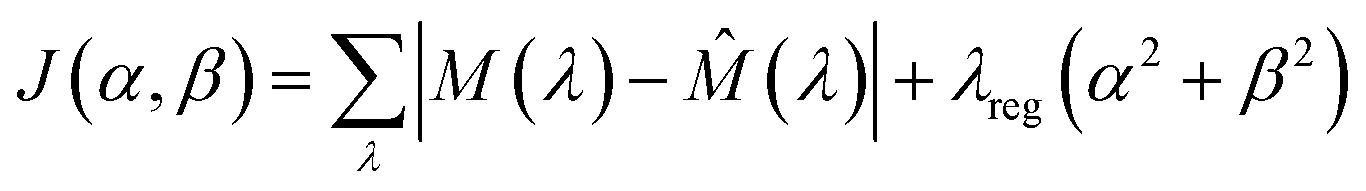DOI:
10.1039/D5SC08710D
(Edge Article)
Chem. Sci., 2026,
17, 9481-9491
Redox-mediator enhanced electrochemiluminescence under non-aqueous conditions
Received
9th November 2025
, Accepted 2nd March 2026
First published on 24th March 2026
Abstract
The introduction of small-molecule redox mediators into aqueous co-reactant electrochemiluminescence (ECL) systems has emerged as an effective strategy to increase signal intensity. Herein, we investigate the influence of a series of neutral iridium(III) complexes (Ir(pmi)3, Ir(ppy)3, Ir(ppz)3 and Ir(ppy)2(acac)) as redox mediators for co-reactant ECL in acetonitrile. Using [Ru(bpy)3]2+ as a benchmark luminophore and tri-n-propylamine (TPrA) as a co-reactant, the redox mediators elicit similar effects in this solvent to those of their sulfonated [Ir(sppy)3]3− and [Ir(sppz)3]3− analogues under aqueous conditions. The Ir(ppz)3 complex was most effective; at a concentration of 100 µM it produced an 11-fold increase in the maximum intensity of the ‘first wave’ ECL of [Ru(bpy)3]2+. The approach was extended to iridium(III) luminophores, where the maximum first-wave ECL intensity of [Ir(piq)2(dm-bpy)]+ was increased by up to 4-fold. As with [Ru(bpy)3]2+, the onset potential of the ECL from this luminophore and the extent to which the intensity was enhanced could be predominantly ascribed to the potential at which the mediator was oxidised. In contrast, the redox mediators were generally not effective at increasing the co-reactant ECL intensity of [Ir(df-ppy)2(dm-bpy)]+ or Ir(piq)2(acac) because the required excitation pathways were either thermodynamically inaccessible or initiated at the same potentials as competing reactions involving the direct electrochemical oxidation of the luminophore. These findings establish that neutral iridium(III) complexes can function as redox mediators in non-aqueous co-reactant ECL systems and provide mechanistic insight for extending redox-mediator-enhanced ECL to new luminophores and applications.
Introduction
The introduction of tri-n-propylamine (TPrA) as a co-reactant for tris(2,2′-bipyridine)ruthenium(II) ([Ru(bpy)3]2+) electrochemiluminescence (ECL)1,2 marked a critical step in the development of this highly sensitive mode of detection.3–5 The reaction can be initiated in either organic or aqueous solution by applying a single electrochemical potential to oxidise both the luminophore and co-reactant (eqn (1)–(5) and (9), where M is the luminophore; TPrA˙+ is a radical cation; TPrA˙ is the neutral α-amino radical, Pr2NC˙HCH2CH3; and P+ is the iminium cation, Pr2N+![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCH2). At high luminophore concentrations, the reaction between the oxidised and reduced luminophore can also generate the excited state (eqn (8)). An alternative pathway in which only the co-reactant is oxidised (eqn (2), (3), (6), (7) and (9)) enables efficient excitation of [Ru(bpy)3]2+ remote from the electrode surface, providing the basis for ECL labelling in microbead-supported assays for clinical diagnostics.6–9 The pathways can be conveniently distinguished by their final reductive, oxidative, or ‘comproportionative’ excitation of the luminophore (eqn (5), (7) and (8), respectively).10
CHCH2). At high luminophore concentrations, the reaction between the oxidised and reduced luminophore can also generate the excited state (eqn (8)). An alternative pathway in which only the co-reactant is oxidised (eqn (2), (3), (6), (7) and (9)) enables efficient excitation of [Ru(bpy)3]2+ remote from the electrode surface, providing the basis for ECL labelling in microbead-supported assays for clinical diagnostics.6–9 The pathways can be conveniently distinguished by their final reductive, oxidative, or ‘comproportionative’ excitation of the luminophore (eqn (5), (7) and (8), respectively).10| | | M+ + TPrA → M + TPrA˙+ | (4) |
| | | M− + TPrA˙+ → M* + TPrA | (7) |
The intensity of co-reactant ECL is dependent on the formation and lifetime of the intermediates responsible for chemi-excitation, and thus influenced by conditions such as electrode materials and the solvent matrix.11 We have shown that the ECL of [Ru(bpy)3]2+ with TPrA under the aqueous conditions typically employed for bioassays8 can be enhanced by small-molecule redox-mediators that serve as electrocatalysts of co-reactant oxidation (eqn (10) and (11), where E is the redox mediator), and alternative intermediates for the oxidative excitation pathway (eqn (12)).10,12–17 These pathways are analogous to those shown in eqn (1), (4), and (8), respectively, but can occur at much lower potentials than those required to oxidise the luminophore, and at greater rates due to the higher concentration of the redox mediator than the luminophore.
| | | E+ + TPrA → E + TPrA˙+ | (11) |
Redox mediators with the requisite properties16,18 were developed by adding sulfonate groups to homoleptic iridium(III) complexes.19–21 This includes [Ir(sppy)3]3−, which enhances the oxidative excitation10 pathway (eqn (2), (3), (6), (7) and (9); M = [Ru(bpy)3]2+) by over an order of magnitude, but also acts as a luminophore via a reductive excitation pathway (eqn (2), (3), (10), (13) and (14); E = [Ir(sppy)3]3−).12,16 The more recently reported phenylpyrazole analogue, [Ir(sppz)3]3−, enabled the same mechanisms of enhancement without concomitant emission from the mediator.18 These studies have focused on the [Ru(bpy)3]2+ luminophore, but were recently extended to the heteroleptic iridium(III) complex [Ir(spbt)2(bpy)]−,22 indicating a broad scope for application, including the highly promising class of iridium(III) luminophores.23–26
Both the optimisation of redox-mediator properties and their application to enhance novel luminophores have been hindered by the need to prepare water-soluble analogues of complexes previously studied in organic solvents.16,18,22 Prior application of redox mediators in annihilation ECL systems27–29 have shown their utility to enhance ECL intensity under non-aqueous conditions. Herein, we explore the enhancement of co-reactant ECL in acetonitrile, enabling comparison of commercially available homoleptic and heteroleptic iridium(III) complexes, without modification, as redox mediators and luminophores. The translation of this redox-mediator-enhanced ECL to non-aqueous conditions will inform the development of novel ECL systems, including highly sensitive detection, multi-colour reporting30–32 and displays.33,34
Experimental section
Chemicals
Acetonitrile was distilled over calcium hydride under a nitrogen atmosphere, and tri-n-propylamine was distilled over potassium hydroxide under reduced pressure. All solutions were degassed with grade 5 argon prior to each electrochemical and ECL experiment. Tetrabutylammonium hexafluorophosphate (TBAPF6; ≥99%) and [Ru(bpy)3](PF6)2 were purchased from Sigma-Aldrich; the facial isomers of Ir(ppy)3, Ir(pmi)3 and Ir(ppz)3 were purchased from Luminescence Technology Corp.; Ir(piq)2(acac) and Ir(ppy)2(acac) were sourced from SunaTech. [Ir(piq)2(dm-bpy)]PF6 and [Ir(df-ppy)2(dm-bpy)]PF6 were synthesised as previously described.35
Spectroscopy
Absorption and ambient temperature photoluminescence spectra were acquired using a Cary 300 Bio UV/vis spectrophotometer (600 nm min−1, 2 nm bandwidth; Agilent) and Cary Eclipse fluorescence spectrophotometer (600 nm min−1, 5 nm bandwidth; Agilent), respectively. Spectra were measured in a quartz cuvette (1 cm pathlength) using 10 µM metal complex in acetonitrile. Low temperature (85 K) spectra were obtained using the Eclipse spectrometer with an OptistatDN Variable Temperature Liquid Nitrogen Cryostat (Oxford Instruments). The metal complex was prepared at 5 µM in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 spectrophotometric grade ethanol:methanol in a custom-made quartz sample holder.36 Samples were cooled to 85 K to avoid damaging the cell near 77 K.35,37 Under our conditions, no discernible difference in the maximum emission wavelengths of [Ru(bpy)3]2+ and Ir(ppy)3 were observed between these temperatures.36 All emission spectra were corrected for changes in instrument sensitivity over the wavelength range based on correction factors established using a quartz-halogen tungsten lamp.
:1 spectrophotometric grade ethanol:methanol in a custom-made quartz sample holder.36 Samples were cooled to 85 K to avoid damaging the cell near 77 K.35,37 Under our conditions, no discernible difference in the maximum emission wavelengths of [Ru(bpy)3]2+ and Ir(ppy)3 were observed between these temperatures.36 All emission spectra were corrected for changes in instrument sensitivity over the wavelength range based on correction factors established using a quartz-halogen tungsten lamp.
Electrochemistry and ECL
While the electrochemical potentials referred to in this paper are strictly reduction potentials, for clarity of explanation we use the terms ‘oxidation potential’ (Eox) and ‘reduction potential’ (Ered) to refer to those corresponding to the oxidation and reduction of the metal complex or co-reactant, as adopted in some prior studies in ECL and related fields.
Electrochemical data were acquired in a quartz-bottomed electrochemical cell equipped with a Teflon cap designed to accommodate a three-electrode configuration. A glassy carbon working electrode, leakless Ag/AgCl reference electrode (model ET 069, eDAQ Australia) and platinum wire counter electrode (CH Instruments) were attached to an Autolab PGSTAT204 potentiostat (Metrohm, Australia). Prior to each measurement, the working electrode was polished with 0.05 µm Al2O3 powder, rinsed with water, and sonicated in acetonitrile for 15 s; the reference electrode was rinsed with water and acetonitrile; and the counter electrode was wiped clean with water and acetonitrile and flamed with a blowtorch. The cell was interfaced with (i) a charge coupled device (CCD) spectrometer (QE Pro, Ocean Optics) with 200 µm slit width, via optical fibre with a collimating lens, or (ii) a photomultiplier tube (model 9828B with A1 amplifier, ET Enterprises), aligned with the working electrode surface. The PMT was operated at 900 V provided by a PM20D power supply via A1 voltage divider (ET Enterprises).
A spooling ECL approach16,38,39 was used to obtain ECL spectra at a series of different applied potentials. The pulse sequence was developed in NOVA software, where 2 s oxidative potentials from 0.5 V to 1.9 V vs. Ag/AgCl were applied in 50 mV intervals, interspersed with 0 V for 5 s. The CCD spectrometer was synchronised with each potential pulse using an HR4000 (Ocean Optics) breakout box. Experiments were performed using 10 µM luminophore and 100 µM redox mediator in freshly distilled acetonitrile with 10 mM TPrA co-reactant and 0.1 M TBAPF6 as supporting electrolyte. The relative standard deviation of the maximum ECL intensity over the potential range was generally below 5% (n = 3). The replicate data was averaged, and the spectra were deconvoluted as described below.
Additional ECL experiments were performed using [Ru(bpy)3]2+ and TPrA, with and without the non-emissive Ir(ppz)3 mediator by measuring the ECL intensity from the luminophore with the PMT during cyclic voltammetry experiments. Potentials between 0 V and 2.0 V were scanned at 0.1 V s−1. The standard deviation of the local maximum of first-wave ECL intensity for 10 µM [Ru(bpy)3]2+ with 100 µM Ir(ppz)3 was 4.6% (n = 6).
Deconvolution of spectra from spooling ECL experiments
In cases where ECL was elicited by both the luminophore and redox mediator, a spectral deconvolution approach based on constrained nonlinear optimisation was implemented using Python code. The core assumption of this method is that the measured spectrum of the mixture can be approximated as a weighted sum of the normalised spectra of the pure components. These weights, represented by two coefficients (α for the luminophore and β for the mediator), indicate the relative contribution of each component to the overall signal. For consistency, the spooling ECL spectra collected from luminophores combined with non-emissive mediators were treated with the same process. In those cases, the integrated intensities of the output luminophore spectra matched those of the input spectra from the reaction mixture.
The optimisation was carried out using the L-BFGS-B algorithm, a variant of the Broyden–Fletcher–Goldfarb–Shanno (BFGS) method that supports simple bound constraints,40,41 which makes it particularly appropriate for spectral analysis, where physical constraints such as non-negative contributions must be enforced. L-BFGS-B is a memory-efficient quasi-Newton method that estimates second-derivative (Hessian) information from gradients, allowing it to converge quickly even for high-dimensional problems. The mixture spectrum, denoted by M(λ), was modelled as:
| | ![[M with combining circumflex]](https://www.rsc.org/images/entities/i_char_004d_0302.gif) (λ) = α × L(λ) + β × E(λ) (λ) = α × L(λ) + β × E(λ) | (15) |
where
L(
λ) and
E(
λ) are the shape-normalised spectra of the luminophore and mediator, respectively, and
(
λ) is the predicted mixture spectrum at wavelength
λ. Shape normalisation removes intensity scaling and isolates spectral features, ensuring that
α and
β reflect only the relative intensities in the mixture, not differences in the total magnitude of the spectra. The optimisation minimised the following objective function:
| | 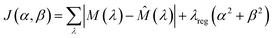 | (16) |
The first term measures the total deviation between the measured and predicted spectra using the sum of absolute differences across all wavelengths, which is less sensitive to outliers than squared error. The second term introduces a regularisation penalty with a small coefficient λreg, typically set to 10−5 to prevent the solution from diverging due to noisy data or overfitting.
Results and discussion
To evaluate the effect of small-molecule redox mediators on ECL with TPrA as a co-reactant in acetonitrile, we selected four iridium(III) complexes (Ir(ppy)3, Ir(ppz)3, Ir(ppy)2(acac) and Ir(pmi)3; Fig. 1), based on the properties found to be essential for the enhancement of ECL in aqueous media.16 These properties included: (i) chemically and electrochemically reversible oxidation at a potential close to that of the irreversible oxidation of TPrA; (ii) no reduction by TPrA˙ (eqn (17)); and (iii) greater triplet excited-state energy than the target luminophore.16
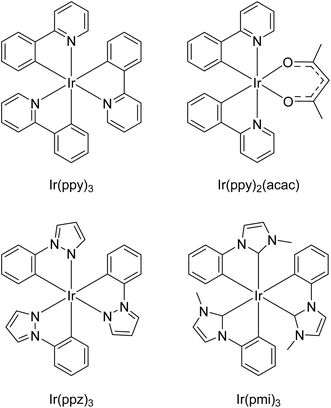 |
| | Fig. 1 Iridium(III) complexes tested as redox mediators. | |
For the luminophores, we selected [Ru(bpy)3]2+ (Fig. 2) as the archetypal metal complex for ECL, Ir(piq)2(acac) and [Ir(piq)2(dm-bpy)]+ as neutral and cationic iridium(III) complexes exhibiting orange-red ECL via either the reductive (TPrA˙) or oxidative (TPrA˙+) excitation pathways, and [Ir(df-ppy)2(dm-bpy)]+, which emits blue-green ECL only via reductive (TPrA˙) excitation following the direct electrochemical oxidation of the luminophore.35
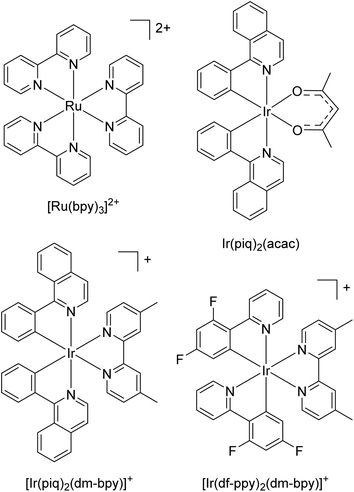 |
| | Fig. 2 Ruthenium(II) and iridium(III) complexes used as luminophores. | |
The spectroscopic and electrochemical properties of the metal complexes selected as redox mediators and luminophores have been well characterised,35,42–44 and are presented in Table 1 and Fig. S1–S5. Two of the redox mediators, Ir(ppy)3 and Ir(ppy)2(acac), exhibit intense green photoluminescence (ϕPL = 0.97 and 0.34, in deaerated 2-MeTHF43,45), whereas Ir(pmi)3 emits weakly in the near-UV (ϕPL = 0.02),42 and Ir(ppz)3 is effectively non-emissive at ambient temperature (ϕPL < 0.01)43 due to thermal deactivation via nonradiative 3MC states. The excited state energies (E0–0) of the redox mediators were higher than those of the luminophores, except for Ir(ppy)3 and Ir(ppy)2(acac) compared to [Ir(df-ppy)2(dm-bpy)]+.
Table 1 Selected spectroscopic and electrochemical properties for the metal complexes designated herein as redox mediators and luminophores
|
|
λ
max
/nm |
λ
max
(85 K)/nm |
E
0–0
/eV |
E
ox
/V (vs. Ag/AgCl) |
E
red
/V (vs. Ag/AgCl) |
|
Metal complexes at 10 µM in acetonitrile at ambient temperature.
Metal complexes at 5 µM in ethanol:methanol (4:1) at 85 K, unless otherwise indicated (sh = shoulder).
Energy gap between the zeroth vibrational levels of the ground and excited states, estimated from the highest energy peak of the low temperature emission spectrum.
Reduction potentials from cyclic voltammetry for metal complexes in acetonitrile with 0.1 M TBAPF6 as supporting electrolyte.
Essentially non-emissive at room temperature.42
Reduction not observed within the electrochemical window of the solvent matrix.
In 2-methyltetrahydrofuran at 77 K.
|
|
Redox mediators
|
| Ir(ppy)3 |
526 |
494, 531 |
2.51 |
0.74, 1.71 |
−2.25 |
| Ir(ppy)2(acac) |
527 |
501, 537 |
2.47 |
0.84, 1.72 |
−2.15 |
| Ir(ppz)3 |
|
410, 435, 459 |
3.02 |
0.82, 1.75 |
|
| Ir(pmi)3 |
384, 405 (ref. 46) |
380g,42 |
3.26 |
0.67, 1.29, 1.43 |
|
|
|
Luminophores
|
| [Ru(bpy)3]2+ |
620 |
581, 629, 690(sh) |
2.14 |
1.30 |
−1.31, −1.51, −1.75 |
| [Ir(piq)2(dm-bpy)]+ |
595, 631 |
581, 594(sh), 631, 687 |
2.14 |
1.24, 1.85 |
−1.44, −1.73, −1.97 |
| Ir(piq)2(acac) |
633 |
603, 654, 717 |
2.06 |
0.83, 1.63 |
−1.72, −1.95 |
| [Ir(df-ppy)2(dm-bpy)]+ |
524 |
450, 482, 508, 518 |
2.76 |
1.58 |
−1.40, −2.05 |
All four mediators were reversibly oxidised (Fig. S4) at a potential similar to that of the irreversible oxidation of the TPrA co-reactant (an Eox ≈ 0.86 ± 0.07 V vs. Ag/AgCl was established in ACN/benzene solution)47 and should not be reduced by TPrA˙ (Eox ≈ −1.7 vs. Ag/AgCl).7,47 In contrast, the luminophores were generally oxidised at higher potentials than TPrA, and would be expected to be reduced by TPrA˙ (eqn (6)). Although not examined in our prior ECL studies in aqueous media, the iridium(III) complexes exhibited subsequent irreversible (or, in the case of Ir(pmi)3, quasi-reversible) oxidations at higher potentials (Fig. S5).
Co-reactant ECL of [Ru(bpy)3]2+ with Ir(ppy)3 or Ir(ppz)3 as a redox mediator
For a comprehensive analysis of the influence of the redox-mediators, we used a spooling ECL approach,16,38,39 where emission spectra were acquired at a series of applied potentials, enabling the generation of contour plots of ECL intensity versus wavelength and potential (Fig. 3a–c). The luminophore and co-reactant concentrations were based on those used in previous investigations of the ECL of iridium(III) complexes.46,48 From these data, the integrated ECL intensity of the luminophore over the applied potential range was extracted (Fig. 3g).
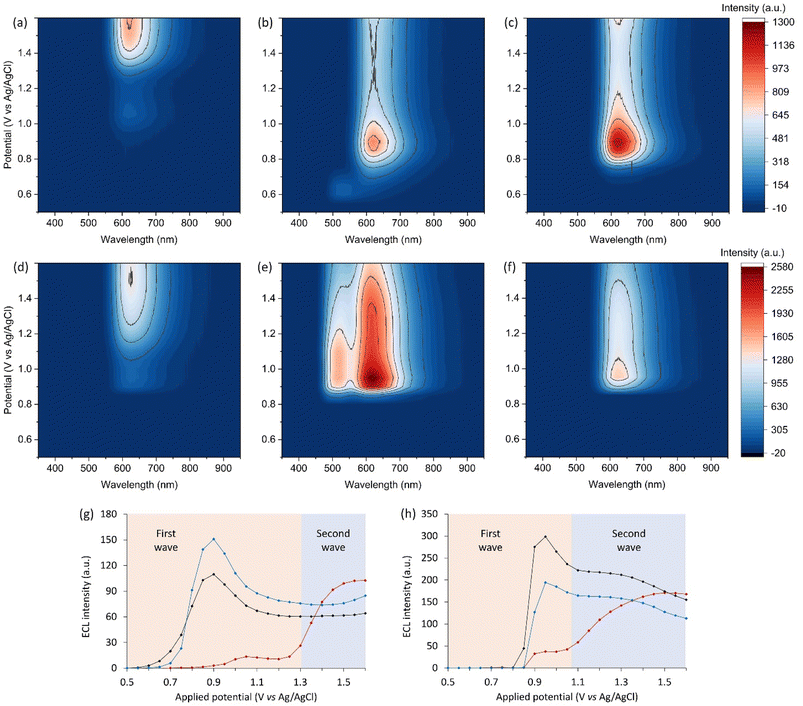 |
| | Fig. 3 (a–f) Contour plots of ECL intensity as a function of wavelength and applied potential, for: 10 µM [Ru(bpy)3]2+ with (a) no redox mediator, (b) 100 µM Ir(ppy)3, or (c) 100 µM Ir(ppz)3 in acetonitrile with 10 mM TPrA and 0.1 M TBAPF6; or 1 µM [Ru(bpy)3]2+ with (d) no redox mediator, (e) 100 µM [Ir(sppy)3]3−, or (f) 100 µM [Ir(sppz)3]3− in aqueous ‘ProCell’ solution containing 180 mM TPrA, 0.1% polidocanol surfactant, and 0.3 M phosphate buffer (pH 6.8).8 (g and h) Comparisons of relative ECL intensity of [Ru(bpy)3]2+ (integrated spectral distribution) without a redox mediator (red plot) or (g) when using Ir(ppy)3 (black plot) or Ir(ppz)3 (blue plot) in acetonitrile; and (h) when using [Ir(sppy)3]3− (black plot) or [Ir(sppz)3]3− (blue plot) in aqueous media, extracted from the data shown in Fig. 3a–c and d–f, respectively. Data in Fig. 3d–f and h is from ref. 18. In the plots for Ir(ppy)3 and [Ir(sppy)3]3− in Fig. 3g and h, respectively, the emission from [Ru(bpy)3]2+ has been deconvoluted from that of the redox mediator (Fig. S6 and S16). The coloured boxes in Fig. 3g and h show the potentials below (red) and above (blue) the Eox of [Ru(bpy)3]2+ in that solvent. | |
Despite the differences in conditions, the change in ECL intensity over the applied potentials with and without the redox mediators in acetonitrile (Fig. 3a–c and g) was similar to that in the aqueous buffer solution used in our prior investigations (e.g., Fig. 3d–f and h).18 In the absence of the redox mediators, the characteristic ‘two waves’ of ECL7 are observed (Fig. 3g and h; red plots): the first at potentials that oxidise only the co-reactant, resulting in the ‘oxidative (TPrA˙+) excitation’ pathway (eqn (2), (3), (6), (7) and (9), where M is [Ru(bpy)3]2+); and the second at potentials that oxidise both the co-reactant and luminophore, where the ‘reductive (TPrA˙) excitation’ pathway (eqn (1)–(5) and (9)) is dominant.
The addition of Ir(ppy)3 (or [Ir(sppy)3]3− in aqueous media) resulted in a new ECL peak matching the characteristic emission from the mediator, and a significant enhancement of the ECL of [Ru(bpy)3]2+, particularly in the first wave (Fig. 3g and h, black plots). The co-reactant ECL of Ir(ppy)3 in organic media (in the absence or presence of the luminophore) occurred only at low potentials, due to efficient oxidative quenching by TPrA˙+ (eqn (18)),38 whereas the co-reactant ECL of [Ir(sppy)3]3− in aqueous solution was still observed at high overpotentials (Fig. S6 and S8). Increases in the ECL of [Ru(bpy)3]2+ were also observed upon addition of Ir(ppz)3 (or [Ir(sppz)3]3−), but without the concomitant emission from the mediator (Fig. 3g and h, blue plots).
| | | M* + TPrA˙+ → M+ + TPrA | (18) |
The ECL intensity of [Ru(bpy)3]2+ in the presence of redox mediators was greatest at the initial onset potentials of the unenhanced first wave. At these potentials, the enhancement exceeded two orders of magnitude (Fig. S9). Even compared to the maximum intensity of the unenhanced first wave (at 1.05 V vs. Ag/AgCl), the addition of Ir(ppy)3 and Ir(ppz)3 increased the peak ECL intensity (measured at 0.90 V vs. Ag/AgCl) by 8.2- and 11-fold, respectively (Fig. S10). In comparison, the same concentrations of [Ir(sppy)3]3− and [Ir(sppz)3]3− enhanced the first wave of [Ru(bpy)3]2+ ECL in aqueous solution by 8.0- and 5.2-fold, respectively, at 0.95 V vs. Ag/AgCl (Fig. 3h).18
Differences in the relative ECL intensities of [Ru(bpy)3]2+ over the applied potential range between the two solvents (Fig. 3g and h) can be attributed to a combination of factors that include the interfacial potentials, the thermodynamic stability of relevant oxidation states of the luminophore and mediators, and the rates of co-reactant radical formation, both the initial heterogeneous oxidation (eqn (2); Fig. S11), and the subsequent rate of deprotonation (eqn (3)).49
Having confirmed that the Ir(ppz)3 mediator was non-emissive under these conditions, we used a simplified experimental approach to examine the influence of luminophore and mediator concentrations, in which the ECL was measured during linear sweep voltammetry. As shown in Fig. 4, the addition of 100 µM Ir(ppz)3 greatly increased the first-wave ECL emission of [Ru(bpy)3]2+ at all concentrations tested (1–200 µM). The enhancement factor for concentrations up to 30 µM [Ru(bpy)3]2+ remained consistent at 11.3 ± 1.2 (Fig. S12) but could not be determined at 100 µM and 200 µM [Ru(bpy)3]2+ because the enhanced ECL intensity exceeded the linear range of the photodetector. Without the redox mediator, 200 µM [Ru(bpy)3]2+ was required to obtain the same first-wave ECL intensity as only 10 µM [Ru(bpy)3]2+ with 100 µM Ir(ppz)3 (Fig. S13). The enhancement factor was found to increase linearly with Ir(ppz)3 up to 200 µM, the highest concentration tested (Fig. S14), which increased the first-wave ECL of 10 µM [Ru(bpy)3]2+ by 20-fold.
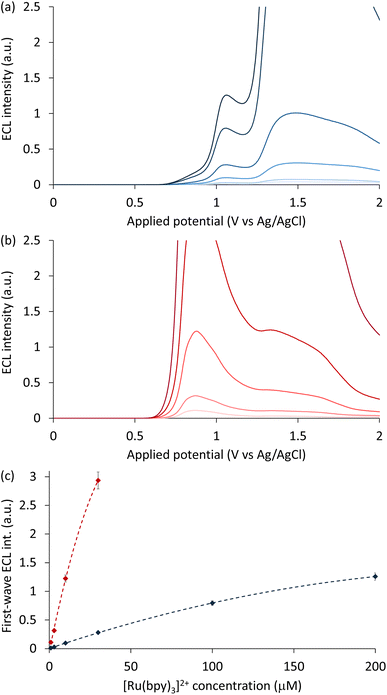 |
| | Fig. 4 (a and b) ECL intensity measured while scanning the applied potential from 0 to 2 V vs. Ag/AgCl, using 1, 3, 10, 30, 100 and 200 µM [Ru(bpy)3]2+ with (a) no redox mediator, or (b) 100 µM Ir(ppz)3. (c) First-wave ECL intensity (local intensity maximum) versus [Ru(bpy)3]2+ concentration with no redox mediator (blue plot) or 100 µM Ir(ppz)3 (red plot). Error bars show ±1 standard deviation for this experimental approach. A log–log plot of this relationship is included in the SI (Fig. S12). | |
The redox-mediator enhancement of ECL involves two pathways, depicted in eqn (10)–(12).10,12–17 The free energy of eqn (12) (ΔGes) can be estimated by eqn (19), where E(M)red is the potential at which the luminophore is reduced, and E(E)ox is the potential at which the enhancer is oxidised.50,51 This excludes the contributions from the electrostatic interactions of the reactants and products, but they are relatively minor and can be reasonably disregarded for a first approximation.
| | | ΔGes (in eV) ≈ (E(M)red – E(E)ox) + E0–0 | (19) |
This indicated the oxidative ([Ir(ppy)3]+ or [Ir(ppz)3]+) excitation to [Ru(bpy)3]2+* (eqn (12)) is slightly endergonic (ΔGes ≈ +0.09 eV and +0.01 eV, respectively), but within the degree of error of the approach (at least 0.1 V).52 We therefore assessed this excitation pathway using mixed annihilation ECL, in which potentials were alternately applied to reduce [Ru(bpy)3]2+ and oxidise the redox mediator (in the absence of co-reactant). The subsequent reaction of the electrogenerated intermediates resulted in intense ECL (Fig. S15a and b), confirming the feasibility of this pathway with both mediators. The mixed annihilation ECL intensity was greater with Ir(ppz)3 than Ir(ppy)3, aligned with their relative enhancement of the co-reactant ECL of [Ru(bpy)3]2+ (Fig. 3g). The electrocatalysis of co-reactant oxidation (eqn (10) and (11)) was examined by comparing cyclic voltammograms of the co-reactant solution with and without the Ir(ppz)3 mediator (Fig. S11a). The mediator increased the rate at which the TPrA was oxidised, although the effect was more subtle than under the previously reported aqueous conditions in which co-reactant oxidation was relatively slow (Fig. S11b).
Both redox mediators reduced the ECL intensity of the second wave (Fig. 3g), which has previously been rationalised for water-soluble analogues as consumption of [Ru(bpy)3]3+ by the mediator (eqn (20)), and TPrA˙ by the oxidised mediator.18
Alternative redox mediators: Ir(ppy)2(acac) and Ir(pmi)3
The photoluminescence of Ir(ppy)2(acac) is bathochromically shifted by ∼5 nm compared to Ir(ppy)3 at both ambient and low temperature (Fig. S2b and S3b), corresponding to slightly lower triplet state energy (Table 1). The oxidation potential of Ir(ppy)2(acac) is approximately 0.10 V more positive than Ir(ppy)3, which has several implications for the mechanisms of enhancement. Firstly, the catalysis of co-reactant oxidation (eqn (10) and (11)) does not occur until higher potentials, resulting in a commensurate shift in the onset of the ECL enhanced by these mediators (Fig. 5a). On the other hand, the redox-mediator oxidative excitation pathway (eqn (12)), is more thermodynamically favourable by 0.10 eV. The combined outcome was lower enhancement of the ‘first wave’ ECL, but less quenching of the ‘second wave’.
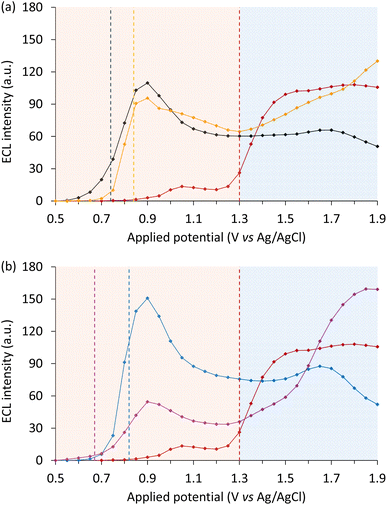 |
| | Fig. 5 ECL intensity (integrated spectrum) across the potential range for (a and b) 10 µM [Ru(bpy)3]2+ without a redox mediator (red plot), or with 100 µM redox mediator: (a) Ir(ppy)3 (black plot) and Ir(ppy)2(acac) (orange plot), or (b) Ir(ppz)3 (blue plot) and Ir(pmi)3 (purple plot), in acetonitrile containing 10 mM TPrA and 0.1 M TBAPF6 extracted from spooling ECL data (Fig. S18). For the plots in Fig. 5a, the emission from [Ru(bpy)3]2+ has been deconvoluted from that of the redox mediators (Fig. S6a and S16). The vertical dashed lines indicate the first Eox of the redox mediators and luminophore. The standard deviation of the ECL intensity at each potential is depicted in Fig. S19. | |
The Ir(pmi)3 complex provides an interesting alternative to Ir(ppz)3 as a non-emissive redox mediator. Although Ir(pmi)3 exhibits a luminescence quantum yield (φPL) of ∼0.02 in 2-MeTHF at ambient temperature42 (whereas φPL of Ir(ppz)3 < 0.01),43 the excited states of both mediators are inaccessible in the ECL co-reactant schemes examined herein, because neither eqn (13) nor (17) are thermodynamically favoured. The oxidation potential of Ir(pmi)3 is 0.16 V less positive than Ir(ppz)3 (Table 1), which could be anticipated to provide a strong enhancement (with an earlier onset) if there is sufficient driving force for the electrocatalysis of co-reactant oxidation (eqn (10) and (11)). On the other hand, the oxidative (Ir(pmi)3+) excitation of [Ru(bpy)3]+ to [Ru(bpy)3]2+* (eqn (12)) should not be energetically feasible (ΔGes ≈ +0.16 eV). Indeed, previous mixed annihilation experiments using Ir(pmi)3 and [Ru(bpy)3]2+ have shown negligible ECL.53 This was verified under our conditions where almost no signal was observed when pulsing 0.1 V beyond the reduction potential of [Ru(bpy)3]2+ and the oxidation potential of Ir(pmi)3 (Fig. S15d).
The enhanced first-wave ECL of [Ru(bpy)3]2+ when using Ir(pmi)3 (measured at 0.9 V vs. Ag/AgCl) was only 0.36-fold that of [Ru(bpy)3]2+ with Ir(ppz)3 (at 0.9 V vs. Ag/AgCl), but still 4.1-fold greater than the local maximum intensity of the unenhanced reaction (at 1.05 V vs. Ag/AgCl). As noted above, the oxidative (Ir(pmi)3+) excitation pathway is not viable, and therefore in this case, the observed enhancement of the first wave can be attributed solely to the electrocatalysis of co-reactant oxidation (eqn (10) and (11)), despite the lower oxidation potential of the mediator than that estimated for the co-reactant (Eox ≈ 0.89 ± 0.04 V vs. Ag/AgCl).7,47 A comparison of the maximum first-wave ECL intensity of [Ru(bpy)3]2+ without a redox mediator and with each mediator in order of increasing oxidation potential is shown in Fig. 6. This potential establishes both the onset of enhancement (eqn (10)) and the relative efficiencies of eqn (11)–(13), with Ir(ppz)3 providing the optimum.
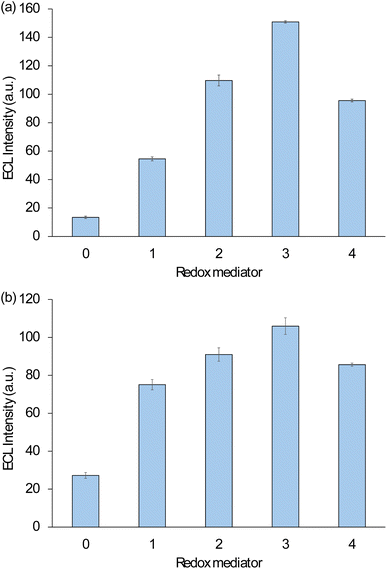 |
| | Fig. 6 Maximum first-wave ECL intensity for (a) [Ru(bpy)3]2+ and (b) [Ir(piq)2(dm-bpy)]+, with (0) no redox mediator, or (1–4) with a redox mediator, in order of increasing oxidation potential: (1) Ir(pmi)3, (2) Ir(ppy)3, (3) Ir(ppz)3, or (4) Ir(ppy)2(acac). ECL intensities were obtained from the data shown in Fig. 5 and 7. Error bars show ±1σ (n = 3). | |
Of the four redox mediators tested, Ir(pmi)3 gave the least enhancement of the first-wave ECL of [Ru(bpy)3]2+, but the greatest increase in intensity once the oxidation potential of the luminophores was attained. This can be rationalised by the chemical reversibility and lower potentials of its subsequent electrochemical oxidations (1.29 V and 1.43 V vs. Ag/AgCl; Table 1), which could diminish the consumption or even increase the generation of [Ru(bpy)3]3+ (eqn (18)). The first of these subsequent oxidations occurs at a similar potential to the oxidation of [Ru(bpy)3]2+, and both correspond to a change in the incline of the ECL intensity with increasing applied potential (Fig. 5b). At 1.90 V vs. Ag/AgCl, this mediator increased the intensity of [Ru(bpy)3]2+ ECL by 1.5-fold. This may have implications for the enhancement of ECL systems where high overpotentials are needed to derive appreciable signal, such as those at the gas/liquid interface of electrochemically generated bubbles.54
Iridium(III) luminophores
[Ir(piq)2(dm-bpy)]+.
The [Ir(piq)2(dm-bpy)]+ complex was selected as a starting point for the exploration of iridium(III) luminophores within this system because of the similarity of its spectroscopic and redox properties to [Ru(bpy)3]2+ (Table 1). [Ir(piq)2(dm-bpy)]+ is, however, marginally easier to oxidise and harder to reduce, with the same triplet state energy, resulting in a thermodynamically more favourable excitation pathway (eqn (12)) with all four oxidised redox mediators, including Ir(pmi)3+, which generated negligible ECL upon reaction with [Ru(bpy)3]+ (Fig. S15d). A mixed annihilation ECL experiment in which potentials were alternately applied to reduce [Ir(piq)2(dm-bpy)]+ and oxidise Ir(pmi)3 (in the absence of co-reactant) resulted in intense ECL (Fig. S20), confirming the feasibility of the excitation pathway involving these species.
All four redox mediators elicited a significant increase in the first-wave ECL intensity of [Ir(piq)2(dm-bpy)]+ (Fig. 7). As observed when using the [Ru(bpy)3]2+ luminophore, the ECL onset was dependent on the oxidation potential of the mediators [i.e. Ir(pmi)3 < Ir(ppy)3 < Ir(ppz)3 < Ir(ppy)2(acac)]. For the mediators with lowest oxidation potentials (Fig. 7a), two intensity maxima were observed within the first-wave region, indicating a different dependence of the multiple, competing excitation and quenching pathways on the applied potential, presumably accentuated by the greater gap in oxidation potential of the mediator and co-reactant. Related effects have previously been observed in the intensity versus applied potential plots of other ECL systems.38,55
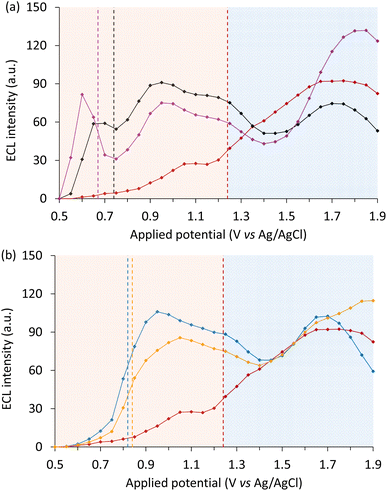 |
| | Fig. 7 ECL intensity (integrated spectrum) across the potential range for (a and b) 10 µM [Ir(piq)2(dm-bpy)]+ without a redox mediator (red plot), or with 100 µM redox mediator: (a) Ir(pmi)3 (purple plot) and Ir(ppy)3 (black plot) and, or (b) Ir(ppz)3 (blue plot) and Ir(ppy)2(acac) (orange plot), in acetonitrile containing 10 mM TPrA and 0.1 M TBAPF6, extracted from spooling ECL data (Fig. S21). Where required, the ECL of [Ir(piq)2(dm-bpy)]+ was deconvoluted from the concomitant emission from the mediator (Fig. S22 and S23). The vertical dashed lines indicate the first Eox of the redox mediators and luminophore. | |
The relative enhancement of the first wave ECL of [Ir(piq)2(dm-bpy)]+ by the four mediators showed a similar trend to that of [Ru(bpy)3]2+ (Fig. 6). Again, Ir(ppz)3 was the most effective; the maximum ECL intensity (at 0.95 V vs. Ag/AgCl) with this mediator was 3.9-fold that of the unenhanced first wave (at 1.05 V vs. Ag/AgCl). Overall, the degree of ECL enhancement of [Ir(piq)2(dm-bpy)]+ was lower than [Ru(bpy)3]2+, which may be due in part to the more efficient oxidative (TPrA˙+) excitation of the Ir(III) luminophore in the absence of redox mediators.
At potentials above the Eox of [Ir(piq)2(dm-bpy)]+, the influence of the redox mediators was similar to that for [Ru(bpy)3]2+ where, following initial signal depletion, the ECL intensity was increased at higher potentials, which was most prominent with Ir(pmi)3 and to a lesser extent Ir(ppy)2(acac).
Ir(piq)2(acac).
The oxidation potential of Ir(piq)2(acac) (Eox = 0.83 V vs. Ag/AgCl) is considerably lower than that of [Ru(bpy)3]2+ or [Ir(piq)2(dm-bpy)]+, and similar to that estimated for TPrA (0.86 ± 0.07 V vs. Ag/AgCl).47 Consequently, although the reductive (TPrA˙) excitation (eqn (1)–(5) and (8)) and oxidative (TPrA˙+) excitation (eqn (2), (4) and (6)–(8)) pathways are both feasible for the Ir(piq)2(acac) luminophore, the electrogenerated intermediates required for these first- and second-wave ECL pathways are generated at the same potentials. Furthermore, as the luminophore is oxidised at potentials similar to those of the redox mediators, it can ‘self-enhance’ the ECL at these low potentials (i.e. through eqn (1), (4) and (8); analogous to eqn (10)–(12)). The combination of these pathways leads to far greater ECL between 0.8 V and 1.5 V than with the other luminophores.
Based on eqn (19), the reactions of the oxidised redox mediators with the reduced luminophore (eqn (12)) to generate the emissive excited state are exergonic (ΔGes < −0.33 eV), but considering the largely opposing influences of the mediators on the oxidative and reductive excitation pathways seen with the above two luminophores, it was not surprising that very little enhancement or even quenching of the already large emission from this system was observed. A small increase (1.2-fold at 1.1 V vs. Ag/AgCl) in the ECL intensity of Ir(piq)2(acac) was provided by the Ir(ppy)2(acac) mediator (orange plot in Fig. 8b), which also induced the least quenching of the second-wave ECL of [Ru(bpy)3]2+ and [Ir(piq)2(dm-bpy)]+ (orange plots in Fig. 5a and 7b, respectively). At high potentials (>1.5 V vs. Ag/AgCl), the ECL of the reactions containing redox mediators rose and then fell with increasing potential, which was again most prominent with Ir(pmi)3 and to a lesser extent Ir(ppy)2(acac).
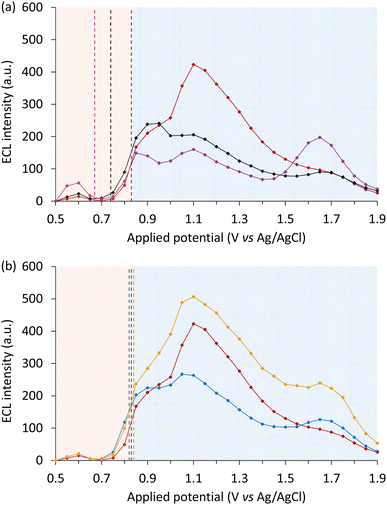 |
| | Fig. 8 ECL intensity (integrated spectrum) across the potential range for (a and b) 10 µM Ir(piq)2(acac) without a redox mediator (red plot), or with 100 µM redox mediator: (a) Ir(pmi)3 (purple plot) and Ir(ppy)3 (black plot) and, or (b) Ir(ppz)3 (blue plot) and Ir(ppy)2(acac) (orange plot), in acetonitrile containing 10 mM TPrA and 0.1 M TBAPF6, extracted from spooling ECL data (Fig. S24). Where required, the ECL of Ir(piq)2(acac) was deconvoluted from the concomitant emission from the mediator (Fig. S25 and S26). The vertical dashed lines indicate the first Eox of the redox mediators and luminophore. Where required, the ECL of Ir(piq)2(acac) was deconvoluted from the concomitant emission from the mediator. ECL contour plots of these data without deconvolution are shown in Fig. S24. | |
[Ir(df-ppy)2(dm-bpy)]+.
The co-reactant ECL of [Ir(df-ppy)2(dm-bpy)]+ was only observed upon its direct electrochemical oxidation (Fig. 9). This complex can be reduced by TPrA˙ (eqn (6)), but the oxidative (TPrA˙+) excitation is endergonic (eqn (7); ΔGes ≈ +0.50 eV). As the redox mediators are oxidised at potentials similar to that of TPrA˙+ their analogous reactions with the reduced luminophore (eqn (12)) also do not populate the excited state (ΔGes > +0.52 eV). Furthermore, the electrocatalysis of TPrA oxidation (eqn (10) and (11)) cannot increase the ECL intensity at potentials below those at which [Ir(df-ppy)2(dm-bpy)]+ is oxidised. At higher potentials, the redox mediators generally quenched the emission, which we attribute to depletion of [Ir(df-ppy)2(dm-bpy)]2+ and TPrA˙ as well as other parasitic side reactions, as discussed for the other luminophores.
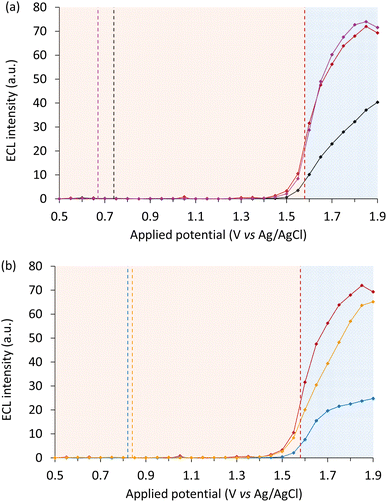 |
| | Fig. 9 ECL intensity (integrated spectrum) across the potential range for (a and b) 10 µM [Ir(df-ppy)2(dm-bpy)]+ without a redox mediator (red plot), or with 100 µM redox mediator: (a) Ir(pmi)3 (purple plot) and Ir(ppy)3 (black plot) and, or (b) Ir(ppz)3 (blue plot) and Ir(ppy)2(acac) (orange plot), in acetonitrile containing 10 mM TPrA and 0.1 M TBAPF6, extracted from spooling ECL data (Fig. S27). Where required, the ECL of [Ir(df-ppy)2(dm-bpy)]+ was deconvoluted from the concomitant emission from the mediator (Fig. S28 and S29). The vertical dashed lines indicate the first Eox of the redox mediators and luminophore. | |
Conclusions
Like their sulfonated derivatives ([Ir(sppy)3]3− and [Ir(sppz)3]3−) in buffered aqueous solution, Ir(ppy)3, Ir(ppz)3, and related complexes can serve as redox mediators to enhance the intensity of the ‘first-wave’ of oxidative-reduction co-reactant ECL in acetonitrile. With the [Ru(bpy)3]2+ luminophore, the mediators had similar effects to those observed in the aqueous systems, although the phenylpyrazole complex (rather than the phenylpyridine) was the most effective enhancer, and the lowering of the ECL onset potential was more pronounced. The Ir(ppz)3 mediator was also an effective enhancer of the distinct first-wave ECL of the [Ir(piq)2(dm-bpy)]+ luminophore, which has similar electrochemical and photophysical properties to the Ru(II) complex. In contrast, the redox mediators were generally not effective at increasing the co-reactant ECL intensity of the Ir(piq)2(acac) or [Ir(df-ppy)2(dm-bpy)]+ luminophores because either the reaction pathways of the first and second wave ECL were initiated at similar potentials, or those of the first wave ECL were not energetically feasible. These findings give insight into the fundamental pathways by which small-molecule redox mediators can enhance co-reactant ECL in organic media. The ability to examine novel luminophores and mediators without the need to synthesise water-soluble analogues removes a considerable bottleneck to explore this chemistry, enabling more rapid fundamental development prior to devising strategies to prepare water soluble derivatives for future applications in biosensing. Moreover, non-aqueous ECL systems have increasingly been used in sensing, exploiting approaches such as bipolar electrochemistry where the sensing and ECL ‘reporter’ reactions can be performed in different solvents,30,32 and in ECL-based light-emitting devices.34
Author contributions
PSF and SJB were responsible for project design. SJB and EMSM undertook the majority of the laboratory experimentation, under the supervision by PSF and EHD. DJH, TUC and PJB prepared some of the metal complexes. FM designed and applied the deconvolution approach using Python code, with assistance and/or supervision by NS, SMJJ and EHD. Funding to support the project was acquired by PSF and TUC. Relevant sections of the manuscript were prepared by SJB and FM. All authors assisted with additional writing and/or editing of the manuscript, which was overseen by PSF.
Conflicts of interest
There are no conflicts of interest to declare.
Data availability
Data are available upon request from the author for correspondence.
Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5sc08710d.
Acknowledgements
This work was funded by the Australian Research Council (DP220100300).
References
- J. B. Noffsinger and N. D. Danielson, Anal. Chem., 1987, 59, 865–868 CrossRef CAS.
- J. K. Leland and M. J. Powell, J. Electrochem. Soc., 1990, 137, 3127–3131 CrossRef CAS.
-
Electrogenerated Chemiluminescence, ed. A. J. Bard, Marcel Dekker, New York, 2004 Search PubMed.
- E. Kerr, E. H. Doeven and P. S. Francis, Curr. Opin. Electrochem., 2022, 35, 101034 Search PubMed.
- E. Villani, K. Sakanoue, Y. Einaga, S. Inagi and A. Fiorani, J. Electroanal. Chem., 2022, 921, 116677 Search PubMed.
- G. F. Blackburn, H. P. Shah, J. H. Kenten, J. Leland, R. A. Kamin, J. Link, J. Peterman, M. J. Powell, A. Shah, D. B. Talley, S. K. Tyagi, E. Wilkins, T.-G. Wu and R. J. Massey, Clin. Chem., 1991, 37, 1534–1539 Search PubMed.
- W. Miao, J.-P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed.
-
E. Faatz, A. Finke, H.-P. Josel, G. Prencipe, S. Quint and M. Windfuhr, in Analytical Electrogenerated Chemiluminescence: From Fundamentals to Bioassays, ed. N. Sojic, The Royal Society of Chemistry, 2019, pp. 443–470 Search PubMed.
- Y. Wang, J. Ding, P. Zhou, J. Liu, Z. Qiao, K. Yu, J. Jiang and B. Su, Angew. Chem., Int. Ed., 2023, 62, e202216525 Search PubMed.
- P. S. Francis, S. Knežević, C. F. Hogan and N. Sojic, ACS Electrochem., 2025, 1, 1006–1013 CrossRef CAS.
-
G. Valenti, A. Fiorani, E. Villani, A. Zanut and F. Paolucci, in Analytical Electrogenerated Chemiluminescence: From Fundamentals to Bioassays, ed. N. Sojic, The Royal Society of Chemistry, 2019, pp. 159–175 Search PubMed.
- E. Kerr, D. J. Hayne, L. C. Soulsby, J. C. Bawden, S. J. Blom, E. H. Doeven, L. C. Henderson, C. F. Hogan and P. S. Francis, Chem. Sci., 2022, 13, 469–477 RSC.
- E. Kerr, S. Knezevic, P. S. Francis, C. F. Hogan, G. Valenti, F. Paolucci, F. Kanoufi and N. Sojic, ACS Sens., 2023, 8, 933–939 CrossRef CAS PubMed.
- A. Fracassa, C. I. Santo, E. Kerr, S. Knezevic, D. J. Hayne, P. S. Francis, F. Kanoufi, N. Sojic, F. Paolucci and G. Valenti, Chem. Sci., 2024, 15, 1150–1158 RSC.
- S. Knezevic, E. Kerr, G. Valenti, F. Paolucci, P. S. Francis, C. F. Hogan, N. Sojic and F. Kanoufi, Electrochim. Acta, 2024, 499, 144677 CrossRef CAS.
- S. J. Blom, N. S. Adamson, E. Kerr, E. H. Doeven, O. S. Wenger, R. S. Schaer, D. J. Hayne, F. Paolucci, N. Sojic, G. Valenti and P. S. Francis, Electrochim. Acta, 2024, 484, 143957 CrossRef CAS.
- J. Cao, J. Ding, Y. Wang and B. Su, Analysis Sensing, 2025, e202500063 CrossRef CAS.
- N. S. Adamson, S. J. Blom, E. H. Doeven, T. U. Connell, C. Hadden, S. Knežević, N. Sojic, A. Fracassa, G. Valenti, F. Paolucci, Y. Wang, J. Ding, B. Su, C. Hua and P. S. Francis, Angew. Chem., Int. Ed., 2024, 63, e202412097 Search PubMed.
- C. Kerzig, X. Guo and O. S. Wenger, J. Am. Chem. Soc., 2019, 141, 2122–2127 CrossRef CAS PubMed.
- B. Pfund, D. M. Steffen, M. R. Schreier, M.-S. Bertrams, C. Ye, K. Börjesson, O. S. Wenger and C. Kerzig, J. Am. Chem. Soc., 2020, 142, 10468–10476 CrossRef CAS PubMed.
- M. R. Schreier, X. Guo, B. Pfund, Y. Okamoto, T. R. Ward, C. Kerzig and O. S. Wenger, Acc. Chem. Res., 2022, 55, 1290–1300 CrossRef CAS PubMed.
- K. S. Manimaran, S. K. Semjanov, K. F. White, J. L. Adcock, E. H. Doeven, D. J. Hayne, P. S. Francis and P. J. Barnard, Chem.–Eur. J., 2025, 31, e202500701 Search PubMed.
- A. Kapturkiewicz, Anal. Bioanal. Chem., 2016, 408, 7013–7033 CrossRef CAS PubMed.
-
S. Laird and C. F. Hogan, in Iridium(III) in Optoelectronic and Photonics Applications, ed. E. Zysman-Colman, John Wiley & Sons, Inc., Chichester, UK, 2017, pp. 359–414 Search PubMed.
- Y. Fu, X. Teng and C. Lu, Trends Anal. Chem., 2023, 167, 117273 CrossRef CAS.
- H. Gao, M. Qian, X. Yang, S. Ma and H. Li, Dyes Pigm., 2025, 232, 112493 CrossRef CAS.
- E. Kerr, E. H. Doeven, G. J. Barbante, C. F. Hogan, D. J. Hayne, P. S. Donnelly and P. S. Francis, Chem. Sci., 2016, 7, 5271–5279 RSC.
- R. Ishimatsu and E. Takano, J. Electroanal. Chem., 2024, 971, 118589 CrossRef CAS.
- N. Ichinohe, R. Otsuka, R. Ishimatsu, M. Kobayashi, J. Mizuno, N. Akino and T. Kasahara, Electrochemistry, 2024, 2, 027004 CrossRef.
- Y.-Z. Wang, C.-H. Xu, W. Zhao, Q.-Y. Guan, H.-Y. Chen and J.-J. Xu, Anal. Chem., 2017, 89, 8050–8056 CrossRef CAS PubMed.
- W. Guo, H. Ding, C. Gu, Y. Liu, X. Jiang, B. Su and Y. Shao, J. Am. Chem. Soc., 2018, 140, 15904–15915 CrossRef CAS PubMed.
- M. R. Moghaddam, S. Carrara and C. F. Hogan, Chem. Commun., 2019, 55, 1024–1027 RSC.
- S. H. Kong, J. I. Lee, S. Kim and M. S. Kang, ACS Photonics, 2018, 5, 267–277 CrossRef CAS.
- K. G. Cho, J. I. Lee, S. Lee, K. Hong, M. S. Kang and K. H. Lee, Adv. Funct. Mater., 2020, 30, 1907936 Search PubMed.
- L. Chen, D. J. Hayne, E. H. Doeven, J. Agugiaro, D. J. D. Wilson, L. C. Henderson, T. U. Connell, Y. H. Nai, R. Alexander, S. Carrara, C. F. Hogan, P. S. Donnelly and P. S. Francis, Chem. Sci., 2019, 10, 8654–8667 RSC.
- L. C. Soulsby, D. J. Hayne, E. H. Doeven, D. J. D. Wilson, J. Agugiaro, T. U. Connell, L. Chen, C. F. Hogan, E. Kerr, J. L. Adcock, P. S. Donnelly, J. M. White and P. S. Francis, Phys. Chem. Chem. Phys., 2018, 20, 18995–19006 Search PubMed.
- C. Mallet, A. Bolduc, S. Bishop, Y. Gautier and W. G. Skene, Phys. Chem. Chem. Phys., 2014, 16, 24382–24390 Search PubMed.
- E. H. Doeven, E. M. Zammit, G. J. Barbante, P. S. Francis, N. W. Barnett and C. F. Hogan, Chem. Sci., 2013, 4, 977–982 RSC.
- M. Hesari and Z. Ding, Nat. Protoc., 2021, 16, 2109–2130 CrossRef CAS PubMed.
- R. H. Byrd, P. Lu, J. Nocedal and C. Zhu, SIAM J. Sci. Comput., 1995, 16, 1190–1208 CrossRef.
- C. Zhu, R. H. Byrd, P. Lu and J. Nocedal, ACM Trans. Math. Softw., 1997, 23, 550–560 CrossRef.
- T. Sajoto, P. I. Djurovich, A. Tamayo, M. Yousufuddin, R. Bau, M. E. Thompson, R. J. Holmes and S. R. Forrest, Inorg. Chem., 2005, 44, 7992–8003 CrossRef CAS PubMed.
- T. Sajoto, P. I. Djurovich, A. B. Tamayo, J. Oxgaard, W. A. Goddard and M. E. Thompson, J. Am. Chem. Soc., 2009, 131, 9813–9822 CrossRef CAS PubMed.
- L. Chen, E. H. Doeven, D. J. D. Wilson, E. Kerr, D. J. Hayne, C. F. Hogan, W. Yang, T. T. Pham and P. S. Francis, ChemElectroChem, 2017, 4, 1797–1808 CrossRef CAS.
- S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704–1711 CrossRef CAS PubMed.
- G. J. Barbante, E. H. Doeven, E. Kerr, T. U. Connell, P. S. Donnelly, J. M. White, T. Lópes, S. Laird, C. F. Hogan, D. J. D. Wilson, P. J. Barnard and P. S. Francis, Chem.–Eur. J., 2014, 20, 3322–3332 CrossRef CAS PubMed.
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335–3340 CrossRef CAS.
- E. Kerr, E. H. Doeven, G. J. Barbante, T. U. Connell, P. S. Donnelly, D. J. D. Wilson, T. D. Ashton, F. M. Pfeffer and P. S. Francis, Chem.–Eur. J., 2015, 21, 14987–14995 CrossRef CAS PubMed.
- R. M. Wightman, S. P. Forry, R. Maus, D. Badocco and P. Pastore, J. Phys. Chem. B, 2004, 108, 19119–19125 Search PubMed.
- A. Kapturkiewicz, J. Nowacki and P. Borowicz, Electrochim. Acta, 2005, 50, 3395–3400 CrossRef CAS.
-
A. Kapturkiewicz, in Analytical Electrogenerated Chemiluminescence: From Fundamentals to Bioassays, ed. N. Sojic, The Royal Society of Chemistry, 2019, pp. 29–58 Search PubMed.
- W. E. Jones Jr. and M. A. Fox, J. Phys. Chem., 1994, 98, 5095–5099 CrossRef.
- E. Kerr, E. H. Doeven, G. J. Barbante, C. F. Hogan, D. Bower, P. S. Donnelly, T. U. Connell and P. S. Francis, Chem. Sci., 2015, 6, 472–479 RSC.
- S. Knežević, J. Totoricaguena-Gorriño, R. K. R. Gajjala, B. Hermenegildo, L. Ruiz-Rubio, J. L. Vilas-Vilela, S. Lanceros-Méndez, N. Sojic and F. J. D. Campo, J. Am. Chem. Soc., 2024, 146, 22724–22735 CrossRef PubMed.
- J. M. Fernandez-Hernandez, E. Longhi, R. Cysewski, F. Polo, H.-P. Josel and L. De Cola, Anal. Chem., 2016, 88, 4174–4178 CrossRef CAS PubMed.
Footnote |
| † These authors contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Fazeleh
Mesgari†
a,
El M. S.
Martin
a,
Egan H.
Doeven
a,
David J.
Hayne
a,
Fazeleh
Mesgari†
a,
El M. S.
Martin
a,
Egan H.
Doeven
a,
David J.
Hayne