
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterogeneous photocatalytic C4 remote fluorosulfonamidation of pyridines
Panjie
Xiang
a,
Kai
Sun
 *a,
Xiaolan
Chen
*a,
Kang
Li
ab,
Xi
Chen
c,
Yunkai
Zhang
a,
Lingbo
Qu
ac and
Bing
Yu
*a
*a,
Xiaolan
Chen
*a,
Kang
Li
ab,
Xi
Chen
c,
Yunkai
Zhang
a,
Lingbo
Qu
ac and
Bing
Yu
*a
aCollege of Chemistry, Zhengzhou University, Zhengzhou 450001, China. E-mail: sunkaichem@zzu.edu.cn; chenxl@zzu.edu.cn; bingyu@zzu.edu.cn
bNational Engineering Research Center of Low-Carbon Processing and Utilization of Forest Biomass, Nanjing Forestry University, Nanjing 210037, China
cInstitute of Chemistry, Henan Academy of Sciences, Zhengzhou 450002, China
First published on 23rd January 2026
Abstract
N-Fluorosulfamoyl pyridinium salts have been ingeniously developed as redox-active precursors for generating fluorosulfamoyl radicals. However, the ubiquitous pyridine scaffold within these compounds, commonplace in numerous pharmaceuticals, has remained being wasted in reported methods only as a radical leaving group. This underutilization presents both a significant challenge and an exciting opportunity for further research in synthetic, medicinal, and materials chemistry. Herein, N-fluorosulfamoyl pyridinium salts were employed as bifunctional reagents to achieve remote and selective C–H functionalization of pyridines through a radical relay process that enables polarity reversal of radical intermediates. By employing ZnIn2S4 as a heterogeneous photocatalyst, this protocol enables the mild and efficient incorporation of both fluorosulfamoyl and pyridyl groups into alkenes, affording synthetically valuable fluorosulfamoyl pyridine derivatives in moderate to good yields. These transformations feature broad substrate scope, good functional group tolerance, and their synthetic utility is further demonstrated through late-stage functionalization of complex bio-relevant molecules. Importantly, the ZnIn2S4 photocatalyst displays exceptional recyclability, retaining high catalytic efficiency over five consecutive cycles. This work represents the first successful application of a heterogeneous photocatalyst compatible with N-fluorosulfamoyl pyridinium salts and N-aminopyridinium salts, effectively overcoming the longstanding challenge of catalyst recovery encountered in previous systems.
Introduction
Since the introduction of sulfur(VI) fluoride exchange (SuFEx) reactions by Sharpless and coworkers,1 sulfonyl fluorides have rapidly gained prominence as highly valuable “clickable” reagents.2 The unique reactivity–stability balance provided by the S–F bond imparts sulfonyl fluorides with rapid and selective nucleophilic reactivity, while simultaneously maintaining exceptional stability under aqueous and physiological conditions.3 The exceptional characteristics confer sulfonyl fluorides with unparalleled versatility, establishing them as indispensable tools not only for the synthesis and modification of small molecules,4 polymers,5 and advanced materials,6 but also for the intricate rational design of selective enzyme inhibitors and activity-based probes (Fig. 1a).7 Despite their remarkable potential, the broader application of sulfonyl fluorides has been limited by the restricted diversity and accessibility of sulfonyl fluoride scaffolds, highlighting the urgent need for novel and efficient synthetic strategies to expand the SuFEx toolbox.8 | ||
| Fig. 1 (a) Significance of sulfonyl fluoride and pyridine; (b) photoredox pathways for C4 functionalization of pyridines; (c) reaction design strategy; (d) the transformation of NFSAP salts. | ||
Pyridines and related nitrogen-containing heterocycles are widely present in biologically active compounds, with pyridine ranking as the most abundant heteroaromatic ring among FDA-approved medications (Fig. 1a).9 In recent years, visible-light-driven photoredox catalysis has significantly advanced the construction of complex molecular frameworks, exploiting unique redox pathways to generate value-added molecules under mild and sustainable conditions.10 In this context, the Minisci reaction, which involves the addition of carbon-centered nucleophilic radicals to protonated azines, remains a fundamental method for functionalizing pyridines (Fig. 1b, path (i)). However, this reaction often faces challenges due to competing regioselectivity at the C2 and C4 positions. For photocatalytic C4 derivatization, pyridines substituted with 4-cyano or 4-triphenylphosphonium groups are particularly prone to ipso substitution after undergoing single-electron reduction (Fig. 1b, path (ii)). Alternatively, C4-selective functionalization can be achieved by introducing a blocking group on the nitrogen of the pyridine, effectively directing the subsequent Minisci-type transformation (Fig. 1b, path (iii)).11 Despite advancements in the field, the photochemical C4 functionalization of pyridine remains underexplored.
Driven by the privileged role of sulfamoyl fluorides in drug discovery and the ubiquity of pyridine cores in functional materials, we set out to develop a site-selective, radical-based fluorosulfonamidation of pyridines that proceeds through pyridinium salts (Fig. 1c). The endeavor confronts a fundamental polarity paradox: the N-centered fluorosulfonamidyl radical (˙NSO2F) is intrinsically electrophilic and therefore favors electron-rich partners, whereas pyridine, whether neutral or in its cationic salt form, is rendered electron-deficient by the strongly electron-withdrawing nitrogen atom.12 This electrophilic-radical/electron-poor-substrate mismatch erects a formidable thermodynamic barrier to conventional direct radical addition.13 To address this issue, we proposed a radical relay strategy to achieve dynamic polarity inversion of the radical species: first converting the electrophilic ˙NSO2F radical into a nucleophilic C-centered radical, which then selectively reacts with the electron-deficient pyridinium salt. In recent years, independent reports by the groups of Wang,14 Liao,15 and Weng16 employed N-fluorosulfamoyl pyridinium salts (NFSAPs) as robust and efficient photoredox-active precursors for generating fluorosulfamoyl radicals, thereby enabling direct radical fluorosulfonamidation of alkenes and heterocycles (Fig. 1d, path (i)). However, the pyridine motif embedded within NFSAPs has been largely underutilized in reported transformations, typically serving only as a transient radical leaving group, rather than being incorporated into the target molecules. Furthermore, existing photocatalytic methodologies predominantly rely on homogeneous transition metal complexes or organic dyes as visible-light absorbers.17 These homogeneous photoredox catalysts frequently suffer from limited compatibility with highly acidic or basic media, strong nucleophiles, electrophiles, or highly reactive radical intermediates, thereby restricting their utility in diverse synthetic settings.18 Additionally, challenges associated with recycling and reusing these catalysts remain unsolved, substantially hindering their potential in sustainable chemical synthesis.
In contrast, heterogeneous catalysts offer several compelling advantages, including facile separation from the reaction medium, enhanced recyclability, and a reduced risk of catalyst contamination in the final products.19 These attributes are particularly advantageous for large-scale and sustainable processes, where catalyst recovery and reusability are critical considerations.20 Among heterogeneous photocatalysts, metal–sulfur compounds, particularly indium zinc sulfide (ZnIn2S4), have attracted considerable attention, owing to their favorable attributes such as non-toxicity, strong visible-light absorption, appropriate bandgap structure, and tunable morphology. Inspired by these advantages, we employed NFSAPs as bifunctional reagents that simultaneously supply the aminosulfonyl fluoride motif and the pyridine core (Fig. 1d, path (ii)). Under ZnIn2S4-photocatalytic conditions, the in situ generated radical ˙NSO2F undergoes addition with an alkene, achieving a polarity switch from electrophilic to nucleophilic character, followed by selective addition to the NFSAP salts in the system. The sequence culminates in remote C4-selective aminosulfonyl fluorination of the pyridine ring. By orchestrating a radical-relay pathway that inverts radical polarity, the strategy neutralizes the intrinsic mismatch between electrophilic radicals and electron-deficient pyridinium salts, offering a powerful new platform for the precision editing of heteroaromatic systems. Importantly, ZnIn2S4 demonstrates exceptional recyclability, maintaining high catalytic activity over at least five consecutive cycles. This feature effectively circumvents the principal limitations associated with catalyst recovery in previous photoredox systems, underscoring the promising potential of ZnIn2S4-based heterogeneous catalysis in sustainable organic synthesis.
Results and discussion
Reaction optimization
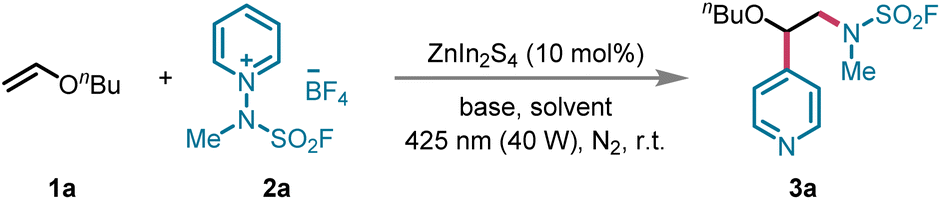
Our investigation began with the selection of n-butyl vinyl ether 1a and N-fluorosulfamoyl pyridinium salt 2a as model substrates to optimize the reaction conditions. The initial attempt employed ZnIn2S4 (10 mol%) as the photocatalyst and Na3PO4 (2.5 equiv.) as the base in CH3OH, resulting in the formation of the desired product 3a in only 14% yield (Table 1, entry 1). To improve the reaction efficiency, we systematically evaluated several solvents, including ethyl acetate (EA), dimethyl sulfoxide (DMSO), dichloromethane (DCM), 1,2-dichloroethane (DCE), acetonitrile (CH3CN), and acetone (entries 2–7). Among these, acetone proved to be the most effective solvent, enhancing the yield of 3a to 57% (entry 7). Subsequently, we investigated the influence of various bases on the reaction, testing a range of inorganic bases such as Na2HPO4, Na2CO3, NaHCO3, and K2HPO4, along with organic bases like 1,4-diazabicyclo[2.2.2]octane (DABCO) and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (entries 8–13). Among these, NaHCO3 emerged as the most efficient, significantly increasing the yield of 3a to 71% (entry 10). We also optimized the reaction time and the amount of NaHCO3, but these adjustments did not result in any further improvements in the yield (entries 14–20). Control experiments conducted in the absence of the photocatalyst, base, or light demonstrated a marked reduction in the yield of 3a, thereby underscoring the indispensable roles of these components in facilitating the transformation (entries 21 and 22). The optimized reaction conditions are as follows: 1a (0.1 mmol), 2a (3.0 equiv.), ZnIn2S4 (10 mol%) as the photocatalyst, NaHCO3 (2.5 equiv.) as the base in acetone (0.1 M), under a nitrogen atmosphere, irradiated by blue light (425 nm) for 3 h.|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | Time (h) | Yield (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (3.0 equiv.), ZnIn2S4 (10 mol%) and solvent (0.1 M) were irradiated with 425 nm blue LED (40 W) at room temperature under N2 atmosphere. Yields were given by 1H NMR using 1,1,2,2-tetrachloroethane as an internal standard based on 1a. b Isolated yield. c 1.5 equiv. of NaHCO3. d 20 equiv. of NaHCO3. e 3.0 equiv. of NaHCO3. f No catalyst or no light. | ||||
| 1 | CH3OH | Na3PO4 | 3 | 14 |
| 2 | EA | Na3PO4 | 3 | 8 |
| 3 | DMSO | Na3PO4 | 3 | Trace |
| 4 | DCM | Na3PO4 | 3 | 7 |
| 5 | DCE | Na3PO4 | 3 | 7 |
| 6 | CH3CN | Na3PO4 | 3 | 20 |
| 7 | Acetone | Na3PO4 | 3 | 57 |
| 8 | Acetone | Na2HPO4 | 3 | Trace |
| 9 | Acetone | Na2CO3 | 3 | 26 |
| 10 | Acetone | NaHCO3 | 3 | 71 (65)b |
| 11 | Acetone | KHCO3 | 3 | 25 |
| 12 | Acetone | DABCO | 3 | 6 |
| 13 | Acetone | DBU | 3 | N.D. |
| 14 | Acetone | NaHCO3 | 1 | 63 |
| 15 | Acetone | NaHCO3 | 2 | 66 |
| 16 | Acetone | NaHCO3 | 4 | 58 |
| 17 | Acetone | NaHCO3 | 5 | 55 |
| 18c | Acetone | NaHCO3 | 3 | 59 |
| 19d | Acetone | NaHCO3 | 3 | 51 |
| 20e | Acetone | NaHCO3 | 3 | 57 |
| 21f | Acetone | NaHCO3 | 3 | N.D. |
| 22 | Acetone | — | 3 | 16 |
Substrate scope
Having optimized the reaction conditions, we next explored the substrate scope, testing a wide range of alkenes and pyridines. The reaction showed excellent reactivity with electron-rich alkenes such as vinyl ethers and enamides, leading to efficient formation of β-aminopyridine derivatives with good C4-regioselectivity. Additionally, the transformation proved to be highly versatile, working well with aliphatic olefins of various chain lengths and cycloolefins, all of which showed good reactivity and compatibility (Scheme 1, 3a–3d). Substrates bearing polar functional groups, including hydroxyl and chloro moieties, were also well tolerated, providing the desired products in good yields (3e and 3f). The methodology was further applied to internal olefins, yielding the desired difunctionalized products 3g and 3h in moderate yields, albeit with slightly reduced efficiency. The approach also demonstrated significant flexibility, allowing the reaction to be extended to vinyl amides, formamides, and carbamates, resulting in products 3i–3l in yields ranging from 37% to 61%. Moreover, pyridine derivatives with electron-donating groups such as 2-methyl, 2-methoxy, 2-phenyl, and 2-benzyl groups, as well as those with electron-withdrawing groups like 2-acetyl and 2-carboxymethyl (2-COOMe), all participated effectively in the reaction, yielding products 3m–3r in moderate to excellent yields (35–68%). Even bicyclic pyridinium substrates gave good results, leading to products 3s and 3t in 96% and 69% yields. The lower yield of 3t relative to 3s is primarily ascribed to the more pronounced steric hindrance of the six-membered ring in 3t compared to the five-membered ring in 3s. This steric discrepancy between the two ring systems directly affects the spatial accessibility of the reaction site, thereby regulating the reaction efficiency (for details see SI). Notably, pyridine bearing a 2-thienyl group underwent concurrent sulfonyl fluorination at both heteroaromatic rings, resulting in product 3u. It was also possible to expand the scope beyond the methyl substituent of N-fluorosulfamoyl pyridinium salt to include other groups, such as ethyl, resulting in the formation of the corresponding product 3v with a yield of 65%. Afterward, we examined the N-methyl-free fluorosulfamoyl pyridinium salt under our standard photocatalytic conditions. This substrate was completely unreactive, which might be caused by the more negative reduction potential (for details, see the SI). To further evaluate the generality of our method, we investigated quinolinium and isoquinolinium salts. Quinolinium salts proved amenable to our standard conditions, delivering the remote quinoline sulfonyl fluoride product 3w in 32% yield. In contrast, isoquinolinium salts failed to react, likely due to the lack of a suitable 4-position reaction site analogous to that in pyridinium and quinolinium systems (for details see the SI). | ||
| Scheme 1 The scope of alkenes and pyridinium salts. Reaction conditions: 1 (0.1 mmol), 2 (3.0 equiv.), ZnIn2S4 (10 mol%), NaHCO3 (2.5 equiv.) and acetone (0.1 M) were irradiated with 40 W blue LED (425 nm) at room temperature under N2 atmosphere for 3 h. a1 (3.0 equiv.), 2 (0.1 mmol); bone-pot synthesis; c1 (0.1 mmol), 2 (6.0 equiv.). | ||
Late-stage functionalization has emerged as a groundbreaking approach in contemporary organic synthesis, providing a streamlined and direct pathway to architecturally intricate molecules with enhanced functionalities.21 This rapid synthesis method for highly complex compounds opens up significant opportunities across various domains, such as drug discovery, advanced materials science, and cutting-edge molecular imaging technologies.19a,22 To further underscore the synthetic versatility of this methodology, we investigated its application in modifying bioactive molecules. A diverse array of drug-derived alkenes, such as DL-menthol, nortropine, L-(−)borneol, and diacetone-D-glucose, were successfully employed as compatible substrates, yielding the desired products 3x–3aa in moderate to high yields. Additionally, bioactive compounds, including ibuprofen piconol, nikethamide, nicotinyl alcohol, loratadine, and pyriproxyfen, were seamlessly integrated into the reaction, furnishing the corresponding products 3ab–3af. To gain deeper insights into the reaction's sensitivity to varying conditions, we conducted a series of experiments to assess its reproducibility. We systematically varied a range of factors, including concentration, oxygen levels, water content, light intensity, and reaction scale, both positively and negatively relative to the standard reaction conditions (see SI for further details). As shown in Scheme 1, it revealed that the inert gas atmosphere had the most significant impact on reaction reproducibility, with optimal performance observed under a nitrogen atmosphere. The reaction also exhibited moderate sensitivity to light intensity. In contrast, factors such as concentration and humidity had minimal influence on the outcome.
Encouraged by the promising experimental results, we further extended our investigation by substituting N-fluorosulfamoyl pyridinium salts with N-benzene sulfonyl pyridinium salts to broaden the substrate scope of the heterogeneous photocatalytic system (Scheme 2). Notably, aminopyridylation of alkenes was achieved using a variety of N-aminopyridinium salts as dual aminating and pyridylating reagents. A wide range of alkyl (e.g., butyl, ethyl, propyl, cyclohexyl), aryl, and distal chloro- or hydroxy-substituted alkenyl ethers, as well as cyclic enol ethers, underwent efficient transformations, leading to the corresponding target products 5a–5g in moderate to good isolated yields. Notably, NaHCO3 is also an effective base for this reaction, affording 5a in 66% yield. Furthermore, vinyl amide derivatives, including N-vinylformamide, N-vinylacetamide, N-vinyl-2-pyrrolidone, and N-vinylcaprolactam, exhibited smooth reactivity under the established photochemical conditions, resulting products 5h–5k in acceptable yields, respectively. Additionally, a series of pyridine substrates with ortho- and meta-substituents on the aromatic ring, such as methyl, methoxy, phenyl, benzyl, and ester groups, were well-tolerated, yielding the desired products 5n–5t in yields ranging from 40% to 74%. Moreover, N-benzene sulfonyl pyridinium salts derived from 2,6-dimethylpyridine, cyclopenta[b]pyridine, and 5,6,7,8-tetrahydroquinoline were also viable substrates, successfully yielding the corresponding target compounds 5u–5w.
 | ||
| Scheme 2 The scope of alkenes and pyridinium salts. Reaction conditions: 1 (0.1 mmol), 2 (3.0 equiv.), ZnIn2S4 (10 mol%), Na3PO4 (2.5 equiv.), and acetone (0.1 M) were irradiated with 40 W blue LED (425 nm) at room temperature under N2 atmosphere for 3 h. aNaHCO3 as base. | ||
To further validate the feasibility of the experimental approach, gram-scale synthesis was performed using a specially designed continuous-flow fixed-bed photoreactor (Fig. 2a). Leveraging the heterogeneous nature of ZnIn2S4 as a photocatalyst, the reactor was loaded with 0.25 g of ZnIn2S4 and 1.26 g of NaHCO3 supported on 50.00 g of commercially available silica microspheres (0.80 mm diameter). The system operated at a flow rate of 3.00 mL min−1, corresponding to a residence time of 100 seconds within a 5.00 mL reactor volume, enabling efficient substrate conversion. Under simultaneous irradiation by two Kessil lamps (427 nm, 40 W each, providing an irradiance of 399.00 nW cm−2 at an 8.00 cm distance), a total of 1.14 g of the desired product 3a was obtained, corresponding to a 65% yield from 6 mmol of substrate 1a. These results confirm the efficacy of the photoreactor design for gram-scale synthesis employing ZnIn2S4 as the photocatalyst. The heterogeneous ZnIn2S4 photocatalyst could be conveniently recovered via centrifugation and reused for at least five cycles without appreciable loss of catalytic activity (Fig. 2b). Scanning electron microscopy (SEM) analysis of the recovered catalyst revealed no significant changes in overall morphology compared to the fresh material, indicating that the primary structural framework remained intact (Fig. S6). However, closer examination revealed small particles on the surface of the recovered catalyst, likely arising from minor deposition of slight agglomeration, or adsorption of trace impurities. While the macroscopic structure is well preserved, these surface modifications could block active sites or disrupt light–matter interactions, which reasonably explains the slight decline in catalytic efficiency upon recycling. This excellent stability was further corroborated by XRD analysis, which showed that the crystal structure remained unchanged after catalytic cycling (Fig. 2c). Collectively, these findings underscore the robust recyclability and structural integrity of ZnIn2S4, highlighting its potential for sustainable applications.
 | ||
| Fig. 2 Synthetic applications. (a) Scale-up experiments. (b) Recycle experiments. (c) XRD comparison of the recovered material, fresh ZnIn2S4 and PDF of ZnIn2S4. (d) Diversification of sulfamoyl fluorides. | ||
The S(VI)–F bond in sulfamoyl fluorides exhibits exceptional synthetic versatility, serving as a crucial functional handle for a variety of SuFEx-mediated transformations. Therefore, we exploited the S–F bond of the synthesized compound 3a to investigate efficient coupling reactions with diverse nucleophiles, including phenols, amines, and imidazole derivatives. As depicted in Fig. 2d, compound 3a underwent nucleophilic substitution with alcohol-based pharmaceuticals such as estrone and sesamol, affording the corresponding products 6a and 6b in isolated yields of 70% and 39%, respectively. Moreover, imidazole and imidazole-containing pharmaceuticals, exemplified by detomidine hydrochloride, served effectively as nucleophiles, enabling the synthesis of products 6c and 6d with high isolated yields of 95% and 90%. Additionally, amine-containing drug molecules, such as phenylpiperazine and vortioxetine, reacted smoothly with the S(VI)–F bond, furnishing products 6e and 6f in excellent isolated yields of 95% and 96%, respectively. Furthermore, the synthetic utility of this approach was demonstrated through a base-mediated elimination of aminosulfonyl fluoride from compound 3a in tetrahydrofuran, affording product 6g with a yield of 58%. These results highlight the practical applicability of the S(VI)–F bond in facilitating diverse and efficient transformations, underscoring its significant potential in pharmaceutical chemistry and the development of novel therapeutic agents.
Mechanism
To gain deeper mechanistic insight into the reaction pathway, a comprehensive series of experiments was performed. Initial control experiments demonstrated that the addition of radical scavengers, specifically (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) and 2,6-di-tert-butyl-4-methylphenol (BHT), under standard conditions completely inhibited the formation of the desired product, implicating radical intermediates in the transformation. Moreover, the introduction of K2S2O8, serving as an electron scavenger, or ammonium oxalate, a hole scavenger, resulted in a marked reduction in the yield of product 3a, underscoring the critical roles of both photogenerated electrons and holes derived from the semiconductor photocatalyst in facilitating the reaction progress. Furthermore, the addition of CuCl, a known single-electron transfer (SET) inhibitor, fully suppressed the reaction, highlighting the indispensable contribution of SET processes in this photocatalytic system (Fig. 3a). Stern–Volmer fluorescence quenching studies further substantiated the mechanistic hypothesis. The excited state of the photocatalyst underwent significant quenching in the presence of compound 2a, while no appreciable quenching was detected with compound 1a, suggesting a selective interaction likely involving SET between the photocatalyst and compound 2a (Fig. 3b). To further elucidate the photochemical nature of the reaction, light “on–off” experiments were conducted. The reaction mixture was subjected to alternating cycles of LED irradiation, switching the light on and off every 20 minutes over a total period of 2 h. As depicted in Fig. 3c, product formation notably increased during the illumination intervals. Interestingly, increases in yield were also observed during the dark periods, implying the involvement of a radical chain propagation mechanism. This conclusion was further supported by the determination of a quantum yield of 14 (for details, see the SI), confirming that radical chain processes significantly contribute to the overall reaction pathway. Subsequently, to evaluate whether the pyridine species formed in situ could act as a substrate, mixtures of compound 2a and 2-phenylpyridine were subjected to the standard conditions. The reaction proceeded exclusively with the pyridinium salt 2a, whereas the pyridine derivative showed no reactivity, indicating that free pyridine does not participate in the transformation (Fig. 3d). The C-4 blocked experiment demonstrated that no desired product was obtained when the N-fluorosulfamoyl pyridinium salt, with a –OMe group at the C-4 position of the pyridine ring, was used (Fig. 3e). Cyclic voltammetry analysis showed that the reduction potential of compound 1a (Ered1/2 = −0.59 V vs. Ag/AgCl) is appropriate for reduction by ZnIn2S4 (Fig. 3f and h). Finally, kinetic isotope effect (KIE) experiments were conducted to identify the rate-limiting step. The observed competitive KIE value of 1.7 suggests that C–H bond cleavage in N-fluorosulfamoyl pyridinium salts is not the rate-determining step of the overall transformation (Fig. 3g). | ||
| Fig. 3 Mechanism studies. (a) Control experiments. (b) Stern–Volmer fluorescence-quenching experiments. (c) Light on–off experiments. (d) Competitive experiment. (e) C-4 blocked salt experiment. (f) Cyclic voltammetry of 2a. (g) Kinetic isotope effect study. (h) Proposed mechanism. | ||
Based on our experimental results and consistent with previous studies,11g a plausible mechanistic pathway is proposed and illustrated in Fig. 3h. Upon visible light irradiation, the photocatalyst ZnIn2S4 is excited to generate electron–hole pairs, producing conduction band (CB) electrons and valence band (VB) holes. The photogenerated electrons in the CB undergo a single electron transfer (SET) process with pyridinium salts, resulting in the formation of free pyridines and sulfamoyl fluoride radical intermediate A. Subsequently, radical intermediate A adds to alkene 1a, affording carbon-centered radical species B. Intermediate B then reacts with another equivalent of pyridinium salts to generate radical intermediate C. The base facilitates deprotonation of intermediate C, which undergoes N-heteroatom bond cleavage to yield the final product while concurrently regenerating radical intermediate A, thus propagating the radical chain reaction. Moreover, to maintain catalyst turnover, an alternative pathway is proposed involving rearomatization and reduction via SET events within the photoredox catalytic cycle. In both pathways, the base plays a crucial role in the deprotonation steps essential for product formation.
In addition, we applied density functional theory (DFT) to calculate the factors governing the catalytic mechanism and regioselectivity during this transformation process. The photocatalytic cycle begins with excitation of the photocatalyst, followed by single-electron transfer to 2a, generating the transient N-pyridyl radical 2a′. Subsequent homolytic N–N bond cleavage releases pyridine while forming the key N-centered radical intermediate A. Computational analysis reveals this step has a moderate activation barrier of 6.40 kcal mol−1 and is strongly exergonic (ΔG = −23.00 kcal mol−1), rendering it effectively irreversible. For radical A, we identified two competing pathways: direct addition to either the alkene 1a or to a second equivalent of the pyridinium 2a. Surprisingly, the attack on 2a is both kinetically and thermodynamically unfavorable, requiring an energy barrier of 27.20 kcal mol−1 (via transition state TSAp) to form the putative intermediate Bp, which lies 12.10 kcal mol−1 higher in energy than A (Fig. 4, red dotted line). Computations reveal that the N-centered radical A instead adds smoothly to the olefin, surmounting a mere 8.10 kcal mol−1 barrier and releasing 10.80 kcal mol−1 to furnish radical B. We also examined the alternative α-position addition pathway; the corresponding transition state (TSAa) lies −11.1 kcal mol−1, which is higher in energy than the transition state for the preferred pathway (TSA). The α C–N bond-forming route is energetically less favorable and does not compete significantly under the reaction conditions (Fig. 4, purple dotted line). For the reaction to proceed, alkyl radical B attacks the pyridinium salt species, first forming the unstable cationic radical intermediate C (a process with a low energy barrier), which then rapidly deprotonates to generate the more stable neutral radical D. This proton transfer step is expected to be barrierless in solution and was not subjected to explicit transition state optimization. The final step of the catalytic cycle involves the homolytic cleavage of the N–N bond, producing the target product 3a while releasing radical intermediate A. Notably, theoretical calculations indicate that the difference in the energy barriers of the radical addition transition states from B to C is the primary controlling factor for the regioselectivity of the reaction. Although the two regioisomers Cp and Co exhibit nearly identical free energies (−30.10 vs. −29.00 kcal mol−1, respectively), the transition state TSBp leading to the C4-intermediate Cp is energetically favored by 2.60 kcal mol−1 over the C2-pathway alternative, showing excellent agreement with the experimentally observed regioselectivity.
 | ||
| Fig. 4 Computational studies. | ||
As depicted in Fig. 5a, the frontier orbitals reveal a decisive energy pattern: the SOMO of radical A sits at −8.81 eV, while the π and π* orbitals of the olefin lie at −6.42 and +0.25 eV, respectively; the analogous orbitals of 2a are located at −9.21 and −3.50 eV. As highlighted in blue in Fig. 5a, the SOMO of radical A is energetically aligned with the occupied π orbital of olefin substrate 1a, resulting in a stabilizing 2-orbital–3-electron interaction that renders the N-centered radical electrophilic toward the olefin. In contrast (red highlight, Fig. 5a), the positively charged pyridinium substrate 2a exhibits substantially lowered orbital energies, with its π* orbital becoming the frontier orbital closest in energy to the SOMO of A. Consequently, these two MOs give a 2-orbital–1-electron interaction, and the radical acts as a nucleophile towards the pyridinium substrate. The interaction strength is controlled by both orbital overlap and the energy gap between participating orbitals: larger overlap and smaller energy gap lead to stronger interactions. The energy difference for olefin 1a is 2.39 eV between interacting frontier orbitals, compared to 5.31 eV for the pyridinium substrate. Furthermore, the π orbital of the olefin exhibits greater localization than the π* orbital of the pyridinium, resulting in superior orbital overlap with the radical. Both factors synergistically favor radical–olefin interactions, rationalizing the observed transition state and intermediate energetics.
 | ||
| Fig. 5 MO diagram and distortion–interaction analysis. (a) Qualitative MO diagram of N-centered radical interaction with the enol ether 1a or the pyridinium salt 2a. Energies are given in eV. (b) Distortion–interaction analysis of TSBo and TSBp. [2a] and [B] indicate the structural distortion energy of radical and pyridinium fragments from the corresponding intermediates 2a and B to each transition state geometry; Natural Population Analysis (NPA) was performed using the NBO 7.0 program to obtain NPA atomic charges. | ||
To establish the intrinsic reactivity of the pyridinium substrate, we conducted a comprehensive Fukui function analysis. The Fukui indices (f0) for C2 and C4 positions of pyridinium salt 2a were calculated as 0.074 and 0.154, respectively, where higher values indicate greater susceptibility to radical attack (for details, see SI). To understand how this intrinsic reactivity manifests in the transition states, natural population analysis (NPA) was performed using NBO 7.0 to map the atomic charge distributions (Fig. 5b). The analysis reveals two critical electronic distinctions between TSBo and TSBp. Regarding the pyridinium nitrogen (N1), TSBo shows a markedly negative charge (−1.18 a.u.), whereas TSBp exhibits a substantially less negative value (−0.16 a.u.) approaching electrostatic neutrality. For the sulfonyl oxygen atoms (O1, O2), both structures retain consistently electron-rich character (−0.88/−0.90 a.u. in TSBo and −0.89/−0.91 a.u. in TSBp). This charge distribution directly correlates with the observed regioselectivity: In TSBp, the weakly negative (electron-poor) N1 engages in favorable electrostatic attraction with the electron-rich sulfonyl O1, facilitated by a 2.96 Å noncovalent contact; this aligns with TSBp's more stabilizing interaction energy (−14.70 kcal mol−1). In contrast, the strongly negative N1 in TSBo (−1.18 a.u.) repels the negatively charged sulfonyl oxygens, and steric hindrance further blocks such contacts, consistent with its weaker interaction energy (−12.10 kcal mol−1).
To quantify these energetic contributions, we performed a distortion–interaction analysis (Fig. 5b) by separating the pyridinium and alkyl fragments while maintaining their transition-state geometries. This decomposition reveals two critical components: the geometric distortion energy required to achieve the transition-state configurations (11.90 kcal mol−1 for TSBoversus 11.60 kcal mol−1 for TSBp) and the subsequent interaction energy between these distorted fragments (−12.10 kcal mol−1 for TSBo compared to −14.70 kcal mol−1 for TSBp). At the electronic energy level, this analysis identifies a 2.80 kcal mol−1 stabilization of TSBp relative to TSBo. Finally, to visualize the origin of the differential interaction energies, non-covalent interaction (NCI) analysis was conducted (Fig. S16). The NCI isosurfaces reveal that the enhanced stabilization in TSBp originates from a distinct O⋯N interaction between the sulfonyl oxygen and pyridinium nitrogen. In TSBo, steric hindrance from the adjacent alkyl chain obstructs this contact, as evidenced by the absence of an NCI isosurface in this region. This analysis provides direct visual evidence that the additional electrostatic attraction in TSBp is responsible for its lower interaction energy. Collectively, these results, from reactivity indices to electronic structure, energy decomposition, and visualization, confirm that the favorable electrostatic/non-covalent interaction in TSBp, enabled by its distinct electronic structure, is the key driver of the observed regioselectivity.
Conclusions
In conclusion, we have developed a pioneering heterogeneous visible-light photocatalytic system utilizing ZnIn2S4 for the efficient difunctionalization of alkenes with N-fluorosulfamoyl salts. This protocol facilitates the simultaneous incorporation of fluorosulfamoyl and pyridyl groups under mild conditions, delivering fluorosulfamoyl pyridine derivatives with broad substrate scope, excellent functional group tolerance, and moderate to good yields. The methodology significantly advances the practical application of the pyridine scaffold, widely prevalent in pharmaceuticals, by enabling rapid molecular complexity enhancement. Moreover, this heterogeneous photocatalytic system is also compatible with N-aminopyridinium salts, further showcasing its versatility. The exceptional recyclability and sustained catalytic performance of ZnIn2S4 over multiple cycles effectively address the challenge of catalyst recovery encountered in previous homogeneous systems. The synthetic utility of this approach is further highlighted by the successful late-stage functionalization of complex bio-relevant molecules, and it leverages SuFEx click chemistry to diversify the resulting products. Overall, this work provides a practical and sustainable platform for constructing structurally complex fluorosulfamoyl and aminopyridyl derivatives, offering significant potential for future applications in pharmaceutical synthesis and chemical biology. Ongoing studies aim to further expand the scope and enhance the versatility of this photocatalytic strategy.Author contributions
Kai Sun and Bing Yu designed and guided this project. Panjie Xiang, Kang Li, and Yunkai Zhang are responsible for the implementation of the experimental work. Kai Sun, Xiaolan Chen, and Xi Chen co-wrote the manuscript. Bing Yu and Lingbo Qu discussed the results and commented on the manuscript.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: synthetic experiments, computational details, characterization data, and spectra. See DOI: https://doi.org/10.1039/d5sc08495d.Acknowledgements
We acknowledge the financial support from the National Natural Science Foundation of China (21971224 and 22171249), the Science & Technology Innovation Talents in Universities of Henan Province (23HASTIT003), the 111 Project (D20003), Natural Science Foundation of Henan Province (242301420006, 252300421245), and Zhongyuan Leading Young Talents in Scientific and Technological Innovation.Notes and references
- J. Dong, L. Krasnova, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2014, 53, 9430–9448 CrossRef CAS PubMed.
- (a) G. Laudadio, A. d. A. Bartolomeu, L. M. H. M. Verwijlen, Y. Cao, K. T. de Oliveira and T. Noël, J. Am. Chem. Soc., 2019, 141, 11832–11836 CrossRef CAS PubMed; (b) M. Magre and J. Cornella, J. Am. Chem. Soc., 2021, 143, 21497–21502 CrossRef CAS PubMed; (c) Q. Feng, T. He, S. Qian, P. Xu, S. Liao and S. Huang, Nat. Commun., 2023, 14, 8278 CrossRef CAS PubMed.
- (a) R. Xu, T. Xu, M. Yang, T. Cao and S. Liao, Nat. Commun., 2019, 10, 3752 CrossRef PubMed; (b) J. E. Erchinger, R. Hoogesteger, R. Laskar, S. Dutta, C. Hümpel, D. Rana, C. G. Daniliuc and F. Glorius, J. Am. Chem. Soc., 2023, 145, 2364–2374 CrossRef CAS PubMed; (c) X. Nie, T. Xu, Y. Hong, H. Zhang, C. Mao and S. Liao, Angew. Chem., Int. Ed., 2021, 60, 22035–22042 CrossRef CAS PubMed.
- (a) S. D. Schimler, M. A. Cismesia, P. S. Hanley, R. D. J. Froese, M. J. Jansma, D. C. Bland and M. S. Sanford, J. Am. Chem. Soc., 2017, 139, 1452–1455 CrossRef CAS PubMed; (b) G. Meng, T. Guo, T. Ma, J. Zhang, Y. Shen, K. B. Sharpless and J. Dong, Nature, 2019, 574, 86–89 CrossRef CAS PubMed; (c) M.-C. Giel, C. J. Smedley, E. R. R. Mackie, T. Guo, J. Dong, T. P. Soares da Costa and J. E. Moses, Angew. Chem., Int. Ed., 2020, 59, 1181–1186 CrossRef CAS PubMed; (d) D.-D. Liang, D. E. Streefkerk, D. Jordaan, J. Wagemakers, J. Baggerman and H. Zuilhof, Angew. Chem., Int. Ed., 2020, 59, 7494–7500 CrossRef CAS PubMed; (e) C. Liu, C. Yang, S. Hwang, S. L. Ferraro, J. P. Flynn and J. Niu, Angew. Chem., Int. Ed., 2020, 59, 18435–18441 CrossRef CAS PubMed; (f) C. J. Smedley, Q. Zheng, B. Gao, S. Li, A. Molino, H. M. Duivenvoorden, B. S. Parker, D. J. D. Wilson, K. B. Sharpless and J. E. Moses, Angew. Chem., Int. Ed., 2019, 58, 4552–4556 CrossRef CAS PubMed.
- (a) S. Li, G. Li, B. Gao, S. P. Pujari, X. Chen, H. Kim, F. Zhou, L. M. Klivansky, Y. Liu, H. Driss, D.-D. Liang, J. Lu, P. Wu, H. Zuilhof, J. Moses and K. B. Sharpless, Nat. Chem., 2021, 13, 858–867 CrossRef CAS PubMed; (b) B. Gao, L. Zhang, Q. Zheng, F. Zhou, L. M. Klivansky, J. Lu, Y. Liu, J. Dong, P. Wu and K. B. Sharpless, Nat. Chem., 2017, 9, 1083–1088 CrossRef CAS PubMed; (c) R. W. Kulow, J. W. Wu, C. Kim and Q. Michaudel, Chem. Sci., 2020, 11, 7807–7812 RSC.
- T. Hmissa, X. Zhang, N. R. Dhumal, G. J. McManus, X. Zhou, H. B. Nulwala and A. Mirjafari, Angew. Chem., Int. Ed., 2018, 57, 16005–16009 CrossRef CAS PubMed.
- (a) T. S.-B. Lou and M. C. Willis, Nat. Rev. Chem., 2022, 6, 146–162 CrossRef CAS PubMed; (b) O. L. Symes, H. Ishikura, C. S. Begg, J. J. Rojas, H. A. Speller, A. M. Cherk, M. Fang, D. Leung, R. A. Croft, J. I. Higham, K. Huang, A. Barnard, P. Haycock, A. J. P. White, C. Choi and J. A. Bull, J. Am. Chem. Soc., 2024, 146, 35377–35389 CrossRef CAS PubMed; (c) J. J. Rojas, R. A. Croft, A. J. Sterling, E. L. Briggs, D. Antermite, D. C. Schmitt, L. Blagojevic, P. Haycock, A. J. P. White, F. Duarte, C. Choi, J. J. Mousseau and J. A. Bull, Nat. Chem., 2022, 14, 160–169 CrossRef CAS PubMed; (d) X. Wu, W. Zhang, G. Sun, X. Zou, X. Sang, Y. He and B. Gao, Nat. Commun., 2023, 14, 5168 CrossRef CAS PubMed; (e) Y. Chen, G. B. Craven, R. A. Kamber, A. Cuesta, S. Zhersh, Y. S. Moroz, M. C. Bassik and J. Taunton, Nat. Chem., 2023, 15, 1616–1625 CrossRef CAS PubMed; (f) A. Narayanan and L. H. Jones, Chem. Sci., 2015, 6, 2650–2659 Search PubMed; (g) W. Chen, J. Dong, L. Plate, D. E. Mortenson, G. J. Brighty, S. Li, Y. Liu, A. Galmozzi, P. S. Lee, J. J. Hulce, B. F. Cravatt, E. Saez, E. T. Powers, I. A. Wilson, K. B. Sharpless and J. W. Kelly, J. Am. Chem. Soc., 2016, 138, 7353–7364 Search PubMed.
- (a) N. L. Frye, C. G. Daniliuc and A. Studer, Angew. Chem., Int. Ed., 2022, 61, e202115593 CrossRef CAS PubMed; (b) P. Wang, H. Zhang, X. Nie, T. Xu and S. Liao, Nat. Commun., 2022, 13, 3370 CrossRef CAS PubMed; (c) A. S. Barrow, C. J. Smedley, Q. Zheng, S. Li, J. Dong and J. E. Moses, Chem. Soc. Rev., 2019, 48, 4731–4758 RSC; (d) D. Zeng, W.-P. Deng and X. Jiang, Natl. Sci. Rev., 2023, 10, nwad123 CrossRef CAS PubMed; (e) W. Zhang, H. Li, X. Li, Z. Zou, M. Huang, J. Liu, X. Wang, S. Ni, Y. Pan and Y. Wang, Nat. Commun., 2022, 13, 3515 CrossRef CAS PubMed.
- (a) C. M. Marshall, J. G. Federice, C. N. Bell, P. B. Cox and J. T. Njardarson, J. Med. Chem., 2024, 67, 11622–11655 CrossRef CAS PubMed; (b) P. W. Xu, Z. Wang, S. M. Guo and A. Studer, Nat. Commun., 2024, 15, 4121 CrossRef CAS PubMed; (c) H. Cao, Q. Cheng and A. Studer, Science, 2022, 378, 779–785 CrossRef CAS PubMed.
- (a) X.-Y. Yu, J.-R. Chen and W.-J. Xiao, Chem. Rev., 2021, 121, 506–561 CrossRef CAS PubMed; (b) M. Zhang, L. Liu, Y. Tan, Y. Jing, Y. Liu, Z. Wang and Q. Wang, Angew. Chem., Int. Ed., 2024, 63, e202318344 CrossRef CAS PubMed; (c) T. Huang, P. G. Du, X. L. Cheng and Y. M. Lin, J. Am. Chem. Soc., 2024, 146, 24515–24525 CrossRef CAS PubMed; (d) P. F. Zhong, J. L. Tu, Y. T. Zhao, N. Zhong, C. Yang, L. Guo and W. J. Xia, Nat. Commun., 2023, 14, 6530 CrossRef CAS PubMed; (e) Y. Xie, R. Zhang, Z.-L. Chen, M. Rong, H. He, S. Ni, X.-K. He, W.-J. Xiao and J. Xuan, Adv. Sci., 2024, 11, 2306728 CrossRef CAS PubMed; (f) J. Kim, M. Kim, J. Jeong and S. Hong, J. Am. Chem. Soc., 2023, 145, 14510–14518 CrossRef CAS PubMed; (g) Y. M. Gao, J. Liu, C. Wei, Y. Li, K. Zhang, L. L. Song and L. C. Cai, Nat. Commun., 2022, 13, 7450 CrossRef CAS PubMed; (h) J. Huang, Q. Gao, T. Zhong, S. Chen, W. Lin, J. Han and J. Xie, Nat. Commun., 2023, 14, 8292 CrossRef CAS PubMed; (i) Z. Zhang, X. Lv, X. Mu, M. Zhao, S. Wang, C. Ke, S. Ding, D. Zhou, M. Wang and R. Zeng, Nat. Commun., 2024, 15, 4445 CrossRef CAS PubMed; (j) M. Wu, Q. Jiang, Q. Tian, T. Guo, F. Cai, S. Tang, J. Liu and X. Wang, CCS Chem., 2022, 4, 3599–3608 CrossRef CAS; (k) E. Le Saux, E. Georgiou, I. A. Dmitriev, W. C. Hartley and P. Melchiorre, J. Am. Chem. Soc., 2023, 145, 47–52 CrossRef CAS PubMed; (l) C. Li, X. Li, J. Li, Z. Wang, D. Ouyang, N. Jiao and S. Song, Nat. Synth., 2025, 5, 36 CrossRef; (m) C. Li, Z. Yan, B. Wang, J. Li, W. Lyu, Z. Wang, N. Jiao and S. Song, Chem, 2024, 10, 628–643 CrossRef CAS; (n) J. Zhang, G. Wu, Y. Zhao, X. Liu, H. Zhu, A. Studer, X. Qi and Q. Cheng, Nat. Synth., 2025, 5, 27 Search PubMed.
- (a) F.-S. He, S. Ye and J. Wu, ACS Catal., 2019, 9, 8943–8960 CrossRef CAS; (b) M. Vellakkaran, T. Kim and S. Hong, Angew. Chem., Int. Ed., 2022, 61, e202113658 Search PubMed; (c) S. Jung, S. Shin, S. Park and S. Hong, J. Am. Chem. Soc., 2020, 142, 11370–11375 CrossRef CAS PubMed; (d) M. Kim, Y. Koo and S. Hong, Acc. Chem. Res., 2022, 55, 3043–3056 CrossRef CAS PubMed; (e) Y. Moon, W. Lee and S. Hong, J. Am. Chem. Soc., 2020, 142, 12420–12429 CrossRef CAS PubMed; (f) J. Kim, Y.-E. Kim and S. Hong, Angew. Chem., Int. Ed., 2024, 63, e202409561 CrossRef CAS PubMed; (g) Y. Moon, B. Park, I. Kim, G. Kang, S. Shin, D. Kang, M.-H. Baik and S. Hong, Nat. Commun., 2019, 10, 4117 CrossRef PubMed.
- J. J. A. Garwood, A. D. Chen and D. A. Nagib, J. Am. Chem. Soc., 2024, 146, 28034–28059 CAS.
- (a) A. Thakur, S. S. Gupta, Sumit, Sachin and U. Sharma, Org. Lett., 2024, 26, 8515–8520 CrossRef CAS PubMed; (b) Y.-L. Su, G.-X. Liu, L. De Angelis, R. He, A. Al-Sayyed, K. S. Schanze, W.-H. Hu, H. Qiu and M. P. Doyle, ACS Catal., 2022, 12, 1357–1363 CrossRef CAS; (c) H. Im, W. Choi and S. Hong, Angew. Chem., Int. Ed., 2020, 59, 17511–17516 CrossRef CAS PubMed; (d) J. Q. Buquoi, J. M. Lear, X. Gu and D. A. Nagib, ACS Catal., 2019, 9, 5330–5335 Search PubMed.
- H. Li, X. Zhang, Z. Wang, C. Sun, M. Huang, J. Liu, Y. Li, Z. Zou, Y. Pan, W. Zhang and Y. Wang, Org. Lett., 2024, 26, 6714–6719 CrossRef CAS PubMed.
- P. Wang, L. Lin, Y. Huang, H. Zhang and S. Liao, Angew. Chem., Int. Ed., 2024, 63, e202405944 CrossRef CAS PubMed.
- T. Xiong, Q.-L. Chen, Z.-Y. Zhang, G. Lu and J. Weng, ACS Catal., 2024, 14, 12082–12092 Search PubMed.
- (a) W. Ou, G. Zhang, J. Wu and C. Su, ACS Catal., 2019, 9, 5178–5183 Search PubMed; (b) J. Dong, Z. Wang, X. Wang, H. Song, Y. Liu and Q. Wang, Sci. Adv., 2019, 5, eaax9955 Search PubMed; (c) J. Djossou, A. Aloia, L. Capaldo, D. D. Snabilié, M. Regnier, J. H. A. Schuurmans, A. Monopoli, B. d. Bruin and T. Noël, Angew. Chem., Int. Ed., 2025, 64, e202508710 Search PubMed.
- (a) Y. Cai, Y. Tang, L. Fan, Q. Lefebvre, H. Hou and M. Rueping, ACS Catal., 2018, 8, 9471–9476 Search PubMed; (b) I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS PubMed; (c) Z. Teng, Z. Zhang, H. Yang, Q. Zhang, T. Ohno and C. Su, Sci. Adv., 2024, 10, eadl5432 Search PubMed; (d) R.-N. Ci, C. Huang, L.-M. Zhao, J. Qiao, B. Chen, K. Feng, C.-H. Tung and L.-Z. Wu, CCS Chem., 2022, 4, 2946–2952 Search PubMed.
- (a) J.-J. Li, J.-Y. Liu, S.-Y. Gao, X. Yang and R. Cao, J. Am. Chem. Soc., 2025, 147, 21754–21763 CrossRef CAS PubMed; (b) P. Nayek, B. Pal and P. Mal, ACS Catal., 2025, 15, 15519–15558 Search PubMed.
- (a) Y. Wu, Z. Liu, Y. Li, J. Chen, X. Zhu and P. Na, Chin. J. Catal., 2019, 40, 60–69 CrossRef CAS; (b) D. Liu, S. Chen, Y. Zhang, R. Li and T. Peng, Appl. Catal., B, 2023, 333, 122805 CrossRef CAS; (c) S. Su, G. Lv, X. Zou, J. Wang, C. Zhou, Y. Chen, J. Shen, Y. Shen and Z. Liu, Green Chem., 2023, 25, 9262–9271 RSC; (d) Z. Sun, Y. Tan, X. Shi, B. Li, X. Wang, J. Wen, L. Huang and W.-M. Lau, ACS Sustainable Chem. Eng., 2024, 12, 1051–1061 CrossRef.
- (a) C.-L. Ji, Y.-N. Lu, S. Xia, C. Zhu, C. Zhu, W. Li and J. Xie, Angew. Chem., Int. Ed., 2025, 64, e202423113 CrossRef CAS PubMed; (b) J. Xu, J. Cao, X. Wu, H. Wang, X. Yang, X. Tang, R. W. Toh, R. Zhou, E. K. L. Yeow and J. Wu, J. Am. Chem. Soc., 2021, 143, 13266–13273 CrossRef CAS PubMed; (c) S. Tong, K. Li, X. Ouyang, R. Song and J. Li, Green Synth. Catal., 2021, 2, 145–155 Search PubMed; (d) Q. Shi, X.-W. Kang, Z. Liu, P. Sakthivel, H. Aman, R. Chang, X. Yan, Y. Pang, S. Dai, B. Ding and J. Ye, J. Am. Chem. Soc., 2024, 146, 2748–2756 Search PubMed.
- (a) S. Y. Guo, Y. P. Liu, J. S. Huang, L. B. W. He, G. C. He, D. W. Ji, B. S. Wan and Q. A. Chen, Nat. Commun., 2024, 15, 6102 CrossRef CAS PubMed; (b) X.-Y. Chen, Y.-N. Li, Y. Wu, J. Bai, Y. Guo and P. Wang, J. Am. Chem. Soc., 2023, 145, 10431–10440 CrossRef CAS PubMed; (c) S. Xu, Y. Ping, M. Xu, G. Wu, Y. Ke, R. Miao, X. Qi and W. Kong, Nat. Chem., 2024, 16, 2054–2065 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |