
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSupramolecular protecting groups can impart prosthetic stereoselectivity to catalytic systems employing unmodified achiral heterogenous catalysts
B. Ivonne.
Vergara-Arenas
 ,
J. Antonio.
Morales-Serna
and
Andrew. J.
Surman
*
,
J. Antonio.
Morales-Serna
and
Andrew. J.
Surman
*
Department of Chemistry, King's College London, 7 Trinity Street, London, SE1 1DB, UK. E-mail: andrew.surman@kcl.ac.uk
First published on 18th February 2026
Abstract
Use of homogeneous catalysis – typically based on scarce precious metals – remains a dominant approach to afford good yields of enantiopure compounds. Combining typical strengths of heterogenous catalysts (low cost, sustainable, recyclable) with those of precious metal-mediated homogenous catalysis (amenability to design for selectivity) is desirable: several approaches have been demonstrated (chiral material surfaces, modification of surfaces with chiral auxiliaries, immobilisation of chiral catalysts), but it remains a challenge. Here we present a systems catalysis approach, with a heterogeneous material providing catalytic activity, and a separate host species controlling access to the catalyst to impart ‘prosthetic’ chiral selectivity. Since this non-covalent analogue to conventional covalent protecting group strategies is modular, the same substrate/host combination may be applied to a range of catalytic surfaces. The potential of this approach to achieve effective kinetic resolution is demonstrated in stereoselective synthesis of the drug (R)-cinacalcet.
Introduction
Catalysis is fundamental to the making of the modern world, with the manufacture of most consumer products involving some form of catalysis. Efficient, selective, asymmetric (chiral) catalysis in particular is paramount in the manufacture of fine chemicals (e.g. pharmaceutical and agrochemical products) and, increasingly, advanced materials. Archetypal stereoselective chemical catalysts consist of well-defined, soluble (homogenous), metal–ligand complexes.1–5 These require careful design and are often difficult to develop/produce, making them expensive. Furthermore, many incorporate precious metals like platinum and palladium,6 deposits of which are limited, leading to sustainability concerns,7 which are only partially addressed by a movement to earth-abundant metals.8 Alternative approaches to homogenous catalysis include selective chiral ‘poisoning’ of catalysts,9 organocatalysis,10–15 reaction inside molecular containers (“Supramolecular Catalysts”),16–27 and the use of enzymes:28–33 all can offer some measure of selectivity, activity, and sustainability, but often to varying degrees beyond a narrow range of reactions, substrates, or conditions.By contrast, heterogeneous (insoluble/solid) catalysts are widely used in industrial chemistry, and typically are relatively cheap, sustainable, stable, recyclable, and easy to produce/obtain.34 However, heterogenous catalysts typically lack precisely-defined sterically-controlled active sites, akin to those which mediate control in homogenous catalysts, making design for (stereo)selectivity challenging.29,35–41
Attempts to achieve “the best of both worlds” – the selectivity of homogenous precious metal complexes, with the low cost and recyclability of heterogenous catalysts – have been numerous, but their success and adoption limited. Approaches to impart selectivity to catalytic surfaces by modifying them have included the tethering otherwise-soluble metal complexes to surfaces,42 or depositing chiral compounds on their surface,29,35–41 have met with some success, but are not widely reported in routine use. In all these approaches, the catalyst – or catalytic surface – is engineered for selectivity: a costly and laborious process, even when successful.
In this work, we explore an alternative: a modular systems approach, where instead of engineering the catalytic species for selectivity, we separate the roles of catalysis and selection (Fig. 1). A non-selective catalyst may be used, and “prosthetic” selectivity (selectivity not mediated by the catalyst) is imparted by molecular recognition of one enantiomer by stoichiometric amounts of a Supramolecular Protecting Group (SPG), preventing its reaction in a manner analogous to a covalently-bound protecting group. Using SPGs to control reactivity, where molecular recognition prevents access of reagents or catalysts to a substrate – or a region of a substrate – has been reported for some time,43 including for regioselectivity43–50 and kinetic resolution reactions,51,52 particularly following a landmark publication by Gibb et al. applying this principal to control a simple, otherwise-unselective, ester hydrolysis reactions.51 Indeed, the approach might be more well-recognised but for the diverse range of terms used to describe it (‘Supramolecular Inhibitors’, ‘Noncovalent Auxiliaries’, ‘Shadow Mask’, among others).53 However we are not aware of examples incorporating this into a catalytic system employing an unselective solid catalyst, which may be recycled.
 | ||
| Fig. 1 Supramolecular Protecting Groups (SPGs) impart prosthetic enantioselectivity to reactions mediated by unmodified achiral solid catalysts. In this work (b) a chiral SPG selectively recognises one enantiomer of substrate (S-enantiomer), allowing only one enantiomer to access catalyst. | ||
Specifically, here we demonstrate how a Supramolecular Protecting Group may be applied to impart stereoselectivity to an otherwise-achiral catalytic system, a reductive amination: a common reaction in pharmaceutical synthesis.54 We employ an unmodified, commercially-available, solid acid catalyst, which is active, but not stereoselective, and an established host molecule (a modified β-cyclodextrin) which provides stereoselectivity by selectively recognising one enantiomer of the starting material selectively, preventing its reaction. We demonstrate this system in kinetic resolution in a model reductive amination reaction, and in the stereoselective synthesis of the drug (R)-cinacalcet, sold under the names Sensipar®, Mimpara®, or Regpara® in enantiopure form,55,56 from a racemic amine.
Results and discussion
Catalysis of model reductive amination by unselective solid catalyst
To establish our approach, we chose a model reductive amination of racemic 1-(1-napthyl)ethylamine ((R/S)-1), initially reacting with an aldehyde (2) to produce an imine (3), which can then be reduced to form the amine product (4) (Scheme 1). | ||
| Scheme 1 Model reductive amination reaction, which may be carried out over a range of temperatures (see SI, Section 2). | ||
The amine, 1, was chosen, since a stereoselective host/SPG for this amine (6-O-triisopropylsilylated β-cyclodextrin, “TIPS-β-CD”, see Fig. 3) is already established by Kida et al.,52 achieving 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 stereoselective recognition of the (S)-1 and kinetic resolution in more simple model reactions at low temperatures (−20 °C). Amberlyst 15, a polymeric resin with strongly acidic sulfonic acid groups, with was chosen as an available, affordable, achiral, and reusable solid acid catalyst.57,58
:1 stereoselective recognition of the (S)-1 and kinetic resolution in more simple model reactions at low temperatures (−20 °C). Amberlyst 15, a polymeric resin with strongly acidic sulfonic acid groups, with was chosen as an available, affordable, achiral, and reusable solid acid catalyst.57,58
Exploring reaction conditions in the absence of TIPS-β-CD, in toluene (see SI, Table S1; protic solvents absent) we found that the imine (3a) formation proceeds at reduced temperatures only in the presence of the Amberlyst 15 catalyst (Brønsted acid) (Table S1, Entries 4 & 5). Furthermore, the subsequent reduction of the imine (without isolation) to yield the amine (4a) product requires the catalyst, even at room temperature (Table S1, Entries 7 & 8): likely as the catalyst acts as a source of H+ (4.7 mmol H+ per g)59 for initiation of the reduction by NaBH4 in an aprotic solvent.60 Taken together, in the low-temperature conditions required for selective recognition of (S)-1 by TIPS-β-CD, the reaction only proceeds when catalysed by Amberlyst 15.
An important advantage of solid catalysts is recyclability. To screen the recyclability of the Amberlyst 15, two reactivation processes were applied to the material after catalysing reaction between 1 and 2a at in our reaction conditions (see SI, Fig. S1). While reuse of the catalyst after simple washing with an organic solvent (CH2Cl2) led to decreased activity in subsequent reactions, an additional wash with dilute aqueous hydrochloric acid maintained the efficiency of the catalyst for at least three subsequent reactions.
SPG-mediated kinetic resolution with an achiral catalyst
Next we introduce the TIPS-β-CD as an SPG in the reaction between racemic (R/S)-1 and benzaldehyde (2a) in presence of Amberlyst 15 at different temperatures, exploring its ability to impart enantioselectivity to the otherwise-unselective system (see Fig. 2, and SI, Table S2). TIPS-β-CD was synthesised in a modification of reported procedures for microwave reaction (see SI, Section 5).61 | ||
| Fig. 2 (a) Model kinetic resolution reactions scheme. [(i) amine 1 (0.1 mmol), aldehyde 2a (0.05 mmol), TIPS-β-CD (0.4 mmol) and toluene (2 mL), 15 min. (ii) Amberlyst 15 (50% mmol), 1 h. (iii) NaBH4 (0.20 mmol), 1 h.] (b) Results for varying temperature of reaction between amine 1 and aldehyde 2a, showing increasing enantioselectivity as temperature decreases. [Conversion of aldehyde 2a to 4a, and % ee of product 4a determined by HPLC (see SI, Section 7), with yield of (R)-4a shaded light, and (S)-4a shaded dark]. | ||
In our initial experiments (all 1 h imine formation; 1 h NaBH4 reduction), the reaction was conducted with 2 equivalents of (R/S)-1 at room temperature (relative to aldehyde 2a), and we obtained 90% conversion observed with 4% enantiomeric excess (ee). This is consistent with observations of mild enantioselectivity in 1/TIPS-β-CD interactions at room temperature.52 Progressively lowering temperature (all reactions: 1 h imine formation, 1 h reduction) led to progressively increasing chiral selectivity in product 4a, up to 90% ee, with only a minimal drop in conversion (to ca. 80%). Decreasing the amount of limiting reagent (aldehyde) to 0.2 eq. at the lowest temperature led to negligible change in selectivity (ee = 91%), and the use of a different aldehyde (2b, 4-nitrobenzaldehyde) provided similarly high yield and selectivity profiles to 2a (see SI, Section 7).
While fresh samples of TIPS-β-CD were used here, the host (used in stoichiometric amounts) can be readily recovered and reused after each reaction. To remove the amine 1, an extraction can be performed using ethyl acetate and an acidic aqueous solution, with the amine retained in the aqueous layer. After two extraction cycles, the TIPS-β-CD was not distinguishable from fresh host (unused as SPG) by 1H NMR (see SI, Section 9).
Exploring selective recognition of amine (S)-1 in reaction conditions
To explore the interaction responsible for the remarkable enantioselectivity observed in model reactions, (S)-1 by TIPS-β-CD (see Fig. 3a), we performed 1H NMR binding titrations with both enantiomers of amine 1. | ||
| Fig. 3 (a) Structure of TIPS-β-CD, with key protons labelled, and (b) schematic representation of stepwise 2:1 complexation of 1 by TIPS-β-CD, to yield 1⊂TIPS-β-CD2 (note: asymmetry omitted in this schematic for illustrative clarity). (c and d) 1H NMR chemical shift of TIPS-β-CD (0.05 M) protons on titration with (S)-1 (c) and (R)-1 (d); lines correspond to fitting stepwise 2:1 binding to all four protons' data simultaneously [d8-toluene, room temperature (ca. 298 K), see SI, Section 6]. | ||
At room temperature, progressive changes in the chemical shift of the well-resolved host proton resonances (H1, H3, H6(a), H6(b), see Fig. 3 and SI Section 6; others are not well-resolved) on guest addition reveals binding with a fast exchange regime, in contrast to reports which observed slow exchange for 1/TIPS-β-CD binding in cyclohexane/benzene.52 This difference may be attributed to inclusion of bulkier solvent molecules (toluene) in the host cavity, analogous to inclusion of benzene in reported XRD-derived structures (no crystallisation was observed in our system, despite many attempts).52
Comparing chemical shift changes at multiple host protons (Fig. 3c and d) reveals the nature of the binding of (S)-1 and (R)-1 is qualitatively distinct. While binding of (S)-1 leads to marked changes in all four protons (suggesting binding deep in the cyclodextrin cavity to perturb H6), binding of (R)-1 affects H6(a) and H6(b) notably less, suggesting a distinct binding conformation for (R)-1 (likely sandwiched more ‘flatly’ between the two hosts) resulting from a poorer ‘fit’ in the host. This is also reflected in the broadening of some (S)-1 aromatic protons on binding TIPS-β-CD, not observed for the corresponding resonances from (R)-1 (see SI, Fig. S3). Rotating Frame Overhauser Effect Spectroscopy (ROESY) NMR spectra of the (S)-1 and (R)-1 recognition by TIPS-β-CD in the same conditions (see SI, Section 6) show interactions between the aromatic protons of 1 and cavity protons (H-3 and H-5) of TIPS-β-CD, with notably more intense cross-peaks with (S)-1 reflecting closer interaction.
Consistent with all other reports of guest binding by TIPS-β-CD binding in other solvents being 2:1 “sandwich-like”, a Job's plot suggests 2:1 host:guest binding (see SI Section 6).52,61–63 We applied a standard stepwise 2:1 binding model to fit binding (a simultaneous fit of the chemical shifts of H1, H3, H6(a), and H6(b) (see Fig. 3b, and SI, Section 6; we note that, since chemical shift changes are not exclusively monotonic responses, the unusual one-step 2:1 binding model applied elsewhere,52 or a 1:1 model, are not appropriate here). The binding constants estimated show modestly increased binding constants for (S)-1 (estimating K11 as 0.22 M−1 and K12 as 2000 M−1), relative to (R)-1 (estimating K11 as 0.08 M−1 and K12 as 1220 M−1), and in both cases these are consistent with 2:1 binding predominating in reaction conditions (i.e. in the absence of large excess of TIPS-β-CD). The small differences between these values reflect the scant selectivity observed in SPG-mediated reactions at room temperature.
Since greater selectivity in our SPG-mediated reactions is observed at −20 °C (−253 K), a series of 1H spectra were acquired at this temperature (see SI, Fig. S8; all resonances broadened at low temperature). While all room-temperature studies manifest fast guest exchange regimes, these spectra suggest slow guest exchange for the second (K12) binding of (S)-1, but fast exchange for the first binding (K11); contrastingly, both binding steps of (R)-1 appear in fast exchange. Furthermore, the disappearance of the resonance corresponding to unbound host on the addition of 1 equivalent of (S)-1 suggest complete guest binding in these conditions. Together these observations explain increased (S)-1 protection at low temperatures, however the mixed fast/slow regime observed for (S)-1 binding prevents fitting under these conditions using established models.
Overall these results support the reaction selectivity we observe being derived from selective binding of (S)-1 by TIPS-β-CD.
Modular substitution of catalyst
An important advantage of our approach of separating the roles of catalytic activity and selectivity is the modular nature of the system. In such modular catalytic systems, it should be possible to vary the catalyst, and maintain selectivity (though activity may vary).To establish this, we performed a series of reactions employing a range of solid acid catalysts (Montmorillonite, Graphene Oxide, MCM-41, SiO2), without further optimisation of conditions. We observe that in all cases where measurable yield is observed, selectivity remains almost constant, at around 90% ee. Very similar results were observed on varying the aldehyde to 2b (see SI, Table S3). This demonstrates the modularity of the system. While selectivity is unaffected, yield varies to a great extent, reflecting the optimisation of conditions/catalyst loading for Amberlyst 15 (Table 1).
| Entry | Aldehyde | Catalyst | Conv.b (%) | % eeb |
|---|---|---|---|---|
| a Reagents: (a) amine 1 (0.1 mmol), aldehyde 2a (0.05 mmol), TIPS-β-CD (0.4 mmol) and toluene (2 mL), 15 min; (ii) solid acid (50% mmol), 1 h; (iii) NaBH4 (0.20 mmol), 1 h at −20 °C. b The conversion of 2a to 4, and % ee of product 4a was determined by HPLC (see SI, Section 8). | ||||
| 1 | 2a | Amberlyst 15 | 78 | 90 |
| 2 | 2a | Graphene oxide | 28 | 87 |
| 3 | 2a | Montmorillonite | 20 | 87 |
| 4 | 2a | MCM-41 | — | — |
| 5 | 2a | SiO2 | — | — |
Stereoselective synthesis of (R)-cinacalcet
To demonstrate the usefulness of our approach to catalytic selectivity, we applied it to the synthesis of the drug (R)-cinacalcet (4c), which is approved for the treatment of secondary hyperparathyroidism and hypercalcaemia.56 In almost all reported syntheses of this drug, enantiopure (R)-1 has been used as a starting material (typically obtained by enzymatic resolution) to ensure only one enantiomer is produced55 (one exceptional report employs another enantiopure feedstock, (R)-tert-butanesulfinamide, to introduce an asymmetric C–N bond).64 Here we use our SPG-mediated approach to perform the stereoselective synthesis from a racemic starting material – the amine (R/S)-1 and an achiral aldehyde (2c) reported in a number of syntheses of (R)-cinacalcet – and achiral Amberlyst 15 as the catalyst for the enantioselective bond-forming step.Following the approach developed in our model reactions, we performed implemented the reaction as shown in Scheme 2. Varying conditions (see SI, Table S4), we find similar responses to temperature: at room temperature no enantioselectivity is observed in the system. Lowering temperature to favour TIPS-β-CD acting as an SPG for (S)-1 (preventing access to catalyst and reagents), we are able to produce (R)-cinacalcet (4c) in good yield and high enantiopurity (up to 94% ee with 80% aldehyde conversion) from the racemic amine.
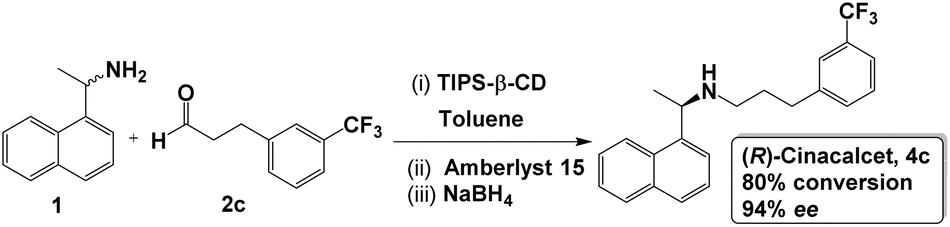 | ||
| Scheme 2 Synthesis of (R)-cinacalcet using SPG to impart selectivity. (i) Amine 1 (0.1 mmol), aldehyde 2c (0.05 mmol), TIPS-β-CD (0.4 mmol) and toluene (2 mL), 15 min; (ii) solid acid (50% mmol), 1 h; (iii) NaBH4 (0.20 mmol), 1 h at −20 °C. | ||
Conclusions and outlook
Our approach of separating the roles of catalytic activity and selectivity in a system, using stoichiometric amounts of an established Supramolecular Protecting Group to impart selectivity to solid catalysts, can produce chiral products (over 90% ee) from racemic feedstocks, employing otherwise-unselective (low-cost and recyclable) solid catalysts. We have also demonstrated that these low-cost catalysts can be recycled, that modular ‘catalyst swapping’ does not hinder selectivity, and that all this can be used in an unprecedented synthesis of the chiral drug (R)-cinacalcet from a racemic starting material. We emphasise that this SPG approach is distinct to Supramolecular Catalysis,16 as molecular recognition is employed to prevent reaction (just as covalent protecting groups are distinct from covalent activation).While we are not aware of previous applications to systems employing solid catalysts, we have noted that the use of Supramolecular Protecting Groups to control reactivity is not a new phenomenon (though nomenclature may vary). Since early reports,43 however, concepts and implementation of catalytic systems for synthesis have developed enormously. Where refining/engineering catalysts (the selectivity and activity of singles species) was long the overarching focus, increasingly catalytic systems are becoming accessible. Whether they employ multiple enzymes in a ‘cascade’,31 chemo–enzymatic combinations,65 or the use of multiple chemical catalysts,66 all these approaches share the division of roles into modules, comprising a (catalytic) system.67 We suggest that the value of SPGs is not as stand-alone alternatives to ‘traditional’ selective catalysts, but promising modules for use in developing more advanced catalytic systems. We hope this demonstration of their use in a catalytic system incorporating a solid catalyst, and multiple steps, will lead to the exploration of more complex catalytic systems.
Author contributions
Conceptualization and methodology were by AJS and JAMS. Investigation, data curation, formal analysis were by BIVA and JAMS. Funding acquisition and writing – review & editing were by AJS, JAMS and BIVA.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5sc08648e.Acknowledgements
BIVA is grateful to Consejo Nacional de Ciencia y Tecnología – México for a postdoctoral fellowship (CONACyT CVU: 556069, support number: 770736 and 800994). JAMS is grateful to the Royal Society (UK) for the International Alumni Grant. AJS thanks both JAMS and the Royal Society for facilitating the taxi journey on which this line of research was conceived in conversation between AJS and JAMS. All authors gratefully acknowledge the support and facilities of King's College, London. We are also grateful to Drs Tom Hicks and James Jarvis to assistance with carrying out NMR spectroscopy.Notes and references
- P. S. Steinlandt, L. Zhang and E. Meggers, Chem. Rev., 2023, 123, 4764–4794 Search PubMed.
- K. T. Mahmudov and A. J. L. Pombeiro, Chem.–Eur. J., 2023, 29, e202203861 CrossRef CAS PubMed.
- X. Xiao, K. Xu, Z.-H. Gao, Z.-H. Zhu, C. Ye, B. Zhao, S. Luo, S. Ye, Y.-G. Zhou, S. Xu, S.-F. Zhu, H. Bao, W. Sun, X. Wang and K. Ding, Sci. China: Chem., 2023, 66, 1553–1633 CrossRef CAS.
- T. Yu, Z. Ding, W. Nie, J. Jiao, H. Zhang, Q. Zhang, C. Xue, X. Duan, Y. M. A. Yamada and P. Li, Chem.–Eur. J., 2020, 26, 5729–5747 CrossRef CAS PubMed.
- Y. G. Shelke, A. Yashmeen, A. V. A. Gholap, S. J. Gharpure and A. R. Kapdi, Chem.–Asian J., 2018, 13, 2991–3013 CrossRef CAS PubMed.
- A. Remmel, Nature, 2022, 606, 448–451 CrossRef CAS PubMed.
- M. Pitts, New Sci., 2011, 209, 26–27 CrossRef.
- S. Shekhar, T. S. Ahmed, A. R. Ickes and M. C. Haibach, Org. Process Res. Dev., 2022, 26, 14–42 Search PubMed.
- J. W. Faller, A. R. Lavoie and J. Parr, Chem. Rev., 2003, 103, 3345–3368 Search PubMed.
- D. W. C. MacMillan, Nature, 2008, 455, 304–308 Search PubMed.
- E. Reyes, L. Prieto and A. Milelli, Molecules, 2023, 28, 271 CrossRef CAS PubMed.
- A. Antenucci, S. Dughera and P. Renzi, ChemSusChem, 2021, 14, 2785–2853 CrossRef CAS PubMed.
- M. P. van der Helm, B. Klemm and R. Eelkema, Nat. Rev. Chem., 2019, 3, 491–508 CrossRef CAS.
- M. C. Holland and R. Gilmour, Angew. Chem., Int. Ed., 2015, 54, 3862–3871 CrossRef CAS PubMed.
- P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138–5175 Search PubMed.
- P. W. M. N. van Leeuwen and M. Reynal, Supramolecular Catalysis: New Directions and Developments, Wiley-VCH, 2021 Search PubMed.
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, Wiley-VCH, 3rd edn, 2022 Search PubMed.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969–984 CrossRef CAS.
- Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed.
- N. Abuhafez, A. Perennes and R. Gramage-Doria, Synthesis, 2022, 54, 3473–3481 Search PubMed.
- B. Tang, J. Zhao, J.-F. Xu and X. Zhang, Chem.–Eur. J., 2020, 26, 15446–15460 CrossRef CAS PubMed.
- S. Kosiorek, N. Rad and V. Sashuk, ChemCatChem, 2020, 12, 2776–2782 Search PubMed.
- S. Funk and J. Schatz, J. Inclusion Phenom. Macrocyclic Chem., 2020, 96, 1–27 CrossRef CAS.
- A. Pappalardo, R. Puglisi and G. Trusso Sfrazzetto, Catalysts, 2019, 9, 630 CrossRef CAS.
- C. Deraedt and D. Astruc, Coord. Chem. Rev., 2016, 324, 106–122 Search PubMed.
- L. Cunningham, Synlett, 2025, 36, 1189–1200 Search PubMed.
- R. Ning and Q.-Q. Wang, Chem. Soc. Rev., 2025, 54, 11105–11140 RSC.
- Z. Song, Q. Zhang, W. Wu, Z. Pu and H. Yu, Front. Bioeng. Biotechnol., 2023, 1129149 CrossRef PubMed.
- K.-Y. Wang, J. Zhang, Y.-C. Hsu, H. Lin, Z. Han, J. Pang, Z. Yang, R.-R. Liang, W. Shi and H.-C. Zhou, Chem. Rev., 2023, 123, 5347–5420 CrossRef CAS PubMed.
- T. Chen, Y. Peng, M. Qiu, C. Yi and Z. Xu, Int. J. Biol. Macromol., 2023, 230, 123206 CrossRef CAS PubMed.
- A. I. Benítez-Mateos, D. Roura Padrosa and F. Paradisi, Nat. Chem., 2022, 14, 489–499 CrossRef PubMed.
- E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner and S. L. Flitsch, Nat. Rev. Methods Primers, 2021, 1, 1–21 Search PubMed.
- D. C. M. Albanese and N. Gaggero, RSC Adv., 2015, 5, 10588–10598 RSC.
- G. Busca, Heterogeneous Catalytic Materials, Elsevier, Amsterdam, 2014 Search PubMed.
- A. Iemhoff, M. Vennewald and R. Palkovits, Angew. Chem., Int. Ed., 2023, 62, e202212015 Search PubMed.
- V. B. Saptal, V. Ruta, M. A. Bajada and G. Vilé, Angew. Chem., Int. Ed., 2023, 62, e202219306 CrossRef CAS PubMed.
- J. Guo, H. Liu, D. Li, J. Wang, X. Djitcheu, D. He and Q. Zhang, RSC Adv., 2022, 12, 9373–9394 RSC.
- L. Zhang, M. Zhou, A. Wang and T. Zhang, Chem. Rev., 2020, 120, 683–733 CrossRef CAS PubMed.
- F. Zaera, Chem. Soc. Rev., 2017, 46, 7374–7398 RSC.
- A. Baiker, Chem. Soc. Rev., 2015, 44, 7449–7464 RSC.
- V. Demers-Carpentier, G. Goubert, F. Masini, R. Lafleur-Lambert, Y. Dong, S. Lavoie, G. Mahieu, J. Boukouvalas, H. Gao, A. M. H. Rasmussen, L. Ferrighi, Y. Pan, B. Hammer and P. H. McBreen, Science, 2011, 334, 776–780 Search PubMed.
- M. P. de Almeida and S. A. C. Carabineiro, ChemCatChem, 2012, 4, 18–29 CrossRef.
- R. Breslow and P. Campbell, J. Am. Chem. Soc., 1969, 91, 3085 Search PubMed.
- P. S. Bols and H. L. Anderson, Angew. Chem., Int. Ed., 2018, 57, 7874–7877 Search PubMed.
- Q. Sun, L. Escobar and P. Ballester, Angew. Chem., Int. Ed., 2021, 60, 10359–10365 CrossRef CAS PubMed.
- A. A. A. Smith, C. L. Maikawa, G. A. Roth and E. A. Appel, Org. Biomol. Chem., 2020, 18, 4371–4375 RSC.
- K. Iizuka, H. Takezawa and M. Fujita, J. Am. Chem. Soc., 2023, 145, 25971–25975 CrossRef CAS PubMed.
- E. Ubasart, O. Borodin, C. Fuertes-Espinosa, Y. Xu, C. García-Simón, L. Gómez, J. Juanhuix, F. Gándara, I. Imaz, D. Maspoch, M. von Delius and X. Ribas, Nat. Chem., 2021, 13, 420–427 CrossRef CAS PubMed.
- R. M. Yebeutchou and E. Dalcanale, J. Am. Chem. Soc., 2009, 131, 2452–2453 CrossRef CAS PubMed.
- R. Pinalli, G. Brancatelli, A. Pedrini, D. Menozzi, D. Hernández, P. Ballester, S. Geremia and E. Dalcanale, J. Am. Chem. Soc., 2016, 138, 8569–8580 CrossRef CAS PubMed.
- S. Liu, H. Gan, A. T. Hermann, S. W. Rick and B. C. Gibb, Nat. Chem., 2010, 2, 847–852 CrossRef CAS PubMed.
- T. Kida, T. Iwamoto, H. Asahara, T. Hinoue and M. Akashi, J. Am. Chem. Soc., 2013, 135, 3371–3374 CrossRef CAS PubMed.
- A. B. Grommet, M. Feller and R. Klajn, Nat. Nanotechnol., 2020, 15, 256–271 CrossRef CAS PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 Search PubMed.
- M. Barniol-Xicota, R. Leiva, C. Escolano and S. Vázquez, Synthesis, 2016, 48, 783–803 CrossRef CAS.
- Mimpara, European Medicines Agency, https://www.ema.europa.eu/en/medicines/human/EPAR/mimpara, accessed 20 May 2024 Search PubMed.
- P. Gupta and S. Paul, Catal. Today, 2014, 236, 153–170 Search PubMed.
- P. Mäki-Arvela, I. L. Simakova and D. Yu. Murzin, Catal. Rev., 2023, 65, 501–568 CrossRef.
- H. B. El-Nassan, Russ. J. Org. Chem., 2021, 57, 1109–1134 CrossRef CAS.
- H. Alinezhad, M. Tajbakhsh and N. Mahdavi, Synth. Commun., 2010, 40, 951–956 CrossRef CAS.
- T. Kida, T. Iwamoto, Y. Fujino, N. Tohnai, M. Miyata and M. Akashi, Org. Lett., 2011, 13, 4570–4573 CrossRef CAS PubMed.
- H. Asahara, T. Kida, T. Iwamoto, T. Hinoue and M. Akashi, Tetrahedron, 2014, 70, 197–203 CrossRef CAS.
- H. Asahara, T. Kida, T. Hinoue and M. Akashi, Tetrahedron, 2013, 69, 9428–9433 CrossRef CAS.
- V. R. Arava, L. Gorentla and P. K. Dubey, Beilstein J. Org. Chem., 2012, 8, 1366–1373 CrossRef CAS PubMed.
- S. González-Granda, L. Escot, I. Lavandera and V. Gotor-Fernández, Angew. Chem., Int. Ed., 2023, 62, e202217713 CrossRef PubMed.
- J. Hou, S. Chevallier-Michaud, M. Jean, L. Favre, D. Hérault and C. Bressy, J. Am. Chem. Soc., 2023, 145, 27236–27241 CrossRef CAS PubMed.
- S. Burgener, S. Luo, R. McLean, T. E. Miller and T. J. Erb, Nat. Catal., 2020, 3, 186–192 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |