
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRemote α- and β-C(sp3)–H alkenylation of amines via visible-light supported paired electrolysis
Siddharth K.
Dave
 and
Sebastian
Stecko
*
and
Sebastian
Stecko
*
Institute of Organic Chemistry, Polish Academy of Sciences, Marcina Kasprzaka Street 44/52, 01-224 Warsaw, Poland. E-mail: sebastian.stecko@icho.edu.pl
First published on 21st January 2026
Abstract
A general method for photoelectrochemical site-selective α- and β-C(sp3)−H alkenylation of amines with vinyl bromides has been developed. Regioselective activation of inert C–H bonds is achieved by intramolecular hydrogen atom abstraction (HAT) by an oxidatively generated aryl radical. Depending on the HAT directing group attached to the amines' N-atom, either 1,5- or 1,6-H-atom transposition occurred, leading to regioisomeric carbon-centered radical species. C-radicals thus formed at the α/β-position of the amines' functionality undergo radical cross-coupling with Ni complex-activated vinyl bromide to provide the corresponding α- or β-functionalized amines. Good functional group tolerance, gram-scale experiments, post-functionalization and demonstration of preparation of key structural scaffolds of the selected naturally occurring compounds and drug candidates greatly highlight the potential applicability of the presented method. The mechanistic experiments demonstrated that the reported protocol consists of a photoelectrochemically induced thiyl radical anodic cycle and an electrochemically driven Ni-catalytic cathodic cycle. The first one is responsible for the generation of CO2˙−, a XAT reagent capable of activation of aryl halide to provide aryl radical species. Cathodic reduction of a Ni(I) intermediate to the Ni(0) complex allows closing the Ni-catalyzed cycle, enabling the execution of the cross-coupling stage leading to a C–H functionalized amine derivative. The developed conditions enable precise synchronization of anodic and cathodic catalytic cycles occurring in the presented variant of paired electrolysis, which has been a significant challenge until now.
Introduction
The allylic amine moiety is a ubiquitous structural motif found in numerous bioactive molecules and naturally occurring compounds like anticancer maclekarpine E,1,2 antimicrobial zanthocapensine2–4 or alkaloid caulophyllumine5 (Fig. 1). Furthermore, allylamines are versatile intermediates and building blocks, with numerous applications in organic synthesis. For instance, alkenylated heterocyclic scaffolds often serve as a direct precursor for the total synthesis of numerous alkaloids (e.g., galipeine,6,7 cuspareine,6 galipinine,6 angustureine,6 dysoxyline,8,9 and methofoline8,9) or heterocyclic drugs (e.g., almorexant,10,11 iferanserin,12 and encainide13,14) presented in Fig. 1. | ||
| Fig. 1 Representative bioactive compounds containing an allylic amine moiety (in green) and bioactive molecules obtained from the corresponding allylamine precursors (in yellow). | ||
Classic methods for allylamine synthesis rely on nucleophilic substitution by amines,15,16 including Tsuji-Trost allylic amination,15,17–19 vinylation of imines,20,21 reductive amination of enals or enones,22 or sigmatropic rearrangement reactions.23–25 Although these are well-established protocols in synthetic organic chemistry, they also exhibit several drawbacks, like the use of stoichiometric amounts of organometallic reagents, insufficient levels of stereocontrol, or the formation of numerous side products. So far, the Pd- or Ir-catalyzed Tsuji-Trost allylic amination reaction seems to be the most efficient strategy to deliver linear or branched allylamines (depending on the metal catalyst used) by reacting amines with various allylic electrophiles.15,17–19 Nevertheless, even this strategy still encounters challenges and drawbacks, particularly in the preparation of α-alkenylated saturated aza-heterocycles.
From the synthetic perspective, direct regio- and stereoselective α-C(sp3)–H alkenylation is regarded as the most attractive strategy for synthesizing the mentioned types of allylamines since it starts with inexpensive, readily available cyclic amines (Scheme 1a).26–28 The classic approaches involve deprotonation by organolithium reagents, followed by transition metal-mediated cross-coupling with vinyl halide reagents (Scheme 1a). An alternative approach is direct C–H bond activation by transition metal complexes followed by coupling with a vinyl reagent (Scheme 1a).28 Alkynes, in addition to vinyl halides, can serve as effective alkenylating agents (Scheme 1a).28–30 Nevertheless, the use of a strong base significantly limits the substrate scope in the first case, whereas the requirement of expensive metal complexes and harsh reaction conditions significantly limits the efficiency of the second process. Moreover, both cases require specific directing groups to achieve regioselective activation of the α-C–H bond, and these groups must be removed in further steps. A less explored approach for the synthesis of complex allylamines relies on functionalization, for instance arylation or alkylation, of allylamine derivatives, e.g., allylamine vinyl bromides, through their reactions with arylating agents (e.g., arylboronic acids)31–33 or alkylating reagents,18a,33 including alkyl radical precursors (e.g., alkyl halides, carboxylic acids, etc.) under photoredox34,35 (Scheme 1b).
 | ||
| Scheme 1 Background and concept design: (a) traditional direct α-C(sp3)–H alkenylation of amines; (b) divergent functionalization of allylamine vinyl bromides; (c) remote α-C(sp3)–H functionalization of amines. | ||
Intensive developments in modern organic photochemistry brought new concepts in the synthesis of allylamines, particularly methods involving the generation and alkenylation of α-aminoalkyl radical species (Scheme 1a). In their pioneering studies, MacMillan and co-workers36,37 demonstrated conditions for Ir-catalysed photoredox vinylation of tertiary N-aryl amines with vinyl sulfones. The key intermediates, α-aminoalkyl radical species, were generated either from amines via an SET/deprotonation sequence or through the decarboxylation of amino acids, mediated by a photoexcited Ir complex acting as a strong oxidant. A few years later, Xie and co-workers38 employed this approach for the total synthesis of maclekarpine E (Fig. 1) and its analogues.
The intramolecular 1,5-hydrogen atom transfer (1,5-HAT) strategy is another method for the generation of α-aminoalkyl radical species (e.g., INT-B).39–45 It has recently gained a lot of attention as a powerful tool for selective C–H bond functionalisation, introducing various functional groups and delivering complex molecules. This method often involves using a substrate with a pre-installed functional group that operates as a source of reactive radical intermediates (e.g., INT-A), serves as a directing group to control the 1,5-HAT process, and acts as a protective group. In 2022, Gevorgyan and his team46 reported a simple and effective method for adding a boron group to the α-C–H bond of 2-iodobenzoyl-protected secondary amines, resulting in the formation of α-amino boronic acids (Scheme 1c(i)). The key step of this process was single-electron transfer (SET) or halogen-atom transfer (XAT) cleavage of the C(sp2)–I bond to generate an aryl radical (e.g., INT-A). This species is capable of subsequent 1,5-hydrogen atom abstraction from the α-C position of secondary amine. The trapping agent, such as B2cat2, then intercepts the resulting alkyl radical (e.g., INT-B) to produce a functionalised product. Soon after, Zhang's group47 demonstrated a visible-light-induced asymmetric C(sp3)–H Cu-catalysed alkynylation of cyclic tertiary amines initiated by the 1,5-HAT process. After this research, both the Wang group48 and the Saget group (Scheme 1c(ii))49 and others developed similar methods, using aryl radicals to start the 1,5-HAT process for the α-C–H functionalisation of secondary amines with Michael acceptors, while Sureshkumar and others reported the conditions for photoredox α-C–H alkylation using [1.1.1]-propellane.50 Several reports also described the α-C–H arylation method through cross-coupling of α-aminoalkyl radicals with heteroarenes. The pool of potent trapping agents that can be used for such radical remote C–H functionalisation includes phosphites (α-C–H phosphonation, Scheme 1c(iii)),51 aldehydes (α-C–H acylation),52 silyl reagents (α-C–H silylation, Scheme 1c(v)),53 imines (α-C–H aminoalkylation) and azides (α-C–H azidation).54 Surprisingly, an analogous concept for α-C–H alkenylation of cyclic amines is almost unknown so far, with only one report on the related aryl radical-mediated α-C(sp3)–H alkenylation of secondary amines by Guo, Zhang, and their co-workers (Scheme 1c(vi)).55 They employed the ate complexes of vinylboronic acid esters and alkoxides as alkenylation agents. This ate complex interacted with the secondary amine substrate to deliver an EDA complex, which underwent photoexcitation to generate an aryl radical from the 2-iodobenzoyl group attached to the amine nitrogen atom. The subsequent 1,5-HAT process produced the desired 2-alkenylated products in excellent yield by intercepting the alkyl radical with the activated alkenylboronic acid. Although this method is quite elegant, some drawbacks related to the employed reaction conditions still leave a space for further improvements and advancements in terms of substrate diversity, cost-effectiveness, and reaction selectivity.
We questioned if vinyl halides, instead of vinyl boronic acid derivatives, could serve as suitable reagents for the aforementioned C–H alkenylation of secondary amines through the 1,5-HAT/cross-coupling sequence facilitated by aryl radicals. Based on the previous reports, we decided to use the 2-halogenbenzoyl group to produce aryl radicals, which should deliver the α-aminoalkyl radical after the 1,5-HAT process; this radical is designed to react with the Ni(II)-alkene complex generated from the vinyl halide and Ni(0). Our primary objective was to establish conditions suitable for the activation of both reaction partners. This phase appeared to be the most challenging, as the Ni(0) complex may activate both the vinyl halide and the aryl halogen moiety of the HAT-directing group. Consequently, our primary task was to develop conditions for the effective activation of the directing group and the ensuing cross-coupling process with activated vinyl halide. The next objective was to synchronise all three steps: aryl radical generation, 1,5-HAT and cross-coupling within the specified sequence. This issue was critical because failing to meet the correct conditions may predominantly lead to undesired homocoupling and dehalogenation products from the starting materials, resulting in a complex reaction mixture.
Results and discussion
Development of reaction conditions
With our hypothesis in mind, we started exploring the feasibility of our proposed alkenylation reaction. We chose 2-iodobenzoyl-protected morpholine 1a and vinyl bromide 2a as model substrates. The reaction between them was performed in the presence of Ni(glyme)Br2 (10 mol%) and 4,4′-di-t-butyl-2,2′-bipyridine (dtbbpy) (12 mol%) which formed the corresponding Ni-complex for the final cross-coupling step.34,56Initially, we examined diverse photochemical conditions for the reductive generation of aryl radical INT-A. In preliminary studies, we adapted the conditions for the generation of aryl radicals described by Doyle57,58 and others59 and the alkenylation of 1a with 2a was conducted using an Ir complex, as a photocatalyst, K3PO4 as a base and TBABr as a source of Br radicals acting as an XAT reagent. The model reaction was performed in MeCN under blue light irradiation (450–460 nm) (Scheme 2, ent. 1a). While complete conversion of 1a took place, only traces of desired product 3aa were detected (as determined by GC analysis), alongside the deiodination product 4a and the homocoupling product of vinyl bromide 2a. Further modifications to these reaction conditions, including variations in the nickel source (e.g., Ni(glyme)Cl2, Ni(cod)2, NiI2, etc.), base (K2CO3, K2HPO4, KH2PO4, Na2CO3, etc.), or solvent(s) (e.g., DMF, MeCN, etc.), failed to enhance the process, with product 4a remaining predominant. Next, we turned our attention to the Leonori studies60 revealing trialkylamines as potent XAT reagents for the generation of aryl radicals from the corresponding aryl halides.49,66 Adapting the Leonori report,60 the model alkenylation reaction was conducted with 4-CzIPN as a photocatalyst and Hünig's base as an XAT reagent.61 Unluckily, the conversion of 1a was quite low (45%) and again merely traces of the expected product were detected (Scheme 2, ent. 2a). Furthermore, any further modifications to the reaction conditions, such as altering the photocatalyst (e.g., 4-DPAIPN,67,68 3-DPA(F)IPN, 4-Cl-CzIPN, and [Ir(dFppy)2(dtbbpy)]PF6) or changing the employed trialkylamine reagent (e.g., Et3N, n-Bu3N, etc.), were unsatisfactory. The same result was obtained when the HAT reagent, aryl radical, was formed via a C(sp2)–I bond cleavage by a silyl radical, as reported by Yang and Guo.69,70 In this case, tris(trimethylsilyl)silane (TTMS) acted as a source of an XAT reagent produced photochemically with or without a photocatalyst (Scheme 2, ent. 3a and 3b). Under both conditions, the competitive reductive dehalogenation of the heterocyclic substrate occurred more rapidly than the expected Ni-catalyzed cross-coupling process. We have also examined the protocol outlined by Sureshkumar,50 wherein 2-iodobenzoyl-protected amines, e.g.1a, with the carbonate anion form an electron donor–acceptor complex (EDA complex), which upon irradiation (440 nm) should undergo photoexcitation and generate the aryl radical (via homolysis of the C–I bond) for the subsequent 1,5-HAT step.
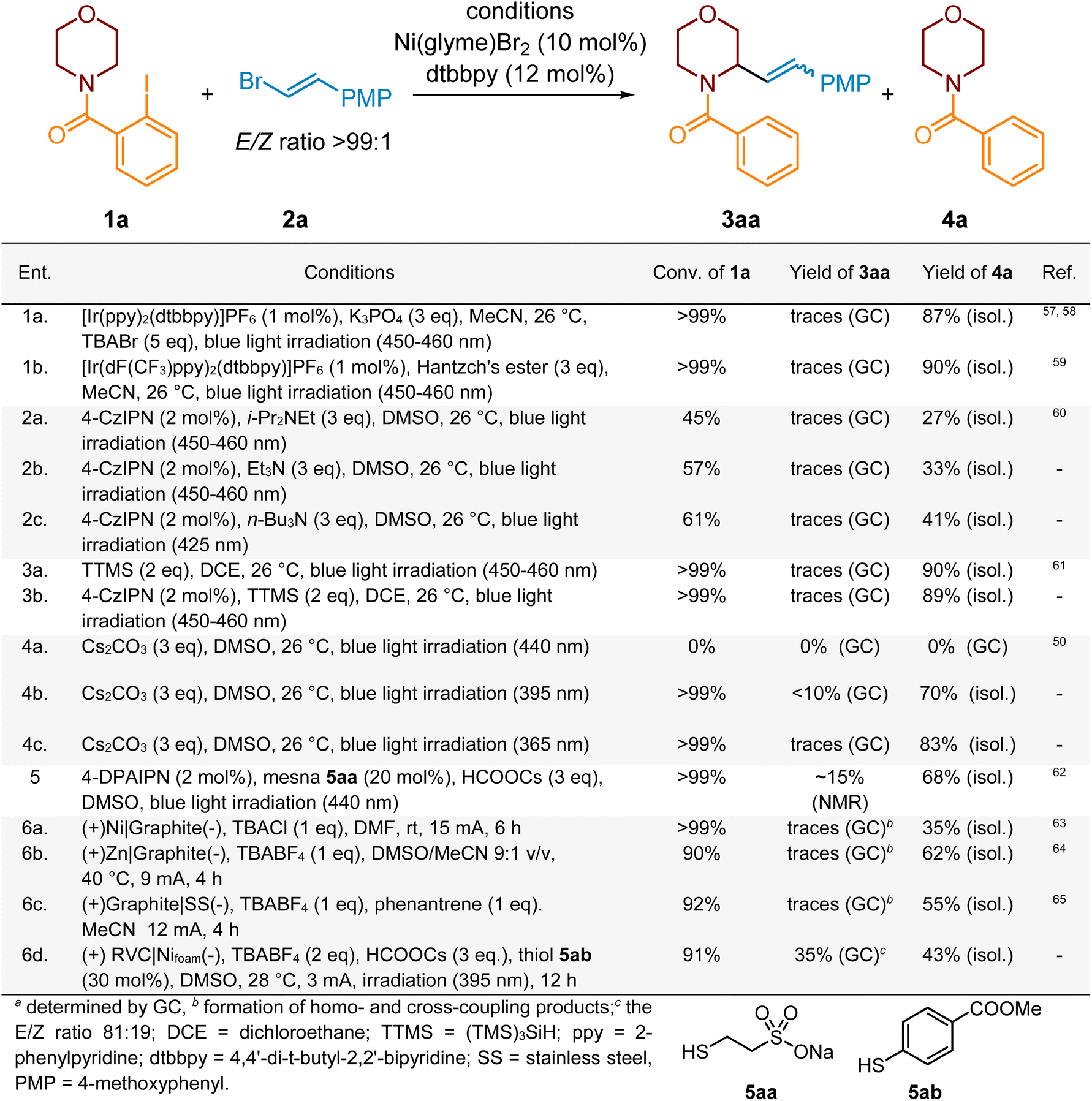 | ||
| Scheme 2 Screening of initial conditions. PMP = 4-methoxyphenyl. | ||
Disappointingly, upon implementing the specified conditions (Scheme 2, ent. 4a), we neither observed the generation of product 3aa nor even product 4a and recovered entire starting material 1a. Our spectroscopic studies confirmed that compound 1a forms the EDA complex with carbonate ions postulated by authors, as indicated by the bathochromic shift of the absorption band for 1a from ca. 290 nm to 332 nm (see SI, Fig. SI-8). Nonetheless, this maximum is evidently beyond the spectrum of the light source employed by the authors (Kessil™ LED lamp 440 nm),71 hence precluding any potential for photoexcitation of the specified complex!
In light of the above observations and evidence, we decided to repeat this experiment using higher-energy light sources. Upon irradiating the reaction mixture with light at a wavelength of 395 nm (Scheme 2, ent. 4b), we observed complete conversion of the substrate 1a, but the yields of the desired product 3aa were below 10%. Similarly, when irradiating with light at a wavelength of 365 nm (Scheme 2, ent. 4c), substrate 1a underwent complete conversion; however only traces of 3aa were noticed by a GC analysis.
The lack of success of a photochemical approach forced us to investigate the generation of the aryl radical for the 1,5-HAT process under electrochemical conditions via either the direct or electromediator-aided pathway (Scheme 2, ent. 6a–d).72–74 Following the protocol for an electroreduction of aryl iodides reported by Wang,63 we electrolyzed the model reaction mixture in an electrochemical cell equipped with a Ni-anode and graphite cathode (Scheme 2, ent. 6a); however mostly we observed the formation of homo- and cross-coupling products. The further modification of reaction conditions, like the change of electrolyte and solvent, did not provide significant improvement. The change of the sacrificial anode material to Zn,64 Al,75 Mg76 or steel65 along with the use of an electromediator, e.g., phenanthrene, provided 10–15% of the desired product along with homo-/cross-coupling side products. An important observation from these experiments was the high conversion of 1a, although the formation of 3aa and deiodination product 4a was not noticed. Instead of this we observe the formation of a mixture of homo- and cross-coupling products of 1a and 2a indicating that the examined electrochemical conditions are capable of efficiently executing Ni-catalytic cycles, plausibly due to reduction of Ni(I) species to the key Ni(0) one.
Finally, we turned our attention to the reduction of aryl halides by a carbon dioxide radical anion (CO2˙−). This radical species is a valuable synthetic intermediate, thanks to extremely negative reduction potential (E1/2(red) (CO2/CO2˙−) = −2.2 V vs. SCE)77 and reactivity that makes it an efficient reductant and carboxylating agent.78 Moreover, it can be easily generated from readily available formic acid salts by photoredox-HAT protocols in the presence of photocatalysts and additional hydrogen transfer reagents, commonly thiols.78
Initially, we employed the protocol for a photoinduced remote Giese-type C–H alkylation of amines in the presence of HCOOCs, reported by Yatham et al.79 Thus, the mixture of model substrates 1a and 2a along with the Ni-complex and HCOOCs in DMSO was irradiated with purple light (390 nm). The starting material 1a was consumed completely after 12 h to provide mostly product 4a along with traces of 3aa and small amounts of cross- or homo-coupling products. Based on the last observation, we conclude that these conditions were not suitable to execute the complete Ni-complex catalytic cycle. Therefore, guided by seminal reports by Ju,80 Wickens68 and Jui81 and our previous experience in the generation of the CO2 anion radical,62 we modified reaction conditions by introducing a photocatalyst (PC), like 4-CzIPN, and thiol 5aa (mesna) to enhance the generation of the required XAT reagent through a thiyl radical-aided HAT from the formate anion (Scheme 2, ent. 5). Additionally, we expected that, generated under reaction conditions, the PC-derived radical anion will also serve as a reductant for Ni(I) species to deliver Ni(0) to close the cross-coupling catalytic cycle. Now, the activation of substrate 1a proceeded efficiently, but product 4a still dominated. The yield of 3aa did not exceed 15%. The replacement of 4-CzIPN with 4-DPAIPN and the use of other thiols than mesna resulted in a slight improvement only. This confirmed our previous suspicions about the inefficient synchronicity of HAT and Ni-catalyzed cross-coupling steps; thus, we hypothesized that better synchronization of the reaction rates of individual processes should break the current impasse and significantly increase the efficiency of the process. Therefore, considering previous observations indicating that electrochemical conditions should enable the effective realization of the nickel catalytic cycle and the substrate 1a activation strategy based on photoinduced-XAT, we decided to combine these strategies and investigate the possibility of alkenylation of compound 1a with vinyl bromide 2a under photoelectrochemical conditions.
In initial photoelectrochemical experiments, the mixture of vinyl bromide 2a and morpholine derivative 1a, containing HCOOCs, thiol 5ab and Ni-complex (Ni(glyme)Cl2 + dtbbpy) in DMSO, was irradiated (395 nm) and electrolyzed (3 mA) for 12 h. The initial electrochemical setup consisted of an undivided electrochemical cell equipped with RVC and Ni(foam) as an anode and a cathode, respectively. TBABF4 was used as an electrolyte. Under these conditions, the expected product 3aa was obtained in 35% yield (conversion of 1a 91%) along with side product 4a (Scheme 2, ent. 6d). This promising result encouraged us for further optimization of this process (Scheme 3).
 | ||
| Scheme 3 Optimization studies on photoelectrochemical C–H alkenylation of amines (selected examples). PMP = 4-methoxyphenyl. | ||
We started the optimization of the photoelectrochemical protocol from examination of the influence of Ni salt on the products' yield. The replacement of Ni(glyme)Cl2 with Ni(glyme)Br2 allowed the product yield to be increased from 35% to 51% (Scheme 3, ent. 2). For other Ni salts, like NiI2, Ni(OTf)2 or Ni(cod)2, the yields were much lower (see SI, Section 4.2.1.1). Next, we focused on the ligand choice. Initially 4,4′-di-tert-butyl-2,2′-bipyridine (dtbbpy) was used; however, further experiments demonstrated that the best results can be achieved when the Ni-complex with the PyBOX ligand was employed (Scheme 3, ent. 3 and SI, Section 4.2.1.2). Initially, 10 mol% loading of Ni salt and 11 mol% of PyBOX ligand were employed; however, we found out that these amounts can be lowered to 5 mol% and 5.5 mol%, respectively, without significant change of SM's conversion and product's yield after the standard 12 hours. A further decrease in the amount of Ni salt to 3 mol% decreased conversion of 1a from >99% to 67% and product yield to 53% after the standard 12 h (see SI, Section 4.2.2.2).
Among various formate salts, HCOOCs was the reagent of choice mostly due to its best solubility in DMSO compared to other formats (see SI, Section 4.2.1.4). The conversions of 1a in the presence of K, Na or Li formate were in the range of 42–66%, and only traces of product 3aa were detected. Interestingly, tetrabutylammonium formate (HCOOTBA) allowed for complete conversion of 1a but the yield of 3aa was less than 40%, indicating that it affected the cross-coupling step (see SI, Section 4.2.1.4).
Initially mesna (5aa) was used as a HAT reagent. However, an examination of other thiols (see SI, Section 4.2.1.3) revealed that methyl 4-mercaptobenzoate (5ab) provides complete conversion of 1a and the highest yield of 3aa (67%). Interestingly, its ortho-isomer delivered product 3aa only in 37% (see SI, Section 4.2.1.3). In the case of the electron-rich aromatic thiols (e.g., 4-mercaptoanisol) and aliphatic thiols (e.g., CySH or methyl mercaptoacetate) the yields of the model product did not exceed 20%, despite over 90% conversion of starting material 1a (see SI, Section 4.2.1.3).
As already mentioned, DMSO resulted in being the solvent of choice which assures proper solubility of all reagents, including TBABF4 (1 equiv.) which served as an electrolyte of choice (see SI, Section 4.2.1.5). The optimal concentration was 0.1 M with respect to substrate 1a. Higher dilution (0.05 M) caused a dramatic decrease in yield of 3aa and an increase in the content of the reduced substrate (4a). In the case of other solvents, like MeCN or DMF, the conversion of 1a was ca. 60% (after 12 h), but the product's yield was <15%, indicating that these solvents strongly affect the cross-coupling step (Scheme 3, ent. 7 and 8) (see SI, Section 4.2.1.6). Furthermore, the solvent mixtures, like DMSO/water or DMSO/MeCN, were examined to improve the yield of the reaction; however, all of these attempts were not successful (see SI, Section 4.2.1.6).
Finally, we focus on an optimization of photochemical and electrochemical conditions. Initial conditions, a constant current electrolysis (3 mA) in the presence of an RVC anode and a Ni foam cathode along with purple light irradiation (395 nm), provided the product 3aa in 67% yield (Scheme 3, ent. 3). The use of light sources with longer wavelength light resulted in a significant decrease in the product's yield, except in the experiment when blue light with a wavelength of 427 nm was applied (ent. 3 and 9–11). In this case, the yield of the product increased up to 72% (ent. 10). The replacement of the RVC electrode with a graphite one resulted in a lower yield (ca. 42%, ent. 13). However, when two graphite electrodes were used instead of RVC and Ni(foam) ones, the model product was obtained in 65% (irradiation at 395 nm) (ent. 14) and 87% (irradiation at 427 nm) (ent. 15) yields. Additionally, we have also checked the influence of the applied current by varying it from 1 to 4 mA (see SI, Section 4.2.3.2). A current of 3 mA was optimal.
During optimization studies, we found out that the efficiency of the cross-coupling process decreased during the experiments due to “poisoning” of the anode by deposition of solid by-products at the electrode surface. To avoid this, we set up a power supply to periodically change the polarization of the electrodes. We examined different polarization changing times, 5, 10, and 15 min, and the 10 min intervals were optimal (3 mA for 10 min and then −3 mA for 10 min). The alternating current electrolysis82 with time intervals less than 60 s was ineffective, and the target product was obtained in yields below 65%. Therefore, irradiation with purple light (395 nm) and electrolysis with polarization change of two graphite electrodes every 10 min resulted in an increase in the product yield up to 65% (Scheme 3, ent. 14). Finally, the change in the light wavelength from 395 nm to 427 nm allowed for further improvement of the process and yielded the product 3aa in 87%.
Finally, among 2-halobenzamides, the iodo-derivative, e.g., 1a, demonstrated superior reactivity and efficiency of the alkenylation process (see SI, Section 4.3.1). In the context of vinyl halides, vinyl bromide 2a emerged as the superior cross-coupling partner, owing to its exceptional combination of stability and reactivity (see SI, Section 4.3.2).
The final optimization studies were devoted to elaborate the optimal amounts of the required reagents. Thus, the reaction of 1a and 2a in a ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.5, in the presence of 1 equiv. of TBABF4, 3 equiv. of HCOOCs, 30 mol% of thiol 5ab, 10 mol% of Ni(glyme)Br2, and 11 mol% of PyBox ligand in DMSO (0.1 M), yielded product 3aa in 87% (GC) as a mixture of E/Z isomers in a ratio of 72:28 (Scheme 3). Aqueous work-up of the post-reaction mixture followed by flash column chromatography delivered 3aa in 78% isolated yield.
:1.5, in the presence of 1 equiv. of TBABF4, 3 equiv. of HCOOCs, 30 mol% of thiol 5ab, 10 mol% of Ni(glyme)Br2, and 11 mol% of PyBox ligand in DMSO (0.1 M), yielded product 3aa in 87% (GC) as a mixture of E/Z isomers in a ratio of 72:28 (Scheme 3). Aqueous work-up of the post-reaction mixture followed by flash column chromatography delivered 3aa in 78% isolated yield.
Substrates' scope
With optimal conditions in hand, we next explored the scope of the aryl radical-triggered remote C(sp3)–H alkenylation protocol with various heterocyclic scaffolds and model vinyl bromide 2a. As shown in Scheme 4, C–H alkenylation of a range of 2-iodobenzoyl-protected saturated heterocycles with different ring sizes proceeded smoothly and provided the desired products (e.g., 3ab, 3al and 3aq) smoothly in 72–75% isolated yield. In contrast, alkenylation of the azetidine ring failed (3as), and deiodinated azetidine benzamide was obtained as a single product, indicating that the geometry of the 4-membered ring along with its rigidity does not allow for executing the 1,5-HAT process since the distance between the radical center and ring's hydrogen atom is too long.Additional heteroatoms in the ring, like oxygen for 1a and 1g or nitrogen in the case of 1d and 1e derivatives, did not have a significant influence, and desired products 3aa, 3ag, 3ad and 3ae, respectively, were obtained in very good yields. Only in the case of alkenylation of thiomorpholine-based benzamide 1c, the desired product 3ac was isolated in lower yield (55%), although the starting material was consumed completely. The product 3ac was accompanied by an increased amount of the deiodinated starting material (∼20–30%), indicating a potent issue with the HAT process. We believe that it could be the effect of the change of the 6-membered ring geometry in thiomorpholine due to the presence of the S atom and elongation of the C–S bond lengths compared to C–C (like for piperidine) or C–O (like for morpholine) that may affect the 1,5-HAT transition state geometry decreasing the efficiency of the H-atom translocation. Of course, the alternate negative interactions of the S atom with the Ni-catalyst, which might affect the cross-coupling cycle, cannot be neglected either.
Notably, the alkenylation of monocyclic 6-membered substrates usually proceeded more efficiently than for the corresponding fused analogues (e.g., 3aavs.3ak or 3abvs.3ah), which were often accompanied by a side benzamide product originating from the deiodination of starting 2-iodobenzamide. However, at the same time, all benzofused products (e.g., 3ah, 3ak, etc.) were formed exclusively as E-isomers, whereas for monocyclic substrates the content of the (Z)-isomer varied from 2 up to 30%. The potent rationalization of this phenomenon is related to a different conformational dynamic of mono-cyclic and fused heterocyclic scaffolds. It should be expected that reducing the degrees of freedom in fused systems should significantly reduce their conformational dynamics, which will potentially translate into more difficulty in obtaining the appropriate geometry of the transition state of the HAT process. As a result, the rate of the H-atom transfer should decrease, thus providing an opportunity for competitive quenching of the aryl radical to form the corresponding benzamide, which is in accordance with our experimental observations.
For heterocyclic substrates, such as 1i and 1j, with two potent positions for H-atom abstraction, the regioselectivity of the HAT reaction was governed by the relative stability of the resulting α-aminoalkyl radical. As presented in Scheme 4, in these cases, the 1,5-HAT step proceeded regioselectively at the C-1 position, resulting in the formation of more stable benzyl-type radical species, and yielded products 3ai and 3aj, respectively. Disappointingly, in both cases the yields did not exceed 40% due to the high content of unwanted deiodinated side benzamides (40–60%).
 | ||
| Scheme 4 The scope of amines for remote α-C(sp3)–H alkenylation of heterocycles 1. | ||
The significance of conformational factors was also strongly manifested in the C–H functionalization of the 5- and 7-membered substrates. As previously mentioned, the alkenylation of the 5-membered pyrrolidine ring (1l) proceeded more efficiently than that of the fused analogue, indolidine (3alvs.3an). Furthermore, the flat nature of an oxazolidinone ring (e.g., 3ao) along with a specific arrangement of the benzoyl group makes the effective orientation of the HAT directing group strongly disfavoured due to an electric repulsion and steric hindrance. As a result, the XAT-generated aryl radical species is incapable of abstraction of any hydrogen atom and is quenched directly, yielding N-benzoylated oxazolidinone only. The replacement of a carbonyl group in the oxazolidinone ring (1o) with a C(sp3)-atom (oxazoline 1p) increased conformational lability of the ring enabling the 1,5-HAT process and yielding compound 3ap in 50% yield (isol.). Nevertheless, the flexibility of the oxazoline 5-membered ring is still not enough to reach the efficient geometry of the HAT-transition state, compared with 6-membered heterocycles; therefore, again, desired product 3ap was accompanied by a type-4 side product. Herein, the high ring lability along with entropic factors decreases the probability of the system adopting the optimal geometry of the HAT-transition state that again results in a decrease in the rate of H-atom abstraction promoting side quenching of aryl radical species.
In the case of the alkenylation of benzoazepan-5-one derivative 1r (product 3ar), the initial steps, the generation of an aryl radical and the subsequent 1,5-HAT process, proceeded noticeably slower than those for azepane derivative 1q and other heterocyclic substrates. As a result, after a standard 12 h, product 3ar was isolated in 39% only and was accompanied by a dehalogenated substrate (ca. 20%) and numerous vinyl bromide homo-coupling products. Additionally, ca. 10–15% of unreacted substrate 1r was recovered. The reaction proceeded well also for more complex amines, like estrone- and cholesterol-derived substrates, delivering products 3at and 3au in good yields and high content of the E-isomer (Scheme 4). In both cases a single diastereoisomer was isolated; however, overlapping of diagnostic signals in NMR spectra did not allow relative configuration to be assigned clearly.
Ring substituted heterocycles, e.g., 4-methylpiperidine 1f, are also suitable substrates for the C–H alkenylation process, as demonstrated in Scheme 4, and, for example, product 3af was yielded in 74%. An intriguing substrate was piperidin-4-one derivative 1v. Under standard conditions, beside expected product 3av (yield 32%), its alkenylation also delivered compound 6 (yield 30%) and a small amount of side-product 4v (Scheme 5). The product 6 plausibly originated from the oxidation of α-aminoalkyl radical intermediate 7 to the corresponding iminium cation 8 followed by a subsequent base-mediated deprotonation (e.g., by the formate anion) at the Cα-position to the carbonyl group to deliver enamide 6. Recently, Yatham and co-workers83 demonstrated synthesis of type-6 cyclic enamides by a closely related approach involving radical translocation followed by Co-assisted dehydrogenation.
 | ||
| Scheme 5 The C–H alkenylation of piperidin-4-one derived benzamide 1v. PMP = 4-methoxyphenyl. | ||
We expected that type 6 enamide could also be formed during the alkenylation of substrate 1w, bearing ester functionality at the C3 position. However, it was not the case, and this product was not detected in the reaction mixture. On the other hand, this reaction led to the formation of two regioisomeric (E)-products 3aw and 3aw’, in a ratio of 91:9 (Scheme 6). The NMR analysis of the crude reaction mixture revealed only traces of (Z)-isomers (<5%). We were not able to separate both regioisomers to confirm the relative configuration. The regioisomeric mixture was also obtained for the alkenylation of 3-methyl-substituted substrate 1x (Scheme 6, 3ax/3ax′ in a ratio of 5:1). Again, the C6-alkenylated product was the major one, and only traces of the corresponding (Z)-isomers were noticed by a 1H NMR analysis. In contrast, piperidin-3-one derivative 1y delivered only C6-alkenylated products 3ay (E/Z-isomer mixture in a ratio of 92:8) in a moderate yield of 46%. For all these cases lower product yields were the effect of side substrate reduction leading to the corresponding benzamides.
 | ||
| Scheme 6 Remote C–H alkenylation of 3-substituted piperidine scaffolds. The E/Z ratio of vinyl bromide 2a was >99:1. PMP = 4-methoxyphenyl. | ||
Next, using a model morpholine scaffold (Scheme 7), we investigated the scope of a HAT directing group capable of promoting a 1,5-HAT reaction. The 2-iodobenzoyl group outperformed the other two examined functional groups, 2-iodobenzyl and 2-iodobenzosulfonyl, in terms of effective radical translocation through the 1,5-HAT reaction. The 2-iodobenzyl group was completely worthless since it did not deliver an aryl radical through XAT by the CO2˙−, under standard reaction conditions. Neither the expected product nor the deiodinated side-product, N-benzyl morpholine, was detected in the crude reaction mixtures by the MS analysis. Consequently, only about all of the starting material 9 could be recovered from these experiments. This led us to the conclusion that the electron-deficient character of the HAT directing group is essential for an efficient cleavage of the C(sp2)–I bond by an XAT reagent.
 | ||
| Scheme 7 The effect of a HAT-directing group on C–H alkenylation of morpholine derivatives with vinyl bromide 2a. | ||
The reaction of 2-iodobenzosulfonamide 10 delivered primarily deiodinated product 11 (∼50%) (Scheme 7). We have also isolated a small quantity of another compound. Its structure was elucidated and indisputably assigned as 12, according to NMR spectra, including COSY and HSQC correlations (see the SI). The installation of an alkenyl fragment at the C3 position of the morpholine ring indicated that initial H-atom translocation proceeded in the 1,6-manner contrary to all so far studied examples. It is well known that, thanks to the longer length of N–SO2 and C–SO2 bonds, sulfamides are capable of promoting 1,6- over 1,5-HAT in acyclic systems.39,41,44,84–87 However, according to the best of our knowledge, it is the first example of such β-C(sp3)–H functionalization for the heterocyclic substrate. The low yield of product 12 (∼15%, (E/Z)-isomers mixture in a ratio of 96:4) with a simultaneous large content of side-product 11 in the reaction mixture is not surprising considering that the 1,6-HAT process proceeds through a 7-membered transition state and the 6-membered structure of the substrate with its conformational preferences should definitely not favour efficient translocation of the H atom. At this point, we hypothesized that increasing the substrates' ring size should facilitate the 1,6-HAT process thanks to the higher flexibility of such rings. Therefore, to prove this assumption, we prepared azepane benzosulfamide 13 and subjected it to the reaction with model vinyl bromide 2a under standard photoelectrochemical conditions. As expected, the reaction proceeded with complete conversion of substrate 13 and yielded compound 14 (only the E-isomer) in 63%. Notably, deep analysis of the crude reaction mixture revealed the presence of small quantities of the 1,5-HAT-derived product 15 but its yield did not exceed 5%.
The above-presented results encouraged us for one more experiment. As already mentioned, among 6-membered heterocycles, the α-C–H alkenylation of the thiomorpholine ring proceeded in a moderate yield (Scheme 4, 3acvs.3aa, 3ab, and 3ad–3af), which was rationalized by a deformation of the 6-membered ring due to the longer length of the C–S bond. On the other side, β-C–H alkenylation of morpholine scaffolds assisted by the 2-iodobenzosulfonyl group yielded product 12 poorly. Therefore, we hypothesized that the replacement of the HAT directing group in the thiomorpholine ring and the combination of both structural effects connected with the presence of the S-atom should enhance the HAT process along with a change of regioselectivity of the C–H functionalization. Indeed, the C–H alkenylation of substrate 16 with model vinyl bromide 2a yielded expected product 17 in 50% along with side product 18 (20%). The increase in yield from 15% for substrate 12 to 50% for product 17 confirmed the above assumption; however, the enhancement of HAT is moderate, indicating that the geometry of the transition state is still not the most efficient one.
Subsequently, we investigated the scope of the vinyl bromides in the reaction with morpholine-derived substrate 1a. The reaction proceeded smoothly for a range of 2-aryl-substituted vinyl bromides bearing both electron-donating and electron-withdrawing groups, indicating very good functional group tolerance (Scheme 8). 2-Aryl vinyl bromides with a nitro group at the phenyl ring did not provide the expected product, plausibly due to potent photoexcitation of the nitro group88–91 followed by side reactions. The cross-coupling of 2-aryl vinyl bromides with a CN group at various positions of phenyl (2v–2x) proceeded less efficiently (products 3bq–3bs) than for other EWG groups, like ester ones (e.g., 2u) which yielded product 3bp in 67%. The reason for this was competitive side radical reactions of the CN group as a radical acceptor,92,93 resulting in low yield of the desired products and a complicated post-reaction mixture.
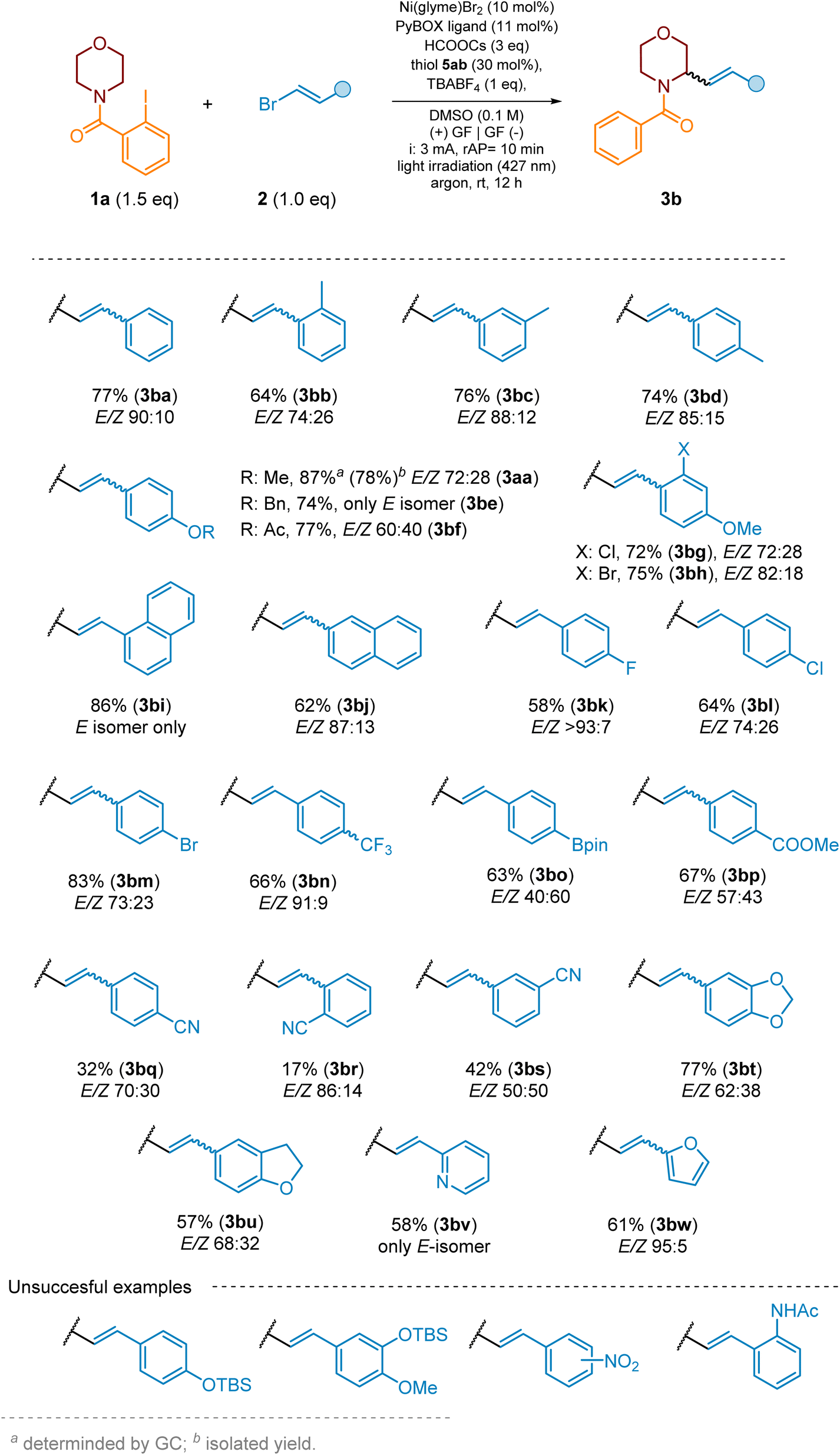 | ||
| Scheme 8 The scope of vinyl bromides for remote C(sp3)–H alkenylation of saturated heterocycles. For the E/Z ratio for starting vinyl bromides please see SI, Section 3.2. | ||
We were delighted to find that 2-aryl vinyl bromide with a boronic acid ester group can also be efficiently coupled with a morpholine ring (63%), providing product 3bo for further functionalization via standard cross-coupling reactions. Finally, we examined the reactivity of several vinyl bromides bearing heterocyclic rings (2y–2ab). As seen in Scheme 8, these reagents were also suitable cross-coupling partners for the investigated reaction sequence, providing desired products (3bt–3bw) in 58–77% yield.
In addition to (E)-vinyl bromides (2), their (Z)-isomers (e.g., 19) may also be used in the examined alkenylation procedure. Scheme 9 shows that for both vinyl bromides, 2a and 19, the product 3aa was obtained as an (E/Z)-isomer mixture, with the (E)-isomer predominating. However, in the case of the (Z)-isomer, the cross-coupling proceeded relatively slower, delivering product 3aa in lower yield after the standard reaction time (12 h). As already mentioned, in contrast to monocyclic amines, the C–H alkenylation of bicyclic heterocycles leads to (E)-products only, suggesting that steric effects might prevent subsequent E-to-Z isomerization. This assumption seems to be supported by other experimental observations presented in Scheme 9. The alkenylation of quinolidine derivative 1h with (E)-vinyl bromide 2a yielded product 3ah in 57% as a single (E)-isomer. An analogous reaction of 1h with (Z)-vinyl bromide 19 proceeded slightly less efficiently (yield 50%) but delivered the same (E)-isomer 3ah.
 | ||
| Scheme 9 Reactivity of isomeric vinyl bromides. The E/Z ratio is >99:1 for vinyl bromide 2a and 1:17.5 for vinyl bromide 19. PMP = 4-methoxyphenyl. | ||
Furthermore, vinyl dibromides, e.g., 20, which served as intermediates in the synthesis of type-2 substrates, were also suitable for the investigated C–H functionalization and delivered the product 3aa again with dominance of the (E)-isomer after the subsequent debromination process under the same reaction conditions. Disappointingly, in contrast to the 2-aryl vinyl bromides presented so far, their sterically more hindered 1-aryl substituted analogues, such as compound 21, were much less efficient cross-coupling partners and yielded only traces of the desired products (e.g., 23). The major product was dehalogenated morpholine benzamide 4a. In addition, the alkenylation of the model substrate 1a with unsubstituted vinyl bromide 22 was unsuccessful and did not furnish the desired 2-vinylated morpholine 24.
The developed protocol could also be successfully applied for acyclic amine derivatives, like compounds 25a and 25b (Scheme 10). For both cases, the alkenylation with the model vinyl bromide 2a delivered regioisomeric products 26 and 27 in 56% and 36% yield, respectively. Regioisomers 26a and 26b were major products indicating that 1,5-HAT toward the formation of a more stable secondary α-aminoalkyl radical is more preferred. Furthermore, so far used 2-aryl vinyl bromides could be replaced by their alkyl-substituted ones. The reactions with simple 2-alkyl vinyl bromides (see SI, Section 7), under standard reaction conditions, were unsuccessful since, in these cases, the rates of side debromination and homocross-coupling reactions were higher than that of the desired XAT/HAT/cross-coupling sequence. Better results were achieved in the case of more complex vinyl bromides, like compound 28. The alkenylation of morpholine derivative 1a with this bromide, under standard conditions, yielded product 29 in 68% as a single diastereoisomer and with (E)-geometry of a double bond, as presented in Scheme 10.
 | ||
| Scheme 10 Remote C–H alkenylation of acyclic amines and alkenylation with 2-alkyl substituted vinyl bromides. | ||
Finally, we questioned whether the disclosed procedure could be done in an enantioselective manner by employing chiral Ni complexes. For this purpose, we replaced the so far used PyBOX ligand by its chiral analogues and attempted to perform the reaction between morpholine 1a and vinyl bromide 2a. However, since we could not achieve suitable separation of all isomeric products 3aa during HPLC analysis of the racemic sample, we replaced the model substrate 1a by pyrrolidine derivative 1l which was then submitted to alkenylation with vinyl bromide 2a. Unfortunately, beside numerous chiral PyBOX ligands, we did not observe any asymmetric induction, resulting in the formation of the racemic (E)– and (Z)-products 3al, although the overall yields were quite high in all studied cases (see SI, Section 10). The same result was obtained when PyBOX ligands were replaced by a series of chiral BOX ligands (see SI, Section 10).
Contrary to unsuccessful realization of enantioselective mode of C–H alkenylation, we focused on photochemical Cu complex-assisted kinetic resolution following the report by Yuan and Yu.94 Under reported conditions, racemic pyrrolidine 3al (E/Z mixture in a ratio of 90:10), in the presence of a chiral Cu-BOX ligand and under blue light irradiation (427 nm), yielded (S,E)-3al and (R,Z)-3al in 45% and 55%, respectively (Scheme 11). Enantiomeric excess for (S,E)-3al was 76%, whereas the optical purity of (R,Z)-3al was 63% ee.
 | ||
| Scheme 11 Photochemical kinetic resolution of pyrrolidine derivative rac-3al (E/Z ratio 90:10) in the presence of a chiral Cu/BOX complex. | ||
Applications
To explore the practicality of this reaction, a scale-up experiment was carried out under the standard reaction conditions. Initially a reaction of 1a and 2a was performed in a 50-mL 3-neck round-bottom flask with angled side arms; however, the desired product 3aa was obtained in a significantly lower yield (57%, 0.86 g starting from 1.0 g (4.7 mmol) of substrate 2a) compared to the same process at the 0.6 mmol scale (see SI, Section 12, Fig. SI-S25). The change of the reactor to a 3-neck round-bottom flask with parallel side arms enhanced the yield to 61%. Finally, placing additional electrodes allowed 1.34 g (84%) of the desired product 3aa (E/Z isomer ratio 90:10) to be obtained, starting from 1.06 g (5.00 mmol) of vinyl bromide 2a and 1.74 g (5.53 mmol) of morpholine derivative 1a (Scheme 12).
 | ||
| Scheme 12 The large-scale synthesis of compound 3aa. | ||
To demonstrate the synthetic potential of the alkenylated heterocycles prepared via our remote C(sp3)–H alkenylation conditions, model product 3aa was subjected to several transformations presented in Scheme 13a. The basic hydrolysis of 3aa yielded free amine 30 in 67%, and its hydrogenation in the presence of Pd(OH)2/C yielded product 31 in 89%. Finally, a sequential reduction of amide functionality and hydrogenation of a double bond allowed for synthesis of morpholine 32 in 79% isolated overall yield. The Wacker oxidation of 3aa delivered ketone 33 in 79%.
 | ||
| Scheme 13 Synthetic application: (A) functional group transformations; (B) post-functionalization of type-3 products; (C) synthesis of galipinine precursor 36; (D) synthetic approach for the preparation of the piperazine fragment of type-41 bioactive molecules. | ||
The tolerance of the functional group, like halides or boronic acid, provides an extraordinary opportunity to use the resulting products for their late-stage functionalization to deliver complex molecular systems. For example, boronic acid ester 3bo was readily coupled with aryl halides, like 4′-bromoacetophenone, to furnish product 34 in 75% yield, as demonstrated in Scheme 13b. Notably, varying the aryl halides allows for the late-stage structure diversification of the core fragment, e.g., 1 and 3, delivering a broad library of products, for instance, for structure–activity relation studies (SARSs), a relatively faster and more efficient way of avoiding time-consuming parallel multistep synthesis. Furthermore, as presented in Scheme 13b, substrate 3bm can be decorated with an additional heterocyclic scaffold through a Cu-catalyzed Ullmann-type coupling reaction with an N-nucleophile, e.g., N-Boc piperazine, yielding product 35 in 67%.
Another example of practical application of our alkenylation protocol is the synthesis of compound 36, which can serve as a precursor of (−)-galipinine 37 (Scheme 13c). The same approach allows for construction of similar tetrahydroquinoline-based alkaloids, like (−)-galipinine (38) and (−)-cuspareine (39). All of them are isolated from the Galipea officinalis shrubby tree, whose trunk bark is used in indigenous folk medicine for its healing properties.6
Moreover, as already presented in Scheme 4 and 8, broad substrate scope regarding the vinyl bromides and heterocyclic scaffolds makes our protocol a suitable synthetic tool for rapid and efficient structure diversification either by the already mentioned post-functionalization or by varying both starting materials to differentiate either the heterocyclic core or the side chain. We demonstrated this by synthesizing the piperazine-containing compound 40b. This functionalized piperazine-based structural motif can be found in numerous naturally occurring compounds or drug candidates,95–98 like type-41 compounds widely investigated as potent dopamine receptor antagonists for the treatment of psychotic disorders.99–102 As shown in Scheme 13d, the already known approach for the synthesis of the core fragment, like compound 40a, was executed in 8–9 steps starting from piperazine-2-carboxylic acid and resulted in an overall yield of 30–33%.99 No doubt, practical application of this reaction sequence for the preparation of a large library of type-40a structures, for instance, for SAR studies, is highly time-consuming which limits its synthetic value. Therefore, we proposed the synthesis of the analogue of 40a, piperazine 40b (R: Cbz), from readily available mono-protected piperazine, and included an installation of the HAT directing group, key C(sp3)–H alkenylation of the heterocyclic ring, and hydrogenation of the double bond. The target product, 40b, was obtained in 65% overall yield.
Mechanism discussion
To determine the essential parameters of the investigated process, we conducted several control experiments disclosed in Scheme 14. | ||
| Scheme 14 Mechanism studies: the exclusion tests and control experiments. | ||
In the absence of irradiation (Scheme 14, ent. 2), only traces of product 3aa were noticed (∼5%) although the conversion of 1a was above 90% due to side processes, like homo-coupling of 1a and cross-coupling of 1a with 2a (Scheme 14, bottom), along with slight amounts of the dehalogenated substrate (4a). These observations allowed us to draw two conclusions. Firstly, the Ni-catalyzed coupling step does not depend on photochemical conditions. Moreover, since only a catalytic amount of Ni salt is used (10 mol%), the formation of the mentioned by-products proves that electrochemical conditions ensure closure of the Ni-catalytic cycle, restoring key Ni(0) species. Secondly, light is plausibly essential to enable the activation of the substrate, e.g., 1a, via the HAT process.
Without electric current, only traces of product 3aa and homo/cross-coupling side products were detected along with dominant side-product 4a (Scheme 14, ent. 3 and 4) regardless of whether blue (427 nm) or purple light (395 nm) was applied. This observation supported the above statement about the essential role of electrolysis in a cross-coupling cycle.
In the control experiment conducted without the addition of formate salt, the conversion of substrate 1a proceeded very slowly, reaching only 9% after 8 hours. The reaction predominantly yielded homo- and cross-coupling products, as detected by mass spectrometry (Scheme 14, ent. 5). Furthermore, the formation of side product 4a did not exceed 5–7% even after 12 hours. These results highlight the pivotal role of formate in activating the starting material and demonstrate the absence of other reactive species in the reaction mixture capable of reducing the C(sp2)–I bond.
When the model reaction was performed without thiol 5ab under blue light irradiation (427 nm, ent. 6) only 38% conversion of substrate 1a was noticed along with a slight amount of product (6%, E/Z is 80:20). On the other side, when a light wavelength was changed from 427 to 395 nm (ent. 7), 75% conversion of 1a was observed but the yield of the desired product 3aa was at the same level (6%, an E/Z ratio of 79:21). The formation of the slight amount of product in both cases confirms the crucial role of thiol in formation of the XAT reagent. At the same time, the formation of the product indicates the operation of an alternative mechanism delivering the CO2˙− intermediate. This could proceed by a mechanism reported by Yatham and coworkers,79 postulating the formation of this radical anion directly from formate salt in the presence of a dimsyl anion. Accordingly, they form an EDA-complex which upon purple light irradiation (390 nm) undergoes excitation to produce intermediate CO2˙−. Indeed, deep analysis of UV spectra of 1a + HCOOCs (SI, Fig. SI-5) allows very slight absorption around 370 and 450 nm to be noticed; however, this excitation is too weak to have any influence on the process and can be neglected.
Finally, as indicated in Scheme 14 (ent. 10), the investigated process is slightly air sensitive; the experiments performed without inert gas yielded product 3aa in ca. 70%, whereas under an argon atmosphere, the same product was obtained in 87% (ent. 1). In contrast, the moisture is not allowed, as indicated by control experiments furnishing only traces of the desired product (ent. 11).
Considering that thiyl radicals may also function as hydrogen atom transfer (HAT) reagents, we conducted control experiments to exclude this possibility. Accordingly, compound 4a was subjected to a reaction with vinyl bromide 2a under the standard reaction conditions (Scheme 14, ent. 12). After irradiation with blue light and passing an electric current for a standard 12 hours, no formation of the expected product 3aa was observed. This result indicates that neither ArS˙ nor CO2˙− acts as a HAT reagent and is incapable of activating our starting material via cleavage of the C(sp3)–H bond.
Deuterium-labelling and crossover experiments indicated that the reaction exclusively underwent intramolecular 1,5-HAT processes, additionally ruling out intermolecular hydrogen atom abstraction (Scheme 15a) and are in agreement with related studies previously reported.47,55,103
 | ||
| Scheme 15 Mechanistic studies: (A) deuterium-labelling and cross-over experiments; (B) cyclic voltamperograms of key reagents; (C) radical trapping experiments with TEMPO; (D) UV-vis spectra of thiol 5ab, its Cs salt and disulfide 5bb with and without addition of HCOOCs in DMSO; (E) kinetic studies of the model reaction without light; (F) kinetic studies of the model reaction (standard conditions). | ||
The addition of a radical trapping reagent, such as TEMPO, effectively inhibited the reaction, and only partial conversion of substrate 1a was observed. Mass spectrometric analysis of the crude reaction mixture revealed the presence of products resulting from the interception of radical species, specifically, aryl radicals and/or α-aminoalkyl radicals (Scheme 15c, SI and Fig. SI-1a) and aryl thiyl radical (Scheme 15b, SI and Fig. SI-1b), by TEMPO. Notably, the expected product 3aa was not detected in the crude reaction mixture (see SI, Section 9.1).
The above disclosed observations, along with additional physicochemical measurements, including UV-vis spectra (Scheme 15d, SI, Section 9.3) and CV measurements (Scheme 15b, SI, Section 9.4), allowed us to propose the plausible mechanism that is shown in Scheme 16. The key step is the formation of aryl thiyl radical 42, a strong HAT reagent,104 capable of H-atom abstraction from formate salt to deliver highly reducing CO2˙− species 49 (E1/2 (CO2/CO2˙−) = −2.2 V).78,80,105,106 Radical anion 49 reduces substrate 1a (Ered = −1.56 V) via the XAT process, furnishing aryl radical 50 capable of the subsequent 1,5-HAT process to afford nucleophilic α-aminoalkyl radical 51. Meanwhile, vinyl bromide 2a undergoes oxidative addition with the Ni(0) complex to form Ni(II)-complex 52. Then, complex 52 intercepts radical 51 to provide Ni(III)-complex 53. The final reductive elimination furnishes product 3aa and Ni(I) intermediate 54. The latter one is then reduced to Ni(0) to close the Ni-catalytic cycle. This final reduction is realized as a cathodic process, which is essential for the efficiency of the investigated reaction sequence. As already mentioned, without electric current, the product yield cannot exceed 10%, which is equal to the Ni catalyst loading.
 | ||
| Scheme 16 Proposed mechanism for α-C(sp3)–H alkenylation of saturated heterocycles. | ||
We have considered two alternative mechanisms to generate thiyl radical 42 presented in Scheme 16. The first mechanism assumes anodic oxidation of thiol 43 (via radical cation 46 following deprotonation, path Aa) or thiolate 44 (path Ab) to thiyl radical 42 which rapidly dimerizes to disulfide 45. Photo-induced homolysis of the S–S bond in 45 delivers aryl thiyl radical 42 which dimerizes again or reacts with formate, delivering intermediate 49. At the same time, the resulting disulfide 45 could take one electron from the cathode, leading to disulfide radical anion 48 which decomposes to the thiyl radical 42 and thiolate 44 (path C). The thiolate anion can be converted again to the thiyl radical at the anode and the latter one dimerizes or acts as a HAT reagent.107,108
On the other hand, it is a well-known fact that arene thiolate anions, formed by thiol deprotonation with a weak base, may absorb visible light and participate in a photoredox cycle.109 Therefore, we considered an alternative mechanism, presented in Scheme 16 (path B), in which irradiation of 44 generates excited thiolate 47, which undergoes a subsequent anodic SET process to deliver thiyl radical 42.
To determine the mechanism of the generation of radical 42, we measured UV-vis absorption spectra to detect the light-absorbing species in the reaction mixture (Scheme 15c and SI, Section 9.3). Neither 1a nor 2a absorbed visible light in the operational range of 390–430 nm, which eliminated them as potent photoactive species (see SI, Fig. SI-5 and SI-6). The addition of HCOOCs to 1a provided a slight bathochromic shift of the band, but still the observed absorption was out of the investigated range of wavelengths (see SI, Fig. SI-6). The UV-vis studies also excluded the thiol 5ab as a photoactive species because its absorbance was also out of the operating light window (λmax 337 nm) (Scheme 15c). As already mentioned, arene thiolate anions may absorb visible light and participate in a photoredox cycle because their HOMO energy is higher than that of the neutral thiol.109 Indeed, UV spectra of deprotonated thiol 5ab, obtained by its treatment with Cs2CO3, showed a bathochromic shift of absorption to ca. 375 nm, along with an increase in absorption intensity (Scheme 15c). Although such absorbance onset overlaps with emission of the employed light source at 395 nm, it does not rationalize why less energetic blue light is also suitable to execute C–H alkenylation efficiently. The answer came after the measurement of the absorption spectrum of thiol 5ab after the addition of HCOOCs as a base instead of Cs2CO3. The recorded spectra showed an almost exact absorption curve with a characteristic red-shifted band at 375 nm (Scheme 15c). However, there was an additional weak band at ca. 425 nm, which was missed in the case of cesium thiolate UV-vis data. Finally, when cesium thiolate and HCOOCs were mixed together, the absorption curve was the same as the one for the thiol/HCOOCs mixture, along with similar green emission as shown in Scheme 15d. The appearance of the mentioned band at ca. 425 nm suggests that thiolate may also interact with formate anion(s) to give a certain type of donor–acceptor complex that is a photoactive species in the reaction. Noteworthily, the formate salt counterion has no influence on spectroscopic properties of UV spectra, and the same curve was obtained for the thiol mixed with Li, Na, K, Cs, and NH4 formates and tetra-n-butylammonium (TBA) formate (see SI, Fig. SI-12).
To confirm the formation of the excited species 47 and elucidate its role in the proposed mechanism, fluorescence quenching experiments were conducted using photoexcited anion 47. The addition of varying amounts of the potent quencher, like substrate 1a, to a mixture of thiol 5ab and HCOOCs in DMSO did not result in a decrease in emission at either 428 nm (excitation at 375 nm) or 527 nm (excitation at 425 nm). Instead, increasing the concentration of 1a led to an enhancement of emission intensity, suggesting that 1a acts as a fluorescence sensitizer or an energy donor, rather than as a quencher. While these findings do not rule out pathway B, they allow us to conclude that the excited anion 47 is not directly involved in the activation of starting materials 1a and 2a (see SI, Section 9.3.2).
As previously demonstrated, the C–H alkenylation of 1a also proceeded when thiol 5ab was replaced by disulfide 5bb, affording product 3aa in 51% yield (Scheme 14, entry 9). Given that disulfides can be readily prepared via oxidative coupling of thiols,110 we hypothesized that similar oxidative coupling could occur under our reaction conditions. Specifically, disulfide 45 is expected to undergo photolysis of its weak S–S bond, generating the key arene thiyl radical 42. Indeed, the MS and GC analyses of crude reaction mixtures from C–H alkenylation experiments conducted in the presence of thiol 5ab revealed the presence of disulfide 5bb, supporting our hypothesis. Furthermore, control experiments in which a DMSO solution of thiol 5ab was electrolyzed in the presence of TBABF4 as electrolyte confirmed the formation of 5bb under reaction conditions, which was isolated in 73% yield (see SI, Section 8.4).
As shown in Scheme 14, entry 2, the reaction performed in the absence of light yielded only trace amounts of the desired product, suggesting that the generation of the thiyl radical from disulfide via cathodic reduction (path C) either does not occur or proceeds at a rate where dimerization of the thiyl radical outpaces H-atom abstraction. Kinetic experiments appear to support the latter scenario. As shown in Scheme 15e, under dark conditions, substrate 1a undergoes slow conversion, forming primarily coupling byproducts. Simultaneously, the concentration of thiol 5ab decreases and stabilizes within approximately 6 hours, while the concentration of disulfide increases to a constant level during the same period. This suggests the establishment of an equilibrium between RSH and RSSR, resulting in insufficient levels of thiyl radical species to promote the designed reaction sequence. Notably, gas chromatography analysis showed only trace amounts (3%) of the desired product after 20 hours.
Upon irradiation of the reaction mixture, the kinetic profile changed significantly (Scheme 15f). In control experiments under blue light irradiation (without formate), the concentration of thiol 5ab rapidly decreased during the first 60 minutes and then gradually increased to a stable level after approximately three hours. Concurrently, the concentration of disulfide 5bb initially increased rapidly, then slightly decreased and stabilized after about three hours.
These observations support two possible hypotheses. The first posits that light irradiation shifts the thiol/disulfide equilibrium by promoting homolytic cleavage of the disulfide S–S bond, thereby generating thiyl radicals. While thiol dimerization continues, photolysis ensures a continuous supply of aryl thiyl radicals, facilitating the generation of CO2˙− and initiating the sequence of XAT, 1,5-HAT, and cross-coupling processes. Alternatively, the second hypothesis suggests that light activates an alternate pathway (such as path B), enabling rapid consumption of thiol, possibly through a rapid anodic oxidation of species 47 to radical 42. Simultaneously, both paths A and C may operate too, converting the excess of the reactive thiyl radicals into disulfide, which serves as a reservoir and retains their effective concentration to initiate all intended transformations, thereby enabling the formation of C(sp3)–H alkenylation products. Unfortunately, due to complexity of the entire system, it is not possible to judge undoubtedly which pathway, A or B, dominates; therefore, it seems to be more appropriate to assume that both paths operate simultaneously along with cathodic transformations of disulfide (path C).
The last issue was the explanation of partial (E/Z)-isomerization of the C–H alkenylation for some products, although, in all examined cases pure (E)-isomers of vinyl bromides were used. Since our protocol does not use any of the common metal-based or organic photocatalysts, we considered two possible scenarios of observed double bond isomerization: (1) direct excitation of a double bond to a diradical (path A) or (2) reactive species-promoted isomerization via either triplet energy transfer (path B)111,112 or an addition/elimination sequence (path C), as shown in Scheme 17a.104 Moreover, the isomerization can either be a primary or secondary process depending on whether it deals with a substrate, vinyl bromide, or product.
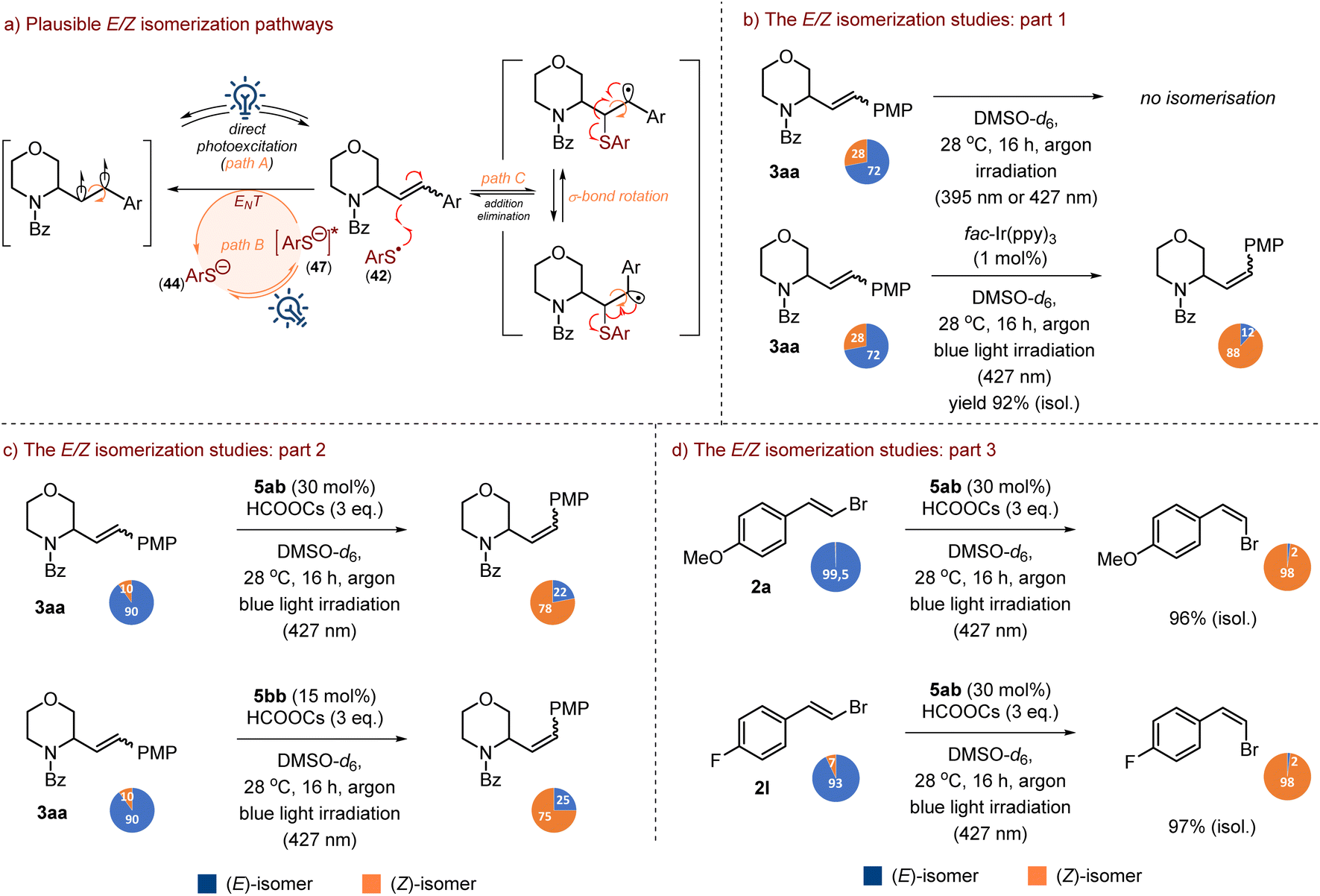 | ||
| Scheme 17 The E/Z isomerization studies. | ||
The initial control experiments, in which it was directly irradiated with either purple (395 nm) or blue light (427 nm) excluded path A, since blue (427 nm) or purple (395 nm) light irradiation of a solution of 3aa (E/Z ratio 72:28) in DMSO did not result in any change in the isomer ratio after 16 hours (Scheme 17b, eqn 1). In contrast, in the control experiment, we have added fac-Ir(ppy)3 to the solution of 3aa in DMSO, and the resulting mixture was irradiated with blue light (427 nm). After 16 hours, an initial (E/Z)-isomer ratio of 72:28 changed leading predominately to a contra-thermodynamic (Z)-isomer (E/Z ratio of 12:88) which was isolated in 92% yield (Scheme 17b, eqn 2).
Based on these two observations, additional control experiments were performed to determine the mechanism (path B or C) and identify active species involved in the isomerization process. In particular, we focused on the arene thiyl radical as a potent isomerisation agent.104 Blue light irradiation (427 nm) of a solution of (E/Z)-3aa, thiol 5ab (30 mol%) and HCOOCs (3 equiv.) in DMSO resulted in a significant change in the (E/Z)-ratio from 90:10 to 22:78, after 16 h, with a dominance of the (Z)-isomer (Scheme 17c, eqn 1). The same outcome was observed when thiol 5ab was replaced by disulfide 5bb (15 mol%). After standard time, 16 h, again the E/Z-isomer ratio changed from 90:10 to 25:75 (Scheme 17c, eqn 2). As can be seen from the data collected in Scheme 4, the tendency for the partial formation of type-(Z)-3 products was noticed only for mono-cyclic substrates 1, which may suggest that E-to-Z isomerisation of a double bond is easier for them due to their less steric hindrance and better accessibility to the double bond compared to fused systems. Furthermore, as disclosed in Scheme 8, the electronic nature of aryl groups of vinyl bromides did not have a significant influence on the (E/Z)-isomer ratio of the product, and partial isomerisation was observed for both electron-rich and electron-poor systems, without any strict trends. This again confirms our statement about the pivotal role of steric factors on the final isomer ratio. As further experiments revealed, the starting (E)-vinyl bromides, e.g., 2a and 2l, can also be isomerized to (Z)-isomers in the presence of thiol and formate salt (Scheme 17d, eqn 1 & 2). The isomerization proceeded smoothly for electron-rich systems, like 2a, and electron-deficient systems, such as bromide 2l.
It is important to emphasize that the isomerization our products, e.g., 3aa, occurred only in the presence of HCOOCs. Otherwise, the (E/Z)-isomer ratio became almost unchanged (see SI, Section 8.2). The same was observed in the cases when the mixture of (E/Z)-3aa and HCOOCs without thiol or disulfide was exposed to blue light irradiation. This corresponds to our UV-vis studies, which revealed that neither thiol 5ab nor disulfide 5bb absorbs the blue light with a wavelength of 427 nm. This again confirms the pivotal role of formate salt in the investigated process, not only as a precursor for the XAT reagent (CO2 radical anion) but also as an essential activator for the generation of the thiyl radical either from thiol or disulfide.
Analysis of the mass spectra of crude reaction mixtures from the isomerization experiments, as depicted in Scheme 17c and d, revealed the presence of very weak mass peaks potentially corresponding to aryl thiyl addition/hydrogen atom transfer (HAT) reaction products. The formation of these species appears to support our hypothesis regarding thiyl radical-induced double bond isomerization via an addition/elimination mechanism (path C) and is consistent with previous reports.104
To assess the plausibility of isomerization proceeding through the path B mechanism, fluorescence quenching experiments involving the excited thiolate species (47) were conducted. Emission spectra recorded upon excitation at 375 nm and 425 nm (see SI, Fig. SI-14) indicated that compound 3aa could act as a potent quencher of the excited species 47, although quenching was relatively weak, especially with excitation at a lower energy wavelength. This observation suggests that a significant interaction between species 47 and the product (or vinyl bromide) under the reaction conditions cannot be excluded, potentially facilitating double bond activation to a diradical species, for example, via an energy transfer process as illustrated in Scheme 17a.
Nevertheless, the limited amount of currently available data, along with the inherent complexity of the reaction system, precludes unequivocal and definitive proof of isomerization via path B. Therefore, we are more inclined to attribute the observed partial isomerization of the products to path C as the most probable mechanism.
Conclusions
In summary, we developed a general and mild photoelectrochemical method for α- or β-C(sp3)–H alkenylation of various readily available monocyclic or benzofused amines using a series of vinyl bromides. The established protocol accommodates a wide variety of amine scaffolds and vinyl bromides, demonstrates tolerance for different functional groups, exhibits adequate E/Z selectivity, and shows slight sensitivity to oxygen. An Ni complex catalyses the installation of an alkenyl fragment, which results in limited compatibility with moisture. Mechanistic studies suggest that the transformation relies on the formation of aryl thiyl radical species, which react with formate to yield CO2˙−. This species acts as an XAT reagent capable of generating an aryl radical from the corresponding aryl iodide. Subsequently, the aryl radical abstracts an H-atom from the amine fragment through intramolecular 1,5-HAT, resulting in the formation of an α-aminoalkyl radical, which is then intercepted by the Ni-alkenyl complex. Furthermore, we demonstrated that substituting the 2-iodobenzoyl HAT-directing group with its sulfonyl analogue facilitates a 1,6-HAT process, leading to β-alkenylated products. This method has shown significant applicability in the construction of complex heterocyclic scaffolds and in the late-stage functionalisation of bioactive molecules. Additionally, the resulting allyl- and homoallyl amine products can be readily transformed into various α- and β-functionalised cyclic amines, thereby facilitating the synthesis of numerous nitrogen-containing heterocyclic natural products and pharmacoactive compounds. Consequently, the reported procedure is anticipated to have numerous applications in synthetic organic chemistry and medicinal chemistry, including drug discovery studies.Methods
In a glove box, an oven dried ElectraSyn vial (A) (10 mL) equipped with a magnetic stir bar was charged with vinyl bromide (0.6 mmol, 1.0 equiv.), 2-halobenzamide (0.66 mmol, 1.1 equiv.), HCOOCs (1.8 mmol, 3.0 equiv.), methyl 4-mercaptobenzoate 5ab (0.12 mmol, 0.2 equiv.) and n-Bu4NBF6 (0.6 mmol, 1.0 equiv.). The vial was closed with a PTFE cap equipped with an anode (graphite) and a cathode (graphite). Another oven dried 5 mL vial (B) was charged with Ni(glyme)Br2 (0.06 mmol, 0.1 equiv.) and PyBOX ligand L8a (0.066 mmol, 0.11 equiv.). A magnetic stir bar was added and vial B was closed with a septa cap. Both vials were taken out from the glove box and kept under argon. Under argon 4 mL dry DMSO was added to both vials; 4 mL to the vial A and 2 mL to the vial B. The mixture in the vial B was continuously stirred at 60 °C for 10 min to prepare the pre–nickel complex. After that, solution of the Ni-complex was transferred from the vial B to the vial A. Now, the vial A was installed on an ElectraSyn stand and electrolysis was started (constant current 3 mA, time 12 h, and polarity of alternating electrodes was changed every 10 min) along with blue light irradiation using a Kessil™ LED lamp (427 nm, 5 cm away, with a cooling fan to keep the reaction temperature at 25 °C) for 12 h. After completion of the reaction, the reaction mixture was transferred to a separatory funnel; the electrodes were rinsed with ethyl acetate (5 mL) and water. The aqueous layer was extracted with ethyl acetate (3 × 20 mL). The combined organics were washed successively with brine (1 × 30 mL) and then dried over MgSO4 or Na2SO4. After removal of the drying agent, solvents and volatiles were removed on a rotary evaporator. The crude product was purified by automated flash chromatography. The obtained product was kept under high vacuum (10−3 bar) at 70 °C in the Kugelrohr apparatus for 10 h to remove the traces of solvents. The progress of the reaction was followed by TLC by using UV (254 nm) detection and vanillin stain. The E/Z-isomer ratio was determined by 1H NMR or GC analysis.Author contributions
Siddharth K. Dave: synthesis of starting materials, optimization studies, scope studies, application studies, characterization of chemical compounds, mechanism studies, manuscript preparation. Sebastian Stecko: project concept, preliminary studies, synthesis of selected compounds, scope studies, UV-vis, CV, fluorescence measurements, data analysis, manuscript preparation, mechanism studies, constructor of the required equipment.Conflicts of interest
The authors declare no conflict of interest.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: all experimental setup and details, materials used, the optimization process of remote C–H alkenylation of amines, mechanistic studies, synthetic procedures, the 1H, 13C, and 19F NMR characterization data of the products, equipment setup, optimisation studies, characterisation of organic compounds, photochemical studies, and copies of 1H, 13C, and 19F NMR spectra. See DOI: https://doi.org/10.1039/d5sc08559d.Acknowledgements
The authors are grateful to the National Science Center of Poland for financial support through a research grant (OPUS 2021/43/B/ST4/019115).References
- A.-j. Deng and H.-l. Qin, Phytochemistry, 2010, 71, 816–822 CrossRef CAS PubMed.
- P. Huang, P. Cheng, M. Sun, X. Liu, Z. Qing, Y. Liu, Z. Yang, H. Liu, C. Li and J. Zeng, Med. Plant Biol., 2024, 3, e020 Search PubMed.
- X. Luo, L. Pedro, V. Milic, S. Mulhovo, A. Duarte, N. Duarte and M.-J. U. Ferreira, Planta Med., 2012, 78, 148–153 CrossRef CAS PubMed.
- X. Luo, D. Pires, J. A. Aínsa, B. Gracia, N. Duarte, S. Mulhovo, E. Anes and M.-J. U. Ferreira, J. Ethnopharm., 2013, 146, 417–422 CrossRef CAS PubMed.
- Y. G. Xia, G. Y. Li, J. Liang, B. Y. Yang, S. W. Lü and H. X. Kuang, Evid. Based. Complement. Alternat. Med., 2014, 2014, 684508 CrossRef PubMed.
- S. G. Davies, A. M. Fletcher, I. T. T. Houlsby, P. M. Roberts, J. E. Thomson and D. Zimmer, J. Nat. Prod., 2018, 81, 2731–2742 CrossRef CAS PubMed.
- I. Jacquemond-Collet, F. Benoit-Vical, A. Valentin, E. Stanislas, M. Mallié and I. Fourasté, Planta Med., 2002, 68, 68–69 CrossRef CAS PubMed.
- A. J. Aladesanmi, C. J. Kelley and J. D. Leary, J. Nat. Prod., 1983, 46, 127–131 CrossRef CAS.
- E. Tojo, M. A. Önür, A. J. Freyer and M. Shamma, J. Nat. Prod., 1990, 53, 634–637 CrossRef CAS.
- S. Dayot, D. Speisky, A. Couvelard, P. Bourgoin, V. Gratio, J. Cros, V. Rebours, A. Sauvanet, P. Bedossa, V. Paradis, P. Ruszniewski, A. Couvineau and T. Voisin, Oncotarget, 2018, 9, 6952–6967 CrossRef PubMed.
- P. Malherbe, E. Borroni, E. Pinard, J. G. Wettstein and F. Knoflach, Mol. Pharm., 2009, 76, 618–631 CrossRef CAS PubMed.
- DrugBank.com: Iferanserin, https://go.drugbank.com/drugs/DB11686 Search PubMed.
- DrugBank.com: Encainide, https://go.drugbank.com/drugs/DB01228 Search PubMed.
- M. J. Antonaccio, A. W. Gomoll and J. E. Byrne, Cardiovasc. Drugs Ther., 1989, 3, 691–710 CrossRef CAS PubMed.
- R. L. Grange, E. A. Clizbe and P. A. Evans, Synthesis, 2016, 48, 2911–2968 CrossRef CAS.
- A. K. Mailyan, J. A. Eickhoff, A. S. Minakova, Z. Gu, P. Lu and A. Zakarian, Chem. Rev., 2016, 116, 4441–4557 CrossRef CAS PubMed.
- S. L. Rössler, D. A. Petrone and E. M. Carreira, Acc. Chem. Res., 2019, 52, 2657–2672 CrossRef PubMed.
- Q. Cheng, H.-F. Tu, C. Zheng, J.-P. Qu, G. Helmchen and S.-L. You, Chem. Rev., 2019, 119, 1855–1969 CrossRef CAS PubMed.
- O. Pàmies, J. Margalef, S. Cañellas, J. James, E. Judge, P. J. Guiry, C. Moberg, J.-E. Bäckvall, A. Pfaltz, M. A. Pericàs and M. Diéguez, Chem. Rev., 2021, 121, 4373–4505 CrossRef PubMed.
- Y. Huang, R.-Z. Huang and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 6571–6576 CrossRef CAS PubMed.
- B. M. Trost, C.-I. Hung and G. Mata, Angew. Chem., Int. Ed., 2020, 59, 4240–4261 CrossRef CAS PubMed.
- O. I. Afanasyev, E. Kuchuk, D. L. Usanov and D. Chusov, Chem. Rev., 2019, 119, 11857–11911 CrossRef CAS PubMed.
- R. A. Fernandes, P. Kattanguru, S. P. Gholap and D. A. Chaudhari, Org. Biomol. Chem., 2017, 15, 2672–2710 RSC.
- Y. Ichikawa, Synlett, 2007, 2007, 2927–2936 CrossRef.
- P.-A. Nocquet, S. Henrion, A. Macé, B. Carboni, J. M. Villalgordo and F. Carreaux, Eur. J. Org Chem., 2017, 2017, 1295–1307 CrossRef CAS.
- R. Karl Dieter and R. R. Sharma, Tetrahedron Lett., 1997, 38, 5937–5940 CrossRef.
- R. K. Dieter, C. M. Topping, K. R. Chandupatla and K. Lu, J. Am. Chem. Soc., 2001, 123, 5132–5133 CrossRef CAS PubMed.
- D. Antermite and J. A. Bull, Synthesis, 2019, 51, 3171–3204 CrossRef CAS.
- E. N. Bahena, S. E. Griffin and L. L. Schafer, J. Am. Chem. Soc., 2020, 142, 20566–20571 CrossRef CAS PubMed.
- L. Li, Y.-C. Liu and H. Shi, J. Am. Chem. Soc., 2021, 143, 4154–4161 Search PubMed.
- A. L. Krasovskiy, S. Haley, K. Voigtritter and B. H. Lipshutz, Org. Lett., 2014, 16, 4066–4069 Search PubMed.
- E. Bisz, M. Koston and M. Szostak, Green Chem., 2021, 23, 7515–7521 RSC.
- R. N. Shakhmaev, A. S. Sunagatullina and V. V. Zorin, Russ. J. Org. Chem., 2014, 50, 322–331 Search PubMed.
- M. Garbacz and S. Stecko, Org. Biomol. Chem., 2021, 19, 8578–8585 RSC.
- M. Garbacz and S. Stecko, Org. Biomol. Chem., 2023, 21, 115–126 Search PubMed.
- A. Noble and D. W. C. MacMillan, J. Am. Chem. Soc., 2014, 136, 11602–11605 CrossRef CAS PubMed.
- Y.-T. Wang, Y.-L. Shih, Y.-K. Wu and I. Ryu, Adv. Synth. Catal., 2022, 364, 1039–1043 CrossRef CAS.
- Z. Qing, H. Cao, P. Cheng, W. Wang, J. Zeng and H. Xie, Org. Chem. Front., 2018, 5, 353–357 RSC.
- M. Nechab, S. Mondal and M. P. Bertrand, Chem.–Eur. J., 2014, 20, 16034–16059 CrossRef CAS PubMed.
- W. Guo, Q. Wang and J. Zhu, Chem. Soc. Rev., 2021, 50, 7359–7377 RSC.
- L. Capaldo, D. Ravelli and M. Fagnoni, Chem. Rev., 2022, 122, 1875–1924 CrossRef CAS PubMed.
- L. Capaldo and D. Ravelli, Eur. J. Org Chem., 2017, 2017, 2056–2071 CrossRef CAS PubMed.
- W. Li and C. Zhu, Radically Initiated Distal C(sp3)–H Functionalization, in Remote C-H Bond Functionalizations, ed. D. Maiti and S. GuinWiley & Sons, New York 2021. Search PubMed.
- S. Sarkar, K. P. S. Cheung and V. Gevorgyan, Chem. Sci., 2020, 11, 12974–12993 Search PubMed.
- L. M. Stateman, K. M. Nakafuku and D. A. Nagib, Synthesis, 2018, 50, 1569–1586 CrossRef CAS PubMed.
- S. Sarkar, S. Wagulde, X. Jia and V. Gevorgyan, Chem, 2022, 8, 3096–3108 CAS.
- R. Guo, H. Xiao, S. Li, Y. Luo, J. Bai, M. Zhang, Y. Guo, X. Qi and G. Zhang, Angew. Chem., Int. Ed., 2022, 61, e202208232 CrossRef CAS PubMed.
- J. Wang, Q. Xie, G. Gao, H. Li, W. Lu, X. Cai, X. Chen and B. Huang, Org. Chem. Front., 2023, 10, 4394–4399 RSC.
- M. G. Pizzio, E. G. Mata, P. Dauban and T. Saget, Eur. J. Org Chem., 2023, 26, e202300616 CrossRef CAS.
- V. Srinivasu, K. Pal, S. Giri and D. Sureshkumar, Org. Lett., 2024, 26, 10328–10333 CrossRef CAS PubMed.
- Z. Lei, W. Zhang and J. Wu, ACS Catal., 2023, 13, 16105–16113 CrossRef CAS.
- X. Lin, H. Huang, F. Yang, Y. Ren, Y. Gao and W. Su, Org. Chem. Front., 2025, 12, 829–837 Search PubMed.
- J. Zheng, H. Zhang, S. Kong, Y. Ma, Q. Du, B. Yi, G. Zhang and R. Guo, ACS Catal., 2024, 14, 1725–1732 Search PubMed.
- H. Zhang, W. Gu, Q. Li, J. Zheng, J. Xiao, Y. Kang, W. Kandegama, X. Qi, G. Zhang and R. Guo, ACS Catal., 2025, 15, 15425–15434 CrossRef CAS.
- J. Lu, K. Yuan, J. Zheng, H. Zhang, S. Chen, J. Ma, X. Liu, B. Tu, G. Zhang and R. Guo, Angew. Chem., Int. Ed., 2024, 63, e202409310 Search PubMed.
- M. Garbacz and S. Stecko, Adv. Synth. Catal., 2020, 362, 3213–3222 CrossRef CAS.
- S. K. Kariofillis, B. J. Shields, M. A. Tekle-Smith, M. J. Zacuto and A. G. Doyle, J. Am. Chem. Soc., 2020, 142, 7683–7689 Search PubMed.
- A. Q. Cusumano, B. C. Chaffin and A. G. Doyle, J. Am. Chem. Soc., 2024, 146, 15331–15344 CrossRef CAS PubMed.
- X.-C. He, K.-R. Li, J. Gao, J.-P. Guan, H.-B. Chen, H.-Y. Xiang, K. Chen and H. Yang, Org. Lett., 2023, 25, 4056–4060 Search PubMed.
- T. Constantin, F. Juliá, N. S. Sheikh and D. Leonori, Chem. Sci., 2020, 11, 12822–12828 Search PubMed.
- Z. Li, W. Yan, X. Zhou, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2025, 12, 4946–4955 Search PubMed.
- D. Kobus-Bartoszewicz, M. Zalewski, K. Melcer, M. Warda and S. Stecko, Adv. Synth. Catal., 2025, 367, e202401326 Search PubMed.
- S. Wang, C. Yang, S. Sun and J. Wang, Chem. Commun., 2019, 55, 14035–14038 Search PubMed.
- Y. Lai, A. Halder, J. Kim, T. J. Hicks and P. J. Milner, Angew. Chem., Int. Ed., 2023, 62, e202310246 Search PubMed.
- A. A. Folgueiras-Amador, A. E. Teuten, M. Salam-Perez, J. E. Pearce, G. Denuault, D. Pletcher, P. J. Parsons, D. C. Harrowven and R. C. D. Brown, Angew. Chem., Int. Ed., 2022, 61, e202203694 Search PubMed.
- K. Mondal, S. Mallik, S. Sardana and M. Baidya, Org. Lett., 2023, 25, 1689–1694 Search PubMed.
- Y. Kwon, J. Lee, Y. Noh, D. Kim, Y. Lee, C. Yu, J. C. Roldao, S. Feng, J. Gierschner, R. Wannemacher and M. S. Kwon, Nature Commun., 2023, 14, 92 Search PubMed.
- A. F. Chmiel, O. P. Williams, C. P. Chernowsky, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2021, 143, 10882–10889 CrossRef CAS PubMed.
- Z. Li, W. Yan, X. Zhou, C. Yang, L. Guo and W. Xia, Org. Chem. Front., 2025, 12, 4946–4955 Search PubMed.
- L. W. Souza, N. D. Ricke, B. C. Chaffin, M. E. Fortunato, S. Jiang, C. Soylu, T. C. Caya, S. H. Lau, K. A. Wieser, A. G. Doyle and K. L. Tan, J. Am. Chem. Soc., 2025, 147, 18747–18759 CrossRef CAS PubMed.
- Kessil Lighting Ltd, Website. Kessil LED lamps Series PR160L specification for Kessil-PR160L-440nm, https://kessil.com/products/science_PR160L.php Search PubMed.
- K. Grudzień, A. Zlobin, J. Zadworny, K. Rybicka-Jasińska and B. Sadowski, Org. Chem. Front., 2024, 11, 5232–5277 RSC.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 Search PubMed.
- S. R. Waldvogel, S. Lips, M. Selt, B. Riehl and C. J. Kampf, Chem. Rev., 2018, 118, 6706–6765 Search PubMed.
- X.-Q. Zhou, P.-B. Chen, Q. Xia, T.-K. Xiong, X.-J. Li, Y.-M. Pan, M.-X. He and Y. Liang, Org. Chem. Front., 2023, 10, 2039–2044 Search PubMed.
- N. Kurono, E. Honda, F. Komatsu, K. Orito and M. Tokuda, Tetrahedron, 2004, 60, 1791–1801 Search PubMed.
- W. H. Koppenol and J. D. Rush, J. Phys. Chem., 1987, 91, 4429–4430 Search PubMed.
- Q. Shen, K. Cao, X. Wen and J. Li, Adv. Synth. Catal., 2024, 366, 4274–4293 CrossRef CAS.
- V. K. Simhadri, R. Sur and V. R. Yatham, Eur. J. Org Chem., 2024, 27, e202400490 Search PubMed.
- C. M. Hendy, G. C. Smith, Z. Xu, T. Lian and N. T. Jui, J. Am. Chem. Soc., 2021, 143, 8987–8992 CrossRef CAS PubMed.
- M. C. Maust, C. M. Hendy, N. T. Jui and S. B. Blakey, J. Am. Chem. Soc., 2022, 144, 3776–3781 Search PubMed.
- L. Zeng, J. Wang, D. Wang, H. Yi and A. Lei, Angew. Chem., Int. Ed., 2023, 62, e202309620 CrossRef CAS PubMed.
- A. Bisoyi, A. Behera, A. R. Tripathy, V. K. Simhadri and V. R. Yatham, J. Org. Chem., 2024, 89, 17818–17823 CrossRef CAS PubMed.
- Z.-Y. Ma, L.-N. Guo, Y. You, F. Yang, M. Hu and X.-H. Duan, Org. Lett., 2019, 21, 5500–5504 Search PubMed.
- A. L. G. Kanegusuku, T. Castanheiro, S. K. Ayer and J. L. Roizen, Org. Lett., 2019, 21, 6089–6095 CrossRef CAS PubMed.
- M. A. Short, J. M. Blackburn and J. L. Roizen, Synlett, 2020, 31, 102–116 Search PubMed.
- M. A. Short, M. F. Shehata, M. A. Sanders and J. L. Roizen, Chem. Sci., 2020, 11, 217–223 RSC.
- P. L. Gkizis, I. Triandafillidi and C. G. Kokotos, Chem, 2023, 9, 3401–3414 CAS.
- J. K. Mitchell, W. A. Hussain, A. H. Bansode, R. M. O'Connor, D. E. Wise, M. H. Choe and M. Parasram, Org. Lett., 2023, 25, 6517–6521 CrossRef CAS PubMed.
- D. E. Wise, E. S. Gogarnoiu, A. D. Duke, J. M. Paolillo, T. L. Vacala, W. A. Hussain and M. Parasram, J. Am. Chem. Soc., 2022, 144, 15437–15442 CrossRef CAS PubMed.
- A. Ruffoni, C. Hampton, M. Simonetti and D. Leonori, Nature, 2022, 610, 81–86 CrossRef CAS PubMed.
- K. Sun, Q.-Y. Lv, Y.-W. Lin, B. Yu and W.-M. He, Org. Chem. Front., 2021, 8, 445–465 RSC.
- C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- H. Zhang, C. Huang, X.-A. Yuan and S. Yu, J. Am. Chem. Soc., 2022, 144, 10958–10967 CrossRef CAS PubMed.
- M. Faizan, R. Kumar, A. Mazumder, Salahuddin, N. Kukreti, A. Kumar and M. V. N. L. Chaitanya, Chem. Biol. Drug Discov., 2024, 103, e14537 Search PubMed.
- R. R. Kumar, B. Sahu, S. Pathania, P. K. Singh, M. J. Akhtar and B. Kumar, ChemMedChem, 2021, 16, 1878–1901 CrossRef CAS PubMed.
- A. F. Brito, L. K. S. Moreira, R. Menegatti and E. A. Costa, Fund. Clin. Pharm., 2019, 33, 13–24 CrossRef CAS PubMed.
- S. Singh, R. Kumar, S. Tripathi, Salahuddin, A. Mazumder and N. Singh, Chem. Biol. Drug Discov., 2025, 105, e70077 CrossRef CAS PubMed.
- R. B. Clark and D. Elbaum, Tetrahedron, 2007, 63, 3057–3065 Search PubMed.
- Patent WO2003082877A1, 2003 Search PubMed.
- Patent WO2005026177A1, 2005 Search PubMed.
- US20050203296A1, 2005 Search PubMed.
- B. Ma, M. Ma, T. Zhang, D. Chen and J. Yang, J. Org. Chem., 2025, 90, 13643–13656 Search PubMed.
- F. Dénès, M. Pichowicz, G. Povie and P. Renaud, Chem. Rev., 2014, 114, 2587–2693 Search PubMed.
- W. Xiao, J. Zhang and J. Wu, ACS Catal., 2023, 13, 15991–16011 CrossRef CAS.
- Y. Dang, J. Han, A. F. Chmiel, S. N. Alektiar, M. Mikhael, I. A. Guzei, C. S. Yeung and Z. K. Wickens, J. Am. Chem. Soc., 2024, 146, 35035–35042 Search PubMed.
- Y. Yuan, J.-C. Qi, D.-X. Wang, Z. Chen, H. Wan, J.-Y. Zhu, H. Yi, A. D. Chowdhury and A. Lei, CCS Chem., 2022, 4, 2674–2685 Search PubMed.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
- C. Liu, K. Li and R. Shang, ACS Catal., 2022, 12, 4103–4109 Search PubMed.
- G. Laudadio, N. J. W. Straathof, M. D. Lanting, B. Knoops, V. Hessel and T. Noël, Green Chem., 2017, 19, 4061–4066 RSC.
- T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
| This journal is © The Royal Society of Chemistry 2026 |