
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural insights into copper and zinc binding to tau protein and the impact of metal binding on amyloid aggregation
Gerardo U.
Juárez-Romero
 a,
Xun
Sun
b,
Juan Atilio
Gerez
c,
Christophe
Den Auwer
d,
Gautier
Landrot
e,
Maarten
Nachtegaal
cf,
Roland
Riek
c,
Jinghui
Luo
b and
Liliana
Quintanar
*ag
a,
Xun
Sun
b,
Juan Atilio
Gerez
c,
Christophe
Den Auwer
d,
Gautier
Landrot
e,
Maarten
Nachtegaal
cf,
Roland
Riek
c,
Jinghui
Luo
b and
Liliana
Quintanar
*ag
aDepartment of Chemistry, Center for Research and Advanced Studies (Cinvestav), 07360 Mexico City, Mexico. E-mail: lilianaq@cinvestav.mx
bDepartment of Biology and Chemistry, Paul Scherrer Institute, 5232 Villigen, Switzerland
cETH Zurich, Department of Chemistry and Applied Biosciences, 8093 Zurich, Switzerland
dUniversité Côte d'Azur, CNRS, Institute de Chimie de Nice, 06108 Nice, France
eSynchrotron SOLEIL, L'Orme des Merisiers, Départementale 128, Saint-Aubin, 91190, France
fCenter for Energy and Environmental Sciences and Center for Photon Science, Paul Scherrer Institute, Swiss Light Source, 5232 Villigen, Switzerland
gCenter for Research in Aging, Cinvestav, Mexico City, 14330, Mexico
First published on 10th February 2026
Abstract
Tau protein is a microtubule-associated protein central to the pathogenesis of Alzheimer's disease (AD) and other tauopathies. While metal ion homeostasis is disrupted in AD, the presence of copper and zinc in neurofibrillary tangles suggests a pathological role for metal–tau interactions. In this study, the metal binding properties of Tau441 were probed using a wide range of spectroscopic tools. Specifically, electron paramagnetic resonance, circular dichroism, nuclear magnetic resonance (NMR) and X-ray absorption spectroscopy (XAS) results point to a single high-affinity Cu2+ binding site within the microtubule-binding domain (MTBD), coordinated by the bis-His motif in R3 (His329/His330), possibly His299 and an oxygen-based ligand. This complex can be reduced resulting in a trigonal Cu+–Tau441 complex involving Cys322, His299 and a third ligand (likely Cys291 or His329/330), as characterized by XAS and NMR. NMR and XAS results indicate the presence of three Zn2+ binding sites: one high-affinity site in the MTBD involving His299, His330, Cys322 and Asp295, and two lower-affinity sites in the N-terminal region, coordinated predominantly by carboxylate and His residues. Moreover, the impact of Cu2+ and Zn2+ ions on the amyloid aggregation of full length Tau441 was evaluated using thioflavin T fluorescence, electrophoresis and transmission electron microscopy. Both metal ions significantly accelerate aggregation, promoting the formation of amyloid fibrils with distinct morphologies. Our study provides valuable structural insights into the copper and zinc binding sites in Tau441 that provide a rational basis to understand the impact of metal ions on amyloid fibril aggregation and morphology. The current study expands the bioinorganic facet of AD and other tauopathies, and it underscores the importance of metal–tau interactions as potential therapeutic targets in these neurodegenerative diseases.
Introduction
Tauopathies are a group of neurodegenerative disorders characterized by the abnormal deposition of the microtubule-associated protein tau in the brain.1 Among these, Alzheimer's disease (AD) is the most prevalent, defined pathologically by the presence of extracellular amyloid-beta (Aβ) plaques and intracellular neurofibrillary tangles (NFTs) composed primarily of hyperphosphorylated tau.2 Increasing evidence suggests that disruption in metal homeostasis, particularly involving copper and zinc, contributes to the pathogenesis of AD and other tauopathies.3,4 Indeed, zinc is increased, and copper is decreased in the AD brain, while blood levels of labile copper, i.e. not bound to ceruloplasmin, are also increased in a subset of AD patients, as compared to healthy ones.5 These metals are essential for normal central nervous system function, as they can regulate different neurotransmitter receptors,6,7 and they are cofactors in several important enzymes. For example, superoxide dismutase 1 requires copper and zinc, dopamine-β-hydroxylase is a copper-dependent enzyme critical in the synthesis of the neurotransmitter norepinephrine8 and zinc plays structural roles in a variety of transcription factors.9 The loss of metal homeostasis can lead to metal accumulation, undesired metal redox activity and metal-induced protein aggregation.5,10,11 X-ray fluorescence analysis has demonstrated the presence of both Cu and Zn in senile plaques of AD patients,12 while metal accumulation has been correlated with Aβ deposits in AD transgenic murine models.13 Indirect evidence for the presence of copper and zinc in NFTs of AD patients has been derived from copper-dependent oxidase activity14 and the use of fluorescence zinc chelators.15The early discovery that Aβ is a metal binding peptide16 and that extracellular amyloid plaques in AD concentrate copper and zinc ions,3 has driven extensive research into the metal binding properties of Aβ peptide in the last two decades.5,17 Copper and zinc can bind Aβ and impact its aggregation, while copper-bound peptides display redox activity that can contribute to the generation of reactive oxygen species, thereby exacerbating neuronal damage.18,19 While the presence of metal ions in NFTs suggests a direct pathological role for metal–tau interactions, the metal binding properties of tau and the impact of metal ions on tau protein misfolding and aggregation have not been investigated as extensively as in the case of Aβ.
Tau protein exists in six major isoforms in the adult human brain, generated through alternative splicing of the microtubule associated protein tau (MAPT) gene, and characterized by the number of N-terminal negatively charged inserts (0N, 1N, or 2N) and the presence of either three (3R) or four (4R) microtubule-binding repeats.20 Tau441 is the longest isoform comprising a 2N4R structure (Scheme 1), and together with 2N3R and 2N4R, represents the most expressed isoforms in the adult brain. The most studied model fragments containing the microtubule binding domain (MTBD) are TauK18,21 encompassing only the MTBD repeats, and TauK32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 that also contains two regions flanking the MTBD (Scheme 1). These models have been used to study microtubule binding as well as protein aggregation. Tau441 undergoes different posttranslational modifications (PTMs)23 under physiological and pathological conditions. Tau phosphorylation, normally regulated by different kinases and phosphatases, is exacerbated in AD. Under physiological conditions, tau stabilizes microtubules and supports axonal transport,24 and it is also located in the nucleus, where it is involved in nuclear maintenance.23 Canonically, tau is an intracellular protein, but recent evidence suggests that tau exists in the extracellular environment under physiological and pathological conditions, although the role that it may play extracellularly remains unclear.25
22 that also contains two regions flanking the MTBD (Scheme 1). These models have been used to study microtubule binding as well as protein aggregation. Tau441 undergoes different posttranslational modifications (PTMs)23 under physiological and pathological conditions. Tau phosphorylation, normally regulated by different kinases and phosphatases, is exacerbated in AD. Under physiological conditions, tau stabilizes microtubules and supports axonal transport,24 and it is also located in the nucleus, where it is involved in nuclear maintenance.23 Canonically, tau is an intracellular protein, but recent evidence suggests that tau exists in the extracellular environment under physiological and pathological conditions, although the role that it may play extracellularly remains unclear.25
 | ||
| Scheme 1 Tau441 and fragments. (Top) Schematic representation of the full-length tau protein, Tau441. Primary sequence of Tau441 includes an N-terminal, with N1 and N2 that are negatively charged inserts, P1 and P2 are the proline-rich region; the microtubule binding domain (MTBD) encompasses the four pseudo-repeats, R1–R4 and finally the C-terminal domain. (Middle) TauK32 and TauK18 are fragment models of the full-length tau and encompass the MTBD. (Bottom) Sequence of the two short peptides of the R2 and R3 domains that contain Cys291, Cys322, His299 and the bis-His motifs His329 and His330. | ||
Tau is an intrinsically disordered protein in solution with a very low transient secondary structure.26 Tau is a protein with a low tendency to aggregate; however, in the presence of cofactors such as heparin and negatively charged polymers, the protein forms aggregates.27 Tau has been observed to aggregate in the presence of metal ions. Copper can induce aggregation of R1–R4 peptides,28 and oligomerization of Tau441 in vitro.29 In triple transgenic (3xTg-AD) mice, dietary copper administration causes increased tau phosphorylation, aggregation and toxicity.30 Zinc accelerates the aggregation of TauK1831 and induces conformational changes and oligomerization of Tau441.32–35 Indeed, early pioneering studies suggest that both copper and zinc ions, may bind to tau. Soragni et al.29 examined the copper binding properties of TauK32 by nuclear magnetic resonance (NMR) spectroscopy and isothermal titration calorimetry (ITC), finding a single copper coordination site with a metal binding affinity in the micromolar range (Kd = 0.7 µM), similar to that of the full-length protein (Kd = 0.5 µM); however, structural characterization of the copper binding site in the full length Tau441 has not been further explored. However, zinc binding to Tau441 has been studied by ITC, revealing that it may have up to four metal-binding sites,33 while ITC and mutagenesis studies of the TauK18 fragment show that this fragment has a single metal-binding site with binding affinity in the micromolar range, and that Cys291 and 322 and His268, 299, 329, 330 and 361 may be important for metal binding.31,36 Although these early studies reveal that tau can bind copper and zinc, the molecular and structural details of these metal–tau interactions have not been elucidated.
Given the fact that metals accumulate in NFTs, understanding the metal binding properties of tau should yield insight into the structural and biochemical basis of metal-induced aggregation. In this study, the metal binding properties of Tau441 were probed using a wide range of spectroscopic tools. Specifically, Cu2+ binding to Tau441 was studied by electron paramagnetic resonance (EPR), circular dichroism (CD), NMR and X-ray absorption spectroscopy (XAS). Reduction of the Cu2+–Tau441 species was probed by EPR and XAS, while the nature of the resulting Cu+–Tau441 complex was studied by XAS and NMR. The Zn2+ binding properties of Tau441 were examined by NMR and XAS. Finally, the impact of Cu2+ and Zn2+ ions on the amyloid aggregation of full length Tau441 was evaluated, while the nature of the resulting aggregates was examined by electrophoresis and transmission electron microscopy. This spectroscopic study provides valuable insights into the copper and zinc binding sites in Tau441 and expands the bioinorganic facet of AD and other tauopathies.
Results
Tau has a single Cu2+ binding site
Cu2+ binding to Tau441 was studied by titrating the protein with 0 to 2 equivalents of the metal ion, followed by electron paramagnetic resonance (EPR) and circular dichroism (CD) spectroscopies (Fig. 1). Having a d9 electronic configuration, Cu2+ ions have an unpaired electron, and EPR spectroscopy can give insights into the chemical environment around the metal ion. EPR spectra of Tau441 with Cu2+ show one set of signals, indicating the presence of a single Cu2+ coordination site (Fig. 1). It should be noted that these signals are distinct from those of Cu2+ in buffer (Fig. S1), and hence, can be assigned to Cu2+ bound to Tau441. The EPR signals of the Cu2+–Tau441 complex (g∥ > g⊥) are indicative of a dx2−y2 ground state, with g∥ = 2.304 and A∥ = 149 × 10−4 cm−1. The second derivative of the EPR spectra in the perpendicular region reveals hyperfine structure, likely corresponding to His nitrogen super-hyperfine couplings (inset Fig. 1). The titration of Tau441 followed by CD (Fig. S2A) results in no signals in the range of 0–1.0 equiv. of Cu2+; hence the Cu2+–Tau441 complex does not give rise to chiral electronic transitions, suggesting that it might have more than two equivalent ligands in the coordination sphere. | ||
| Fig. 1 Tau441 has a single Cu2+ binding site. Titration of 0.2 mM Tau441 in 20 mM MOPS buffer at pH 7.4 with Cu2+ ions (with 0, 0.25, 0.5, 0.75, 1.0 and 1.5 equiv., represented by lines from light gray to black), followed by electron paramagnetic resonance (EPR) spectroscopy; the inset shows the second derivative of the EPR spectra. | ||
To elucidate the regions of Tau441 involved in Cu2+ coordination, TauK18 and the peptide fragments R2 and R3 were studied. TauK18 encompasses residues 244–372 of the full-length protein, containing the four pseudo-repeats of the microtubule binding domain (MTBD). The R2 and R3 peptides correspond to residues 290–300 and 321–331, respectively, and contain residues that have been previously identified as putative ligands for Cu2+ binding to TauK32, namely His residues 299, 329 and 330.29 Each fragment was titrated with 0–2 equiv. of Cu2+ and followed by CD and EPR (Fig. S2C–F); the CD signals that arise from Cu binding to R2 and R3 fragments saturate below 2 equiv., while EPR shows the presence of one main Cu2+ species that is the same at 1 and 2 equiv. For TauK18, no CD signals were observed (Fig. S2B), as in the case of Tau441, indicating that the metal–tau complex is not optically active. A comparison of the EPR and CD spectra of Cu2+ bound to Tau441, TauK18 and the R2 and R3 fragments at 1 equivalent clearly shows that only TauK18 replicates the Cu2+ coordination site in Tau441 (Fig. 2A and B). Although R2 and R3 display complex metal–peptide speciation (Fig. S2C–F), none of the observed species resemble the Cu2+ site in Tau441. Consistently, the Cu K-edge X-ray absorption near edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra for the Cu2+ complexes with Tau441 and TauK18 are identical, while that of R3 are quite different (Fig. 2B–D). While all XANES spectra (Fig. 2C) show a white line at 8990 eV that is a characteristic edge value for tetragonal square-planar Cu2+ centers (typically known as type 2 Cu centers),37,38 distinct differences are observed in the edge shape and EXAFS spectra of the protein complexes (red and black in Fig. 2D), as compared to the R3 peptide (blue in Fig. 2D). EXAFS yields information about the local structure and nearby atoms around the metal center; hence, the EXAFS spectra shown in Fig. 2D clearly indicate that the coordination sphere around the Cu2+ ions in TauK18 and Tau441 is practically the same, while that of the Cu2+–R3 peptide complex is completely different.
 | ||
| Fig. 2 Spectral comparisons of Tau441 and fragments with Cu2+. EPR (A), CD (B), Cu K-edge XANES (C), EXAFS and FT (D) spectra of Cu2+ bound to Tau441 (black), TauK18 (red), TauR3 (blue) and TauR2 (purple) fragments; all with 1 equiv. of Cu2+ in 20 mM MOPS buffer at pH 7.4. | ||
Overall, R2 and R3 peptide fragments cannot reproduce the Cu2+ binding features of Tau441, while the shorter protein TauK18 can bind this metal ion in a similar fashion to Tau441. Hence, we can conclude that metal binding to tau peptide fragments is different from that to the full protein, underscoring the importance of studying metal binding to physiologically and pathologically relevant forms of tau.
Cu2+ binding to Tau441 involves amino acids in R2 and R3
The Cu2+ coordination site in Tau441 was explored at single-residue resolution by 1H–15N Heteronuclear Multiple Quantum Coherence (HMQC) NMR, which displays cross-peaks for each amide group in the protein, and thus, provides multiple probes to locate the Cu2+-binding site. Having an unpaired electron, Cu2+ causes paramagnetic relaxation effects (PREs), resulting in the differential broadening of the amide cross-peak signals by bond-mediated or through space interactions, providing a means of mapping metal-binding sites in the protein. A titration of Tau441 with Cu2+ was followed by 1H–15N HMQC spectroscopy. While addition of 0.01 equiv. of the metal ion causes a drastic broadening of all amide resonances, addition of 0.005 equiv. causes moderate broadening of certain residues of the protein, as compared to that of Tau441 with no metal ion (Fig. 3A and B). A ratio of signal intensities before and after the addition of the metal ion (I/I0) (Fig. 3C) allows for the identification of two regions that are affected by copper binding. The most affected region encompasses residues between 275 and 330 (R2 and R3 domains), containing His299, His329 and His330, which could be participating in copper binding. Other regions affected by copper are in the C-terminal domain, namely: residues 370–380 that contain His374, and the proline-rich domain encompassing residues 210–230. However, these residues are likely not involved in metal binding, since TauK18 lacks the C-terminal domain, and still yields a similar copper complex to Tau441, as assessed by EPR, CD and XAS (Fig. 2). Signal broadening in the 370–380 region might be due to electrostatic interaction between the C-terminal and the R2/R3 domains, which would cause PREs due to the proximity to the copper site. However, the proline rich domain has a propensity to form a transitory secondary structure, which could be affected by metal binding.26,39 | ||
| Fig. 3 R2 and R3 domains are involved in Cu2+ binding to Tau441 protein. (A) Overlay of 1H–15N HMQC NMR spectra of Tau441 100 µM with no metal ion (black), and with 0.005 (red) and 0.01 (green) equiv. of Cu2+; a zoomed image of the region of 1H–15N HMQC (8.00–8.52 1H ppm and 119.5–122.5 15N ppm) spectra is shown in (B). (C) I/I0 profiles of the 1H–15N HMQC NMR signals of assigned residues at different concentrations of Cu2+: 0.005 equiv. (red bars) and 0.01 equiv. (green bars); circles of dotted black lines show the patches due to the PREs of Cu2+. (D) Chemical shift perturbations (CSPs) for the HMQC signals of assigned residues of Tau441 with 0.005 equiv. of Cu2+. | ||
However, individual chemical shift perturbations show some changes in the following regions: 52–63, 82–89 and 104–132 in the N-terminal domain; 190–226 in the proline-rich region proximal to the microtubule binding domain; the region flanking the R3 region (285–326); 339–372, 386–410 and 434–435 of the C-terminal regions (Fig. 3D). Of all these, residues 285–326 are within the region that displays the most line broadening due to PREs, where metal binding is likely to occur, hence the chemical shift perturbations at 285–326 are likely due to copper binding, while changes in all regions might reflect conformational changes caused by metal binding to Tau441.
Overall, the NMR results reveal that the copper binding site of Tau441 is in the MTBD, within the R2 and R3 domains. Residues in the R2 and R3 domains, namely His299 and the bis-His motifs His329 and His330, are putative ligands for the copper site. Furthermore, long-distance interactions exist between the C-terminus and the copper site of Tau441, as has been previously observed for monomeric tau in the absence of metal ions.26
Structural features of the Cu2+–Tau441 complex
To gain insights into the coordination sphere of Cu2+ bound to the full-length tau, EPR simulation and EXAFS fitting were performed. The EPR simulation for the Cu2+–Tau441 complex is consistent with having an axial Cu2+ center with g∥ = 2.304 and A∥ = 149 × 10−4 cm−1 (448 MHz) (Table 1). The parallel copper hyperfine value is at the lower end of the range of A values for typical tetragonal square-planar type 2 Cu2+-sites (120–200 × 10−4 cm−1).40 The copper hyperfine value is indicative of the interaction of the electron spin with the nuclear spin of the metal ion, and hence, correlates with the covalency of the Cu2+-complex: a lower value is indicative of larger covalency, i.e. more delocalization of the spin density toward the ligands. Hence, the A∥ value for Cu2+ bound to Tau441 suggests increased covalency in the site. The g∥ and A∥ values of the Cu2+–Tau441 complex indicate a 2N2O equatorial coordination sphere, according to Peisach–Blumberg correlations (Fig. S3).41 Consistently, the fine structure in the second derivative of the perpendicular region of the EPR spectrum (Fig. 4A) suggests the presence of super hyperfine couplings arising from non-equivalent nitrogens in the coordination sphere. This super hyperfine pattern can be simulated using two or three non-equivalent nitrogen-based ligands (Table 1 and Fig. 4A, red and blue lines, respectively), with N(His)A values that are typical for imidazole ligands.42–44 These results suggest that the Cu2+–Tau441 complex contains at least two nitrogen-based ligands that are likely His residues.| 2N | 3N | |
|---|---|---|
| a A values are in MHz units. | ||
| g x | 2.066 | 2.068 |
| g y | 2.078 | 2.078 |
| g z | 2.304 | 2.304 |
| Cu A x | 5 | 10 |
| Cu A y | 20 | 20 |
| Cu A z | 448 | 448 |
| N1(His) A x | 42 | 41 |
| N1(His) A y | 37 | 36 |
| N1(His) A z | 37 | 36 |
| N2(His) A x | 34 | 35 |
| N2(His) A y | 39 | 40 |
| N2(His) A z | 34 | 35 |
| N3 A x | — | 33 |
| N3 A y | — | 28 |
| N3 A z | — | 28 |
| g-Strainx | 0.012 | 0.014 |
| g-Strainy | 0.012 | 0.0095 |
| g-Strainz | 0.020 | 0.019 |
 | ||
| Fig. 4 Spectral simulation of the Cu2+–Tau441 complex. EPR simulations for the spectrum of Cu2+–Tau441 (A) with 2N (red line) and 3N (blue line) coordination spheres using the parameters listed in Table 1; the inset shows the second derivative of the perpendicular region. Fitting of EXAFS (B) and FT (C) of Cu2+–Tau441 (black trace) with 2N2O (red dotted line) and 3N1O (blue dotted trace) coordination spheres using the parameters listed in Table 2. | ||
To gain insights into the local coordination environment of the Cu2+–Tau441 complex, EXAFS spectra were collected (Fig. 2D and 4B, C black trace) and simulated. EXAFS provides information about the local coordination structure, its nature and distances from the metal center. Considering that EPR results suggest a 2N or 3N coordination sphere for the Cu2+–Tau441 complex, likely with His ligands, simulations with two or three nitrogens in the coordination sphere were tested. As for the other ligands, sulfurs from Cys or oxygen-based ligands (water, Asp or Glu) were considered, and hence, the following first coordination spheres were probed: 2N2S, 2N1S1O, 2N2O, 3N1S and 3N1O. Overall, Bond Valence Sum (BVS) analysis45 reveals a larger formal charge on the metal center for the coordination models that include sulfur-based ligands (Table S1), achieving values closer to +2 only with the coordination models that include oxygen-based ligands. Moreover, Debye–Waller values for the fits that include sulfur-based ligands (especially 2N2S) are higher (Table S1), further supporting the exclusion of Cu–S interactions. A comparison of the 2N2O and 3N1O fits to the experimental data (Fig. 4B, C and Table 2) shows that they both yield an adequate fit, with reasonable Cu–N distances (1.92–2.02 Å).46 In both cases, the Cu–O distances are in the range of 1.88–2.00 Å, which would be consistent with having oxygen-based ligands from a carboxylate ligand, i.e. Glu or Asp residues, or water molecules.47 Moreover, the scattering pattern at larger distances (R + Φ > 2 Å) that is evident in the FT of the EXAFS (Fig. 4C) is better fitted by including the carbon and nitrogen atoms of His imidazole rings (Table 2). Both, the 2N2O and the 3N1O models yield a reasonable EXAFS fit of the experimental data, although the 3N1O model better fits the scattering patterns at larger distances with a lower R-factor (Fig. 4B and C). Nonetheless, it would be risky to discard one of the two possible coordination models, given the limitations of EXAFS to discern scattering from atoms within a ΔZ = ±1 range, i.e. to distinguish between nitrogen and oxygen scatterers in the coordination sphere. Hence, from EXAFS data, it can be concluded that the Cu2+ site in Tau441 involves two or three His ligands.
| 2N2O | 3N1O | ||||||
|---|---|---|---|---|---|---|---|
| Path | N | R (Å) | σ 2 (Å2) | Path | N | R (Å) | σ 2 (Å2) |
| a Coordination numbers have been fixed. The number of long-distance Cu–C and Cu–N scattering paths was fixed to the number of His ligands. Numbers in brackets are the estimated uncertainties in the last digit. b R-Factor filtered between 1.1 and 3.1 Å in FT. | |||||||
| Cu–N | 2 | 1.92(1) | 0.006(3) | Cu–N | 3 | 2.02(2) | 0.004(2) |
| Cu–O | 2 | 2.00(1) | 0.003(2) | Cu–O | 1 | 1.88(3) | 0.005(4) |
| Cu–C(His) | 2 | 2.92(2) | 0.003 | Cu–C(His) | 3 | 2.98(2) | 0.003 |
| Cu–C(His) | 2 | 2.80(3) | 0.003 | Cu–C(His) | 3 | 2.82(2) | 0.003 |
| Cu–N(His) | 2 | 3.86 | 0.003 | Cu–N(His) | 3 | 3.79(5) | 0.003 |
| Cu–C(His) | 2 | 3.74 | 0.003 | Cu–C(His) | 3 | 3.55(5) | 0.008(4) |
| S 0 2 = 0.9, E0= −6.03 eV, Rfactorb = 17.87%, BVS = 2.24 | S 0 2 = 0.85, E0 = −4.47 eV, Rfactorb = 7.97%, BVS = 2.26 | ||||||
The spectroscopic features (EPR and EXAFS spectra) of Cu2+ bound to Tau441 are strikingly similar to those reported for Cu2+ bound to human carbonic anhydrase II (HCA II),48–52 for which X-ray crystallography indicates a binding site involving a bis-His motif, and possibly a third or fourth N or O ligand.52 Here, a comparison to HCA II might be useful, since the Cu2+ binding site in HCA II is highly sensitive to the substitution of the exogenous ligand. Cu2+ bound to HCA II displays an EPR signal with g = 2.320 and A = 156 × 10−4 cm−1 that changes drastically upon binding of a sulphide ligand,48 yielding EPR parameters that are quite far from those observed for Cu2+ bound to Tau441. This is consistent with ruling out a Cys residue acting as a fourth ligand in the Cu2+–Tau441 complex. However, the EXAFS spectrum of Cu2+ bound to HCA II was fitted with 2 Cu–N interactions at 1.98 Å that were assigned to the bis-His motif, and 2 Cu–N/O interactions at 1.88 Å; while multiple-scattering analysis shows that the second coordination sphere is best fit by considering 2 or 3 His ligands, with the participation of His64 and the bis-His motif of HCA II. Similarly, the inclusion of multiple-scattering from His ligands is needed to best fit the EXAFS spectrum of Cu2+ bound to Tau441, while the Cu–N distances are in the same range (Table 2). The striking similarity of the spectroscopic features of Cu2+ bound to HCA II and the Cu2+–Tau441 complex further supports the conclusion that a bis-His motif participates in copper binding to Tau441.
Together, EPR and XAS results indicate that Cu2+ bound to Tau441 is a covalent square planar type 2 site encompassing two or three His ligands in its coordination sphere. While it is not possible to discern between 2N and 3N equatorial coordination spheres, it is likely that the oxygen-based ligands would arise from Asp or Glu residues, while the participation of Cys ligation can be discarded. NMR results suggest that His299 from R2, and the bis-His motif (His329/His330) from R3 could be the nitrogen-based ligands in the Cu2+–Tau441 complex. NMR experiments also point to a possible participation of Asp295 or Asp314, which could act as oxygen-based ligands. Future studies using site-directed mutagenesis will be needed to probe the role of these residues in Cu2+ binding to Tau441.
Chemical reduction of the Cu2+–Tau441 complex
Given that the principal location of Tau441 is intracellular, where copper ions are in their reduced form, it would be of great interest to probe Cu+ binding to Tau441. For this purpose, chemical reduction of the Cu2+–Tau441 complex was attempted using ascorbic acid and/or dithiothreitol (DTT) as reducing agents (see text in the SI and Fig. S4).Full reduction of the complex was achieved using 2 mM DTT and 50 mM ascorbic acid, assuring that the pH of the copper–protein solution was kept at 7.4. Spin quantitation of the EPR spectrum for the reduced sample indicated 2% of residual Cu2+ species (Fig. 5A), while the XANES spectrum displayed the typical feature at 8982 eV associated with the electric dipole-allowed 1s → 4p transition for Cu+ ions and its position and intensity are correlated with the coordination pattern of Cu+ (Fig. 5B).37 The intensity and shape of the Cu+ feature peak suggest that the Cu+–Tau441 complex would be best represented as a tri-coordinated complex.37,53 The FT of the EXAFS spectrum for the Cu+–Tau complex is shifted towards longer metal–ligand distances with respect to those for the Cu2+–Tau complex (Fig. 5C and D), consistent with having a lower metal oxidation state. EXAFS fitting for the spectra of the Cu+–Tau complex was performed considering nitrogen and/or sulfur-based ligands (Table S2 and Fig. S5), revealing that the best results were achieved with a mixture of both types of ligands. The two possibilities for a tri-coordinate Cu+ complex were considered, i.e. 1N2S and 2N1S coordination spheres (Fig. 5E, F and Table 3), revealing that both models fit adequately (Fig. 5C and D), although BVS analysis for the 1N2S model yields a metal formal charge (+1.13) that is closer to the true value, as compared to that of the 2N1S model. These fits (Table 3) yield Cu–S interactions in the range of 2.24–2.27 Å and Cu–N distances in the range of 1.88–2.15 Å; the former falls within the range for known Cu–S distances for Cys coordination in Cu+ metalloproteins, while the latter is consistent with distances reported for His ligation.47 The second sphere contribution in the FT was best fit considering the remote carbons of Cys and His residues (Table 3). Overall, these results support a tri-coordinate model for the Cu+–Tau441 complex with one His and one Cys, while the third ligand could be a second His or Cys residue.
 | ||
| Fig. 5 Spectral comparison of Cu2+–Tau441 and Cu+–Tau441 complexes. EPR (A), Cu K-edge XANES (B), EXAFS (C) and FT (D) of Cu2+–Tau441 (black) and Cu+–Tau441 (light blue) complexes. The inset in (B) shows the feature at 8982 eV associated with the electric dipole-allowed 1s → 4p transition for Cu+ ions. (E) EXAFS fit and (F) FT for the spectrum of the Cu+–Tau441 complex with 1N2S (blue dotted line) and 2N1S (pink dotted line) coordination spheres using the parameters listed in Table 3. | ||
| Path | 1N2S | 2N1S | ||||
|---|---|---|---|---|---|---|
| N | R (Å) | σ 2 (Å2) | N | R (Å) | σ 2 (Å2) | |
| a Coordination numbers have been fixed. The number of paths of imidazole rings was fixed to the number of His ligands. Numbers in brackets are the estimated uncertainties in the last digit. b R-Factor filtered between 1.0 and 4.0 Å in FT. | ||||||
| Cu–S | 2 | 2.238(4) | 0.003(1) | 1 | 2.267(25) | 0.006(8) |
| Cu–N | 1 | 1.881(23) | 0.005(3) | 2 | 2.151(20) | 0.002(2) |
| Cu–C(His) | 2 | 2.714(2) | 0.002 | 4 | 2.788(22) | 0.002 |
| Cu–C(Cys) | 2 | 3.079(15) | 0.002 | 1 | 3.063(28) | 0.002 |
| S 0 2 = 0.95, E0 = −4.47 eV, Rfactorb = 13.26%, BVS = 1.11 | S 0 2 = 0.95, E0 = 8.57 eV, Rfactorb = 13.09%, BVS = 0.76 | |||||
Preliminary NMR studies of the Cu+–Tau441 complex were performed using an excess of ascorbic acid (166 equiv.) (Fig. S6); under these conditions, 71% reduction of the complex was achieved, as assessed by EPR (Fig. S6B). Nevertheless, NMR analysis (Fig. S6C and D) reveals a distinct result with respect to that observed for the Cu2+–Tau441 complex (Fig. 3C and D). Cu+ is a diamagnetic metal ion, and hence, no signal broadening due to paramagnetic relaxation effects would be observed upon reduction of the Cu2+ complex; consistently, the I/I0 shows very little changes (Fig. S6C). However, individual chemical shift perturbations show some changes in the MTBD region; specifically at residues: I278, I297, Q307, V318, G326, I328 and H329 (Fig. S6D). The most affected residues in the R2/R3 region are I297 and Q307, which flank His299, a residue that also acts as a ligand for the Cu2+–Tau441 complex; hence His299 is likely to act as a ligand in the Cu+–Tau441 complex. Although the NMR signals for the two Cys residues of Tau441 (Cys291 and Cys322) cannot be discerned, chemical shift perturbations around Cys322 are quite prominent, while residues around Cys291, such as G292, S293, K294 and N296, are not perturbed (Fig. S6D); hence Cys322 is more likely to be acting as a ligand in the Cu+–Tau441 complex than Cys291. Additional chemical shift perturbations in all regions might reflect conformational changes caused by metal binding to Tau441.
Altogether, XAS and NMR results indicate that Tau441 can bind Cu+ ions and that the Cu+–Tau441 complex is tri-coordinate with one His and one Cys, likely His299 and Cys322, while the third ligand could be Cys291 or a His from the bis-His motif. Future studies using site-directed mutagenesis and direct Cu+ binding assays with Tau441 are needed to further elucidate the nature of the Cu+ binding site in Tau441.
Tau441 has multiple Zn2+ binding sites
Zn2+ binding to Tau441 was probed at single-residue resolution by NMR, by titrating the protein with different equivalents of Zn2+, followed by 1H–15N HMQC spectroscopy (Fig. 6A). Minor changes are observed upon addition of 0.1 equiv. of the metal ion (Fig. 6A, left panel), while larger changes in different regions of the NMR spectra become evident upon addition of 0.3 equiv. (Fig. 6A, middle panel), including the Gly (15N chemical shifts of 105–112 ppm) and His (15N chemical shifts of 118–124 ppm) regions. Finally, after addition of 1.5 equiv. of Zn2+, most protein residue signals are drastically perturbed (Fig. 6A, right panel). An analysis of individual chemical shift perturbations (CSPs) shows that the addition of 0.1 equiv. of the metal ion impacts the C-terminal and the microtubule binding domain (light blue region in Fig. 6B), while the rest of the protein remains mostly unchanged. At 0.3 equiv., the changes at the C-terminal and the MTBD become even more evident, while large perturbations are also observed at the N-terminal region, encompassing residues 43–135 that include His121 (orange bars in Fig. 6B). A couple of residues in the proline-rich region 210–230 known to form a transient secondary structure,26,39 are also affected upon addition of 0.3 equiv. of Zn; however, they are unlikely to participate directly in metal binding and the CSP effects may reflect an indirect effect of Zn binding to the local structure. Finally, the N-terminal residues are the most affected region upon addition of 1.5 equiv. of Zn2+ (purple bars and region in Fig. 6B). | ||
| Fig. 6 Domains involved in the Zn2+ coordination sites. (A) Overlays of 1H–15N HMQC NMR spectra of Tau441 100 µM in the absence (black) and in the presence of 0.1 equiv. (light blue), 0.3 equiv. (orange) and 1.5 equiv. (light blue) of Zn2+; (B) analysis of the chemical shift perturbations (CSPs) for the HMQC 1H–15N signals of assigned residues of tau with 0.1 equiv. (blue), 0.3 equiv. (orange) and 1.5 equiv. (purple) of Zn2+. Blue circles highlight perturbed residues with 0.1 equiv. of zinc and purple circles highlight residues perturbed with 1.5 equiv. of zinc. | ||
A more detailed analysis of the NMR spectra reveals that at 0.1 equiv. of Zn2+ the most affected residues are Gly120 (Fig. 7A), His94 (Fig. 7B), Ala119, His121, Ile297, Val318 and Ile328 (Fig. 7C). These changes are mapped onto the protein sequence in Scheme 2 (in light blue color), where it is evident that His94 and Ala119/Gly120/His121 flank a region that is rich in Glu residues, which together with His94 and His121 could act as ligands for Zn2+. However, Ile297 is in the R2 domain, within the sequence DNIKH, that contains Asp295 and His299, which could also act as Zn anchoring ligands. Finally, Val318 and Ile328 are in the R3 region, near the bis-His motif (His329/His330), which is also known to bind zinc. It should be noted that all these residues are further perturbed upon addition of 0.3 equiv. of Zn2+ (Scheme 2, in orange), where the most drastic changes include the disappearance of the Val318 signal and large displacements of Ile297 and 328 (Fig. 7C), which are residues in the vicinity of the DNIKH motif in R2, Cys322 and the bis-His motif in R3. Since no further changes are observed in these residues at the R2/R3 region upon addition of 1.5 equiv. of Zn (light blue region in Fig. 6B), these anchoring residues might constitute a Zn binding site (labeled as site 1 in Scheme 2) that gets saturated at >1 equiv. of the metal ion. In contrast, drastic changes are observed for the Gly120 (Fig. 7A), His94 (Fig. 7B) and Ala119 (Fig. 7C) signals, which shift significantly at 0.3 equiv. and continue to change at 1.5 equiv. Moreover, the signals for Gly100 and Gly109 (Fig. 7A), Glu96 and Glu99 (Fig. 7B) shift significantly at 0.3 and 1.5 equiv. of zinc or even disappear, as is the case for Glu99 and Gly100. Overall, these results suggest that the region flanked by His94 and His121, containing glutamate residues like Glu96 and Glu99, may constitute a zinc anchoring site (labeled as site 2 in Scheme 2) that gets populated starting from 0.1 and up to 1.5 equiv. of the metal ion.
 | ||
| Fig. 7 Residues involved in the Zn2+ coordination sites. A zoomed image of the Gly (A), His (B) and Ile–Val (C) regions of 1H–15N HMQC overlaid spectra of Tau441 (black) with 0.1 equiv. (blue), 0.3 equiv. (orange) and 1.5 equiv. (purple) of Zn2+. | ||
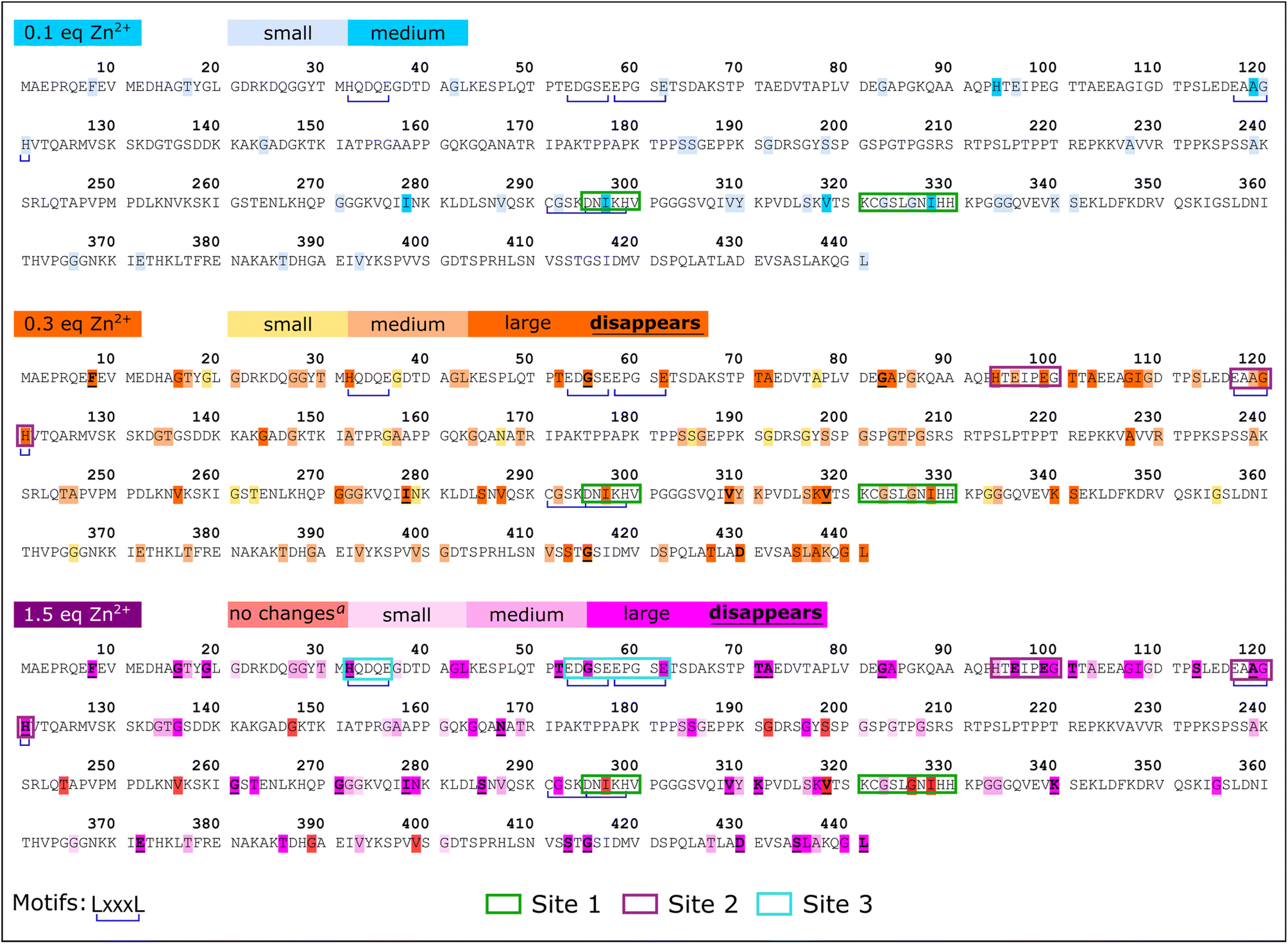 | ||
| Scheme 2 Mapping of the NMR perturbations of Tau441 with Zn2+. Mapping of the chemical shift perturbations of the sequence of tau protein with 0.1 equiv. (blue), 0.3 equiv. (orange) and 1.5 equiv. (magenta) of Zn2+. aNo changes in the CS between 0.3 and 1.5 equiv. of Zn2+. Motifs LxxxL are underlined in blue. Coordination sites are highlighted in green (site 1), purple (site 2) and light blue (site 3). | ||
At 0.3 equiv. of Zn2+, several additional residues at the N-terminal domain of the protein are affected, namely Gly55 (Fig. 7A) and Glu62 (Fig. 7B) disappear, while Gly42 (Fig. 7A), His32 (Fig. 7B) and Leu43 (Fig. 7C) show a large shift at 0.3 equiv., which further changes at 1.5 equiv. of zinc. Mapping these and other shifts onto the sequence of the protein (Scheme 2, orange and purple), it becomes evident that these changes occur in a region containing several putative ligands for zinc; for example, His32 is contained within the sequence HQDQEGDTD, while Glu62 is located in the EDGSEEPGSE motif, both containing several glutamates and aspartates that could act as anchoring ligands for zinc. This region likely constitutes a third zinc binding site of lower affinity, labeled as site 3 in Scheme 2.
In summary, NMR results reveal the presence of at least three putative binding sites for zinc in Tau441: site 1 likely involves the DNIKH motif in R2 containing Asp295 and His299, and Cys322 and the bis-His motif (His329/His330) in R3; site 2 encompasses the region flanked by His94 and His121 at the end of the N2 insert, containing several Glu residues like Glu96 and Glu99; while site 3 is located at the N-terminal domain, in the N1-insert, involving the HQDQEGDTD and EDGSEEPGSE motifs. It should be noted that all these sites contain motifs of the type LxxxL, where L is a putative ligand for Zn2+ (Scheme 2 blue underline); this type of zinc binding motif is known to exist in zinc binding proteins, such as zinc fingers and zinc transporters.54 Examples of identified LxxxL motifs that may be involved in Zn2+ binding to Tau441 are DNKIH in site 1, EAAGH in site 2, and HQDQEGDTD or EDGSEEPGSE in site 3. Finally, it should be noted that several other regions of the protein are affected upon zinc binding that are not located in the vicinity of residues that could act as zinc ligands (Scheme 2), but that might correspond to protein regions that suffer conformational changes upon metal binding.
To gain insights into the nature of the Zn2+ binding sites of Tau441, Zn K-edge XANES (Fig. 8A) and EXAFS (Fig. 8B and C) spectra were collected for the protein with 1 equiv. (magenta), 2 equiv. (purple) and 3 equiv. (dark blue) of the metal ion. The spectrum for Zn2+ in MOPS buffer was collected as a control (light blue traces in Fig. 8); the edge intensity in its XANES spectrum (Fig. 8A) is typical of an octahedral site of Zn2+ (a d10 configuration has no pre-edge feature, regardless of the geometry).55 In contrast, the white line intensity and fine structure observed around 9670 eV in the XANES spectra for the protein with 1 and 2 equiv. of Zn2+ reveal that the protein bound Zn2+ species have a tetrahedral geometry, while the XANES spectrum for 3 equiv. shows a peak shoulder at 9682 eV that arises from a mixture of Zn2+ in buffer and Tau441-bound Zn2+ species (Fig. 8A). Consistently, the EXAFS spectra (Fig. 8B) for the protein with 1 and 2 equiv. of Zn2+ exhibit similar features, while that of 3 equiv. seems to be a combination of Tau441-bound Zn2+ species and free Zn2+ in buffer solution. Specifically, the double feature centered at 4 Å−1 in the EXAFS spectra (Fig. 8B) is most evident at 1 and 2 equiv. of the metal ion; this feature is typical of Zn2+ bound to a bis-His motif, as observed previously for Zn2+ binding to the beta-amyloid peptide.56 The FT of the EXAFS spectra for the protein with 1 and 2 equiv. of Zn2+ (Fig. 8C) shows slight differences in the short metal–ligand distance region, which could not be adequately modelled by the fits. The best EXAFS fits for Tau441 with 1 and 2 equiv. of zinc were achieved assuming a mixture of species: one with the 2N1S1O coordination sphere (site 1) and a second species (site 2) with 2N2O (Fig. S7 and Table S3); however, determining the percentage of each of these two species is difficult, since a variation in the percentage of the species with a sulfur ligand is compensated for by the metal–ligand distances. A BVS analysis45 (Table S3) reveals that the fits that do not consider any contribution from a Zn–S interaction yield a formal charge for the metal center that is significantly below +2.0 (+1.47 or +1.51, Table S3), supporting the hypothesis that there might be a Cys ligand at site 1. However, the EXAFS fits that yield a formal charge of the metal center closer to +2.0 are those having an average coordination number of 0.3 for the S ligand, i.e. having 30% of the 2N1S1O species (site 1), with Zn–S distances in the range of 2.25–2.28 Å. The Zn–O interactions at 2.12 Å are likely due to Asp or Glu residues, while Zn–N interactions at 1.96 Å likely correspond to His ligands; indeed, scattering at larger distances can be fitted by including carbon and nitrogen atoms of an imidazole ring (Table S3). These results are consistent with reported Zn–S distances from Cys coordination, which are greater with respect to Zn–N or Zn–O distances arising from His or Glu ligation, respectively.57 Hence, XAS results would be consistent with having a coordination sphere for site 2 with two His and two carboxylate residues, while site 1 likely involves two His residues, one carboxylate and one Cys; the latter could be either Cys291 from R2 or Cys322 from R3, both regions being highly affected by zinc, as observed by NMR (Scheme 2). However, better characterization of the Zn binding sites in Tau441 would be achieved by site-directed mutagenesis and/or by studying fragments or mutants that could help isolate the different Zn species.
 | ||
| Fig. 8 Tau441–Zn2+ binding sites at different equivalents. Titration of Tau441 protein (0.8 mM) in 20 mM MOPS buffer at pH 7.4, with Zn2+ (0 to 3 equiv.), followed by XAS. Comparison of the Zn K-edge normalized XANES (A), EXAFS (B), and FT (C) spectra of Tau441 with 1 (magenta), 2 (purple), and 3 (blue) equivalents of Zn2+. The spectrum of ZnSO4 in buffer (turquoise) is included for comparison. | ||
In summary, a combination of NMR and XAS studies reveals that Tau441 is likely to have up to three tetrahedral binding sites for Zn2+, one of them in the R2/R3 region likely encompasses His299 from R2, the bis-His motif (His329/His330) from R3, and Cys291 or Cys322. The other Zn2+ binding sites are located in the N-terminal domain, and they seem to involve mostly His, Asp and Glu residues in LxxxL motifs for zinc binding (Scheme 2).
Cu2+ and Zn2+ ions impact the aggregation of Tau441 protein
The impact of Cu2+ and Zn2+ ions on the amyloid aggregation of Tau441 protein was evaluated by Thioflavin T (ThT) fluorescence assays. ThT is a dye whose fluorescence emission centered at 482 nm increases upon intercalation into the β-sheet structure of amyloid aggregates; hence, an increase in the fluorescence intensity reports on the growth of amyloid fibrils.58 Heparin was added in a 1:4 ratio to achieve Tau441 amyloid aggregation within 20 hours; control experiments with Tau441 without heparin do not display any aggregation in this time frame (Fig. S8). In the absence of metal ions, ThT fluorescence increased significantly over the course of 20 h, displaying a lag phase of 1.2 hours before a sigmoidal growth of Tau441 amyloid fibrils (Fig. 9A and D, black trace). Transmission electron microscopy (TEM) analysis at the end point of the assay shows the formation of long helical Tau441 aggregates (Fig. 9B), as previously observed.21 In the presence of 1 equiv. of Cu2+ ions, the increase of ThT fluorescence is accelerated and no lag time is observed (Fig. 9A, light blue trace), suggesting that the metal ion accelerates the first steps of amyloid aggregation. Moreover, the ThT trace in the presence of 1 equiv. of Cu2+ displays exponential growth of Tau441 amyloid fibrils at a rate that is approximately four times higher than in the absence of metal ions (Fig. S9). TEM analysis at the end point shows the formation of long and intercrossed Tau441 fibrillar aggregates that seem to be less helical than those in the absence of copper (Fig. 9B). Upon addition of 2 or 4 equiv. of Cu2+ ions, the exponential growth behavior is conserved; however the maximum intensity of ThT fluorescence is severely diminished (Fig. 9A, darker blue traces). Being a paramagnetic metal ion, Cu2+ is known to quench fluorescence if bound in the vicinity; hence, decreased ThT fluorescence does not necessarily translate into less aggregation. Indeed, when Cu2+ ions are added to Tau441 fibrils that were grown in the absence of metal ions, the ThT fluorescence of the fibrils is quenched significantly, while TEM still shows the presence of amyloid aggregates (Fig. S10).
 | ||
| Fig. 9 Cu2+ and Zn2+ ions impact the aggregation of Tau441 protein. Aggregation assays of Tau441 protein (25 µM) in 20 mM MOPS buffer at pH 7.4, in the presence of heparin (6.25 µM), and different equivalents of Cu2+ (A) or Zn2+ (D) ions, followed by ThT fluorescence at 37 °C. SDS-PAGE analysis of the samples at the endpoint (20 h) of the ThT aggregation assays (A and D) with Cu2+ (B) or Zn2+ (E) ions, performed in the presence (right) or absence (left) of 2-mercaptoethanol (BME) as the reducing agent. (C) Electron microscopy images of the amyloid aggregates of Tau441 protein, obtained at the endpoint (20 h) of the ThT aggregation assays, in the absence of metal ions (top panel), and with 1 equiv. of Cu2+ (mid panel) or Zn2+ (bottom panel). | ||
SDS-PAGE analysis of Tau441 aggregates at the end point of the ThT fluorescence assay was performed. Analysis of Tau441 amyloid aggregates in the absence of metal ions reveals mostly monomeric species (band at around 60 kDa) and a small amount of oligomers (band between 150 and 250 kDa) (Fig. 9C, left). Tau441 aggregates formed in the presence of copper ions show mostly high molecular weight species (band at MW > 250 kDa) and almost no monomers, while the band associated with low molecular weight oligomers is more evident at 1 equiv. of Cu2+. These bands are no longer observed when the SDS-PAGE analysis is performed in the presence of a reducing agent, such as β-mercaptoethanol (BME) (Fig. 9C, right), suggesting that these species are linked via disulfide bridges.
Similarly to the case of Cu2+ ions, Tau441 aggregation in the presence of Zn2+ ions shows increased ThT fluorescence with no lag time (Fig. 9D, light purple trace), suggesting that Zn2+ ions also accelerate oligomerization. Moreover, the ThT traces in the presence of 0.5 and 1 equiv. of Zn2+ display exponential growth of Tau441 amyloid fibrils at a rate that is approximately four and three times higher than in the absence of metal ions, respectively (Fig. S9). Upon increasing the Zn2+ equivalents,2,4,10 the lag phase is still abolished, but the final ThT intensities decrease, and the aggregation kinetics show biphasic behavior (Fig. 9D, dark purple traces). It should be noted that the final ThT fluorescence with 0.5 and 1 equiv. of Zn2+ is significantly higher than that of Tau441 with no metal ions, suggesting the formation of more amyloid aggregates. However, protein quantification of soluble tau in the supernatant of the aggregation reactions reveals similar residual amounts of soluble Tau441 (about 10%) regardless of the presence of Zn2+ ions. TEM analysis at the end point with 1 equiv. of Zn2+ shows the formation of abundant and shorter Tau441 fibrillar aggregates than those in the absence of metal ions (Fig. 9B). Thus, the increased ThT fluorescence observed in the presence of zinc might be due to a larger fluorescence response of ThT to the short fibrils observed by TEM, which are small straight filaments and lack the helicity of Tau441 fibrils with no metal ions.
SDS-PAGE analysis of Tau441 aggregates in the presence of Zn2+ shows the presence of mostly monomers and a small amount of oligomers (Fig. 9E). The band associated with low molecular oligomers (between 150 and 250 kDa) peaks in intensity at 0.5 equiv. of Zn2+ (Fig. 9E, left). All oligomeric species are no longer observed when the SDS-PAGE analysis is performed under reducing conditions (Fig. 9C, right), suggesting that these species are linked via disulfide bridges. In contrast to the case of copper, no significant amount of high molecular weight species (above 250 kDa) is observed in the aggregates grown with Zn2+, the low molecular oligomer species seems to be more favored, and the aggregate is mostly composed of monomers, regardless of the amount of metal ions added (band between 50 and 75 kDa). These observations point to a metal-bridging mechanism, where monomers and some trimers could be bridged by Zn2+ ions.
Overall, these results suggest that both Cu2+ and Zn2+ ions accelerate the amyloid aggregation of Tau441, while having distinct effects on the morphology of the aggregates. Copper-induced aggregation of Tau441 yields amyloid fibrils that are intercrossed and less helical than those of the protein with no metal ions, while Zn2+ ions favor the formation of short fibrils.
Discussion
Copper and zinc are essential metals that perform important functions in the central nervous system. In this study, we demonstrate that Tau441 can bind Cu2+, Cu+ and Zn2+ ions, at the microtubule binding domain (MTBD); while Zn2+ can also bind at the N-terminal region (Fig. 10A). The MTBD region provides residues that act as anchoring ligands for metal ions: His299 and Cys291 from R2, Cys322 and a bis-His motif in R3. Spectroscopic results indicate that, while the Cu2+ site has a square planar geometry coordinated to two or three His residues (Fig. 10B), the reduced Cu+ site is tri-coordinated involving His299, Cys322 and a third ligand, likely Cys291 or His329/330 (Fig. 10C), and the Zn2+ site is tetrahedral and likely also includes a Cys and an Asp residue in its coordination sphere (Fig. 10D). Structural models for these metal binding sites, as built by Alphafold 3.0,59 confirm some of these features, depicting a Cu2+ ion bound to His299 and His329/His330 from the bis-His motif, a Cu+ ion coordinated to His299, Cys322 and His 330, and a Zn2+ binding site with His299, His330 and Cys322. Although having distinct geometries, the copper and zinc binding sites in Tau441 share some His and Cys ligands; thus, these metal ions could be competing for binding to the MTBD region. It is interesting to note that several proteins related to AD display sites with the ability to bind Cu2+, Cu+ and Zn2+ ions; these include the highest affinity copper-binding site (component 3) in the octarepeat region of the prion protein (PrP),60,61 the E2 domain site of the amyloid precursor protein (APP)62–64 and the N-terminal region of the Aβ peptide.17 This feature, together with the fact that the homeostasis of these metal ions is affected in AD,3,4 strongly suggests an important interplay of copper and zinc in this neurodegenerative disease that remains to be elucidated. The structural features of the metal binding sites in Tau441 and their potential physiological and pathological implications are discussed below. | ||
| Fig. 10 Metal Binding properties of Tau441 and AD-related post-translational modifications. (A) Cu2+, Cu+ and Zn2+ coordination sites in Tau441 are identified by blue, green and gray circles, respectively. The location of Tau441 AD related PTMs is shown above: phosphorylation (red), acetylation (blue) and ubiquitination (black) sites. Lys residues that undergo both ubiquitination and acetylation are marked in purple. Coordination modes (derived from spectroscopic results) and structural models generated by Alphafold 3.0 for the Cu2+ (B), Cu+ (C) and Zn2+ (site 1) (D) binding sites in Tau441 are presented. For the Alphafold 3.0 models, only the R2 and R3 repeats were used, putative metal binding residues are shown in green, the PHF6 sequence is in dark blue, and Lys residues that interact with heparin are in cyan. | ||
Copper binding properties of Tau441
Tau441 displays a single Cu2+ binding site. Spectroscopic comparison of Cu2+ bound to TauK18 shows that it reproduces the metal site in the full-length Tau441, consistent with having a coordination sphere provided only by the MTBD. In contrast, the spectroscopic features of Cu2+ bound to peptide fragments modelling R2 and R3 and those reported for the separate R1, R2, R3 and R4 repeats28,65 cannot replicate the metal coordination site of the full-length protein. The fact that metal binding properties of tau peptide fragments are different from those of Tau441 underscores the importance of studying metal binding to the full protein and to physiologically and pathologically relevant forms of tau.Having a multi-His Cu2+ binding site, Tau441 would be expected to have a higher binding affinity (with a Kd in the low nM range), similar to other multi-His Cu2+ sites, such as: the component 3 in the PrP with a Kd = 10 nM,60 and the E2 domain of the amyloid precursor protein (APP) with a Kd = 28 nM,62 both proteins displaying four-His Cu2+ sites. However, ITC studies reveal a Kd = 0.5 µM for Cu2+ binding to Tau441.29 The fact that Tau441 is an intrinsically disordered protein and that the bis-His motif and His299 are 30 residues apart, may explain its 10× lower Cu2+ binding affinity as compared to those systems. While the His residues acting as ligands for Cu2+ in the E2 site of APP are also many residues apart (>50), they are provided by three alpha helix bundles that impose a specific geometry for a tetragonal copper site.63 However, while the multi-His Cu site in the PrP is located in its intrinsically disordered octarepeat region, each His ligand is provided by an octapeptide with sequence PHGGGWGQ,60 where the Pro residues play an important role in facilitating a protein conformation that is amenable to the coordination of the four His residues to copper. Moreover, having the His residues supplied by the same protein chain and spaced by only 8 residues provides an important chelate effect to achieve high Cu binding affinity. It is important to note that, in the MTBD domain of Tau441, the four repeats are each linked by a PGGG motif, similar to that of the PrP repeats. Specifically, the PGGG motif present between the R2 and R3 repeats may provide structural flexibility for copper binding to His299 in R2 and the bis-His motif in R3; however, the distance between these His ligands is greater (30 residues) than that between the His ligands in component 3 of the PrP (8 residues), which would contribute to the lower Cu binding affinity displayed by Tau441.
Beyond metal binding affinity, the fact that Tau441 displays a multi-His Cu2+ binding site warrants a comparison with other proteins containing multi-His Cu centers in terms of biological relevance and function. In the case of PrP, component 3 is responsible for stabilizing the cis conformation of the protein that is key to its neuroprotective role.60,66 For APP, copper binding to the E2 domain increases the thermal stability (Tm) of its α-helical structure.63 A multi-His Cu site found in the S100A12, a protein with a prominent role in regulating inflammatory processes and immune responses,67 appears to play a role in stabilizing the multimeric structure of the protein.68 The phenoxazinone synthase (PHS), a protein naturally present in Streptomyces antibioticus bacteria,69 has up to five Cu coordination sites, and one of them is a multi-His type 2 Cu site that plays a role in stabilizing the hexameric form of the protein to achieve greater catalytic activity.70 In all these systems, the multi-His Cu2+ sites appear to play a structural stabilizing role in the protein. Recent reports indicate the presence of extracellular tau under both physiological and pathological conditions.23,25,71,72 Given that the oxidation state of copper in the extracellular milieu is primarily +2, Cu2+ binding to Tau441 could stabilize a protein conformation that might be relevant to its function in the extracellular environment. For example, extracellular tau has been shown to interact with membranes, and R2 and R3 appear to be the key domains for this interaction, particularly involving hydrophobic residues located between His299 and His329;73 since copper binding occurs in this region, copper binding to His299 and the bis-His motif could be favoring a protein conformation where such hydrophobic residues located between these His ligands are more readily available for interaction with the lipid membrane. Indeed, NMR studies found that Cu2+ binding to TauK32 impacts the region encompassing such hydrophobic residues.29 If this were the case, Cu binding to the MTBD of Tau441 would be playing a structural role in facilitating the interaction of tau with lipid membranes.
However, bis-His motifs are known to facilitate the formation of Cu complexes with a labile coordination position. For example, the CuB site in cytochrome c oxidase (CcO),74 which activates oxygen in conjunction with an Fe heme a site, involves a bis-His motif and a nearby His residue, leaving a vacant position for O2 binding. Similarly, the bis-His site in HCA II also displays a fourth labile ligand that can accommodate an exogenous ligand.49 This raises the possibility that Cu2+ bound to the bis-His motif and His299 in tau may also display an open coordination position for exogenous ligand binding or for the formation of ternary complexes with proteins that could provide a fourth ligand. Tau is known to interact with other His-containing proteins, such as PrP and Aβ.75–77 Hence, it would not be unreasonable to propose that such protein–protein interactions may be mediated by Cu binding through the formation of ternary complexes, as it has been demonstrated in the case of PrP and Aβ.78,79
Cu+ interaction with Tau441 was studied by chemically reducing the Cu2+–Tau441 complex. Upon reduction, Cu+ remains bound to the MTBD using His299, Cys322 and a third ligand that can be a His or a Cys residue from the R2 or R3 domain. One of the key features of the Cu2+ binding site of Tau441 is that it contains a bis-His motif, which is known to stabilize Cu+ binding80 and is present in several important copper binding proteins, including the copper transporter Ctr1p and the Aβ peptide.17,81 In the case of Ctr1p, the bis-His motif is thought to bind Cu+ and to be auxiliary to its high affinity Cu2+ ATCUN binding site (Amino-Terminal site of Copper and Nickel);81 while in Aβ, the bis-His motif is thought to play a role in both Cu2+ and Cu+ binding. The bis-His (His13/14) of Aβ, together with His6, can yield a multi-His Cu2+ binding site that displays a lower binding affinity, as compared to that of the main N/terminal site;17 while Cu+ binding to the bis-His motif is known to yield a linear bi-coordinate Cu+ site.81 In contrast, this does not seem to be the case for Tau441, where the bis-His motif (His329/330) participates as a bidentate ligand for Cu2+ binding but apparently not for Cu+. This is possibly due to the presence of nearby Cys residues that can coordinate to the Cu+ ion; hence, only one or two His residues remain coordinated to the metal ion upon reduction.
Given that ascorbic acid alone does not completely reduce the Cu2+–Tau441 complex, its reduction potential must be lower than that of ascorbic acid (E° < 52 mV). The reduction potential of the Cu2+–Tau441 complex can be thermodynamically related to the reduction potential of free copper ions and the relative binding affinity of the protein for Cu2+ and Cu+ ions (Scheme S1); from this thermodynamic cycle, a lower limit for the dissociation constant of the Cu+–Tau441 complex can be estimated to be Kd >32 µM. This Cu+ binding affinity is strikingly low, considering the proposed coordination sphere for the Cu+–Tau441 complex. Indeed, Cu+ binding sites with two Cys and one His, as those found in cytosolic copper chaperones Sco1 and Sco2,82,83 display Kd values in the pM to fM range. Tricoordinate type 1 sites with two His and one Cys display Kd in the pM range,82,84,85 while de novo designed sites with the same coordination set have Kd values in the fM range.86 However, it should be considered that these metalloproteins contain Cu+ binding motifs, such as CXXXC or CXXXH, in a well-structured region with a nearby His residue. In contrast, Tau441 is an intrinsically disordered protein and the residues that may be anchoring Cu+ are 8 to 30 amino acid residues apart, leading to a lower binding affinity for Cu+.
Zinc binding properties of Tau441
In contrast to copper, zinc binding to Tau441, not only happens in the MTBD region, but it also occurs in the N-terminal domain, specifically at the N1 and N2 inserts (Fig. 10A). The nature of the N-terminal sites is evidenced by NMR, allowing for the identification of zinc binding motifs of the type LxxxL, that provide coordinating residues for Zn2+ (Scheme 2) in a similar fashion to those found in zinc finger proteins. The N-terminal zinc binding sites (sites 2 and 3) likely involve LxxxL motifs with a combination of His and carboxylate (Glu or Asp) anchoring ligands. In contrast, the MTBD site (site 1) is the only zinc binding site in Tau441 that may have a contribution from a Cys ligand, involving also two His from the R2 and R3 repeats. Cys and His residues constitute the most frequent ligands in high affinity Zn2+ binding sites in metalloproteins,87 while Asp and Glu constitute anchoring residues for sites with lower zinc binding affinity.57 Hence, one would expect sites 2 and 3 to display a lower affinity for zinc than that of site 1. Consistently, ITC studies at different temperatures of Zn2+ binding to Tau441 indicate the presence of one high affinity zinc binding site (Kd ∼ 0.5 µM), likely associated with site 1 in the MTBD region, and three additional zinc binding sites with lower affinity (Kd ∼ 170 µM)33 which might correspond to the more oxygen-rich complexes in sites 2 and 3. It is interesting to note that the N-terminal region of Tau441 is involved in its interaction with proteins involved in synaptic transmission, such as synapsin-1 and synaptotagmin-1, and with annexin A5, a calcium-dependent phospholipid binding protein.88 Hence, the identification of zinc binding sites in the N-terminal domain of Tau441 prompts to question if zinc may be playing a role in modulating the interaction of tau with some of its protein partners where the interaction involves the N-terminal region.The highest affinity Zn2+ binding site in Tau441 protein likely involves His299, His330 and Cys322 (Fig. 10C), all residues located within the MTBD; this suggests a physiological role for zinc binding in regulating the interaction of Tau441 with alpha- and beta-tubulin.24 Indeed, a recent report suggests a role for zinc in the regulation of cargo transport by microtubules (MTs) and their binding to microtubule-associated proteins (MAPs), including tau protein.89 The study by Qin et al. demonstrated that binding of tau, and other MAPs (such as DCX and MAP2C), to microtubules is disrupted by the addition of Zn2+, due to a direct interaction of the metal ion with the MTs with a Kd in the low nanomolar range. Structural analysis and modelling identified a putative Zn2+ binding site in alpha-tubulin, potentially involving residues H192, E196, E417, E420, E423, and D424. The residue pairs His192/Glu196 and Glu420/Asp424 are part of LxxxL motifs that are provided by two different alpha-helices, making them amenable to a tetrahedral Zn coordination site. Interestingly, these carboxylic residues are also engaged in binding of tau, as they display electrostatic interactions with the Lys residues present in the MTBD. Tau441 is a zinc binding protein with a high nM affinity (500 nM), which is one order of magnitude lower than the binding affinity of MTs for Zn (low nM range);89 hence, even though it would be tempting to propose that zinc binding to Tau441 may be playing a role in the Zn-mediated disruption of the interaction of tau with MTs, this is likely not the case. However, a role for the Zn–Tau441 interaction in the assembly of MTs cannot be discarded, since it has been proposed that Cys291 and/or Cys322 may form disulfide bonds with Cys347 in alpha-tubulin and/or Cys131 in beta-tubulin that contribute to stabilizing the tau–MT interaction.90 Given that Cys322 is a putative ligand for Zn2+ in Tau441, a regulatory role for zinc in the function of tau during microtubule assembly would be plausible.
Beyond its cytoplasmic localization and interaction with microtubules, tau protein is also known to localize to the nucleus where it can interact with DNA, histones, RNA and nuclear proteins, potentially playing a role in DNA stabilization, chromatin organization and gene expression.91,92 The repeats R1–R4 in the MTBD of tau are involved in its binding to DNA, as this region is rich in Lys and Arg residues, known to interact electrostatically with negatively charged DNA.93,94 The nucleus is a zinc-rich cellular compartment;95 hence, the Zn binding site 1 in tau could be populated, potentially modulating its interaction with DNA. Indeed, it has been reported that the binding mode of tau with DNA changes in the presence of zinc, specifically, going from minor groove to major groove binding for the Zn-bound protein. Such an effect could reflect a change in the conformation of tau in its metal-bound form. While studying how the conformation of Tau441 would be impacted by zinc binding would require a significant investment in structural studies, an AlphaFold 3.0-built model of Zn-bound tau (Fig. 10B) displays the Lys residues in the R3 repeat aligned on one side of an alpha-helix, a conformation that would facilitate the electrostatic interaction with DNA.
Metal–tau interactions: aggregation, toxicity and pathology
Tauopathies are characterized by the accumulation of amyloid fibrils composed entirely of self-assembled tau protein, displaying distinct morphologies and fibrillar structures in various types of tauopathies.96 In AD, the NFTs are composed of two types of fibrils: paired helical filaments (PHFs) and straight filaments (SFs), sharing a structural core consisting of residues V308–F378, located in the R3/R4 region.97 The core is composed of eight β-sheets that include the hexapeptide motif VQIVYK (PHF6, residues 306–311) and is considered essential for tau self-assembly.98 The PHF6 sequence is located in R3, between anchoring residues that bind copper and zinc (Fig. 10); hence, metal binding could promote a conformation that favors intermolecular interactions between PHF6 motifs, facilitating core formation. Indeed, copper- and zinc-induced Tau441 oligomerization was previously demonstrated by dynamic light scattering (DLS).29,32,33 Copper is known to promote fibril formation of R2 and R3 peptide fragments,28 while Zn2+ binding to TauK18 accelerates its aggregation in the presence of heparin.31 However, the lack of structural insights into the metal binding properties of Tau441 has prevented an understanding of the mechanism by which metal ions impact tau aggregation.Our study demonstrates that Cu2+ and Zn2+ ions accelerate the amyloid aggregation of Tau441 in the presence of heparin. Structural models of copper and zinc bound to tau (Fig. 10) show that the Lys residues in R3 are aligned in an alpha helix motif, an arrangement that, upon interaction with heparin, a sulfated polysaccharide,99 could facilitate the alignment of metal-bound monomers in spatial proximity along the polysaccharide chain. Such interactions would favor the formation of oligomers and protofibrils, which would explain the suppression of the lag phase observed in the metal-induced Tau441 aggregation assays. Tau441 amyloid fibers obtained in the presence of copper are intercrossed and less helical than those formed in the absence of metal ions, while zinc induces the formation of short fibrils. Such morphological differences could arise from the distinct copper and zinc coordination sites (Fig. 10). CryoEM analysis of AD amyloid fibrils indicates that Cys322 is part of the fibril core, possibly forming intermolecular disulfide bridges that facilitate growth along the fibril axis.97 Zinc binding to Tau441 involves Cys322, and it is likely to interfere with intermolecular disulfide bridging (as observed by SDS-PAGE, Fig. 9E), preventing elongation of the fibril and yielding shorter fibrils, as observed by TEM analysis (Fig. 9B). In contrast, since copper does not coordinate Cys322, it would not be expected to interfere with intermolecular disulfide bridging and the extent of fibril growth; indeed, copper-induced aggregates have an important component of disulfide-bridged oligomers (Fig. 9C), and have similar length to amyloid fibrils grown in the absence of metal ions (Fig. 9B). This is consistent with a cryoEM study of the truncated tau dGAE (297–391), showing that aggregates grown in the presence of copper display intermolecular disulfide bridges using Cys322, while zinc does not.100 However, cryoEM analysis of PHFs shows that His330 is facing inwards in the amyloid fibril core, while His329 is facing outwards to the solvent-exposed area of the fibril.97 In monomeric Tau441, copper coordinates to both His residues (Fig. 10); hence, upon fibril formation the copper ion would have to release His330, and it would likely remain bound to His329 on the outer side of the fibril; in such an arrangement, copper would likely recruit other solvent-exposed ligands. A structural analysis of the fibril core region V308–F378,97 reveals that beyond His329, His362, Glu338 and Glu342 are other putative metal anchoring ligands facing the outer side of the fibril core. Hence, a His329-anchored copper ion could recruit any of these ligands from another fibril to complete its coordination sphere, possibly disturbing the inter-fibril interactions that cause the characteristic helicity of PHFs. This would explain why copper-induced aggregates, while preserving their fibril length, are inter-crossed and display decreased helicity. Further characterization of metal-bound aggregates of Tau441 should help probe these hypotheses and gain insight into the impact of metals on amyloid fibril morphology.
Disturbed metal ion homeostasis and aberrant metal–protein interactions associated with aggregation are distinct features of several neurodegenerative disorders.5 In the case of AD, the homeostasis of copper, zinc and iron is perturbed, and these essential metals accumulate in the NFTs.14,15 Specifically, levels of labile copper (not bound to ceruloplasmin) are elevated in blood plasma, while the concentration of extracellular tau is increased by five-fold in cerebrospinal fluid (CSF) of AD patients,101–103 raising the possibility for a potentially toxic extracellular tau–copper interaction that could lead to increased oligomerization and aggregation; although, further studies are needed to determine the pathological consequences of copper-bound tau. However, the role of zinc in pathological models of tau has been explored. A study using SH-SY5Y cells expressing TauΔK280, a pathological mutation associated with frontotemporal dementia (FTD-17), revealed that Zn2+ markedly enhances apoptosis and toxicity induced by ΔK280 fibrillation.36 Furthermore, in a Drosophila tauopathy model with TauR406W, both increased dietary zinc and genetic manipulation of zinc transporters leading to increased metal uptake, which led to increased tau toxicity.34 In both models, mutation of Cys291 and Cys322 to Ala abrogated the effects of zinc in tau toxicity, strongly supporting a pathological role for zinc–tau interactions.34,36
Moreover, it is interesting to note that the 3xTg-AD mouse model, encompassing the P301L mutation in tau, displays disruption of copper and zinc homeostasis.104 Pro301 is important for the conformational flexibility of the loop connecting His299 and the bis-His motif (329/330), which are important anchoring sites for both copper and zinc binding to Tau441. Hence, it is plausible that the ability of Tau441 to bind Cu and Zn could be impacted by the P301L mutation, contributing to metal dyshomeostasis in the 3xTg-AD model. Certainly, murine models of AD could represent an excellent platform to further explore the pathological consequences of metal–tau interactions.
Finally, tau protein undergoes several physiological and pathological post-translational modifications (PTMs),23 including phosphorylation, acetylation and ubiquitination. Some of the PTMs of tau observed in AD patients105 (Fig. 10A) occur in regions of the protein engaged in metal coordination and could impact the metal binding properties of tau. Specifically, PTMs at the R3 domain could be impacting copper and zinc binding to the MTBD, while phosphorylation at Ser113 could affect zinc binding at the N-terminal. Hence, there may be an important interplay between PTMs and metal binding to tau, as it has been observed for alpha-synuclein, the Parkinson's protein.106 Further research is needed to elucidate the interplay between all these processes.
Concluding remarks
This spectroscopic study provides detailed structural characterization of the Cu2+, Cu+ and Zn2+ binding properties of Tau441 and explores the impact of metal binding on tau amyloid aggregation. A comparison of the metal binding properties of Tau441 with those of tau fragments underscores the importance of studying metal binding to full-length tau. Tau441 displays a single high-affinity Cu2+ binding site located in the microtubule-binding domain (MTBD), involving a bis-His motif in R3 (His329/His330) and His299 in R2 (Fig. 10). Reduction of the Cu2+–Tau441 complex yields a Cu+ site that is tri-coordinated involving His299, Cys322 and a third ligand, likely Cys291 or His329/330. Tau441 also exhibits multiple Zn2+ binding sites: a high-affinity site in the MTBD involving the bis-His motif, His299 and Cys322, and two lower-affinity sites in the N-terminal region (Fig. 10). The fact that both copper and zinc bind to the MTBD region suggests a physiological role for metal binding in regulating tau interaction with tubulins and/or microtubule assembly that deserves further investigation.Being an intrinsically disordered protein, metal binding to Tau441 could play a structural role, stabilizing specific conformations to facilitate tau protein–protein and/or protein–nucleic acid interactions that may be physiologically and/or pathologically relevant. In this study, it was found that both copper and zinc, can accelerate the amyloid aggregation of Tau441, promoting distinct fibrillar morphologies that are consistent with the metal coordination models derived from spectroscopic studies. These findings support a pathogenic role for aberrant metal–tau interactions that is consistent with the scenario observed in AD, which includes metal accumulation in NFTs, increased extracellular tau and altered metal homeostasis. Finally, the metal binding sites identified here are located close to important sites for posttranslational modifications, including phosphorylation and ubiquitination; hence, an important perspective is to study the potential interplay between metal binding to Tau441 and its PTMs, both in health and disease. The current study expands the bioinorganic facet of AD and other tauopathies, and it opens new avenues to further explore the nature of metal–tau interactions, while it underscores their importance as potential therapeutic targets in these neurodegenerative diseases.
Author contributions
G. U. J. R.: investigation, writing – original draft; X. S.: investigation, methodology; J. G.: investigation, conceptualization, methodology, funding acquisition, writing – review and editing; C. D. A.: conceptualization, methodology, funding acquisition, writing – review and editing; G. L.: investigation, writing – review and editing; M. N.: methodology, writing – review and editing; R. R.: resources, supervision, writing – review and editing; J. L.: conceptualization, methodology, funding acquisition, project administration, supervision, writing – review and editing; L. Q.: conceptualization, methodology, funding acquisition, project administration, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The supporting data have been provided as part of the supplementary information (SI). Supplementary information: Materials and methods section; EPR spectra comparison of Tau441 and buffer MOPS with Cu2+; titration of Tau441 and different fragments with Cu2+ followed by CD and EPR; Peisach–Blumberg diagram of the EPR parameters for the different fragments of Tau441 with Cu2+; parameters of the EXAFS fits of the Cu2+–Tau441 complex with different coordination spheres; reduction of the Cu2+–Tau441 complex section; EPR, XANES and EXAFS spectra for reduced Cu2+–Tau441 complexes; EXAFS fitting of the Cu+–Tau441 complex considering only first coordination sphere interactions; parameters for the EXAFS fits for the Cu+–Tau441 complex considering only first coordination sphere interactions; 1H–15N HMQC NMR spectra of the Cu+–Tau441 complex; EXAFS fitting and FT of Tau441 with 1 and 2 equiv. of Zn2+; parameters of the best fits for Tau441 with 1 and 2 equiv. of Zn2+; comparison of the Tau441–metal ion aggregation assays with and without heparin; fitting curves of the aggregation kinetics of Tau441 with metal ions; quenching effect in ThT fluorescence intensity upon the addition of Cu2+; thermodynamic scheme for the reduction of copper–tau complexes. Spectroscopic data will be made available upon request from the authors. See DOI: https://doi.org/10.1039/d5sc08604c.Acknowledgements
This research was supported by a Consolidation Grant from the Leading House for the Latin American Region, the University of Saint Gallen (to L. Q., J. G. and J. L.). This research was also supported by the National Council for Humanity, Science and Technology in Mexico (CONAHCYT) through CF-2023-I-2844 (to L. Q.), PhD fellowship to G. U. J. R. (1078671), and the Scientific Cooperation Program with France ECOS-NORD grant #321222/M21S01 (to L. Q. and C. D. A.). This research was also supported by a UC-Cinvestav Alianza MX grant. The authors thank the Swiss Light Source (SLS, Villigen PSI Switzerland) for the provision of beamtime at the Super XAS beamline for preliminary studies. The authors acknowledge the use of the Stanford Synchrotron Radiation Light Source (SSRL); SLAC National Accelerator Laboratory is supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (P30GM133894). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. The authors would like to thank Nils Schuth and the Berlin Electron Storage Ring Society for Synchrotron Radiation (BESSY) for collection of preliminary XAS data. We acknowledge SOLEIL for provision of synchrotron radiation facilities and we would like to thank Emiliano Fonda and Guillaume Alizon for assistance in using beamline SAMBA under proposal 20231494; the TEM microscopy unit of National Experimental Services Laboratories (LaNSE) and Lourdes Rojas-Morales for technical microscopy assistance; Geiser Cuellar for acquisition of the mass spectra to characterize the peptide fragments used in this study; Dr Enrique Pinzón (ICAT-UNAM) for manufacturing sample holders for XAS measurements in SOLEIL.References
- B. C. Creekmore, R. Watanabe and E. B. Lee, Neurodegenerative Disease Tauopathies, Annu. Rev. Pathol.: Mech. Dis., 2024, 19, 345–370 CrossRef CAS PubMed.
- C. A. Lane, J. Hardy and J. M. Schott, Alzheimer's disease, Eur. J. Neurol., 2018, 25(1), 59–70 CrossRef CAS PubMed.
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, Copper, iron and zinc in Alzheimer's disease senile plaques, J. Neurol. Sci., 1998, 158(1), 47–52 Search PubMed.
- A. I. Bush, Copper, zinc, and the metallobiology of Alzheimer disease, Alzheimer Dis. Assoc. Disord., 2003, 17(3), 147–150 CrossRef PubMed.
- Y. Posadas, V. E. López-Guerrero, T. Arcos-López, R. I. Sayler, C. Sánchez-López and J. Segovia, et al., The role of d-block metal ions in neurodegenerative diseases, Comprehensive Inorganic Chemistry III, 2023, pp. 575–628 Search PubMed.
- P. Q. Trombley, L. J. Blakemore and B. J. Hill, Zinc modulation of glycine receptors, Neuroscience, 2011, 186, 32–38 CrossRef CAS PubMed.
- N. D'Ambrosi and L. Rossi, Copper at synapse: Release, binding and modulation of neurotransmission, Neurochem. Int., 2015, 90, 36–45 CrossRef PubMed.
- R. A. Scott, R. J. Sullivan, W. E. DeWolf, R. E. Dolle and L. I. Kruse, The copper sites of dopamine β-hydroxylase: an X-ray absorption spectroscopic study, Biochemistry, 2002, 27(15), 5411–5417 CrossRef PubMed.
- J. E. Coleman, Zinc Proteins: Enzymes, Storage Proteins, Transcription Factors, and Replication Proteins, Annu. Rev. Biochem., 1992, 61(1), 897–946 CrossRef CAS PubMed.
- L.-L. Chen, Y.-G. Fan, L.-X. Zhao, Q. Zhang and Z.-Y. Wang, The metal ion hypothesis of Alzheimer’s disease and the anti-neuroinflammatory effect of metal chelators, Bioorg. Chem., 2023, 131, 106301 CrossRef CAS PubMed.
- R. Bai, J. Guo, X.-Y. Ye, Y. Xie and T. Xie, Oxidative stress: The core pathogenesis and mechanism of Alzheimer’s disease, Ageing Res. Rev., 2022, 77, 101619 CrossRef CAS PubMed.
- L. M. Miller, Q. Wang, T. P. Telivala, R. J. Smith, A. Lanzirotti and J. Miklossy, Synchrotron-based infrared and X-ray imaging shows focalized accumulation of Cu and Zn co-localized with β-amyloid deposits in Alzheimer's disease, J. Struct. Biol., 2006, 155(1), 30–37 CrossRef CAS PubMed.
- H. Wang, M. Wang, B. Wang, M. Li, H. Chen and X. Yu, et al., Immunogold labeling and X-ray fluorescence microscopy reveal enrichment ratios of Cu and Zn, metabolism of APP and amyloid-β plaque formation in a mouse model of Alzheimer’s disease, Metallomics, 2012, 4(10), 1113–1118 CrossRef CAS PubMed.
- L. M. Sayre, G. Perry, P. L. Harris, Y. Liu, K. A. Schubert and M. A. Smith, In situ oxidative catalysis by neurofibrillary tangles and senile plaques in Alzheimer's disease: a central role for bound transition metals, J. Neurochem., 2000, 74(1), 270–279 CrossRef CAS PubMed.
- S. W. Suh, K. B. Jensen, M. S. Jensen, D. S. Silva, P. J. Kesslak and G. Danscher, et al., Histochemically-reactive zinc in amyloid plaques, angiopathy, and degenerating neurons of Alzheimer's diseased brains, Brain Res., 2000, 852(2), 274–278 Search PubMed.
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. Bacarra and D. M. Romano, et al., Dramatic aggregation of Alzheimer abeta by Cu(II) is induced by conditions representing physiological acidosis, J. Biol. Chem., 1998, 273(21), 12817–12826 Search PubMed.
- P. Faller and C. Hureau, Bioinorganic chemistry of copper and zinc ions coordinated to amyloid-beta peptide, Dalton Trans., 2009, 1080–1094 RSC.
- S. K. Singh, V. Balendra, A. A. Obaid, J. Esposto, M. A. Tikhonova and N. K. Gautam, et al., Copper-mediated beta-amyloid toxicity and its chelation therapy in Alzheimer’s disease, Metallomics, 2022, 14(6), mfac018 CrossRef PubMed.
- E. Falcone and C. Hureau, Redox processes in Cu-binding proteins: the “in-between” states in intrinsically disordered peptides, Chem. Soc. Rev., 2023, 52(19), 6595–6600 RSC.
- G. Lee, N. Cowan and M. Kirschner, The primary structure and heterogeneity of tau protein from mouse brain, Science, 1988, 239(4837), 285–288 CrossRef CAS PubMed.
- S. Barghorn, J. Biernat and E. Mandelkow, Purification of recombinant tau protein and preparation of Alzheimer-paired helical filaments in vitro, Methods Mol. Biol., 2005, 299, 35–51 CAS.
- M. D. Mukrasch, M. von Bergen, J. Biernat, D. Fischer, C. Griesinger and E. Mandelkow, et al., The “jaws” of the tau-microtubule interaction, J. Biol. Chem., 2007, 282(16), 12230–12239 Search PubMed.
- C. Parra Bravo, S. A. Naguib and L. Gan, Cellular and pathological functions of tau, Nat. Rev. Mol. Cell Biol., 2024, 25(11), 845–864 CrossRef CAS PubMed.
- P. Barbier, O. Zejneli, M. Martinho, A. Lasorsa, V. Belle and C. Smet-Nocca, et al., Role of Tau as a Microtubule-Associated Protein: Structural and Functional Aspects, Front. Aging Neurosci., 2019, 11, 204 CrossRef CAS PubMed.
- Y. Wei, M. Liu and D. Wang, The propagation mechanisms of extracellular tau in Alzheimer's disease, J. Neurol., 2022, 269(3), 1164–1181 CrossRef PubMed.
- M. D. Mukrasch, S. Bibow, J. Korukottu, S. Jeganathan, J. Biernat and C. Griesinger, et al., Structural polymorphism of 441-residue tau at single residue resolution, PLoS Biol., 2009, 7(2), e34 CrossRef PubMed.
- H. Zheng, H. Sun, Q. Cai and H. C. Tai, The Enigma of Tau Protein Aggregation: Mechanistic Insights and Future Challenges, Int. J. Mol. Sci., 2024, 25(9), 4969 CrossRef CAS PubMed.
- S. Ahmadi, S. Zhu, R. Sharma, B. Wu, R. Soong and R. Dutta Majumdar, et al., Aggregation of Microtubule Binding Repeats of Tau Protein is Promoted by Cu2+, ACS Omega, 2019, 4(3), 5356–5366 CrossRef CAS PubMed.
- A. Soragni, B. Zambelli, M. D. Mukrasch, J. Biernat, S. Jeganathan and C. Griesinger, et al., Structural characterization of binding of Cu(II) to tau protein, Biochemistry, 2008, 47(41), 10841–10851 CrossRef CAS PubMed.
- M. Kitazawa, D. Cheng and F. M. Laferla, Chronic copper exposure exacerbates both amyloid and tau pathology and selectively dysregulates cdk5 in a mouse model of AD, J. Neurochem., 2009, 108(6), 1550–1560 CrossRef CAS PubMed.
- Z. Y. Mo, Y. Z. Zhu, H. L. Zhu, J. B. Fan, J. Chen and Y. Liang, Low micromolar zinc accelerates the fibrillization of human tau via bridging of Cys-291 and Cys-322, J. Biol. Chem., 2009, 284(50), 34648–34657 CrossRef CAS PubMed.
- S. Ahmadi, S. Zhu, R. Sharma, D. J. Wilson and H. B. Kraatz, Interaction of metal ions with tau protein. The case for a metal-mediated tau aggregation, J. Inorg. Biochem., 2019, 194, 44–51 Search PubMed.
- A. Y. Roman, F. Devred, D. Byrne, R. La Rocca, N. N. Ninkina and V. Peyrot, et al., Zinc Induces Temperature-Dependent Reversible Self-Assembly of Tau, J. Mol. Biol., 2019, 431(4), 687–695 CrossRef CAS PubMed.
- Y. Huang, Z. Wu, Y. Cao, M. Lang, B. Lu and B. Zhou, Zinc binding directly regulates tau toxicity independent of tau hyperphosphorylation, Cell Rep., 2014, 8(3), 831–842 CrossRef CAS PubMed.
- G. G. Moreira, J. S. Cristovao, V. M. Torres, A. P. Carapeto, M. S. Rodrigues and I. Landrieu, et al., Zinc Binding to Tau Influences Aggregation Kinetics and Oligomer Distribution, Int. J. Mol. Sci., 2019, 20(23), 5979 CrossRef CAS PubMed.
- J. Y. Hu, D. L. Zhang, X. L. Liu, X. S. Li, X. Q. Cheng and J. Chen, et al., Pathological concentration of zinc dramatically accelerates abnormal aggregation of full-length human Tau and thereby significantly increases Tau toxicity in neuronal cells, Biochim. Biophys. Acta, Mol. Basis Dis., 2017, 1863(2), 414–427 CrossRef CAS PubMed.
- L. S. Kau, D. J. Spira-Solomon, J. E. Penner-Hahn, K. O. Hodgson and E. I. Solomon, X-ray absorption edge determination of the oxidation state and coordination number of copper. Application to the type 3 site in Rhus vernicifera laccase and its reaction with oxygen, J. Am. Chem. Soc., 2002, 109(21), 6433–6442 CrossRef.
- A. Gaur, W. Klysubun, B. Soni, B. D. Shrivastava, J. Prasad and K. Srivastava, Identification of different coordination geometries by XAFS in copper(II) complexes with trimesic acid, J. Mol. Struct., 2016, 1121, 119–127 CrossRef CAS.
- S. Jeganathan, M. von Bergen, H. Brutlach, H. J. Steinhoff and E. Mandelkow, Global hairpin folding of tau in solution, Biochemistry, 2006, 45(7), 2283–2293 CrossRef CAS PubMed.
- M. A. McGuirl and D. M. Dooley, Copper Proteins with Type 2 Sites, Encyclopedia of Inorganic and Bioinorganic Chemistry, 2005 Search PubMed.
- J. Peisach and W. E. Blumberg, Structural implications derived from the analysis of electron paramagnetic resonance spectra of natural and artificial copper proteins, Arch. Biochem. Biophys., 1974, 165(2), 691–708 CrossRef CAS PubMed.
- M. Iwaizumi, T. Kudo and S. Kita, Correlation between the hyperfine coupling constants of donor nitrogens and the structures of the first coordination sphere in copper complexes as studied by nitrogen-14 ENDOR spectroscopy, Inorg. Chem., 2002, 25(10), 1546–1550 CrossRef.
- L. Rivillas-Acevedo, R. Grande-Aztatzi, I. Lomelí, J. E. García, E. Barrios and S. Teloxa, et al., Spectroscopic and Electronic Structure Studies of Copper(II) Binding to His111 in the Human Prion Protein Fragment 106–115: Evaluating the Role of Protons and Methionine Residues, Inorg. Chem., 2011, 50(5), 1956–1972 CrossRef CAS PubMed.
- M. E. Tovar-Ramirez, N. Schuth, O. Rodriguez-Meza, T. Kroll, G. Saab-Rincon and M. Costas, et al., ATCUN-like Copper Site in βB2-Crystallin Plays a Protective Role in Cataract-Associated Aggregation, Inorg. Chem., 2023, 62(27), 10592–10604 CrossRef CAS PubMed.
- H. H. Thorp, Bond valence sum analysis of metal-ligand bond lengths in metalloenzymes and model complexes, Inorg. Chem., 2002, 31(9), 1585–1588 CrossRef.
- N. Binsted, R. W. Strange and S. S. Hasnain, Constrained and restrained refinement in EXAFS data analysis with curved wave theory, Biochemistry, 2002, 31(48), 12117–12125 Search PubMed.
- M. Bazayeva, C. Andreini and A. Rosato, A database overview of metal-coordination distances in metalloproteins, Acta Crystallogr., Sect. D: Struct. Biol., 2024, 80(Pt 5), 362–376 CrossRef CAS PubMed.
- J. S. Taylor, P. Mushak and J. E. Coleman, Electron spin resonance studies of carbonic anhydrase: transition metal ions and spin-labeled sulfonamides, Proc. Natl. Acad. Sci. U. S. A., 1970, 67(3), 1410–1416 CrossRef CAS.
- J. S. Taylor and J. E. Coleman, Electron Spin Resonance of Metallocarbonic Anhydrases, J. Biol. Chem., 1971, 246(22), 7058–7067 Search PubMed.
- J. S. Taylor and J. E. Coleman, Electron Spin Resonance of 63Cu and 65Cu Carbonic Anhydrases, J. Biol. Chem., 1973, 248(3), 749–755 CrossRef CAS.
- G. Tabbi, A. Magri and E. Rizzarelli, The copper(II) binding centres of carbonic anhydrase are differently affected by reductants that ensure the redox intracellular environment, J. Inorg. Biochem., 2019, 199, 110759 Search PubMed.
- W. L. Nettles, H. Song, E. R. Farquhar, N. C. Fitzkee and J. P. Emerson, Characterization of the Copper(II) Binding Sites in Human Carbonic Anhydrase II, Inorg. Chem., 2015, 54(12), 5671–5680 CrossRef CAS PubMed.
- D. Poger, C. Fillaux, R. Miras, S. Crouzy, P. Delangle and E. Mintz, et al., Interplay between glutathione, Atx1 and copper: X-ray absorption spectroscopy determination of Cu(I) environment in an Atx1 dimer, J. Biol. Inorg. Chem., 2008, 13(8), 1239–1248 CrossRef PubMed.
- S. Barber-Zucker, B. Shaanan and R. Zarivach, Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family, Sci. Rep., 2017, 7(1), 16381 CrossRef.
- L. Giachini, G. Veronesi, F. Francia, G. Venturoli and F. Boscherini, Synergic approach to XAFS analysis for the identification of most probable binding motifs for mononuclear zinc sites in metalloproteins, J. Synchrotron Radiat., 2010, 17(1), 41–52 Search PubMed.
- V. Minicozzi, F. Stellato, M. Comai, M. Dalla Serra, C. Potrich and W. Meyer-Klaucke, et al., Identifying the minimal copper- and zinc-binding site sequence in amyloid-beta peptides, J. Biol. Chem., 2008, 283(16), 10784–10792 Search PubMed.
- M. Laitaoja, J. Valjakka and J. Janis, Zinc coordination spheres in protein structures, Inorg. Chem., 2013, 52(19), 10983–10991 Search PubMed.
- M. Biancalana and S. Koide, Molecular mechanism of Thioflavin-T binding to amyloid fibrils, Biochim. Biophys. Acta, Proteins Proteomics, 2010, 1804(7), 1405–1412 CrossRef CAS PubMed.
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green and A. Pritzel, et al., Accurate structure prediction of biomolecular interactions with AlphaFold 3, Nature, 2024, 630(8016), 493–500 CrossRef CAS PubMed.
- E. D. Walter, M. Chattopadhyay and G. L. Millhauser, The affinity of copper binding to the prion protein octarepeat domain: evidence for negative cooperativity, Biochemistry, 2006, 45(43), 13083–13092 Search PubMed.
- E. D. Walter, D. J. Stevens, M. P. Visconte and G. L. Millhauser, The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes, J. Am. Chem. Soc., 2007, 129(50), 15440–15441 Search PubMed.
- T. R. Young, T. L. Pukala, R. Cappai, A. G. Wedd and Z. Xiao, The Human Amyloid Precursor Protein Binds Copper Ions Dominated by a Picomolar-Affinity Site in the Helix-Rich E2 Domain, Biochemistry, 2018, 57(28), 4165–4176 CrossRef CAS PubMed.
- S. O. Dahms, I. Konnig, D. Roeser, K. H. Guhrs, M. C. Mayer and D. Kaden, et al., Metal binding dictates conformation and function of the amyloid precursor protein (APP) E2 domain, J. Mol. Biol., 2012, 416(3), 438–452 Search PubMed.
- G. K. Kong, J. J. Adams, H. H. Harris, J. F. Boas, C. C. Curtain and D. Galatis, et al., Structural studies of the Alzheimer's amyloid precursor protein copper-binding domain reveal how it binds copper ions, J. Mol. Biol., 2007, 367(1), 148–161 CrossRef CAS PubMed.
- C. Bacchella, S. Gentili, D. Bellotti, E. Quartieri, S. Draghi and M. C. Baratto, et al., Binding and Reactivity of Copper to R1 and R3 Fragments of tau Protein, Inorg. Chem., 2020, 59(1), 274–286 CrossRef CAS PubMed.
- K. M. Schilling, L. Tao, B. Wu, J. T. M. Kiblen, N. C. Ubilla-Rodriguez and M. J. Pushie, et al., Both N-Terminal and C-Terminal Histidine Residues of the Prion Protein Are Essential for Copper Coordination and Neuroprotective Self-Regulation, J. Mol. Biol., 2020, 432(16), 4408–4425 Search PubMed.
- O. V. Moroz, G. G. Dodson, K. S. Wilson, E. Lukanidin and I. B. Bronstein, Multiple structural states of S100A12: A key to its functional diversity, Microsc. Res. Tech., 2003, 60(6), 581–592 CrossRef CAS PubMed.
- S. Ghosh, V. Garcia, K. Singewald, S. M. Damo and S. Saxena, Cu(II) EPR Reveals Two Distinct Binding Sites and Oligomerization of Innate Immune Protein Calgranulin C, Appl. Magn. Reson., 2018, 49(11), 1299–1311 CrossRef CAS.
- J. C. Freeman, P. G. Nayar, T. P. Begley and J. J. Villafranca, Stoichiometry and spectroscopic identity of copper centers in phenoxazinone synthase: a new addition to the blue copper oxidase family, Biochemistry, 1993, 32(18), 4826–4830 CrossRef CAS PubMed.
- A. W. Smith, A. Camara-Artigas, M. Wang, J. P. Allen and W. A. Francisco, Structure of phenoxazinone synthase from Streptomyces antibioticus reveals a new type 2 copper center, Biochemistry, 2006, 45(14), 4378–4387 CrossRef CAS PubMed.
- G. Di Natale, G. Sabatino, M. F. M. Sciacca, R. Tosto, D. Milardi and G. Pappalardo, Abeta and Tau Interact with Metal Ions, Lipid Membranes and Peptide-Based Amyloid Inhibitors: Are These Common Features Relevant in Alzheimer’s Disease?, Molecules, 2022, 27(16), 5066 CrossRef CAS PubMed.
- C. M. Karch, A. T. Jeng and A. M. Goate, Extracellular Tau levels are influenced by variability in Tau that is associated with tauopathies, J. Biol. Chem., 2012, 287(51), 42751–42762 CrossRef CAS PubMed.
- N. Ait-Bouziad, G. Lv, A. L. Mahul-Mellier, S. Xiao, G. Zorludemir and D. Eliezer, et al., Discovery and characterization of stable and toxic Tau/phospholipid oligomeric complexes, Nat. Commun., 2017, 8(1), 1678 CrossRef PubMed.
- A. Pezeshk, J. Torres, M. T. Wilson and M. C. Symons, The EPR spectrum for CuB in cytochrome c oxidase, J. Inorg. Biochem., 2001, 83(2–3), 115–119 Search PubMed.
- E. De Cecco, L. Celauro, S. Vanni, M. Grandolfo, E. Bistaffa and F. Moda, et al., The uptake of tau amyloid fibrils is facilitated by the cellular prion protein and hampers prion propagation in cultured cells, J. Neurochem., 2020, 155(5), 577–591 CrossRef CAS PubMed.
- G. T. Corbett, Z. Wang, W. Hong, M. Colom-Cadena, J. Rose and M. Liao, et al., PrP is a central player in toxicity mediated by soluble aggregates of neurodegeneration-causing proteins, Acta Neuropathol., 2020, 139(3), 503–526 CrossRef CAS PubMed.
- M. Manczak and P. H. Reddy, Abnormal interaction of oligomeric amyloid-beta with phosphorylated tau: implications to synaptic dysfunction and neuronal damage, J. Alzheimer's Dis., 2013, 36(2), 285–295 CAS.
- Y. Posadas, L. Parra-Ojeda, C. Perez-Cruz and L. Quintanar, Amyloid beta Perturbs Cu(II) Binding to the Prion Protein in a Site-Specific Manner: Insights into Its Potential Neurotoxic Mechanisms, Inorg. Chem., 2021, 60(12), 8958–8972 CrossRef CAS PubMed.
- A. Smart, K. Singewald, Z. Hasanbasri, R. D. Britt and G. L. Millhauser, Identifying the copper coordination environment between interacting neurodegenerative proteins: A new approach using pulsed EPR with 14N/15N isotopic labeling, J. Biol. Chem., 2025, 301(3), 108311 CrossRef CAS PubMed.
- R. A. Himes, G. Y. Park, G. S. Siluvai, N. J. Blackburn and K. D. Karlin, Structural studies of copper(I) complexes of amyloid-beta peptide fragments: formation of two-coordinate bis(histidine) complexes, Angew. Chem., Int. Ed., 2008, 47(47), 9084–9087 CrossRef CAS PubMed.
- S. Schwab, J. Shearer, S. E. Conklin, B. Alies and K. L. Haas, Sequence proximity between Cu(II) and Cu(I) binding sites of human copper transporter 1 model peptides defines reactivity with ascorbate and O2, J. Inorg. Biochem., 2016, 158, 70–76 CrossRef CAS PubMed.
- J. T. Rubino and K. J. Franz, Coordination chemistry of copper proteins: how nature handles a toxic cargo for essential function, J. Inorg. Biochem., 2012, 107(1), 129–143 CrossRef CAS PubMed.
- L. Banci, I. Bertini, S. Ciofi-Baffoni, I. Leontari, M. Martinelli and P. Palumaa, et al., Human Sco1 functional studies and pathological implications of the P174L mutant, Proc. Natl. Acad. Sci. U. S. A., 2007, 104(1), 15–20 CrossRef CAS.
- J. Liu, S. Chakraborty, P. Hosseinzadeh, Y. Yu, S. Tian and I. Petrik, et al., Metalloproteins containing cytochrome, iron-sulfur, or copper redox centers, Chem. Rev., 2014, 114(8), 4366–4469 CrossRef CAS PubMed.
- S. M. Berry, M. Ralle, D. W. Low, N. J. Blackburn and Y. Lu, Probing the role of axial methionine in the blue copper center of azurin with unnatural amino acids, J. Am. Chem. Soc., 2003, 125(29), 8760–8768 CrossRef CAS PubMed.
- J. S. Plegaria, M. Duca, C. Tard, T. J. Friedlander, A. Deb and J. E. Penner-Hahn, et al., De novo design and characterization of copper metallopeptides inspired by native cupredoxins, Inorg. Chem., 2015, 54(19), 9470–9482 CrossRef CAS PubMed.
- S. F. Sousa, A. B. Lopes, P. A. Fernandes and M. J. Ramos, The Zinc proteome: a tale of stability and functionality, Dalton Trans., 2009, 7946–7956 RSC.
- K. Stefanoska, A. Volkerling, J. Bertz, A. Poljak, Y. D. Ke and L. M. Ittner, et al., An N-terminal motif unique to primate tau enables differential protein-protein interactions, J. Biol. Chem., 2018, 293(10), 3710–3719 CrossRef CAS PubMed.
- T. F. Minckley, L. A. Salvagio, D. H. Fudge, K. Verhey, S. M. Markus and Y. Qin, Zn2+ decoration of microtubules arrests axonal transport and displaces tau, doublecortin, and MAP2C, J. Cell Biol., 2023, 222(8), e202208121 CrossRef CAS PubMed.
- M. Martinho, D. Allegro, E. Etienne, C. Lohberger, A. Bonucci and V. Belle, et al., Structural Flexibility of Tau in Its Interaction with Microtubules as Viewed by Site-Directed Spin Labeling EPR Spectroscopy, Methods Mol. Biol., 2024, 2754, 55–75 CrossRef CAS PubMed.
- A. Anton-Fernandez, L. Valles-Saiz, J. Avila and F. Hernandez, Neuronal nuclear tau and neurodegeneration, Neuroscience, 2023, 518, 178–184 CrossRef CAS PubMed.
- I. Sotiropoulos, M. C. Galas, J. M. Silva, E. Skoulakis, S. Wegmann and M. B. Maina, et al., Atypical, non-standard functions of the microtubule associated Tau protein, Acta Neuropathol. Commun., 2017, 5(1), 91 CrossRef PubMed.
- Y. Wei, M. H. Qu, X. S. Wang, L. Chen, D. L. Wang and Y. Liu, et al., Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation, PLoS One, 2008, 3(7), e2600 CrossRef PubMed.
- A. Sultan, F. Nesslany, M. Violet, S. Begard, A. Loyens and S. Talahari, et al., Nuclear tau, a key player in neuronal DNA protection, J. Biol. Chem., 2011, 286(6), 4566–4575 CrossRef CAS PubMed.
- T. Kambe, T. Tsuji, A. Hashimoto and N. Itsumura, The Physiological, Biochemical, and Molecular Roles of Zinc Transporters in Zinc Homeostasis and Metabolism, Physiol. Rev., 2015, 95(3), 749–784 CrossRef CAS PubMed.
- S. S. Oakley, M. B. Maina, K. E. Marshall, Y. K. Al-Hilaly, C. R. Harrington and C. M. Wischik, et al., Tau Filament Self-Assembly and Structure: Tau as a Therapeutic Target, Front. Neurol., 2020, 11, 590754 CrossRef PubMed.
- A. W. P. Fitzpatrick, B. Falcon, S. He, A. G. Murzin, G. Murshudov and H. J. Garringer, et al., Cryo-EM structures of tau filaments from Alzheimer's disease, Nature, 2017, 547(7662), 185–190 CrossRef CAS PubMed.
- M. von Bergen, P. Friedhoff, J. Biernat, J. Heberle, E. M. Mandelkow and E. Mandelkow, Assembly of tau protein into Alzheimer paired helical filaments depends on a local sequence motif (306VQIVYK311) forming beta structure, Proc. Natl. Acad. Sci. U. S. A., 2000, 97(10), 5129–5134 CrossRef CAS PubMed.
- N. Sibille, A. Sillen, A. Leroy, J. M. Wieruszeski, B. Mulloy and I. Landrieu, et al., Structural impact of heparin binding to full-length Tau as studied by NMR spectroscopy, Biochemistry, 2006, 45(41), 12560–12572 CrossRef CAS PubMed.
- S. Lovestam, F. A. Koh, B. van Knippenberg, A. Kotecha, A. G. Murzin and M. Goedert, et al., Assembly of recombinant tau into filaments identical to those of Alzheimer’s disease and chronic traumatic encephalopathy, eLife, 2022, 11, e76494 CrossRef CAS PubMed.
- N. R. Barthelemy, F. Fenaille, C. Hirtz, N. Sergeant, S. Schraen-Maschke and J. Vialaret, et al., Tau Protein Quantification in Human Cerebrospinal Fluid by Targeted Mass Spectrometry at High Sequence Coverage Provides Insights into Its Primary Structure Heterogeneity, J. Proteome Res., 2016, 15(2), 667–676 CrossRef CAS PubMed.
- K. Samgard, H. Zetterberg, K. Blennow, O. Hansson, L. Minthon and E. Londos, Cerebrospinal fluid total tau as a marker of Alzheimer's disease intensity, Int. J. Geriatr. Psychiatry, 2010, 25(4), 403–410 CrossRef PubMed.
- K. Blennow, H. Hampel, M. Weiner and H. Zetterberg, Cerebrospinal fluid and plasma biomarkers in Alzheimer disease, Nat. Rev. Neurol., 2010, 6(3), 131–144 CrossRef CAS PubMed.
- D. Ciavardelli, A. Consalvo, V. Caldaralo, M. L. Di Vacri, S. Nisi and C. Corona, et al., Characterisation of element profile changes induced by long-term dietary supplementation of zinc in the brain and cerebellum of 3xTg-AD mice by alternated cool and normal plasma ICP-MS, Metallomics, 2012, 4(12), 1321–1332 CrossRef CAS PubMed.
- N. Kyalu Ngoie Zola, C. Balty, S. Pyr Dit Ruys, A. A. T. Vanparys, N. D. G. Huyghe and G. Herinckx, et al., Specific post-translational modifications of soluble tau protein distinguishes Alzheimer's disease and primary tauopathies, Nat. Commun., 2023, 14(1), 3706 CrossRef CAS PubMed.
- N. Gonzalez, T. Arcos-Lopez, A. Konig, L. Quintanar, M. Menacho Marquez and T. F. Outeiro, et al., Effects of alpha-synuclein post-translational modifications on metal binding, J. Neurochem., 2019, 150(5), 507–521 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |