022, 56, 14409–14417 Search PubMed.
- L. Zhao, R. Pu, S. Deng, L. Lin, D. Mantzavinos, R. Naidu, C. Fang and Y. Lei, Sep. Purif. Technol., 2025, 359, 130459 CrossRef CAS.
- M. Pierpaoli, M. Szopińska, B. K. Wilk, M. Sobaszek, A. Łuczkiewicz, R. Bogdanowicz and S. Fudala-Książek, J. Hazard. Mater., 2021, 403, 123606 Search PubMed.
- H. Zhao, J. Gao, G. Zhao, J. Fan, Y. Wang and Y. Wang, Appl. Catal., B, 2013, 136–137, 278–286 CrossRef CAS.
- A. L. Tasca, J. N. Uwayezu, I. Carabante and J. Kumpiene, J. Environ. Chem. Eng., 2025, 13, 117044 CrossRef CAS.
- L. Bu, S. Zhu and S. Zhou, Chemosphere, 2018, 195, 236–244 CrossRef CAS PubMed.
- Q. Zhuo, J. Wang, J. Niu, B. Yang and Y. Yang, Chem. Eng. J., 2020, 379, 122280 CrossRef CAS.
- Y. Liu, X. Fan, X. Quan, Y. Fan, S. Chen and X. Zhao, Environ. Sci. Technol., 2019, 53, 5195–5201 Search PubMed.
- F. Asadi Zeidabadi, E. Banayan Esfahani, R. Moreira, S. T. McBeath, J. Foster and M. Mohseni, Environ. Res., 2024, 246, 118103 CrossRef CAS PubMed.
- H. Olvera-Vargas, Z. Wang, J. Xu and O. Lefebvre, Chem. Eng. J., 2022, 430, 132686 CrossRef CAS.
- H. Lin, J. Niu, S. Ding and L. Zhang, Water Res., 2012, 46, 2281–2289 Search PubMed.
- Q. Zhuo, M. Luo, Q. Guo, G. Yu, S. Deng, Z. Xu, B. Yang and X. Liang, Electrochim. Acta, 2016, 213, 358–367 CrossRef CAS.
- H. Lin, J. Niu, S. Liang, C. Wang, Y. Wang, F. Jin, Q. Luo and Q. Huang, Chem. Eng. J., 2018, 354, 1058–1067 CrossRef CAS.
- J. Niu, H. Lin, J. Xu, H. Wu and Y. Li, Environ. Sci. Technol., 2012, 46, 10191–10198 CrossRef CAS PubMed.
- T. X. H. Le, H. Haflich, A. D. Shah and B. P. Chaplin, Environ. Sci. Technol. Lett., 2019, 6, 504–510 CrossRef CAS.
- Q. Zhuo, S. Deng, B. Yang, J. Huang and G. Yu, Environ. Sci. Technol., 2011, 45, 2973–2979 CrossRef CAS PubMed.
- Y. Liang, A. Wang, S. Liang, K. Sun, R. Xie, C. Zheng, S. Zhang, C. Tang, D. Cheng, J. Wang, Q. Huang and H. Lin, Environ. Sci. Technol., 2025, 59, 4745–4755 Search PubMed.
- C. Gomri, D. Krzyzanowski, M. Rivallin, F. Zaviska, E. Petit, M. Semsarilar and M. Cretin, J. Environ. Chem. Eng., 2024, 12, 113495 Search PubMed.
- C. Zhang, J. Tang, C. Peng and M. Jin, J. Mol. Liq., 2016, 221, 1145–1150 CrossRef CAS.
- Q. Zhuo, Q. Xiang, H. Yi, Z. Zhang, B. Yang, K. Cui, X. Bing, Z. Xu, X. Liang, Q. Guo and R. Yang, J. Electroanal. Chem., 2017, 801, 235–243 CrossRef CAS.
- Z. Niu, Y. Wang, H. Lin, F. Jin, Y. Li and J. Niu, Chem. Eng. J., 2017, 328, 228–235 Search PubMed.
- Q. Zhuo, X. Li, F. Yan, B. Yang, S. Deng, J. Huang and G. Yu, J. Environ. Sci., 2014, 26, 1733–1739 Search PubMed.
- R. G. H. Marks, K. Kerpen, D. Diesing and U. Telgheder, J. Electroanal. Chem., 2021, 895, 115415 Search PubMed.
- L. Saleh, M. Remot, Q. B. Remaury, P. Pardon, P. Labadi, H. Budzinski, C. Coutanceau and J.-P. Croué, Chem. Eng. J., 2024, 489, 151255 Search PubMed.
- S. Li, Q. Wang, L. Lyu and J. Leng, Microchem. J., 2025, 213, 113789 Search PubMed.
- Z. Meng, M. K. Wilsey, C. P. Cox and A. M. Müller, J. Catal., 2024, 431, 115403 Search PubMed.
- A. B. Nienhauser, M. S. Ersan, Z. Lin, F. Perreault, P. Westerhoff and S. Garcia-Segura, J. Environ. Chem. Eng., 2022, 10, 107192 CrossRef CAS.
- Y. Luo, O. S. Awoyemi, S. Gopalan, A. Nolan, F. Robinson, J. Fenstermacher, L. Xu, J. Niu, M. Megharaj, R. Naidu and C. Fang, J. Cleaner Prod., 2023, 433, 139691 Search PubMed.
- T. Stahl, H. F. T. Klare and M. Oestreich, ACS Catal., 2013, 3, 1578–1587 Search PubMed.
- Y. Li, N. Che, N. Liu and C. Li, Chem. Eng. J., 2023, 478, 147443 CrossRef CAS.
- J. Han, M. Zhao, K. Wu, Y. Hong, T. Huang, X. Gu, Z. Wang, S. Liu and G. Zhu, Chem. Eng. J., 2023, 470, 144270 CrossRef CAS.
- J. Zhu, Y. Chen, Y. Gu, H. Ma, M. Hu, X. Gao and T. Liu, J. Hazard. Mater., 2022, 422, 126953 Search PubMed.
- X. Zhang, H. Wu, Y. Wang, Y. Wang, Y. Deng, Y. Zhu, S. Zhang, H. Rijnaarts, H. Bruning, J. Qin, Q. Lin, Z. Ni and R. Qiu, Water Res., 2025, 268, 122625 Search PubMed.
- K. Hughes, M. Pineda, S. Omanovic and V. Yargeau, Sci. Total Environ., 2024, 908, 168415 CrossRef CAS PubMed.
- L. Li, R. Guo, J. Gao, J. Liu, Z. Zhao, X. Sheng, J. Fan and F. Cui, J. Hazard. Mater., 2023, 450, 131028 CrossRef CAS PubMed.
- J. Cui, P. Gao and Y. Deng, Environ. Sci. Technol., 2020, 54, 3752–3766 CrossRef CAS PubMed.
- S. Yin, J. J. Calvillo Solís, C. Sandoval-Pauker, D. Puerto-Diaz and D. Villagrán, J. Hazard. Mater., 2025, 491, 137943 CrossRef CAS PubMed.
- H. Liu, J. Han, J. Yuan, C. Liu, D. Wang, T. Liu, M. Liu, J. Luo, A. Wang and J. C. Crittenden, Environ. Sci. Technol., 2019, 53, 11932–11940 Search PubMed.
- S. D. Tan, R. Y. Wang, K. M. Wang, Z. L. Yang, Y. J. Chen and Y. Y. Zhang, Nat. Water, 2025, 3, 15 Search PubMed.
- R. Doi, M. Yasuda, N. Kajita, K. Koh and S. Ogoshi, J. Am. Chem. Soc., 2023, 145, 11449–11456 Search PubMed.
- J. Liu, D. J. Van Hoomissen, T. Liu, A. Maizel, X. Huo, S. R. Fernández, C. Ren, X. Xiao, Y. Fang, C. E. Schaefer, C. P. Higgins, S. Vyas and T. J. Strathmann, Environ. Sci. Technol. Lett., 2018, 5, 289–294 CrossRef CAS.
- J. F. King and B. P. Chaplin, Curr. Opin. Chem. Eng., 2024, 44, 101014 CrossRef.
- H. Wu, J. Wang, E. Du, T. Liu, M. Liu, H. Guo and W. Chu, Water Res., 2025, 279, 123499 CrossRef CAS PubMed.
- M. Zhang, Y. Li, X. Tian, L. Dai, G. Wang, Z. Lei, G. Ma, Q. Zuo, M. Li, M. Zhao and J. Ren, Water, Air, Soil Pollut., 2024, 236, 42 CrossRef.
- L. Saleh, Z. Lin, M. S. Ersan, C. Coutanceau, P. Westerhoff and J.-P. Croué, Chemosphere, 2024, 363, 142879 CrossRef CAS PubMed.
- H. Ma, G. Kong, C. Chen, Z. Guo, J. Huang, S. Kuang, J. Zhang and Y. Kang, Water Res., 2025, 277, 123302 CrossRef CAS PubMed.
- F. A. Zeidabadi, P. Abbasi, E. B. Esfahani and M. Mohseni, J. Hazard. Mater., 2025, 484, 136624 CrossRef CAS PubMed.
- C. B. Ong, A. W. Mohammad, L. Y. Ng, E. Mahmoudi, S. Azizkhani and N. H. Hayati Hairom, Process Saf. Environ. Prot., 2017, 112, 298–307 CrossRef CAS.
- M. M. Mian, J. Zhu, X. Jiang and S. Deng, Front. Environ. Sci. Eng., 2025, 19, 2095–2201 Search PubMed.
- M. Priyadarshini, A. Ahmad, M. S. Mahtab, S. U. Khan, I. H. Farooqi and N. Pérez, Groundw. Sustain. Dev., 2025, 30, 101465 CrossRef.
- C. Chen, Q. Ma, F. Liu, J. Gao, X. Li, S. Sun, H. Yao, C. Liu, J. Young and W. Zhang, J. Hazard. Mater., 2021, 419, 126452 CrossRef CAS PubMed.
- A. Yousefi, F. R. Omi, L. Yang, S. O. Ganiyu, A. Ullah, M. G. El-Din and M. Sadrzadeh, J. Environ. Manage., 2025, 379, 124818 CrossRef CAS PubMed.
- A. Román Santiago, P. Baldaguez Medina and X. Su, Electrochim. Acta, 2022, 403, 139635 CrossRef.
- Z. Wan, L. Cao, W. Huang, D. Zheng, G. Li and F. Zhang, Environ. Sci. Technol. Lett., 2022, 10, 111–116 CrossRef.
- H. Shi, S.-Y. Chiang, Y. Wang, Y. Wang, S. Liang, J. Zhou, R. Fontanez, S. Gao and Q. Huang, Sci. Total Environ., 2021, 788, 147723 CrossRef CAS PubMed.
- Á. Soriano, D. Gorri and A. Urtiaga, Water Res., 2017, 112, 147–156 CrossRef PubMed.
- A. P. Trzcinski and K. H. Harada, Sci. Total Environ., 2024, 927, 172184 CrossRef CAS PubMed.
- A. P. Trzcinski and K. Harada, Chemosphere, 2023, 323, 138268 CrossRef CAS PubMed.
- H. D. Hieu Nhu, D. V. Tri, T. L. Luu, J. Trippel and M. Wagner, Sep. Purif. Technol., 2025, 362, 131908 CrossRef CAS.
- C. Gomri, E. Makhoul, F. N. Koundia, E. Petit, S. Raffy, M. Bechelany, M. Semsarilar and M. Cretin, Nanoscale Adv., 2025, 7, 261–268 RSC.
- U. Rao, Y. Su, C. M. Khor, B. Jung, S. Ma, D. M. Cwiertny, B. M. Wong and D. Jassby, Environ. Sci. Technol., 2020, 54, 10668–10677 CrossRef CAS PubMed.
- Z. Meng, T. Taseska, M. K. Wilsey and A. M. Müller, RSC Adv., 2025, 15, 8287–8292 RSC.
- A. P. Trzcinski and K. Harada, Environ. Sci. Pollut. Res., 2024, 31, 19946–19960 CrossRef CAS PubMed.
- K. Kim, P. Baldaguez Medina, J. Elbert, E. Kayiwa, R. D. Cusick, Y. Men and X. Su, Adv. Funct. Mater., 2020, 30, 2004635 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |